

Abstract
Alphaviruses, including Chikungunya (CHIKV) and Venezuelan equine encephalitis virus (VEEV), are among the leading causes of recurrent epidemics all over the world. Alphaviral nonstructural protein 1 (nsP1) orchestrates the capping of nascent viral RNA via its S‐adenosyl methionine‐dependent N‐7‐methyltransferase (MTase) and guanylyltransferase activities. Here, we developed and validated a novel capillary electrophoresis (CE)‐based assay for measuring the MTase activity of purified VEEV and CHIKV nsP1. We employed the assay to assess the MTase inhibition efficiency of a few adenosine analogs and identified 5‐iodotubercidin (5‐IT) as an inhibitor of nsP1. The antiviral potency of 5‐IT was evaluated in vitro using a combination of cell‐based assays, which suggest that 5‐IT is efficacious against CHIKV in cell culture (EC50: 0.409 µm).
Keywords: adenosine analog, alphavirus inhibitor, CE‐based MTase assay, Chikungunya virus, nsP1

Abbreviations
5‐IT, 5‐iodotubercidin
BGE, background electrolyte
CE, capillary electrophoresis
CHIKV, Chikungunya virus
DAPI, 4',6‐diamidino‐2‐phenylindole
EEEV, Eastern equine encephalitis virus
ELISA, enzyme‐linked immunosorbent assay
GIDP, guanosine 5′‐[β,γ‐imido]triphosphate
GT, guanylation
GTase, guanylyltransferase
MALDI‐TOF, matrix‐assisted laser desorption‐time of flight spectrometer
MTase, methyltransferase
nsP1, nonstructural protein 1
NW, New World
OW, Old World
PFU, plaque‐forming units
PVA, poly‐vinyl alcohol
qRT‐PCR, quantitative reverse transcription polymerase chain reaction
SAH, S‐adenosyl homocysteine
SAM, S‐adenosyl methionine
VEEV, Venezuelan equine encephalitis virus
Emergence and re‐emergence of mosquito‐borne viral pathogens including alphaviruses such as Chikungunya (CHIKV) and Venezuelan equine encephalitis virus (VEEV) represent a significant health threat to the human population worldwide. Alphaviruses are classified based on their geographical distribution into New World (NW) and Old World (OW) alphaviruses 1. OW alphaviruses are generally arthrogenic and include CHIKV and Sindbis virus, while NW alphaviruses such as VEEV, Eastern equine encephalitis virus (EEEV), and Western equine encephalitis virus are characterized by an encephalitogenic phenotype 1, 2. Alphaviruses replicate to very high titers in cell lines of mosquito and vertebrate origins and possess capability of storage in lyophilized form and their potential of spread in aerosol form makes them potential bioweapons 3. Thus, these viruses are recognized as category B priority pathogens by the U.S. government. VEEV has caused major epidemics in several parts of the world involving a large number of equids and humans 4. CHIKV has been documented to cause disease since 1953 but its explosive outbreak in 2005 in La Rèunion Island involved an overwhelming number of 300 000 reported cases 5. Since 2005, it has caused recurrent outbreaks in Asia and other parts of the world. The lack of effective and safe antiviral agents against alphaviruses including CHIKV and VEEV makes the pursuit of compounds with antiviral activity against these viruses highly imperative.
In alphaviruses, individual nonstructural proteins (nsPs) are produced post‐translationally after processing of a nonstructural polyprotein and they play an indispensable role in the alphaviral life cycle 2. Capping of the viral genome is a vital step in the alphaviral life cycle as this cap structure plays numerous biological roles including the protection of mRNA from cellular exonucleases and efficient recognition of the RNA by eukaryotic translation initiation factor 4E (eIF4E) for the initiation of translation 6. Additionally, this cap structure also prevents recognition by members of the host innate immune pathways such as MDA5 and/or RIG‐I 7. Alphaviruses use a completely distinct molecular mechanism of capping of viral mRNA from the host cell. Cellular capping enzymes typically methylate GTP only after its transfer to the 5′ end of the RNA. In contrast, the alphaviral capping enzyme, nonstructural protein 1 (nsP1) caps viral RNA in two major steps: It first methylates GTP by transferring the methyl group from S‐adenosyl methionine (SAM), which acts as the methyl donor, to GTP and the methylated m7GMP moiety gets covalently attached to the nsP1 enzyme transiently during a guanylation (GT) step 8. S‐adenosyl homocysteine (SAH) is generated as a coproduct of this methyltransferase (MTase) step of capping reaction. The amino‐terminal of nsP2, another replication protein of alphavirus, harbors the RNA triphosphatase activity 9. It contributes to the capping reaction of alphaviruses by cleaving the γ,β‐triphosphate bond specifically at the 5′ end of the nascent RNA prior to the transfer of m7GMP to the nascent viral RNA by nsP1 in the guanylyltransferase (GTase) step. Mutational analyses of the protein and reverse genetics studies involving live virus showed that the abrogation of alphaviral nsP1 activity is detrimental to the viral growth proving that nsP1 capping activity is an essential step during viral replication 10.
Viruses within the alphavirus superfamily have been shown to replicate in membrane invaginations (spherules) in the host cell. nsP1 is also a vital component of the replication complex as it anchors the ensemble of nsPs and other replication factors on the membrane primarily through an amphipathic helix and secondarily through palmitoylation sites on the protein 11, 12, 13. The three‐dimensional structure of alphavirus nsP1 from any member of the alphavirus genus remains elusive. In the absence of the 3D structure of nsP1, the development of enzymatic assays for the screening or development of potent inhibitors targeting replication protein nsP1 is required. It will provide a platform for the identification and characterization of inhibitors of this viral enzyme for treating diseases caused by alphaviral infection. Lately, there has been a surge in the development of novel assays for nsP1 enzymatic activity to accentuate the pursuit of novel antiviral compounds. Previous efforts for the development of the assays for the biochemical characterization and inhibition assays against nsP1 were focussed on the GT step of the nsP1 enzymatic reaction 14, 15, 16. Gel‐based approach for inhibition study used a fluorescently labeled GTP (GTP‐ATTO) for detection or western blot/ELISA uses anti‐m3G/m7G‐cap monoclonal antibodies for quantification of nsP1 enzymatic activity 14, 15, 16. These immuno‐based assays target GT activity inhibition, utilize expensive antibodies, and are labor‐intensive and susceptible to the identification of false positives/false negatives. A fluorescence polarization (FP)‐based assay has also been developed for nsP1, which is used for screening of small molecules against CHIKV nsP1. This assay is based on monitoring the displacement of fluorescently labeled GTP analog by an inhibitory molecule 17. The screening potential of this FP‐based assay is restricted to the compounds competing for the GTP‐binding site on the enzyme.
Nucleosides/nucleoside analogs represent a major class of antiviral drugs 18. Nucleoside analogs have been shown to display antiviral activity against several medically important viruses including HIV, hepatitis B virus, and herpesvirus 19, 20. Sinefungin and other SAM/nucleoside analogs have been shown to inhibit viral MTase activity and consequently abrogate viral replication in vitro in the members of flavivirus family including the Zika virus, West Nile virus, and Dengue virus 21, 22, 23, 24. Regarding alphaviruses, broad‐spectrum antiviral agents ribavirin and favipiravir, have been shown to have in vitro anti‐VEEV and anti‐CHIKV activity, respectively 25, 26. Additionally, a cytosine analog, D‐(‐)‐carbodine, showed moderate in vivo efficacy against VEEV 27. More recently, a modified nucleoside analog, β‐d‐N4‐hydroxycytidine, which was previously shown to be effective against hepatitis C virus 28 and human coronavirus 29, was found to inhibit CHIKV and VEEV replication in cell culture, suggesting that employing nucleoside analogs for alphaviral alleviation is a rewarding approach 30, 31.
