

Abstract
Molecular mimicry has been proposed as a pathogenetic mechanism for autoimmune disease, as well as a probe useful in uncovering its etiologic agents. The hypothesis is based in part on the abundant epidemiological, clinical, and experimental evidence of an association of infectious agents with autoimmune disease and observed cross‐reactivity of immune reagents with host ‘self’ antigens and microbial determinants. For our purpose, molecular mimicry is defined as similar structures shared by molecules from dissimilar genes or by their protein products. Either the molecules' linear amino acid sequences or their conformational fits may be shared, even though their origins are as separate as, for example, a virus and a normal host–self determinant. An immune response against the determinant shared by the host and virus can evoke a tissue‐specific immune response that is presumably capable of eliciting cell and tissue destruction. The probable mechanism is generation of cytotoxic cross‐reactive effector lymphocytes or antibodies that recognize specific determinants on target cells. The induction of cross‐reactivity does not require a replicating agent, and immune‐mediated injury can occur after the immunogen has been removed—a hit‐and‐run event. Hence, the viral or microbial infection that initiates the autoimmune phenomenon may not be present by the time overt disease develops. By a complementary mechanism, the microbe can induce cellular injury and release self antigens, which generate immune responses that cross‐react with additional but genetically distinct self antigens. In both scenarios, analysis of the T cells or antibodies specifically engaged in the autoimmune response and disease provides a fingerprint for uncovering the initiating infectious agent.—Oldstone, M. B. A. Molecular mimicry and immune‐mediated diseases. FASEB J. 12, 1255–1265 (1998)
Keywords: autoimmune disease, microbial agent, insulin‐independent diabetes mellitus, sequence sharing
Abbreviations
- MHC
major histocompatibility complex
- MS
multiple sclerosis
- IDDM
insulin‐independent diabetes mellitus
- AS
ankylosing spondylitis
- TCR
T cell recognition
- HBVP
hepatitis B virus polymerase
- MG
myasthenia gravis
- AChR
acetylcholine receptor
- GAD
glutamate decarboxylase
- CTL
cytotoxic T lymphocyte
- IL
interleukin
Autoimmune diseases are multifactorial. In addition to a genetic link and, in several instances, a major histocompatibility complex (MHC)2 disease association (1–3), other factors (including environmental ones) must be involved. For example, there is strong clinical/epidemiological evidence that monozygotic twins show a striking discordance for several autoimmune diseases like insulin‐dependent diabetes, multiple sclerosis, and HLA‐B27‐associated ankylosing spondylitis (4–6). In addition, when the antibody or T cell receptor associated with disease has been available for structural analysis, primarilysomatic mutations, suggesting an antigen‐driven process rather than germline mutations, have been found (7, 8). Other evidence comes from observations that human viral infections are often associated with autoimmune responses (reviewed in ref 9). Hence, newly formed autoimmune responses or those already present are enhanced after infection by a wide variety of human DNA and RNA viruses. In addition, experimental acute and persistent infections with either DNA or RNA viruses have induced, accelerated, or enhanced autoimmune responses and caused autoimmune disease. Perhaps the best‐known instance occurs during evaluation of the New Zealand mouse model (10). In this system of genetically defined mice that spontaneously develop autoimmune disease, persistent infection with a DNA virus like polyoma or an RNA virus like lymphocytic choriomeningitis virus speeds up the time when antibodies to DNA, red blood cells, and other autoantigens appear. Thus, the kinetics and severity of the resulting autoimmune disease are dramatically accelerated. More germane is the NZW mouse, which normally does not develop autoimmune response or disease but contains the necessary gene(s) to develop the anti‐DNA and other autoimmune responses and disease and does so after viral infection. Others have reported epidemiologic and serologic correlations between viral infections and autoimmune diseases like multiple sclerosis (MS) (11, 12), insulin‐dependent diabetes mellitus (IDDM) (13, 14), bacterial infections, and ankylosing spondylitis (AS) (15). For example, coxsackie B virus and rubella virus have been linked with IDDM; in a few instances, Coxsackie B virus has been directly isolated from pancreatic tissues of individuals with acute IDDM (14, 16). Inoculation of the isolated virus into mice produced diabetes, thereby fulfilling Koch's postulates (16). Several viruses have been associated with MS (17–20), and there is abundant epidemiologic evidence of a close association of viral infection with exacerbations of this demyelinating disease (11, 12, 20). Robust epidemiologic evidence correlates the occurrence of certain bacterial infections such as Klebsiella pneumoniae, Shigella flexneri, Salmonella, and Yersinia with the initial onset of acute Reiter's syndrome, followed by AS in HLA B27+ individuals (15, 21). Last, there are transgenic models in which mice expressing an autoimmune gene and/or its T cell recognition (TCR)/antibodies required to develop autoimmune disease do not do so unless there is an environmental switch, i.e., movement from a pathogen‐free animal facility to a standard vivarium (22, 23).
Microbial agents or viruses can induce autoimmune responses and diseases by a variety of mechanisms, several of which may occur during an infection. For example, proteins of certain infectious agents have a proliferative effect on unique lymphocyte subsets and hence act as polyclonal activators. Viruses can preferentially infect/destroy a particular T cell subset, leading to an imbalance in the immune response. In other instances, infectious agents can up‐regulate Th.1 cytokines, thereby increasing selected expression of molecules such as MHC glycoproteins, as well as activation and costimulatory molecules such as B7.1/CD28. Several microbial agents have been found to encode superantigens that can selectively activate subset(s) of T cells (24–26). In other cases, viruses may break selftolerance by de novo release of self epitopes presumably by direct injury caused by the virus or secondarily by the virus‐specific T cells generated. Either way, damage can occur by a procedure called epitope spreading (27, 28). Microbes can also direct the release of cytokines and chemokines, important regulators of immune responses. These molecules can act as growth, differentiation, or chemotactic factors for different Th populations and regulate expression of MHC class I and class II molecules. Thus, an infectious agent could release a cytokine‐like interferon that induces the expression of MHC determinants. In addition, microbes—especially viruses—can infect and selectively replicate in unique lymphocyte subsets and, by their presence, activation, or replication, cause immunosuppression or immunoenhancement. Last, microbes can contain chemical structures that mimic normal host ‘self’ proteins, an event termed molecular mimicry (Fig. 1 ). Several possibilities are listed in Table 1 . Of these, molecular mimicry is the focus of this review. Whereas there is heightened suspicion that infectious agents and molecular mimicry play a role in several autoimmune diseases, direct evidence has been hard to come by. There are a number of reasons for this discrepancy: for instance, the organism initiating the autoimmune process may be cleared by the host immune response, thus removing our ability to isolate it. However, the initiated immune response would continue the process against cross‐reactive ‘self’ determinant, a ‘hit‐and‐run’ scenario.
Figure 1.
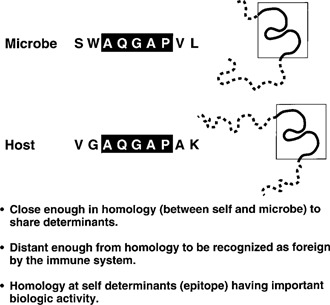
Illustration of the molecular mimicry concept along with associated guidelines. The sharing of a linear amino acid sequence or a conformation fit between a microbe and a host ‘self’ determinant is the initial stage of molecular mimicry. Autoimmunity may occur if the host immune response against the microbe cross‐reacts with the host's ‘self’ sequence and if the host sequence comprises a biologically important domain, i.e., the encephalitogenic site of a myelin protein incriminated in CNS or demyelinating disease, the component of the acetylcholine receptor important for synapse formation, or the hypervariable region of MHC molecule (HLAB27) associated with disease.
Table 1.
Infectious agents can trigger autoimmune disease by activation of autoreactive T cells via:
| 1. | Up‐regulation of Th‐1 cytokines and/or other selected molecules: MHC, B7.1/CD28 |
| 2. | Preferential infection/destruction of CD4 T cell subset |
| 3. | Virus‐encoded super antigens |
| 4. | De novo by self epitopes released secondary to virus‐specific, T cell‐mediated damage: epitope spreading |
| 5. | Cross‐reactive with a viral epitope(s): molecular mimicry |
DEVELOPMENT OF MOLECULAR MIMICRY CONCEPT
The initial clues that molecular mimicry might play a role in autoimmune diseases were found in the early 1980s associated with the development and use of monoclonal antibody technology. It was first noted while generating monoclonal antibodies that the phosphoprotein of measles virus and a protein of herpes simplex virus type 1 cross‐reacted with an intermediate filament protein of human cells (29; Fig. 2 ). The intermediate filament protein was identified as vimentin, and the monoclonal antibody against measles virus phosphoprotein was shown to react with an antigenic determinant on the intermediate filament molecule different from the reaction caused by antibody to herpes simplex virus protein. A number of similar cross‐reactivities between viral proteins and host ‘self’ determinants were subsequently recorded. The observations were firmly established as a result of a collaborative studywith the laboratories of Abner Notkins, Hilary Koprowski, and our own that documented roughly 5% of monoclonal antibodies made against 15 different kinds of viruses cross‐reacted with ‘host–self determinants’ when over 800 independent monoclonal antibodies were studied (30, 31; Fig. 3 ). In these experiments, monoclonal antibodies made against representative DNA and RNA viruses such as herpes simplex, cytomegalo, Epstein‐Barr, vaccinia virus, myxoviruses, paramyxoviruses, arenaviruses, flaviviruses, orthoviruses, rhabdoviruses, coronaviruses, and human retroviruses were studied. On the basis of needing five to six amino acids to induce a monoclonal antibody response, the probability of 20 amino acids occurring in six identical residues between two proteins is 206 or 1 in 128,000,000. Others showed that T lymphocytes obtained from the spinal fluids of patients with viral infections or with MS cross‐reacted with a specific viral determinant as well as a specific host myelin protein.
Figure 2.
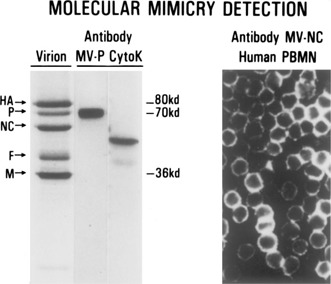
Left panel reproduces data that first suggested the molecular mimicry concept. Monoclonal antibody to measles virus P protein (MV‐P) was found to react with cytokeratin (cytoK; see ref 29 for details). The right panel shows that a monoclonal antibody to measles virus nucleoprotein also reacts with molecule(s) on the surface of T lymphocytes (see ref 31 for details).
Figure 3.
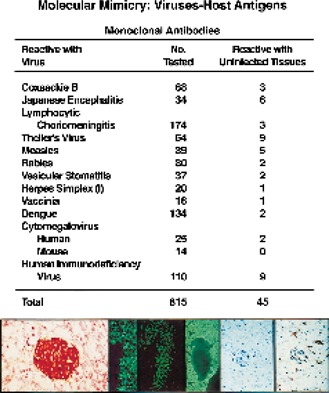
Update on combined data from studies of molecular mimicry of the reactivity of monoclonal antiviral antibodies with normal mouse tissue (see refs 30, 31). Lower panels document reactivity of several antiviral monoclonal antibodies that react with host tissues or cells. Left panel: antibody to herpes simplex virus 1 GP reacts with β cells of the islets of Langerhans or their product. Center panel shows monoclonal antibodies to measles virus, Japanese encephalitis virus, and coxsackie B4, which react with growth hormone cells of the anterior lobe of the pituitary, hippocampal neurons, and myocardium (ventricle only and not smooth muscle around the vessel). Right panel displays a monoclonal antibody to HIV GP41 that reacts with astrocytes and the specificity control where addition of HIV GP41 peptide to the monoclonal antibody blocks staining (see Yamada et al., ref 33).
Biologic evidence was obtained over the next few years in several animal models indicating that molecular mimicry was more than an epiphenomenon (reviewed in ref 9). An early observation used myelin basic protein and allergic encephalomyelitis. The myelin basic protein sequences that cause allergic encephalitis are known and the encephalitogenic site of 8–10 amino acids has been mapped for several different animal species. With the use of computer‐assisted analysis (32), several viral proteins were uncovered that showed significant homology with the encephalitogenic site of myelin basic protein, including fits between the myelin basic protein and nucleoprotein of the hemagglutinin of influenza virus, core protein of adenovirus, EC‐LF2 protein of Epstein‐Barr virus, and hepatitis B virus polymerase as well as others. However, the best fit occurred between the myelin basic protein encephalitogenic site in the rabbit and hepatitis B virus polymerase (HBVP). It was then shown that inoculation of HBVP peptide peripherally into rabbits caused perivascular infiltration localized to the central nervous system, reminiscent of disease induced by inoculation of whole myelin basic protein or the peptide component of the encephalitogenic site of myelin basic protein (34; Fig. 4 ). Further, a specific immune response to both cellular and humoral to myelin basic protein occurred. Thereafter, several other animal models were developed showing cross‐reactivity between a microbe and self antigen leading to disease. For example, coxsackie B3 virus shared sequence homology with heart myosin or other myocardial proteins (i.e., GAP junction), and experimental manipulations in experimental animals led to myocardial necrosis and myocarditis (35–37). Another early example of sequence homologies, immune cross‐reactivity, and disease in animals was autoimmune uveitis with cross‐reactivity between S‐antigen and viral peptides (38).
Figure 4.
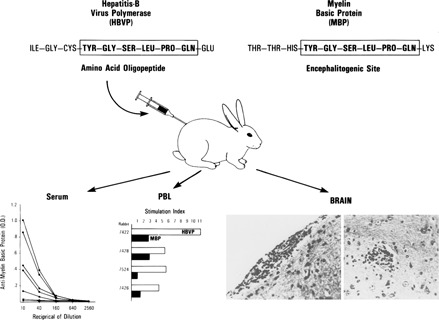
Experimental demonstration that molecular mimicry could cause disease. New Zealand rabbits inoculated with 10 amino acid peptide from hepatitis B virus polymerase generated specific T (proliferation) and B (antibody) lymphocyte responses to myelin basic proteins. Rabbits developed histopathologic evidence of lesion of allergic encephalomyelitis (see ref 34 for details).
CORRELATION OF MOLECULAR MIMICRY WITH HUMAN DISEASE
The next step, and the most difficult to definitively prove, was showing the relevance of molecular mimicry to naturally occurring human disease. Various correlations ranging from reasonably convincing to less so have been published. Table 2 lists a number of such correlations. Several examples are selected here for a more detailed discussion.
Table 2.