In this study, a nonradioactive capillary electrophoresis (CE)‐based assay has been developed that measures the MTase activity of the alphaviral nsP1 capping enzyme independently of its GT activity. To achieve this, VEEV nsP1 (full‐length) and a C‐terminal truncated construct of CHIKV nsP1 were cloned, recombinantly expressed and purified. Using this CE‐based assay, the influence of various reaction parameters on the methylation reaction of VEEV and CHIKV nsP1 enzyme was determined. H37A mutant of VEEV nsP1 was designed and used to examine the formation of m7GTP during the alphaviral capping reaction using a different CE method. Additionally, the assay was used to characterize known inhibitors of alphavirus nsP1, sinefungin, and aurintricarboxylic acid (ATA). Using this assay, the possibility of using adenosine analogs for modulation of nsP1 MTase activity was investigated. One of the compounds was found to inhibit the MTase activity of both VEEV and CHIKV nsP1. The adenosine analog inhibitor identified using this assay was also validated for nsP1 inhibition using an orthogonal nonradioactive enzyme‐linked immunosorbent assay (ELISA) assay. Further, the antiviral efficacy of the identified inhibitor was evaluated in vitro using cell‐based assays. Intriguingly, the compound showed significant inhibition of CHIKV in cell culture‐based studies, asserting the reliability of the developed assay. This novel CE‐based MTase assay and new approach of using adenosine analogs to target alphavirus nsP1 may promote the identification of novel therapeutics against alphaviruses.
Materials and methods
Cell line, virus strain, and compounds
Vero cells were procured from National Centre for Cell Science (NCCS), Pune, India, and were maintained at 37 °C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% inactivated FBS (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). A recent clinical isolate of CHIKV (strain no.: 119067; GenBank: KY057363.1) was propagated in Vero cells using standard viral adsorption techniques, quantified by standard plaque assays and stored at −80 °C 32, 33. SAM, SAH, GTP, UMP, caffeine, sinefungin, ATA, and m7GTP were purchased from Sigma (St. Louis, MO, USA). 2‐Chloroadenosine, 3‐Deazaadenosine, and 3‐Deazaneplanocin A were from cayman chemicals (Ann Arbor, MI, USA) and dissolved in 100% dimethyl sulfoxide (DMSO). 5‐Iodotubercidin (5‐IT), also a product of cayman chemicals, was supplied in 100% ethanol.
Cloning, expression, and purification of CHIKV nsP1 and VEEV nsP1
For cloning of VEEV nsP1 (amino acid residues 1‐535), PCR‐amplified DNA containing the VEEV nsP1 gene, which was a kind gift from Richard J. Kuhn (Purdue University, IN, USA), was taken as a template for PCR amplification. PCR amplification was done with primer pair: sense, 5′‐ GATTCCATATGGAGAAAGTTCACGTTGACATCGAGGAA‐3′; and antisense, 5′‐ GCAT CTCGAGTTAGGCCCCAGCCTCTTGTAACATCAA‐3′. VEEV nsP1 gene was then ligated in‐frame with a hexa‐histidine tag at N‐terminal in a pET‐28c plasmid. Positive clones were subsequently transformed into expression host Rosetta, and the culture was grown overnight in terrific broth supplemented with kanamycin and chloramphenicol at 37 °C and was used as an inoculum for a 1000 mL secondary culture. This secondary culture was grown at 37 °C till OD600 reached 0.4 and was then shifted to 18 °C. At 0.7 OD600, the cultures were induced with 0.4 mm isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) and were allowed to grow for another 16 h at 18 °C. Cells were harvested after incubation by centrifugation and analyzed using 12% SDS/PAGE. MTase/GT mutants were generated using the PCR‐based method for site‐directed mutagenesis using specific primers. CHIKV nsP1 (amino acid residues 1–509) was cloned and purified as described previously 16.
The cell pellet harvested from bacterial culture was resuspended in 50 mL binding buffer (50 mm Tris buffer, pH 7.3, 20 mm imidazole, and 100 mm KCl). 1 mm phenylmethylsulfonyl fluoride (PMSF), 100 µL Halt protease inhibitor (Thermo Fisher Scientific), and 0.01 mg·mL−1 DNase (Promega, Madison, WI, USA) were added to the resuspended cell pellet. Cell pellet was incubated at 4 °C for 10 min and was lysed using a French press system (Constant Systems Ltd, Daventry, UK). The cell lysate was centrifuged to separate insoluble cellular debris from soluble fraction containing recombinant protein. The clarified supernatant was loaded onto nickel‐nitrilotriacetic acid (Ni‐NTA) beads (Qiagen, Germantown, MD, USA) in a gravity flow column and incubated for 30 min at 4 °C and flow‐through was collected. Column was washed with buffers containing increasing concentrations of imidazole. Recombinant nsP1 protein was eluted with 250 mm imidazole in 50 mm Tris buffer, pH 7.3, and 100 mm KCl. Different fractions were analyzed using 12% SDS/PAGE. The identity of the protein was checked with matrix‐assisted laser desorption‐time of flight spectrometer (MALDI‐TOF) after digestion with trypsin. Fractions containing recombinant protein were mixed and dialyzed against dialysis buffer (50 mm Tris buffer, pH 7.0, 20 mm KCl, 2 mm DTT, and 2.5 mm MgCl2). VEEV nsP1 (H37A and D63A) mutants were expressed and purified with Ni+2 affinity chromatography similar to the expression and purification of the wild‐type (wt) protein. The dialyzed protein was frozen in liquid nitrogen and stored at −80 °C. The protein was thawed when needed and concentrated to the required concentration with amicon centrifugal filters (Millipore, Burlington, MA, USA).
CE methods
The products of the nsP1 MTase reaction are SAH and m7GTP. In this study, two distinct methods for the detection of SAH and the detection of m7GTP have been developed. Detection of SAH was performed with a fused‐silica capillary, while poly‐vinyl alcohol (PVA)‐coated capillary was used for the detection of m7GTP.
Method 1: Fused‐silica capillary
CE analysis was done using an Agilent 7100 CE system (Agilent Technologies, Santa Clara, CA, USA) fitted with a diode‐array detector (DAD) detection system. The electrophoretic separation for SAM and SAH was done using an extended light path fused‐silica capillary [64.5 cm (56 cm effective length) × 50 µm internal diameter]. The sample was injected hydrodynamically by applying pressure of 50 mbar for 10 s using autosampler followed by application of 30 kV voltage for separation of analytes. The capillary was thermostated at 20 °C. The sample vials were stored in a sample tray at 8 °C. Detection was done with a DAD coupled to a UV detection system at the cathodic end of the capillary at the wavelength of 260 nm. At the beginning of each day, capillary was conditioned using the following sequence: (1) 5‐min rinse with CE‐grade water, (2) 10‐min rinse with NaOH, (3) 5‐min rinse with water, and (4) 10‐min rinse with running buffer. Between two runs, the capillary was rinsed for 100 s with running buffer. At the end of each day, the capillary was flushed for 5 min with water and subsequently for 5 min with N2, and was taken out of the instrument.
Method 2: PVA capillary
For examining the formation of m7GTP by H37A mutant, PVA‐coated capillary (Agilent Technologies) [64.5 cm (56 cm effective length) × 50 µm internal diameter] was employed. The coating on the capillary suppresses electro‐osmotic flow drastically and reverse voltage polarity was applied across the ends of the coated capillary to facilitate the rapid migration of negatively charged species such as GTP. The capillary was thermostated at 20 °C. The sample vials were stored in a sample tray at 8 °C. To achieve baseline separation of the product m7GTP, a voltage of 30 kV was applied. The sample to be analyzed was introduced hydrodynamically in the capillary using 50 mbar pressure for 10 s using autosampler. PVA capillary was flushed with water for 200 s and buffer with 200 s prior to the first run of the day. Between two successive runs, capillary was rinsed with the running buffer for 100 s. 254 nm wavelength was used for the detection and quantification of the analytes.
MTase reaction
MTase reaction was carried out in a final reaction volume of 20 µL in 50 mm Tris buffer, pH 7.0, 1 mm DTT, 10 mm/20 mm KCl, 4 mm GTP or GIDP (Guanosine 5′‐[β,γ‐imido]triphosphate), 0.3 mm SAM, and 20 µm of VEEV nsP1/10 µm CHIKV nsP1 and incubated at 30 °C. For detection of m7GTP using the H37A mutant of VEEV nsP1, MTase reaction was the same except increased concentration of SAM (1 mm) was used in the reaction owing to the higher detection limit of m7GTP. MTase reaction was also done with the D63A mutant of VEEV nsP1 to be used as a negative control. For following the time‐course study, reaction was stopped at different time points by adding acetonitrile in 1 : 2 (vol/vol) ratio. The mixture was vortex‐mixed for 15 s, and protein was precipitated for 20 min at 18 500 g. Supernatant was transferred to sample vials for CE analysis. All reactions were done in duplicate.
CE‐based nsP1 inhibition assays
The compounds were dissolved in DMSO, and 5‐IT was dissolved in ethanol. Typical inhibition assay reaction was carried out in reaction volume of 20 µL in 50 mm Tris buffer, pH 7.0, 1 mm DTT, 10 mm KCl, 4 mm GTP or GIDP, 0.3 mm SAM, and 20 µm of VEEV nsP1 or 10 µm CHIKV nsP1. Sinefungin and ATA were tested at 7 different concentrations around reported half‐maximal inhibitory concentration (IC50) value of each for VEEV nsP1 34. The reaction mixture was incubated at 30 °C for 2 h. 3‐Deazaneplanocin A, 2‐Chloroadenosine, 3‐Deazaadenosine, and 5‐IT were tested at 50 µm concentration. Control reaction mixtures were incubated along with inhibition assays containing corresponding vehicle concentration. Each compound was tested in duplicate.
nsP1 ELISA‐based assay
GT reaction was performed in 50 mm Tris buffer pH 7.3, 5 mm DTT, 10 mm NaCl, 2 mm MgCl2, 5 mm DTT, 100 μm GTP, and 100 μm SAM with 20 μm protein at 25 °C for 30 min. Detection of CHIKV nsP1 GT activity was done with an ELISA assay as reported earlier 16. Briefly, reaction mixtures were transferred to 96‐well high binding ELISA plate (Greiner Bio‐One, Kremsmünster, Austria) after the reaction was stopped by the addition of an equal volume of 2% SDS and 5 mm EDTA followed by incubation for 1 h at 25 °C. Wells were washed twice with TBST buffer (20 mm Tris pH 7.5, 150 mm NaCl, 0.1% Tween‐20), blocked in 3% skim milk in TBST for 1 h, washed again, and incubated at 25 °C for 1 h with 1 : 5000 anti‐m7G cap monoclonal antibody (Merck Millipore, Burlington, MA, USA). Subsequently, 1 : 10 000 HRP‐conjugated anti‐mouse IgG1 secondary antibody (Affymetrix eBioscience, San Diego, CA, USA) was added to the wells and incubated at 25 °C for 1 h after washing with TBST. Finally, for the detection of immunoreactive protein, 100 μL of TMB/H2O2‐ELISA substrate (GeNei) was added to each well of the plate and incubated and the reaction was stopped by the addition of an equal volume of 1 N H2SO4 after color development. The absorbance readings at 450 nm were obtained using multi‐mode plate reader Cytation 3 (BioTek Instruments, Inc., Winooski, VT, USA). The inhibitor was added to the reaction mixture and subjected to the ELISA procedure as described. Wells containing reaction mixture with vehicle control were used as positive control, while wells without protein were taken as background control. Each concentration was tested in duplicate.
Data analysis
Standard curves for SAH and m7GTP were obtained by plotting the corrected peak areas of SAH and m7GTP against their concentrations. Peak integration and data acquisition were done using the Agilent CE Open Lab analysis software associated with the instrument. The corrected peak area of SAH was taken by subtracting the peak area obtained in the blank assay, which contains all the components of reaction mixture except the enzyme. The progress of the reaction was quantified as the increase in the area of the peak corresponding to SAH. Internal standards were used to compensate for the variability arising due to differences in injection volume. It also takes care of minor fluctuations in migration time between different CE runs. Internal standard improves the precision in quantification of the analyte and acts as a standard for corrected migration time. Caffeine was used as an internal standard to assess the product formation by normalizing the value of peak area of SAH against the peak area of caffeine, while UMP was used to normalize the m7GMP peak area.
Data values were normalized with peak area of the internal standard for each run. The average of two duplicate readings and standard deviations was calculated using Microsoft Excel. Percentage inhibition was calculated using the following equation:
where x is the corrected peak area of the product in the presence of inhibitor, while blank is the peak area when only the substrate is present in the reaction mixture. IC50 values of sinefungin and ATA were calculated using graphpad prism ® software (GraphPad, San Diego, CA, USA). Nonlinear regression was used for curve fitting and equation for the sigmoidal dose response (variable slope) was used to interpolate values for determining IC50 values.
Cell viability assay
Cytotoxicity of 5‐IT was determined in Vero cells prior to the evaluation of its antiviral efficacy. Standard MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide) assay was used to assess the cell viability profile of 5‐IT against Vero cells. Briefly, Vero cells were seeded in a 96‐well plate and incubated overnight for attachment. The next day, cells were treated with different concentrations of 5‐IT along with 0.1% ethanol as the positive control. Cells were incubated for 24 h prior to the addition of MTT. Plates were then incubated for 4 h at 37 °C followed by the addition of DMSO to dissolve the resulting formazan crystals. Finally, absorbance was measured at 570 nm using Cytation 3 multi‐mode plate reader (BioTek Instruments, Inc.). Each concentration was tested in triplicate.
Viral‐yield reduction assay
To evaluate the antiviral efficacy of the compound in vitro, plaque‐reduction assay was performed to quantify the change in viral titer in the presence of compound. Vero cells were seeded in a 24‐well plate and incubated overnight for attachment. When cells were 80% confluent, they were infected with CHIKV at MOI = 1 along with varying concentrations of the compound in the DMEM medium with 2% FBS. After 24 h, supernatants of compound‐treated/vehicle‐treated cells were harvested and stored at −80 °C. Viral‐induced cytopathic effects (CPE) were observed with light microscope (Carl Zeiss, Oberkochen, Germany) 48 hpi (hours postinfection). For time‐of‐addition studies, cells were pretreated 2 h before infection with 0.75 µm compound. Cell monolayer was washed with PBS, and viral infection was done at an MOI of 1. For posttreatment and simultaneous treatment of the cells, 5‐IT was added to the media at 0, 1, 2, 3, 4, and 6 h postinfection. Cell culture supernatant was harvested at 24 h after infection for the determination of viral titer.
Titer determination was performed with conventional plaque‐forming assay 35, 36. Briefly, 10‐fold dilutions of different viral stocks were prepared in DMEM medium with 2% FBS and were used to infect cell monolayer for 1.5 h at 37 °C. Infected cells were washed, overlaid with 1% carboxymethyl cellulose (CMC; Sigma) in minimum essential medium (MEM) with 2% FBS, and incubated for 48 h. Overlay media was removed, and cells were fixed and stained with 10% formaldehyde‐1% crystal violet solution for counting of plaques. Virus titer was indicated as plaque‐forming units per milliliter (PFU per mL). Each concentration was tested in triplicate. Concentration at which compound is able to reduce viral titer by 50% compared to vehicle control (EC50) was calculated with GraphPad Prism (GraphPad Software).
Immunofluorescence assay
CHIKV‐infected monolayer of Vero cells was incubated in the presence of different concentrations of 5‐IT for 36 h prior to being processed for indirect immunofluorescence assay. Monolayers of cells were fixed with acetone–methanol (1 : 1) for 15 min followed by washing with DPBS. Cells were subjected to 0.5% Triton X‐100 for 10 min to facilitate permeabilization and washed with DPBS. Cells were then incubated for 1 h with 1 : 250 mouse anti‐alphavirus IgG2a monoclonal antibody that recognizes the alphavirus group surface antigen (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and were subsequently washed 3 times with DPBS before incubation for another 1 h in 1 : 500 goat anti‐mouse secondary antibody conjugated with fluorescein isothiocyanate (FITC; Sigma). Cell nuclei were counterstained with 4',6‐diamidino‐2‐phenylindole (DAPI). Image acquisition was done on EVOS FL imaging system (Thermo Fisher Scientific) using both DAPI and GFP channels. Images were processed by evos fl software (Waltham, MA, USA).