Compilation of several diseases reported to be possibly associated with mechanism of molecular mimicry
| Disease | Cross‐reactive immune responses | References |
|---|---|---|
| Primary biliary cirrhosis | Pyruvate dehydrogenase complex Anti‐mitochondrial response HLA DRa Microorganisms: E. coli, S. cerevisial | 63–66 |
| Rasmussen's encephalitis | Antiglutamate receptor (GLUR3) response Microorganisms | 66, 67 |
| Multiple sclerosis | Myelin proteins Viruses: corona, measles, mumps, EBV, herpes, etc. | 18, 19, 47, 68–71 |
| Myasthenia gravis | Acetylcholine receptor Neurofilaments Herpes | 39, 72, 73 |
| Guillain‐Barre | Peripheral nerve Gangliosides Campylobacter jejuni | 74–76 |
| Insulin‐dependent diabetes | Islet antigens (GAD 65, proinsulin carboxypeptidase H) Coxsackie B Rotaviruses Hepatitis C, herpes, rhino‐, hanta‐, and faviviruses Retroviral | 40, 77–80 |
| Myocardial (heart) | ADP/ATP carrier protein Cardiac myosin Myocardial cell surface protein Coxsackie B3, streptococci | 35–37, 81, 82 |
| Celiac | Gliadin Enterocyte surface molecules Adenoviruses Enteric microbes | 83, 84 |
| Graft vs. host transplantation | Aminopeptidase N HLA molecule Cytomegalovirus | 85, 86 |
| Pemphigus | Desmosomes VH‐CDR3 | 87 |
| Herpes stomal keratitis | Herpes simplex Corneal antigen(s) | 88 |
| Autoimmune uveitis | S‐antigen IRBP Viruses | 38, 89 |
| Peptic ulcer/gastric CA | H. pylori Self antigens on gastric mucosa | 90 |
| Pathogenesis in: | ||
| HIV | 91–94 | |
| Lyme disease | 95 | |
| Streptococcal | 82, 96 | |
| Syphilis | 97 | |
| Prion protein disease | 98 | |
| Trypanosoma | 99–102 |
The large majority of patients with the autoimmune disease myasthenia gravis (MG) have antibodies against the acetylcholine receptor (AChR). Purification of antibodies from MG patients using the human AChR α‐subunit amino acids 157 to 170 provided IgG antibodies that bound to native AChR and inhibited the binding of α‐bungarotoxin to the receptor (39). The human AChR α‐subunit amino acids 160–167 showed specific immunologic cross‐reactivity with a shared homologous domain on herpes simplex virus glycoprotein D, residues 286–293, by both specific binding and inhibition assays. Antibodies to the human AChR α‐subunit bound to herpes simplex virus‐infected cells. The data of immunologic cross‐reactivity of the AChR ‘self epitope’ with herpes simplex virus and the presence of cross‐reactive antibodies in the sera of MG patients suggest that this virus may be associated with the initiation of some cases of myasthenia (39).
IDDM results from autoimmune destruction of insulin‐making beta cells in the islets of Langerhans believed to be caused by autoreactive T lymphocytes. Clinical studies have shown a high correlation with immune response to glutamate decarboxylase (GAD) and patients progressing to or having IDDM. Epidemiologic evidence has correlated IDDM with several viral infections, including coxsackie B virus, which has been shown, albeit rarely, by Koch's postulates to be associated with cases of acute IDDM (13, 14, 16). A component of GAD amino acids 247–279 was shown to be recognized by patients with increased risk of IDDM and to share sequence similarity with the P2‐C protein of coxsackie B virus. Evaluation of patients with IDDM, of high risk to develop IDDM, or healthy controls showed that peripheral blood mononuclear cells from 4 of 16 newly diagnosed IDDM patients (25%) or 7 of 15 patients (47%) with high risk of developing IDDM (having islet cell autoantibodies with IDDM) proliferated to both GAD and coxsackie B viral peptides; however, none of the 13 healthy controls did (40, 41), suggesting apossible association for a virus like coxsackie B with IDDM.
Recently, Honeyman et al. (41) reported that a dominant peptide (aa 805 to 820) from IA‐2 islet protein, which is a molecular target of pancreatic islet autoimmunity in type 1 diabetes in humans, had 56% identity and 100% similarity for 9 aa with a sequence in VP7, a major immunogenic protein of human rotavirus. The contiguous sequence of VP7, had 75% identity and 92% similarity over 12 aa with a known T cell epitope in GAD. Further, the IA‐2 immunodominant peptide showed significant identity with hepatitis C virus and hemophilus influenza. Three other distinct IA‐2 peptides shown 71 to 100% similarity over 7 to 12 aa to herpes, rhino‐, hanta‐, and flaviviruses.
One of the interesting and still controversial findings for an association of microbes with disease by the mechanism of molecular mimicry occurs with AS and bacterial infections (42–46). Epidemiologic, bacteriologic, and immunologic evidence is firm for a correlation between AS, the hypervariable region of HLA B27, and several bacterial infections including those of S. flexneri, Yersinia entercolytica, and K pneumoniae. Over 90% of individuals developing AS have the HLA B27 haplotype. In a S. flexneri epidemic of about 150,000 cases in Finland in the 1940s, 344 infected individuals developed Reiter's syndrome; of these, 82 went on to develop AS. Amino acid sequence homology between enteric bacteria associated with Reiter's syndrome and thus AS with the hypervariable region of HLA B27 was observed (Fig. 5 ). When transgenic mice were created to express human HLA B27 and challenged with cross‐reactive peptides derived from proteins of such bacteria, immune responses occurred with inflammatory cells localized to joints and vertebral columns in approximately 40% of inoculated mice (M. B. A. Oldstone, S. Shyp, P. Peterson, and D. LaFace, unpublished observations; Fig. 5). Patients with AS showed antibodies in their circulation that cross‐reacted with both the hypervariable region of HLA‐B27 as well as a closely homologous sequence from K pneumoniae nitrogenase (43, 44). Subsequently, a transgenic rat model was developed in which expression of HLA‐B27 was associated with a clinical and histopathologic picture reminiscent of AS (45). AS‐like disease occurred in rats in a normal vivarium and did not occur in rats housed in germ‐free conditions (23). When germ‐free rats were colonized with bacteria, they developed arthritis (46). Besides molecular mimicry, other investigators (23, 46) raised the possibility that HLA B27 may be associated with delayed or disordered clearance of the associated bacterial pathogens.
Figure 5.
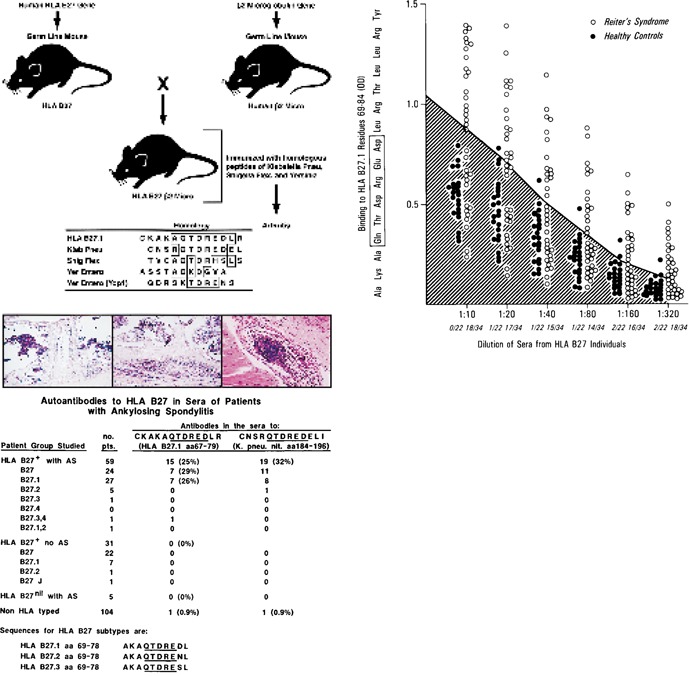
A) Sequence sharing between the hypervariable region of HLA B27 molecule and sequences from bacteria epidemiologically associated with causing Reiter's syndrome, a disease that often precedes HLA B27‐associated ankylosing spondylitis. Inoculation of the Shigella flexneri peptide (center panel) or Klebsiella pneumoniae peptide (right panel) into transgenic mice expressing HLA B27 human β2 microglobulin and human CD8 leads to inflammatory response in joints and the vertebral column. Antibodies to HLA B27 are also found in these mice, but not in other mice given non‐cross‐reactive peptides. The control mice also fail to demonstrate inflammatory lesions (left panel). B, C) Correlation of antibodies to HLA B27 and K. pneumoniae nitrogenase peptide in patients with Reiter's disease (B) or ankylosing spondylitis (C) (for details, see refs 43, 44).