Quantitative reverse transcription polymerase chain reaction (qRT‐PCR)
To quantify the intracellular viral RNA, cells were treated with the compound as described in the above‐mentioned section. Cells were lysed and RNA was extracted using Trizol as described in the manufacturer’s instructions. Subsequently, cDNA was generated using M‐MLV Reverse Transcriptase (Promega), random hexamers, and 1 μg of RNA as recommended by the manufacturer. Reverse‐transcribed DNA was used to amplify actin and viral E1 genes employing KAPA SYBR fast universal qPCR kit on Quantstudio real‐time PCR system (Applied Biosystems, Carlsbad, CA, USA) following manufacturer’s protocol. The primer sequences used were as follows: E1 forward: 5′‐AAGTACACTGTGCAGCTGAGT‐3′; E1 reverse: 5′‐GCATAGCACCACGATTAGAATC‐3'; actin forward: 5′‐ATTGCCGACAGGATGCAGAA‐3′ and actin reverse: 5′‐GCTGATCCACATCTGCTGGAA‐3'; (IDT, USA). Melting curve analysis was performed to ascertain the specificity of the product. ΔΔCt method was used for the relative quantification of actin and E1, and the experiment was undertaken in triplicate.
Results
nsP1 purification
VEEV nsP1 gene was cloned in E. coli. with a fused hexa‐histidine tag at the N terminus, and the construct was confirmed by Sanger sequencing. The recombinant protein was purified to homogeneity using Ni‐NTA affinity chromatography. H37A and D63A, the two mutants of VEEV nsP1, were generated by site‐directed mutagenesis, and recombinant proteins were purified using the same protocol for enzymatic characterization (Fig. 1A). Purified protein samples were analyzed on 12% SDS/PAGE gel for homogeneity (Fig. 1B and 1C). The identity of purified VEEV nsP1 wt protein was further confirmed by MALDI‐TOF after trypsin digestion (data not shown). A C‐terminal truncated form of CHIKV nsP1 (residues 1‐509), which has been recently characterized by our laboratory, was also purified 16. The MTase enzymatic activity of purified nsP1 proteins from VEEV and CHIKV was characterized using the developed nonradioactive CE‐based method.
Figure 1.
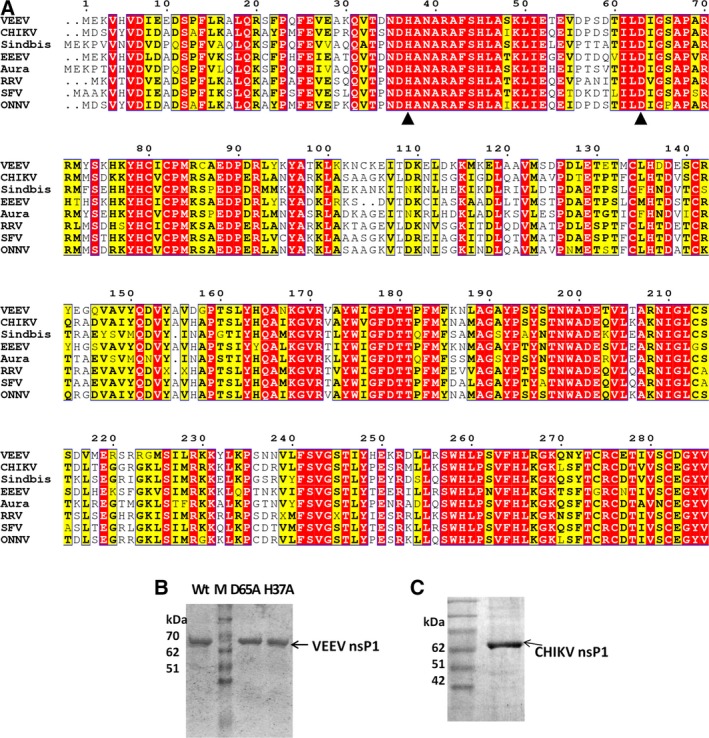
Alphaviral nsP1 sequence comparison and purified recombinant nsP1. (A) Sequence alignment of nsP1 from different alphaviruses was done using Clustal Omega 37. Graphical output was generated using ESPript 38. The conserved region from 29 to 259 amino acids (amino acid residues in VEEV nsP1) referred to as the ‘core’ region based on sequence homology is considered as the putative MTase‐GTase domain 39, 40. The region spanning ~ 200 amino acids downstream of this core region is also essential for nsP1 MTase and GTase activities 10, 12, 41. Invariable residues are shown in red background, while similar residues are in yellow background. Asp‐63 and His‐37 of VEEV nsP1, vital for MTase and GT activities, respectively, were mutated in this study and are denoted with ▲ under them. RRV, Ross River virus; SFV, Semliki Forest virus; ONNV, O’nyong‐nyong virus. (B) and (C) SDS/PAGE analysis of purified recombinant VEEV and CHIKV nsP1.
Optimization of CE running conditions
Running buffer or background electrolyte (BGE) was optimized to achieve good separation of analytes for performing CE‐based nsP1 MTase assay. The main purpose was to resolve the SAM and SAH peaks. SAM is a substrate, and SAH is one of the products in the MTase reaction of nsP1. In the initial trials to resolve analytes, 50 mm phosphate buffer, pH 2.5, 200 mm glycylglycine buffer, pH 8.4, 20 mm Tris buffer, pH 7.0, and 150 mm phosphate buffer, pH 1.5, were tried out before using 50 mm borate buffer, pH 9.3, as the BGE. CE runs with buffers other than borate suffered from either poor baseline separation, peak broadening, or current leakage during each run. Complete separation of SAM and SAH was achieved with the use of borate buffer. Additionally, the two diastereomeric forms of SAM (S,S and R,S) were distinguished from each other during CE analysis (Fig. 2). Furthermore, injection parameters, namely injection mode (electrokinetic or hydrodynamic injection), pressure, time, and running voltage, were optimized as hydrodynamic pressure injection with 50 mbar pressure for 10 s and 30 kV running voltage (normal polarity).
Figure 2.
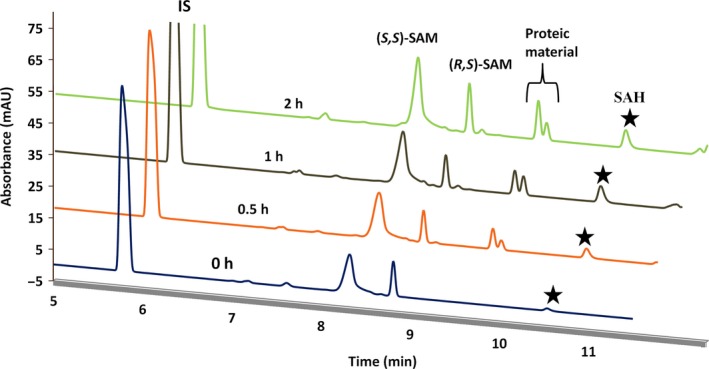
Typical electropherogram of nsP1 MTase reaction products. Overlay of CE‐electropherograms obtained for MTase enzymatic activity of VEEV nsP1 at indicated time points (0, 0.5, 1, and 2 h). MTase reaction was performed with reaction conditions as discussed in the methodology section. The separation was performed in borate buffer, pH 9.3, with 10% acetonitrile; pressure injection with 50 mbar for 10 s; applied voltage: 30 kV; extended light path fused‐silica capillary, 64.5 cm length (56 cm to the detector) and 50 µm internal diameter. Black stars indicate the SAH peak. Absorbance was measured at 260 nm. IS, internal standard (caffeine).
CE run using borate buffer alone was able to achieve complete baseline separation of SAM and SAH but suffered from poor migration time reproducibility. Addition of an organic modifier (acetonitrile) as an additive to the BGE enhanced both the resolution and migration time reproducibility of the peaks. Methanol was also tested as an organic modifier but could not improve the reproducibility of migration time. The percentage of acetonitrile in the separation buffer was optimized to 10% to strike a balance between migration time and resolution. The other substrate, GTP, could not be detected within 11 min of CE run time under the given CE running conditions (Fig. 2).
When reaction buffer was injected without deproteinization of the reaction mixture, protein components adhered to the capillary wall affecting migration time in the subsequent runs. Using trichloroacetic acid for deproteinization of the reaction mixture resulted in peak deformation essentially due to a change in matrix component because of low pH. Therefore, acetonitrile (2 : 1, v/v) was used to precipitate protein before injecting it for analysis by CE. Acetonitrile in the sample buffer gives rise to transient pseudo‐isotachophoresis where the salt acts as a leading ion, and acetonitrile functions as a terminating ion to provide the high field strength for sample concentration and band sharpening improving the overall separation efficiency 42, 43. Under these analysis conditions, we could detect SAH in concentrations as low as 0.5 µm using extended light path fused‐silica capillary.