To apply a greater degree of specificity and a more stringent approach to the study of molecular mimicry and potential associated diseases (32), Wucherpfennig and Strominger (47) as well as Hammer et al. (48) imposed a structural requirement for molecular mimicry searches. Wucherpfennig and Strominger used the known structure for MHC class II disease‐associated molecules with peptide binding and TCR for a known immunodominant myelin basic protein peptide. By database search, a panel of 129 peptides from microbes were obtained that matched the molecular mimicry motif, and these were tested on several T cell clones obtained from the cerebral spinal fluid of multiple sclerosis patients (47). Eight peptides (seven of viral and one of bacterial origin) were found to efficiently activate three of these clones, whereas only one of the eight peptides would have been identified as an appropriate molecular mimic by sequence alignment. These observations indicated that a single T cell receptor could recognize several distinct but structurally related peptides from multiple pathogens, suggesting more permissivity for TCR than previously appreciated, an observation supported by other recent reports (49–51). Hammer et al. (48) analyzed the possible peptide motifs that bound to HLA‐DR β chain associated with autoimmune disease. The influence of single key residues was tested by using site‐directed mutations. A selection of peptides binding to rheumatoid arthritis‐linked DR allotypes was shown to be critical for position aa 71 of DR β and 57 of DQ β (48). Extension of these findings to IDDM (47, 48) suggested that IDDM ‘autoimmune’ peptide would bear a negative charge at P9 in order to bind preferentially to diabetes‐associated DQ allele. In toto, these findings should assist in identification of potential peptides (self? viral? microbe?) associated with the pathogenesis of autoimmune disease. One variation on this theme to help explain MHC linkage with autoimmune disease and infectious agents is that MHC‐derived self peptides prominent in selecting the T cell repertoire may be mutated and/or share immunologic cross‐reactive epitopes with microbial antigens. Others (51) have shown that a six amino acid peptide can sensitize myelin basic protein‐specific T cells to cause EAE and have used this information to identify viral and bacterial proteins (peptides) that share similar amino acids. Of interest will be experiments using such viral or microbial agents to initiate/potentiate or perhaps block the autoimmune EAE disease.
USING MOLECULAR MIMICRY MODELS TO DISSECT THE PARAMETERS REQUIRED FOR THE ACTIVATION AND ASSOCIATION OF VIRUS‐INDUCED AUTOIMMUNE DISEASE
To dissect the pathogenetic mechanisms by which viral infection can break tolerance or unresponsiveness to self and the sequential events involved in the autoreactive cascade that leads to autoimmune disease, we and others have designed and exploited transgenic mouse models. An illustration of a model we developed for analysis of virus‐induced IDDM is shown in Fig. 6 , with the model described in refs 52 and 53. The cardinal observations uncovered through the analysis of this model are as follows. 1) Lymphocytes in the periphery are ignorant or unresponsive to the ‘self’ protein, but can be activated in the absence of viral infection by coexpressing cytokines like interferon‐γ or activation molecules such as B7.1 in the target (β cells) area. 2) Infection by the same virus from which the ‘self’ protein was used as a transgene or a closely related virus that both cross‐reacts and induces sufficient effector cytotoxic T lymphocytes (CTLs) also causes the autoimmune disease. 3) If the transgene is expressed only in the target tissue, the autoimmune disease occurs rapidly, with kinetics of disease occurring by 10–4 days. Such positively selected high affinity effector T cells kill beta cells in the islets of Langerhans. The effector cell is a CD8 CTL and killing is dependent on perforin and interferon‐γ. However, if the transgene is also expressed in the thymus, then the kinetics for initiation of disease after viral infection is prolonged. 4) Depending on the MHC haplotype, disease takes between 1 and 7 months to occur. 5) Whereas the rapidonset autoimmune disease uses high affinity effector CTL, in contrast to the slow‐onset autoimmune disease, the majority of high affinity T cells have been selected out in the thymus (negative selection) and only low affinity cells are available in the periphery to participate in causing the autoimmune disease. 6) For low affinity CD8+ T cells to produce disease, they require help from CD4, but such help is not necessary for the rapid‐onset disease. 7) Expression of Th.2 cytokine such as interleukin 4 (IL‐4) retards the autoimmune diabetes. Table 3 tabulates these findings (details can be found in refs 52–60; M. G. von Herrath, B. Coon, H. Mazarguil, J. E. Gairin, and M. B. A. Oldstone, unpublished results).
Figure 6.
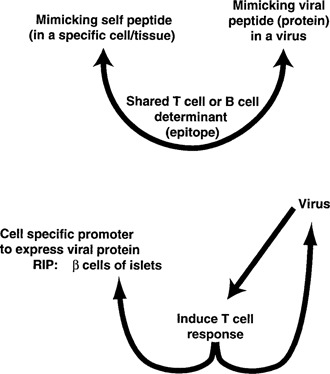
Molecular mimicry transgenic model designed to study insulin‐dependent diabetes mellitus (IDDM). The viral gene product is placed in the β cells of the islets of Langerhans using the rat insulin promoter and transgenic technology. In a second scenario, the transgene is inserted and expressed in the thymus as well as the β cells of the islets. Because the transgene (viral gene) is inserted in the germline of the host and passed to subsequent progeny, it is in essence a host ‘self’ gene. Using both models allows the study of the mechanisms and kinetics by which immunologic tolerance is broken, peripheral low affinity (transgene expressed in the thymus) or high affinity (transgene not expressed in the thymus) effector T cells are activated, home to the islets, and cause autoimmune disease. These models allow the design of therapeutic approaches to prevent or reverse the disease process at different steps along the way.
Table 3.
Lessons for virus‐induced IDDM learned from the molecular mimicry transgenic model
|
These observations allow the design of therapeutic approaches to halt the virus‐induced IDDM. When the cytokine profile in the islets of Langerhans milieu was changed from a Th.1 to a Th.2 phenotype (γ‐interferon to IL‐4, TGF‐β), IDDM was blocked even though this therapy had no demonstrable effect on precursor frequency of the effector CD8+ CTL (58; R. Mueller‐Hoenger, M. G. von Herrath, X. Zhang, G. Patstone, T. Krahl, B. Coon, M. B. A. Oldstone, and N. Sarvetnick, unpublished results). A second successful therapeutic approach was to design a peptide that could bind at high affinity to the MHC allele involved in the IDDM (59; M. G. von Herrath, B. Coon, H. Mazarguil, J. E. Gairin, and M. B. A. Old‐stone, unpublished results). IDDM did not occur by this means; CD4+ and CD8+ T cells did not accumulate in the islets, but were found around them (peri‐islitis). Further, the precursor frequency of the effector CD8+ T cells was diminished by several‐fold, below a level required to cause IDDM. Another successful approach was to abort the expression of MHC class I molecule by coexpressing the E3 transcriptional complex of adenovirus in β cells of the islets of Langerhans (60). The focal reduction of MHC class I expression in the islets was associated with a normal precursor frequency of CD8+ CTLs in the spleen and blood. However, effector T cells did not localize in the islets, destruction of islets did not occur, and IDDM did not develop.
In addition to uncovering the pathogenetic mechanisms and providing therapeutic design to control the autoimmune disease, the transgenic model also proved efficacious for analysis of ‘self’ determinants that may be associated with or potentiate the autoimmune disease. In the transgenic model used (Fig. 6), since the specific amino acids required to bind to the MHC allele associated with the IDDM, as well as their binding motif, are known and fit within the MHC groove and residue recognition by TCR (59, 61, 62), a database search not dissimilar from the approach used by Wucherpfennig and Strominger (47) was undertaken. Twenty‐one host self peptides were uncovered, synthesized, and studied for their direct binding to the Db MHC allele. Those showing high affinity binding (by competition assay, <100 nM) were then selected. As the effector CD8 T cell (antiviral) (anti‐‘self’) was known, the ability of the peptide to stimulate the auto(viral) T cell and cause it to proliferate, release cytokines, or lyse a ‘self’ peptide‐coated target was determined and the ability of such activated T cells to transfer disease was monitored (M. B. A. Oldstone, J. E. Gairin, and M. G. von Herrath, unpublished results). Thus, there is the opportunity to obtain autoreactive T cells to ‘self’ antigen and use this information to uncover the infectious agent that initiated (was associated with) the autoimmune process.