We also optimized the BGE and separation parameters for CE analysis of products of MTase reaction of H37A VEEV nsP1 mutant using PVA‐coated capillary. We evaluated the efficiency of various concentrations of MES buffer, pH 6.4, for separating m7GTP from other reaction components in the MTase assay reaction mixture. m7GTP could be identified in the reaction mixture using 75 mm MES buffer as BGE and using the injection and running parameters mentioned in the methodology section. 75 mm MES buffer offered baseline separation of the product, m7GTP from proteic material, and remaining GTP or GDP present in the reaction mixture.
The two CE methods were validated for SAH and m7GTP detection under the optimized CE analysis conditions with respect to limit of quantitation, RSD of migration time, and linearity (Table 1). Calibration curves of the analytes (SAH and m7GTP) were generated using different concentrations of the analytes. With the use of internal standards, linearity of the calibration curve for corrected migration time (R2) of SAH was found to be 0.9998, while for m7GTP detection using PVA‐coated capillary, the R2 for corrected migration time was calculated to be 0.9865.
Table 1.
Validation parameters for the two CE methods: limit of detection, limit of quantitation, linearity of calibration curve, and reproducibility of migration time.
| Analyte | SAHa | m7GTPb |
|---|---|---|
| Limit of detection (LOD) | 0.5 µm | 5 µm |
| Limit of quantification (LOQ) | 1.5 µm | 15 µm |
| Linearity of calibration curve; R2 | 0.9998 | 0.9865 |
| Mean value of migration time (min) SD (n = 12) | 10.04 0.06 | 10.02 0.06 |
| RSD of migration time (%) | 0.61 | 0.54 |
| Total separation time | 11 min | 12 min |
RSD, Relative standard deviation; SD, Standard deviation.
Using extended path‐length fused‐silica capillary.
Using PVA coated capillary.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Optimization of reaction conditions for MTase activity
For preliminary characterization of the MTase reaction using the CE‐based method, VEEV nsP1 (full‐length) and C‐terminal truncated CHIKV nsP1 were incubated at different concentrations with the reaction mixture for different time periods. Enzyme activity for VEEV nsP1 (20 µm) in 30 min and CHIKV nsP1 (10 µm) could be detected in 45 min using 300 µm SAM. Methyl acceptor GTP was included in the reaction at a saturated concentration (4 mm). Lower concentrations of enzymes/substrates did not yield detectable product within the assay time. This concentration of enzyme and substrates was used for all subsequent experiments. Next, the MTase activity of VEEV nsP1 and CHIKV nsP1 was assayed for determining the optimal reaction parameters viz. pH, magnesium ion concentration, temperature, and salt (KCl) concentration.
The analysis for the temperature dependence of alphavirus nsP1 MTase activity demonstrated a bell‐shaped profile for the temperature dependence of the activity with maximum activity at 30 °C (Fig. 3A). Next, for determining the pH maximal of the MTase activity of VEEV nsP1 and CHIKV nsP1, activity was performed at pH ranging from 6.5 to 8.5. Both the enzymes showed a pH maximal corresponding to physiological pH 7 (Fig. 3B). Thus, the MTase reaction is favoured at physiological conditions that mimic the cellular environment inside the host cell where viral RNA capping takes place.
Figure 3.
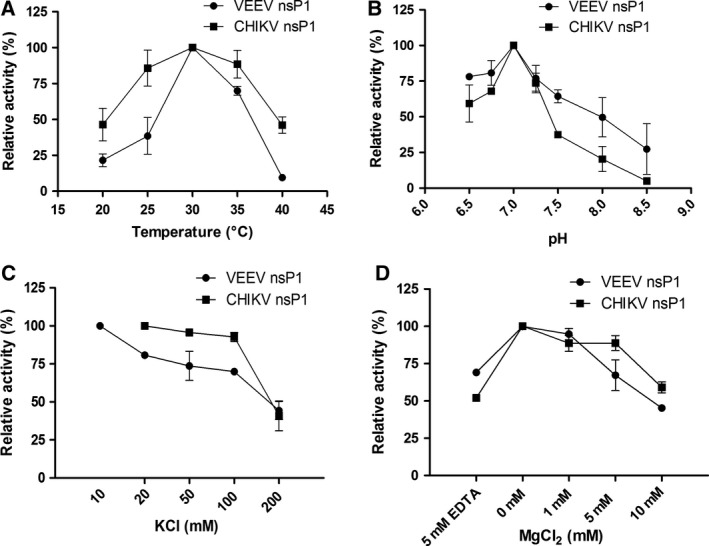
Determination of optimal MTase reaction conditions for VEEV (●) and CHIKV nsP1 (■). The optimal MTase reaction conditions were determined by individually varying (A) temperature, (B) pH, (C) KCl concentration, and (D) magnesium ion concentration, while the other three parameters were kept at optimal levels. Activity was calculated as an increase in the corrected peak area of SAH in the CE electropherogram. For each parameter, the optimal activity observed was taken as 100% and the relative activity was calculated at other conditions. Data points represent mean values and error bars are standard deviations from duplicate experiments.
For CHIKV nsP1, increasing the concentration of KCl in the final assay buffer to 50 and 100 mm had a negligible effect on the MTase activity. Above 100 mm KCl there was a steep reduction in enzymatic activity for both VEEV and CHIKV nsP1 (Fig. 3C). The MTase activity of VEEV and CHIKV nsP1 as a function of magnesium concentration was evaluated using the CE method. A gradual decline in the enzymatic activity with an increase in Mg+2 ion concentration was observed (Fig. 3D). This observation is in agreement with the earlier findings reported for alphaviral nsP1 that the enzyme does not require magnesium ions for its MTase activity 34, 44, 45. Taken together, under the conditions employed in the enzymatic assay, VEEV and CHIKV nsP1 exhibited the highest enzymatic activity at pH 7.0, at KCl concentration 10/20 mm, and at 30 °C in the absence of magnesium ion.
Time‐course MTase reaction
A time‐course experiment where GTP was taken as the methyl acceptor in the presence and absence of magnesium ion was performed with optimal buffer conditions. Intriguingly, it was observed that in the absence of metal ion the MTase reaction with GTP proceeds linearly for 2 h as the SAH formation showed a linear range. In contrast, when magnesium ion, which acts as a cofactor in the subsequent GT reaction, was present, the progress curve became nonlinear after 1 h (Fig. 4A). It is speculated that the subsequent GT reaction that guanylate nsP1 might affect the activity of the enzyme by product inhibition. Hence, to decouple the MTase and GT reactions catalyzed by the same enzyme, a nonhydrolyzable analog of GTP with a β‐γ nonhydrolyzable bond, GIDP was used (Fig. 4B and 4C). The use of GIDP excludes the possibility of the guanylation of nsP1 that can interfere with the linearity of SAH formation over the reaction time. Thus, for subsequent assay development and testing of inhibitors GIDP was used instead of GTP as the methyl acceptor and the reaction time was maintained at 2 h. A typical electropherogram of the MTase nsP1 reaction assay mixture at different time points is shown in Figure 2. nsP1 mutant D63A (MTase‐deficient) of VEEV nsP1 was taken as a negative control and no increase in peak area corresponding to SAH with time in the MTase reaction mixture was observed.
Figure 4.
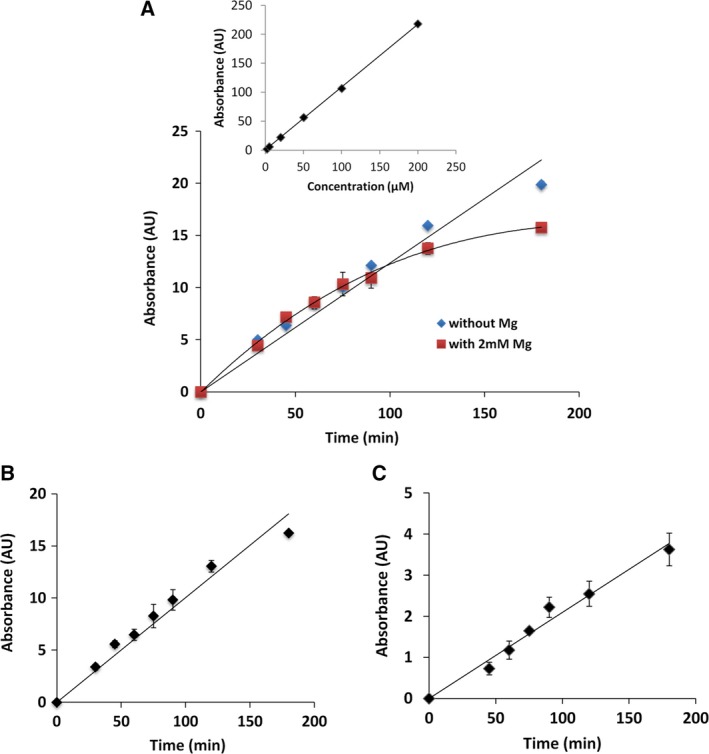
Time‐course of the MTase reaction of VEEV and CHIKV nsP1. (A) A 6‐point calibration curve was generated to demonstrate the linearity of SAH detection using optimized CE method. Corrected peak area plotted against concentrations of SAH show a linear curve in the concentration range 2–200 µm. Effect of magnesium in the reaction buffer on the linearity of product formation was assessed. Reaction progression was measured by the increase in corrected peak area of SAH with caffeine as the internal standard. MTase reaction was carried out with SAM as the methyl donor and GIDP or GTP as the methyl group acceptor and incubated at 30 °C for 3 h. Reaction samples were collected at different time points, and reactions were stopped with the addition of acetonitrile. (B) and (C), SAH formation showed a linear range for 2 h under given reaction conditions with both VEEV nsP1 and CHIKV nsP1 when GIDP was used as a methyl acceptor. Data points represent mean values, and error bars are standard deviations from duplicate experiments. AU, arbitrary units.
Identification of m7GTP using H37A VEEV nsP1 mutant
H37A mutant of nsP1 has been shown to be deficient in the formation of nsP1‐m7GMP intermediate covalent complex by nsP1 GT enzyme reaction 34. To further characterize the molecular mechanism of RNA capping reaction by alphaviral nsP1, the formation of m7GTP by mutant protein was analyzed. To achieve this, H37A mutation was engineered in VEEV nsP1 and purified H37A mutant was used for enzyme characterization. It was hypothesized that the m7GTP, which is otherwise formed transiently during the nsP1 capping reaction and covalently attaches itself to the alphavirus nsP1 protein as m7GMP moiety, could be observed in the reaction mixture when the MTase reaction is carried out with H37A mutant. The formation of m7GTP was analyzed with the help of a PVA‐coated capillary using reverse voltage polarity at the capillary ends (Fig. 5A). Reaction samples were also spiked with m7GTP for positive identification of the product. UMP was taken as the internal standard during m7GTP analysis. Time‐course reaction with H37A mutant in the presence of SAM and GTP showed a linear formation of m7GTP over a 150‐min period (Fig. 5B). The peak corresponding to m7GTP was absent when 10 µm sinefungin or 100 µm ATA was added to the MTase reaction mixture further confirming that the H37A mutant of nsP1 indeed produced m7GTP.
Figure 5.
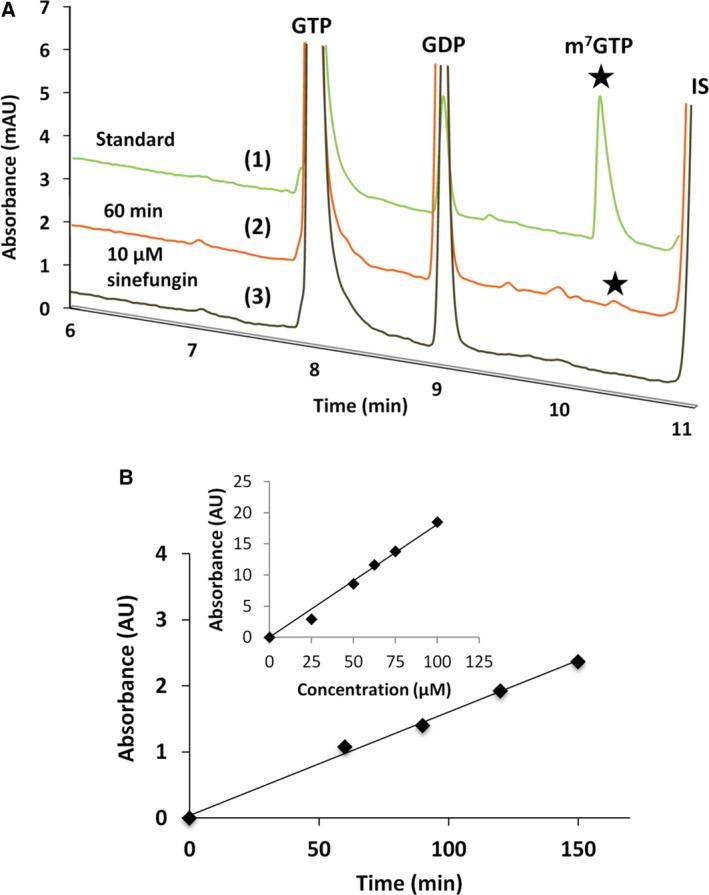
Detection of m7GTP by precapillary CE method using PVA capillary. (A) Overlay of CE‐electropherograms illustrating m7GTP formation by MTase activity of H37A mutant of VEEV nsP1 detected with PVA capillary. Black stars indicate the m7GTP peak. Electropherograms showing the separation of (1) authentic standards of GTP (with contaminating GDP) and m7GTP to determine standard migration time of each analyte, (2) MTase reaction products after methylation reaction was carried out for 60 min, (3) methylation reaction product with sinefungin in the reaction mixture under conditions and substrate concentrations mentioned in the methodology section. Separation of the analytes was performed using pressure injection with 50 mbar pressure for 10 s at −30 kV voltage using 75 mm MES buffer, pH 6.4, in a PVA capillary, 64.5 cm length (56 cm to the detector) and 50 µm internal diameter. (B) Calibration curve of m7GTP detection using optimized CE method is linear over the concentration range 25–100 µm. Linearity of m7GTP formation as a function of time as analyzed by CE with UMP as the internal standard. Absorption was measured at 254 nm. IS, internal standard (UMP).
Inhibition of VEEV and CHIKV nsP1 MTase activity
To validate the established assay for SAH detection, inhibition efficiency of two known inhibitors of nsP1, sinefungin and ATA, was evaluated against nsP1 (from VEEV and CHIKV), and their respective IC50 values for both the enzymes were calculated (Fig. 6). Sinefungin and ATA are known inhibitors of VEEV nsP1. IC50 values of sinefungin and ATA for VEEV nsP1 and CHIKV nsP1 are shown in Table 2. The IC50 values of sinefungin and ATA calculated by the CE‐based nsP1 MTase assay are very similar to the ones reported earlier 34. The observed slight differences in the IC50 values of sinefungin and ATA for VEEV nsP1 can be attributed to the differential folding of the protein owing to the different purification protocols or the different concentrations of substrates used in CE‐based assay. This validates the utility of the CE‐based nsP1 MTase assay for screening inhibitors against alphaviral nsP1 MTase. The inhibition efficiency of these two inhibitors has not been evaluated for MTase activity of CHIKV nsP1 previously.
Figure 6.
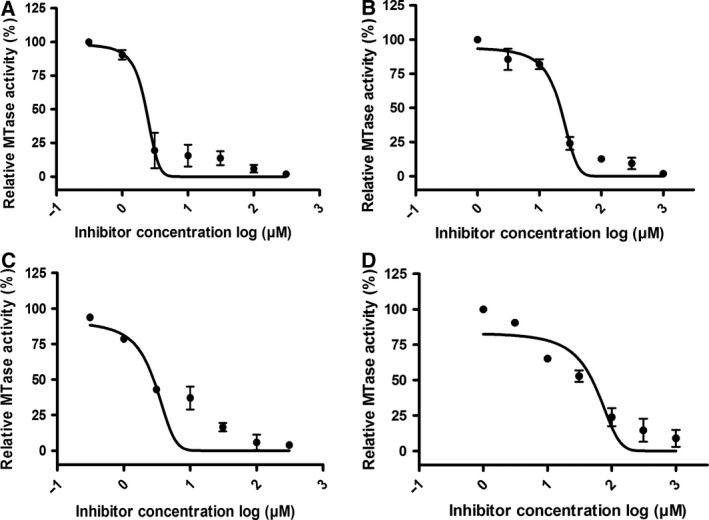
Dose‐dependent inhibition obtained for VEEV nsP1 (A, B) and CHIKV nsP1 (C, D) by inhibitors: sinefungin (A, C) and ATA (B, D). MTase reaction was carried out by incubating the enzyme, SAM, and GIDP for 2 h with indicated concentrations of the inhibitors at reaction conditions discussed in the methodology section. The relative MTase activity is obtained by analyzing the reaction mixture with CE and quantifying the corrected peak area of SAH formed during the reaction relative to the vehicle control. IC50 values of the inhibitors were calculated by curve fitting in graph pad prism and are illustrated in Table 2. Data points represent mean values and error bars are standard deviation generated from duplicate experiments.