In summary, molecular mimicry is but one mechanism by which autoimmune diseases can occur in association with infectious agents. The concept of molecular mimicry remains a viable hypothesis for framing questions and approaches to uncover the initiating infectious agent as well as recognizing the ‘self’ determinant, understanding the pathogenetic mechanism(s) involved, and designing strategies for treatment and prevention of autoimmune disorders. The availability of computer databanks, structural information on specific MHC alleles, MHC mapping to particular autoimmune diseases, and the ability to identify the anchoring and flanking sequences of a peptide that binds to that MHC allele or bind to the TCR provide the opportunity to critically evaluate and identify microbial causes of autoimmune diseases. The application and use of transgenic models designed to evaluate molecular mimicry offer the opportunity to understand the sequence of events leading to the pathology and design of specific and unique therapies in order to reverse or prevent the autoimmune process and disease.
ACKNOWLEDGMENT
This is publication number 11542‐NP from the Department of Neuropharmacology, The Scripps Research Institute, La Jolla, California. This work was supported in part by USPHS grants AG04342 and AI09484 and by a grant from the Juvenile Diabetes Foundation (JDFI 995005).
Oldstone, M. B. A. Molecular mimicry and immune‐mediated diseases. FASEB J. 12, 1255–1265 (1998)
REFERENCES
- 1. Theofilopoulos, A. (1995) The basis of autoimmunity. II. Genetic predisposition. Immunol. Today 16, 150–159 [DOI] [PubMed] [Google Scholar]
- 2. Merriman, T. R. , and Todd, J. A. (1995) Genetics of autoimmune disease. Curr. Opin. Immunol. 7, 786–792 [DOI] [PubMed] [Google Scholar]
- 3. Brewerton, D. , Caffrey, M. , Hart, F. , James, D. , Nichols, A. , and Sturrock, R. (1973) Ankylosing spondylitis and HLA B27. Lancet i, 904–907 [DOI] [PubMed] [Google Scholar]
- 4. Green, A. (1990) The role of genetic factors in development of IDDM. Curr. Top. Microbiol. Immunol. 164, 3–17 [DOI] [PubMed] [Google Scholar]
- 5. Ebers, G. , Bulman, D. , Sadovnik, A. , Paty, D. W. , Warren, S. , Hader, W. , Murray, T. J. , Seland, T. P. , Duquette, P. , Grey, T. , et al. (1987) A population‐based study of multiple sclerosis in twins. New Engl. J. Med. 315, 1638–1642 [DOI] [PubMed] [Google Scholar]
- 6. Eastmond, C. J. , and Woodrow, J. C. (1977) Discordance for ankylosing spondylitis in monozygotic twins. Ann. Rheum. Dis. 36, 360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scott, B. B. , Sadigh, S. , Andrew, E. M. , Maini, R. N. , and Mageed, R. A. (1994) Affinity maturation and isotype switch in clonally related anti‐erythrocyte autoantibodies. Scand. J. Immunol. 40, 16–21 [DOI] [PubMed] [Google Scholar]
- 8. Jury, K. M. , Loeffler, D. , Eiermann, T. H. , Ziegler, B. , Boehm, B. O. , and Richter, W. (1996) Evidence for somatic mutation and affinity maturation of diabetes associated human autoantibodies to glutamate decarboxylase. J. Autoimmun. 9, 371–377 [DOI] [PubMed] [Google Scholar]
- 9. Oldstone, M. B. A. (1989) Molecular mimicry as a mechanism for the cause and as a probe uncovering etiologic agent(s) of autoimmune disease. Curr. Top. Microbiol. Immunol. 145, 127–135 [DOI] [PubMed] [Google Scholar]
- 10. Tonietti, G. , Oldstone, M. B. A. , and Dixon, F. J. (1970) The effect of induced chronic viral infections on the immunologic diseases of New Zealand mice. J. Exp. Med. 132, 89–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kurtzke, J. F. (1993) Epidemiologic evidence for multiple sclerosis as an infection. Clin. Microbiol. Rev. 6, 382–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Panitch, H. S. (1994) Influence of infection on exacerbations of multiple sclerosis. Ann. Neurol. 36, S25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gamble, D. R. (1980) The epidemiology of insulin‐dependent diabetes with particular reference to the relationship of virus infection to its etiology. Epidemiol. Rev. 2, 49–70 [DOI] [PubMed] [Google Scholar]
- 14. Notkins, A. L. , and Yoon, J.‐W. (1984) Virus‐induced diabetes mellitus In Concepts in Viral Pathogenesis (Notkins A. L. and Oldstone M. B. A., eds) pp. 241–247, Springer‐Verlag, New York: [Google Scholar]
- 15. Gilliland, B. , and Mannik, M. (1986) Ankylosing spondylitis and Reiter's syndrome (pp. 1986–1988) and rheumatoid arthritis (pp. 1977–1986) In Harrison's Principles of Internal Medicine (Harrison T. R., ed) 10th Ed, McGraw Hill, New York: [Google Scholar]
- 16. Yoon, J.‐W. , Austin, M. , Onodera, T. , and Notkins, A. L. (1989) Virus‐induced diabetes mellitus: isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 300, 1173–1179 [DOI] [PubMed] [Google Scholar]
- 17. Challoner, P. B. , Smith, K. T. , Parker, J. D. , MacLeod, D. L. , Coulter, S. N. , Rose, T. M. , Schultz, E. R. , Bennett, J. L. , Garber, R. L. , Chang, M. , Schad, P. A. , Stewart, P. M. , Nowinski, R. C. , Brown, J. P. , and Burmer, G. C. (1995) Plaque‐associated expression of human herpesvirus 6 in multiple sclerosis. Proc. Natl. Acad. Sci. USA 92, 7440–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Talbot, P. J. , Paquette, J. S. , Ciurli, C. , Antel, J. P. , and Ouellet, F. (1996) Myelin basic protein and human coronavirus 229E cross‐reactive T cells in multiple sclerosis. Ann. Neurol. 39, 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Banki, K. , Colombo, E. , Sia, F. , Halladay, D. , Mattson, D. H. , Tatum, A. H. , Massa, P. T. , Phillips, P. E. , and Perl, A. (1994) Oligodendrocyte‐specific expression and autoantigenicity of transaldolase in multiple sclerosis. J. Exp. Med. 180, 1649–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sibley, W. A. (1995) Clinical viral infections and multiple sclerosis. Lancet ii, 1313–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geczy, A. F. , and Yap, J. (1979) HLA B27 Klebsiella and AS. Lancet i, 719 [DOI] [PubMed] [Google Scholar]
- 22. Goverman, J. , Woods, A. , Larson, L. , Weiner, L. P. , Hood, L. , and Zaller, D. M. (1993) Transgenic mice that express a myelin basic protein‐specific T cell receptor develop spontaneous autoimmunity. Cell 72, 551–560; Zaller, D. M., and Sloan, V. S. (1996) Transgenic mouse models of experimental autoimmune encephalomyelitis. Curr. Top. Microbiol. Immunol. 206, 15–28 [DOI] [PubMed] [Google Scholar]
- 23. Taurog, J. D. , Richardson, J. A. , Croft, J. T. , Simmons, W. A. , Zhou, M. , Fernandez‐Sueiro, J. L. , Balish, E. , and Hammer, R. E. (1994) The germfree state prevents development of gut and joint inflammatory disease in HLA‐B27 transgenic mice. J. Exp. Med. 180, 2359–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mcken, M. , Urban, J. , and Shevach, E. (1992) Infection breaks T‐cell tolerance. Nature (London) 359, 79 [DOI] [PubMed] [Google Scholar]