Table 2.
Calculated IC50 values of sinefungin and ATA for VEEV nsP1 and CHIKV nsP1.
| Inhibitors | IC50 (VEEV nsP1) | IC50 (CHIKV nsP1) | IC50 (VEEV nsP1)a |
|---|---|---|---|
| Sinefungin | 2.31 ± 0.32 µm | 2.94 ± 0.74 µm | 1.4 ± 0.58 µm |
| ATA | 21.86 ± 2.56 µm | 49.82 ± 14.6 µm | 56.9 ± 1.59 µm |
Values reported by Li et al. 34.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Using the developed CE‐based nsP1 MTase assay, a few adenosine analogs were evaluated for their potency to inhibit the MTase enzyme activity of VEEV nsP1 and CHIKV nsP1. A fixed (50 µm) concentration of each inhibitor candidate, 2‐Chloroadenosine, 3‐Deazaadenosine, 3‐Deazaneplanocin A, and 5‐IT, was used to assess their nsP1 inhibition efficiency. Among the compounds tested against nsP1, only 5‐IT showed effective inhibition of the nsP1 enzyme (Fig. 7A). An indirect ELISA assay was also performed which measures the m7GMP‐nsP1 adduct formation. It detects m7GMP covalently bound to the nsP1 protein formed as a result of the GT activity of nsP1 using anti‐m7‐GMP antibodies 16. This assay also showed a concentration‐dependent reduction in CHIKV nsP1 GT activity in the presence of the inhibitor with an IC50 value of 11.25 µm confirming CHIKV nsP1 GT activity inhibition by 5‐IT (Fig. 7B).
Figure 7.
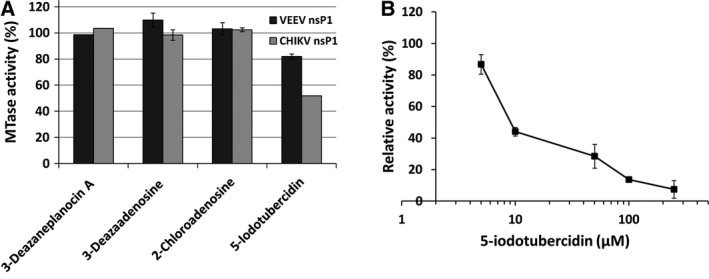
Inhibitory effect of adenosine analogs on the MTase activity of nsP1. (A) Inhibitors were tested at 50 µm concentration in the MTase reaction mixture of VEEV and CHIKV nsP1, and reaction was carried out as discussed in the methodology section. Relative MTase activity for VEEV nsP1 (black bars) and CHIKV nsP1 (gray bars) was calculated using vehicle control as 100%. (B) CHIKV nsP1 activity inhibition in the presence of 5‐IT was determined by an ELISA assay in terms of the amount m7GMP‐nsP1 adduct formation by anti‐m7‐GMP antibodies, and the absorbance was measured at 450 nm. Data points represent mean values of relative activity, and error bars are standard deviations from duplicate experiments.
Anti‐CHIKV activity of 5‐IT
The effect of 5‐IT on cell viability was examined on Vero cells. 5‐IT exerted a weak cytotoxic effect on cells at concentrations above 2 µm at 24 h (Fig. 8A). To remove the possibility of 5‐IT mediated cytotoxicity, concentrations below 1.5 µm were used for investigating the antiviral activity of 5‐IT. To test the in vitro antiviral efficacy of 5‐IT, CHIKV‐infected cells were grown in the presence of varying concentrations of 5‐IT for 24 h and viral titers were quantified using plaque‐forming assay. 5‐IT displayed significant inhibition of infectious progeny virus with an EC50 of 0.409 µm (Fig. 8B). 5‐IT exhibited compound‐mediated inhibition of CHIKV‐induced CPE, which were monitored 48 h after infection (Fig. S1). Anti‐CHIKV activity of 5‐IT was further confirmed by CHIKV immunostaining assay. Anti‐alphavirus antibody was used to evaluate CHIKV reduction in cell culture in the presence of 5‐IT. Fluorescence was barely detectable in infected cells treated with 1 µm compound as compared to untreated cells confirming the reduction of CHIKV antigen expression in the treated cells (Fig. 8C).
Figure 8.
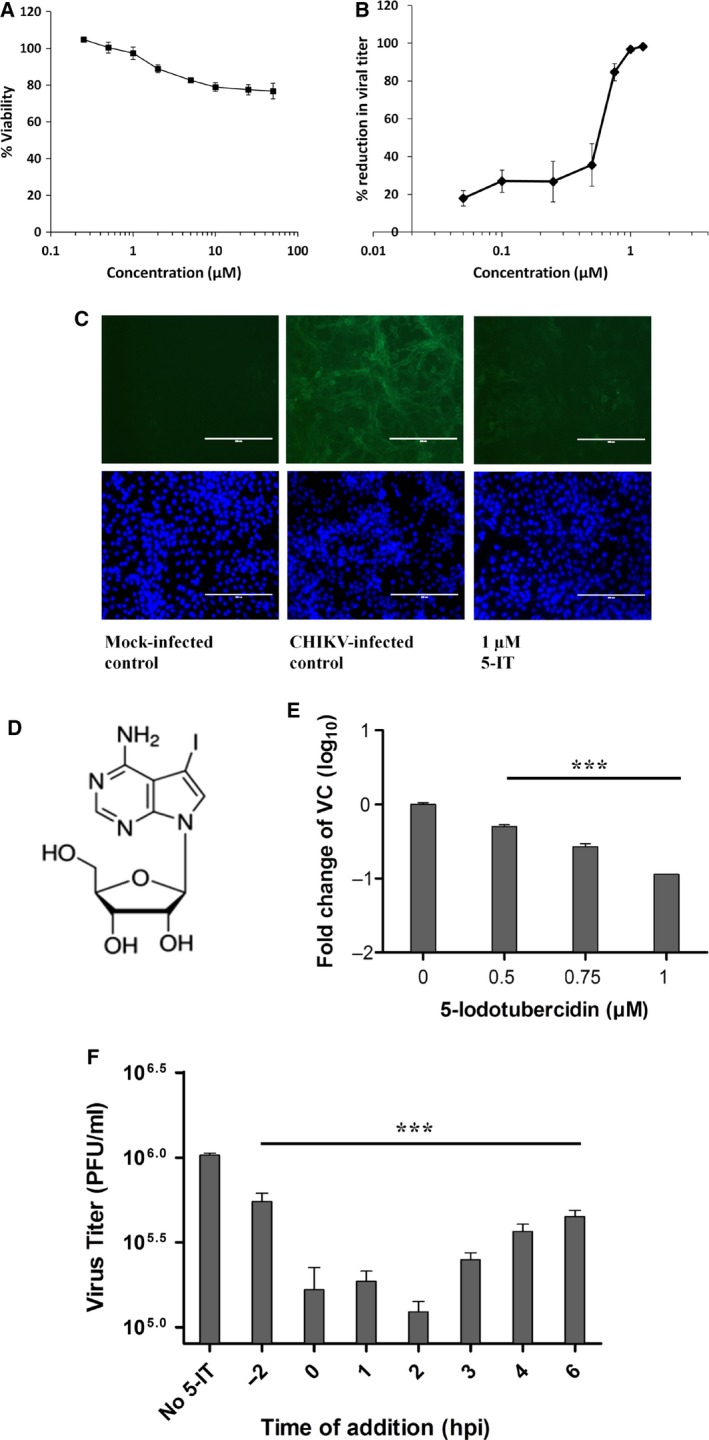
Anti‐CHIKV activity of 5‐IT. (A) Monolayers of Vero cells were incubated with indicated concentrations of 5‐IT for 24 h to evaluate cytotoxicity. MTT reagent was added, and readings were taken at 570 nm. (B) Dose–response curve of 5‐IT against CHIKV. Subconfluent Vero cell monolayer was infected with CHIKV (strain no. 119067) at MOI of 1 along with varying concentrations of 5‐IT. 24 hpi culture supernatant was collected and subjected to viral titer determination using plaque assay. Percent reduction in viral titer was calculated with titer in vehicle‐treated cells as 100%. (C) Immunofluorescence assay for the assessment of CHIKV inhibition by 5‐IT. Cells were processed at 36 hpi, incubated sequentially with primary anti‐alphaviral antibody and FITC‐conjugated secondary antibody, and then counterstained with DAPI. This is a representative micrograph of multiple assessments. Scale bar, 200 µm. (D) Chemical structure of 5‐IT. (E) Reduction in intracellular viral RNA as assessed by qRT‐PCR. Fold reduction in viral RNA in treated cells relative to the control is plotted. Actin: endogenous control; CHIKV E1: target gene. (F) CHIKV viral titer (PFU/ml) at 24 hpi when 0.75 µm 5‐IT was added at indicated time points. Cells were infected with CHIKV at an MOI of 1 and viral titer was measured with standard plaques assay. Data points represent mean values and error bars are standard deviations from triplicate experiments. Significance of difference between viral RNA or PFU per mL in treated cells and vehicle control was measured by one‐way ANOVA test and Dunnett’s post‐test. ***, P < 0.001.