- 25. Conrad, B. , Weissmahr, R. N. , Boni, J. , Arcari, R. , Schupbach, J. , and Mach, B. (1997) A human endogenous retroviral superantigen as candidate autoimmune gene in type I diabetes. Cell 90, 303–313 [DOI] [PubMed] [Google Scholar]
- 26. Soos, J. M. , Schiffenbauer, J. , Torres, B. A. , and Johnson, H. M. (1997) Superantigens as virulence factors in autoimmunity and immunodeficiency diseases. Med. Hypotheses 48, 253–259 [DOI] [PubMed] [Google Scholar]
- 27. Sercarz, E. E. , Lehmann, P. V. , Ametani, A. , Benichou, G. , Miller, A. , and Moudgil, K. (1993) Dominance and crypticity of T cell antigenic determinants. Annu. Rev. Immunol. 11, 729–766 [DOI] [PubMed] [Google Scholar]
- 28. Miller, S. D. , Vanderlugt, C. L. , Begolka, W. S. , Pao, W. , Yauch, R. L. , Neville, K. L. , Katz‐Levy, Y. , Carrizosa, A. , and Kim, B. S. (1997) Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nature Med. 3, 1133–1136 [DOI] [PubMed] [Google Scholar]
- 29. Fujinami, R. S. , Oldstone, M. B. A. , Wroblewska, Z. , Frankel, M. E. , and Koprowski, H. (1983) Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad. Sci. USA 80, 2346–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shrinivasappa, J. , Saegusa, J. , Prabhakar, B. , Gentry, M. , Buchmeier, M. , Wiktor, T. , Koprowski, H. , Oldstone, M. , and Notkins, A. (1986) Molecular mimicry: frequency of reactivity of monoclonal antiviral antibodies with normal tissues. J. Virol. 57, 397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bahmanyar, S. , Srinivasappa, J. , Casali, P. , Fujinami, R. S. , Oldstone, M. B. A. , and Notkins, A. L. (1987) Antigen mimicry between measles virus and human T lymphocytes. J. Infect. Dis. 156, 526–527 [DOI] [PubMed] [Google Scholar]
- 32. Oldstone, M. B. A. (1987) Molecular mimicry and autoimmune disease. Cell 50, 819–820 [DOI] [PubMed] [Google Scholar]
- 33. Yamada, M. , Zurbriggen, A. , Oldstone, M. B. A. , and Fujinami, R. S. (1991) Common immunologic determinant between human immunodeficiency virus type 1 gp41 and astrocytes. J. Virol. 65, 1370–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fujinami, R. , and Oldstone, M. B. A. (1985) Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science 230, 1043–1045 [DOI] [PubMed] [Google Scholar]
- 35. Neu, N. , Rose, N. R. , Beisel, K. W. , Herskowitz, A. , Gurri‐Glass, G. , and Craig, S. (1987) Cardiac myosin induces myocarditis in genetically predisposed mice. J. Immunol. 139, 3630–3636 [PubMed] [Google Scholar]
- 36. Gauntt, C. J. , Tracy, S. M. , Chapman, N. , Wood, H. J. , Kolbeck, P. C. , Karaganis, A. G. , Winfrey, C. L. , and Cunningham, M. W. (1995) Coxsackievirus‐induced chronic myocarditis in murine models. Eur. Heart J. 16, 56–58 [DOI] [PubMed] [Google Scholar]
- 37. Schulze, K. , and Schultheiss, H. P. (1995) The role of the ADP/ATP carrier in the pathogenesis of viral heart disease. Eur. Heart J. 16, 64–67 [DOI] [PubMed] [Google Scholar]
- 38. Singh, V. K. , Kalra, H. K. , Yamaki, K. , Abe, T. , Donoso, L. A. , and Shinohara, T. (1990) Molecular mimicry between a uveitopathogenic site of S‐antigen and viral peptides: induction of experimental autoimmune uveitis in Lewis rats. J. Immunol. 144, 1282–1287 [PubMed] [Google Scholar]
- 39. Schwimmbeck, P. L. , Dyrberg, T. , Drachman, D. B. , and Oldstone, M. B. A. (1989) Molecular mimicry and myasthenia gravis. J. Clin. Invest. 84, 1174–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Atkinson, M. A. , Bowman, M. A. , Campbell, L. , Darrow, B. L. , Kaufman, D. L. , and MacLaren, N. K. (1994) Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin‐dependent diabetes. J. Clin. Invest. 94, 2125–2129; Tian, J., Lehmann, P. V., and Kaufman, D. L. (1994) T cell cross‐reactivity between Coxsackievirus and glutamate decarboxylase is associated with a murine diabetes susceptibility allele. J. Exp. Med. 180, 1979–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Honeyman, M. C. , Stone, N. L. , and Harrison, L. C. (1998) T‐cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA‐2: potential for mimicry with rotavirus and other environmental agents. Mol. Med. 4, 231–239 [PMC free article] [PubMed] [Google Scholar]
- 42. Khare, S. D. , Luthra, H. S. , and David, C. S. (1996) Role of HLA‐B27 in spondyloarthropathies. Curr. Top. Microbiol. Immunol. 206, 85–97 [DOI] [PubMed] [Google Scholar]
- 43. Schwimmbeck, P. , Yu, D. T. , and Oldstone, M. B. A. (1987) Autoantibodies to HLA‐B27 in sera of patients with ankylosing spondylitis and Reiter's syndrome. J. Exp. Med. 166, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schwimmbeck, P. , and Oldstone, M. B. A. (1989) Klebsiella pneumoniae and HLA B27 associated diseases of Reiter's syndrome and ankylosing spondylitis. Curr. Top. Microbiol. Immunol. 145, 45–67 [DOI] [PubMed] [Google Scholar]
- 45. Hammer, R. E. , Maika, S. D. , Richardson, J. A. , Tang, J.‐P. , and Taurog, J. D. (1990) Spontaneous inflammatory disease in transgenic mice expressing HLA‐B27 and human β2m: an animal model of HLA‐B27‐associated human disorders. Cell 63, 1099–1112 [DOI] [PubMed] [Google Scholar]
- 46. Rath, H. C. , Herfarth, H. H. , Ikeda, J. S. , Grenther, W. B. , Hamm, T. E., Jr. , Balish, E. , Taurog, J. D. , Hammer, R. E. , Wilson, K. H. , and Sartor, R. B. (1996) Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA‐B27/human beta2 microglobulin transgenic rats. J. Clin. Invest. 98, 945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wucherpfennig, K. W. , and Strominger, J. L. (1995) Molecular mimicry in T‐cell mediated autoimmunity: viral peptides activate human T‐cell clones specific for myelin basic protein. Cell 80, 695–705; Wucherpfennig, K. W., and Strominger, J. L. (1995) Selective binding of self peptides to disease‐associated major histocompatibility complex (MHC) molecules: a mechanism for MHC‐linked susceptibility to human autoimmune diseases. J. Exp. Med. 181, 1597–16017534214 [Google Scholar]
- 48. Hammer, J. , Gallazzi, F. , Bono, E. , Karr, R. W. , Guenot, J. , Valsasnini, P. , Nagy, Z. A. , and Sinigaglia, F. (1995) Peptide binding to HLA‐DR4 molecules: correlation with rheumatoid arthritis association. J. Exp. Med. 181, 1847–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. García, K. C. , Degano, M. , Stanfield, R. L. , Brunmark, A. , Jackson, M. R. , Peterson, P. A. , Teyton, L. , and Wilson, I. A. (1996) The aαβ T cell receptor structure at 2. 5 Å and its orientation in the TCR‐MHC complex. Science 274, 209–219 [DOI] [PubMed] [Google Scholar]
- 50. Kersh, G. J. , and Allen, P. M. (1996) Essential flexibility in the T‐cell recognition of antigen. Nature (London) 380, 495–498 [DOI] [PubMed] [Google Scholar]
- 51. Gautam, A. M. , Lock, C. B. , Smilek, D. E. , Pearson, C. I. , Steinman, L. , and McDevitt, H. O. (1994) Minimum structural requirements for peptide presentation by major histocompatibility complex class II molecules: implications in induction of autoimmunity. Proc. Natl. Acad. Sci. USA 91, 767–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oldstone, M. B. A. , Nerenberg, M. , Southern, P. , Price, J. , and Lewicki, H. (1991) Virus infection triggers insulin‐dependent diabetes mellitus in a transgenic model: role of anti‐self (virus) immune response. Cell 65, 319–331 [DOI] [PubMed] [Google Scholar]
- 53. von Herrath, M. , Dockter, J. , and Oldstone, M. B. A. (1994) How virus induces a rapid or slow onset insulin‐dependent diabetes mellitus in a transgenic model. Immunity 1, 231–242 [DOI] [PubMed] [Google Scholar]
- 54. von Herrath, M. G. , Guerder, S. , Lewicki, H. , Flavell, R. , and Oldstone, M. B. A. (1995) Co‐expression of B7.1 and viral (self) transgenes in pancreatic β‐cells can break peripheral ignorance and lead to spontaneous autoimmune diabetes. Immunity 3, 727–738 [DOI] [PubMed] [Google Scholar]
- 55. von Herrath, M. G. , Dockter, J. , Nerenberg, M. , Gairin, J. E. , and Oldstone, M. B. A. (1994) Thymic selection and adaptability of cytotoxic T lymphocyte responses in transgenic mice expressing a viral protein in the thymus. J. Exp. Med. 180, 1901–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. von Herrath, M. G. , and Oldstone, M. B. A. (1997) Interferon‐γ is essential for destruction of β cells and development of insulin‐dependent diabetes mellitus. J. Exp. Med. 185, 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee, M.‐S. , von Herrath, M. , Reiser, H. , Oldstone, M. B. A. , and Sarvetnick, N. (1995) Sensitization to self(virus) antigen by in situ expression of murine interferon‐γ. J. Clin. Invest. 95, 486–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. von Herrath, M. G. , Dyrberg, T. , and Oldstone, M. B. A. (1996) Oral insulin treatment suppresses virus‐induced antigen‐specific destruction of β cells and prevents autoimmune diabetes in transgenic mice. J. Clin. Invest. 98, 1324–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hudrisier, D. , Mazarguil, H. , Laval, F. , Oldstone, M. B. A. , and Gairin, J. E. (1996) Binding of viral antigens to major histocompatability complex class I H‐2Db molecules is controlled by dominant negative elements at peptide non‐anchor residues: implications for peptide selection and presentation. J. Biol. Chem. 271, 17829–17836 [DOI] [PubMed] [Google Scholar]
- 60. von Herrath, M. G. , Efrat, S. , Oldstone, M. B. A. , and Horwitz, M. S. (1997) Expression of adenoviral E3 transgenes in β cells prevents autoimmune diabetes. Proc. Natl. Acad. Sci. USA 94, 9808–9813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Oldstone, M. B. A. , Lewicki, H. , Borrow, P. , Hudrisier, D. , and Gairin, J. E. (1995) Discriminated selection among viral peptides with the appropriate anchor residues: implications for the size of the cytotoxic T‐lymphocyte repertoire and control of viral infection. J. Virol. 69, 7423–7429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gairin, J. E. , and Oldstone, M. B. A. (1992) Design of high‐affinity major histocompatibility complex‐specific antagonist peptides that inhibit cytotoxic T‐lymphocyte activity: implications for control of viral disease. J. Virol. 66, 6755–6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baum, H. (1995) Mitochondrial antigens, molecular mimicry and autoimmune disease. Biochim. Biophys. Acta. 1271, 111–121 [DOI] [PubMed] [Google Scholar]
- 64. Baum, H. , Davies, H. , and Peakman, M. (1997) Molecularmimicry in the MHC: hidden clues to autoimmunity? Immunol. Today 18, 252–253 [DOI] [PubMed] [Google Scholar]
- 65. Butler, P. , Hamilton‐Miller, J. , Baum, H. , and Burroughs, A. K. (1995) Detection of M2 antibodies in patients with recurrent urinary tract infection using an ELISA and purified PBC specific antigens. Evidence for a molecular mimicry mechanism in the pathogenesis of primary biliary cirrhosis? Biochem. Mol. Biol. Int. 35, 473–485 [PubMed] [Google Scholar]
- 66. Rogers, S. W. , Andrews, P. I. , Gahring, L. C. , Whisenand, T. , Cauley, K. , Crain, B. , Hughes, T. E. , Heinemann, S. F. , and McNamara, J. O. (1994) Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis. Science 265, 648–651 [DOI] [PubMed] [Google Scholar]
- 67. McNamara, J. O. , Patel, M. , He, X. P. , Janumpalli, S. , and Whitney, K. D. (1996) Glutamate receptor autoimmunity in Rasmussen's encephalitis. Cold Spring Harbor Symp. Quant. Biol. 61, 327–332 [PubMed] [Google Scholar]
- 68. Steinman, L. , and Oldstone, M. B. A. (1997) More mayhem from molecular mimics. Nature Med. 3, 1321–1322 [DOI] [PubMed] [Google Scholar]
- 69. Talbot, P. J. (1997) Virus‐induced autoimmunity in multiple sclerosis: the coronavirus paradigm. Adv. Clin. Neurosci. 7, 215–233 [Google Scholar]
- 70. Soldan, S. S. , Berti, R. , Salem, N. , Secchiero, P. , Flamand, L. , Calabresi, P. A. , Brennan, M. B. , Maloni, H. W. , McFarland, H. F. , Lin, H. C. , et al. (1997) Association of human herpes virus 6 (HHV‐6) with multiple sclerosis: increased IgM response to HHV‐6 early antigen and detection of serum HHV‐6 DNA. Nature Med. 3, 1394–1397 [DOI] [PubMed] [Google Scholar]
- 71. Hemmer, B. , Fleckenstein, B. T. , Vergelli, M. , Jung, G. , Mc‐Farland, H. F. , Martin, R. , and Wiesmueller, K.‐H. (1997) Identification of high potency microbial and self ligands for a human autoreactive class II‐restricted T cell clone. J. Exp. Med. 185, 1651–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Marx, A. , Wilisch, A. , Schultz, A. , Gattenlohner, S. , Nenninger, R. , and Muller‐Hermelink, H. K. (1997) Pathogenesis of myasthenia gravis. Virchows Arch. 430, 355–364 [DOI] [PubMed] [Google Scholar]
- 73. Richman, D. P. , and Agius, M. A. (1994) Acquired myasthenia gravis. Neurol. Clin. 12, 273–284 [PubMed] [Google Scholar]
- 74. Hafer‐Macko, C. , Hsieh, S. T. , Li, C. Y. , Ho, T. W. , Sheikh, K. , Cornblath, D. R. , McKhann, G. M. , Asbury, A. K. , and Griffin, J. W. (1996) Acute motor axonal neuropathy: an antibody‐mediated attack on axolemma. Ann. Neurol. 40, 635–644 [DOI] [PubMed] [Google Scholar]
- 75. van der Meche, F. G. , and van Doorn, P. A. (1995) Guillain‐Barre syndrome and chronic inflammatory demyelinating polyneuropathy: immune mechanisms and update on current therapies. Ann. Neurol. 37, S14–S31 [DOI] [PubMed] [Google Scholar]
- 76. Yuki, N. , Tagawa, Y. , and Handa, S. (1996) Autoantibodies to peripheral nerve glycosphingolipids SPG, SLPG, and SGPG in Guillain‐Barraé syndrome and chronic inflammatory demyelinating polyneuropathy. J. Neuroimmunol. 70, 1–6 [DOI] [PubMed] [Google Scholar]
- 77. MacLaren, N. K. , and Alkinson, M. A. (1997) Insulin‐dependent diabetes mellitus: the hypothesis of molecular mimicry between islet cell antigens and microorganisms. Mol. Med. Today 3, 76–83 [DOI] [PubMed] [Google Scholar]
- 78. Karges, W. J. , Ilonen, J. , Robinson, B. H. , and Dosch, H. M. (1995) Selfand non‐selfantigen in diabetic autoimmunity: molecules and mechanisms. Mol. Aspects Med. 16, 79–213 [DOI] [PubMed] [Google Scholar]
- 79. Solimena, S. , and DeCamilli, P. (1995) Coxsackieviruses and diabetes. Nature (London) Med. 1, 25–26; Baum, H., Brusic, V., Choudhuri, K., Cunningham, P., Vergani, D., and Peakman, M. (1995) MHC molecular mimicry in diabetes. Nature Med. 1, 388. [DOI] [PubMed] [Google Scholar]
- 80. Rudy, G. , Stone, N. , Harrison, L. C. , Colman, P. G. , McNair, P. , Brusic, V. , French, M. B. , Honeyman, M. C. , Tait, B. , and Lew, A. M. (1995) Similar peptides from two beta cell autoantigens, proinsulin and glutamic acid decarboxylase, stimulate T cells of individuals at risk for insulin‐dependent diabetes. Mol. Med. 1, 625–633 [PMC free article] [PubMed] [Google Scholar]
- 81. Huber, S. A. , Moraska, A. , and Cunningham, M. (1994) Alterations in major histocompatibility complex association of myocarditis induced by coxsackievirus B3 mutants selected with monoclonal antibodies to group A streptococci. Proc. Natl. Acad. Sci. USA 91, 5543–5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Krisher, K. , and Cunningham, M. W. (1985) Myosin: a link between streptococci and heart. Science 227, 413 [DOI] [PubMed] [Google Scholar]
- 83. Kagnoff, M. F. (1989) Celiac disease: adenovirus and alpha gliadin. Curr. Top. Microbiol. Immunol. 145, 67–78 [DOI] [PubMed] [Google Scholar]
- 84. Tuckova, L. , Tlaskalova‐Hogenova, H. , Farre, M. A. , Karska, K. , Rossmann, P. , Kolinska, J. , and Kocna, P. (1995) Molecular mimicry as a possible cause of autoimmune reactions in celiac disease? Antibodies to gliadin cross‐react with epitopes on enterocytes. Clin. Immunol. Immunopathol. 74, 170–176 [DOI] [PubMed] [Google Scholar]
- 85. Fujinami, R. S. , Nelson, J. A. , Walker, L. , and Oldstone, M. B. A. (1988) Sequence homology and immunologic cross‐reactivity of human cytomegalovirus with HLA‐DR β chain: a means for graft rejection and immunosuppression. J. Virol. 62, 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Naucler, C. S. , Larsson, S. , and Moller, E. (1996) A novel mechanism for virus‐induced autoimmunity in humans. Immunol. Rev. 152, 175–192 [DOI] [PubMed] [Google Scholar]
- 87. Gilbert, D. , Courville, P. , Brard, F. , Joly, P. , Petit, S. , Bernardi, E. , Schoofs, A. R. , Lauret, P. , and Tron, F. (1997) A complementarity‐determining region peptide of an anti‐desmosome autoantibody may interact with the desmosomal plaque through molecular mimicry with a cytoplasmic desmoglein 1 sequence. Eur. J. Immunol. 27, 1055–1060 [DOI] [PubMed] [Google Scholar]
- 88. Zhao, Z.‐S. , Granucci, F. , Yeh, L. , Schaffer, P. , and Cantor, H. (1997) Molecular mimicry by herpes simplex virus type 1: autoimmune disease after viral infection. Science 79, 1344–1347 [DOI] [PubMed] [Google Scholar]
- 89. Thurau, S. R. , Diedrichs‐Mohring, M. , Fricke, H. , Arbogast, S. , and Wildner, G. (1997) Molecular mimicry as a therapeutic approach for an autoimmune disease: oral treatment of uveitis patients with an MHC‐peptide crossreactive with autoantigen first results. Immunol. Lett. 57, 193–201 [DOI] [PubMed] [Google Scholar]
- 90. Appelmelk, B. J. , Simoons‐Smit, I. , Negrini, R. , Moran, A. P. , Aspinall, G. O. , Forte, J. G. , DeVries, T. , Quan, H. , Verboom, T. , Maaskant, J. J. , et al. (1996) Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect. Immun. 64, 2031–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Silvestris, F. , Williams, R. C., Jr. , and Dammacco, F. (1995) Autoreactivity in HIV‐1 infection: the role of molecular mimicry. Clin. Immunol. Immunopathol. 75, 197–205 [DOI] [PubMed] [Google Scholar]
- 92. Bisset, L. R. (1994) Molecular mimicry in the pathogenesis of AIDS: the HIV/MHC/mycoplasma triangle. Med. Hypotheses 43, 388–396 [DOI] [PubMed] [Google Scholar]
- 93. Dalgleish, A. G. (1995) Autoimmune mechanisms of depletion of CD4 cells in HIV infection. Br. J. Haematol. 91, 525–534 [DOI] [PubMed] [Google Scholar]
- 94. Marchalonis, J. J. , Lake, D. F. , Schluter, S. F. , Dehghanpisheh, K. , Watson, R. R. , Ampel, N. M. , and Galgiani, J. N. (1995) Autoantibodies against peptide‐defined epitopes of T‐cell receptors in retrovirally infected humans and mice. Adv. Exp. Med. Biol. 383, 211–222 [DOI] [PubMed] [Google Scholar]
- 95. Aberer, E. , Brunner, C. , Suchanek, G. et al. (1989) Molecular mimicry and lyme borreliosis: a shared antigenic determinant between borrelia burgdorferi and human tissue. Ann. Neurol. 26, 732–737 [DOI] [PubMed] [Google Scholar]
- 96. Froude, J. , Gibofsky, A. , Buskirk, D. R. , Khanna, A. , and Zabriskie, J. B. (1989) Streptococcus and human tissue: a model of molecular mimicry and autoimmunity. Curr. Top. Microbiol. Immunol. 145, 5–26 [DOI] [PubMed] [Google Scholar]
- 97. Baugh, R. E. , Jiang, A. , Abraham, R. , Ottmers, V. , and Musher, D. M. (1996) Molecular mimicry between an immunodominant amino acid motif on the 47‐kDa lipoprotein of treponema pallidum (tpp47) and multiple repeats of analogous sequences in fibronectin. J. Immunol. 157, 720–731 [PubMed] [Google Scholar]
- 98. Ebringer, A. , Thorpe, C. , Pirt, J. , Wilson, C. , Cunningham, P. , and Ettelaie, C. (1997) Bovine spongiform encephalopathy: is it an autoimmune disease due to bacteria showing molecular mimicrywith brain antigens? Env. Health Persp. 105, 1172–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Van Voorhis, W. , Schlekewy, L. , and Trong, H. (1991) Molecular mimicry by Trypanosoma cruzi: the F1‐160 epitope that mimics mammalian nerve can be mapped to a 12‐amino acid peptide. Proc. Natl. Acad. Sci. USA 88, 5993–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Petry, K. , Voisin, P. , Baltz, T. , and Labouesse, J. (1987) Epitopes common to trypanosomes (T. cruzi, T. dionisii and T. vespertilionis (Schizotrypanum)): astrocytes and neurons. J. Neuroimmunol. 16, 237–252 [DOI] [PubMed] [Google Scholar]
- 101. Cunha‐Neto, E. , Coelho, V. , Guilherme, L. , Fiorelli, A. , Stolf, N. , and Kalil, J. (1996) Autoimmunity in Chagas disease. Identification of cardiac myosin‐B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas cardiomyopathy patient. J. Clin. Invest. 98, 1709–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ferrari, I. , Levin, M. J. , Wallukat, G. , Elies, R. , Lebesgue, D. , Chiale, P. , Elizari, M. , Rosenbaum, M. , and Hoebeke, J. (1995) Molecular mimicry between the immunodominant ribosomal protein PO of Trypanosoma cruzi and a functional epitope on the human beta 1‐adrenergic receptor. J. Exp. Med. 182, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]