The ability of 5‐IT to inhibit viral RNA inside the infected cells was assessed by qRT‐PCR. Treatment of infected cells with the compound led to a dose‐dependent reduction in the intracellular viral RNA relative to mock‐treated cells. Viral RNA was gradually reduced in treated cells above 0.5 µm concentration of the compound, and an approximately 9‐fold reduction was observed at 1 µm (Fig. 8D and 8E). This complements the data from plaque‐reduction assay. Taken together, the results of plaque‐reduction assay, qRT‐PCR, and immunofluorescence indicate that 5‐IT inhibits CHIKV replication in cell culture.
To identify the stage of viral replication affected by 5‐IT, it was added to the infected cell monolayer at different time points (Fig. 8F). Pretreatment of the cells with 5‐IT showed minimal inhibitory effect against CHIKV infection. Addition of the compound at 4 hpi or 6 hpi did not show a marked reduction of CHIKV viral titer. 5‐IT was most efficient when added 0‐2 hpi, suggesting that it inhibits an early, postentry step of the viral replication cycle. The time dependence of the antiviral efficiency of the compound is in agreement with its anti‐nsP1 activity 1.
Discussion
Alphaviruses use an unconventional mechanism of RNA capping, distinct from their host counterpart where the transfer of methyl group from SAM to GTP occurs before the transguanylation step (transfer of GTP to RNA). Alphavirus capping enzyme, nsP1 adds a type 0 cap structure at 5′‐end of the nascent RNA 8. As RNA capping is a critical point in the viral life cycle, nsP1 is a promising candidate for the development of therapeutic intervention against alphaviruses. MTase activity of recombinant nsP1 has been demonstrated with nsP1 from different alphaviruses including SINV and VEEV 34, 45. Previous efforts to detect MTase activity of nsP1 were based on the detection of radioactive product which albeit are sensitive and more reliable than their alternatives, are time‐consuming and cumbersome. Radiolabels also cause environmental, health, cost, waste, and regulatory issues. Another drawback of using this technique is that the SAH produced during the reaction inhibits the activity of most SAM‐dependent methyltransferases (MTases).
Recently, as an alternative to the radioactive assays, a number of colorimetric and fluorometric assays have been developed and commercialized for the characterization of MTase enzymes. These are coupled assays wherein the SAH generated in the MTase reaction is subsequently acted upon by other enzymes included in the assay mixture. The final products of these coupled assays are then quantified by colorimetric or fluorometric detection. The coupled‐assay approach is indirect and circumvents the problem of SAH accumulation, which can act as the potent inhibitor of the MTases 46, 47, 48, 49. These enzyme assays, however, when used for inhibitor screening suffer from artefacts arising mainly because of the promiscuity of several compounds that interfere with the enzyme coupling system by off‐target inhibition 50. Inhibitors identified through such a screening strategy require a secondary assay for hit validation before being considered for follow‐up studies.
The CE method developed in this study holds several advantages such as simple sample preparation, low sample requirement, and low cost. It also eliminates the need for expensive and potentially hazardous radioactive materials. Moreover, the assay relies on the direct detection of SAH generated in the MTase reaction, thereby minimizing any possible interference from inhibitor compounds. This reported assay allows the rapid detection of MTase activity of nsP1 capping enzyme. Whole CE analysis takes 13 min including the buffer run between two runs and multiple samples can be analyzed at once with an automated injection procedure without any manual intervention in between consecutive runs. This assay can be used for batch screening of inhibitors and can be modified and adapted for fully automated in‐line CE assays. Separation of m7GTP from the reaction mixture of H37A mutant using PVA capillary has been demonstrated in this study. Alanine substitution at this position disrupts the formation of the phosphoramide bond between nsP1 and m7GMP, and this supports the hypothesis that the alphaviral capping enzyme methylates GTP before it is hydrolyzed to GMP and transferred to the 5’‐end of RNA unlike host capping enzymes 51.
nsP1, being an attractive drug target, can be effectively perturbed to feed the drug discovery pipeline for anti‐alphaviral therapeutics. Sequence alignment reveals that the putative MTase domain or ‘core region’ of nsP1 [spanning from 28–259 amino acids in VEEV nsP1; 39, 40] is highly conserved within the alphavirus superfamily. The capping enzymes of alphaviruses share a high level of sequence homology, and thus, the development of nsP1 inhibitors can warrant pan‐alphavirus therapeutics. Moreover, viruses from alphavirus‐like superfamily, which includes several plant and animal viruses such as bamboo mosaic virus, brome mosaic virus, tobacco mosaic virus, alfalfa mosaic virus, and hepatitis E virus, carry out capping of viral RNA using a mechanism similar to nsP1 52, 53, 54, 55, 56. This nonradioactive assay provides a robust and versatile platform for screening and identification of novel inhibitors against these viral capping enzymes and other similar SAM‐dependent MTases. Developed CE‐based assay for MTase activity of nsP1 is based on the general detection method of SAH formed during the MTase reaction of alphavirus nsP1. Under our assay conditions, SAH accumulation in the MTase reaction was linear for at least 2 h. This is in agreement with the reports on VEEV and for SINV nsP1 34, 45. The same detection range was used for the characterization of inhibitors of nsP1. Out of the tested compounds, only 5‐IT showed efficient inhibition of both VEEV and CHIKV nsP1 and was further taken up for cell culture‐based viral inhibition studies.
5‐IT is a derivative of tubercidin (7‐deaza‐adenosine) and a known adenosine kinase inhibitor. It has been shown to possess antitumor activity 57, 58. The antiseizure activity of 5‐IT has been demonstrated in vivo 59. This compound is also being studied for its potential as a therapeutic agent to treat epilepsy using mice model 60, 61. The role of cellular kinases in alphavirus replication remains poorly understood. A recent study that profiled the cellular kinome following CHIKV infection found a number of kinases to be undergoing changes in terms of abundance or activity 62. To our knowledge, adenosine kinase was not implicated in CHIKV infection in this or any other study. Thus, host adenosine kinase seems to be not involved in alphavirus infection. Time‐of‐drug addition studies indicate that 5‐IT affects an early, postentry replication step of the viral life cycle. Though extensive studies are required to identify other possible cellular or viral targets of 5‐IT, at this stage it is reasonable to believe that antiviral effects of 5‐IT are most likely, at least partially, can be attributed to the inhibition of alphaviral nsP1. Further studies are also needed for screening and improving the inhibition potency of compounds similar to 5‐IT. In the absence of the structure of nsP1, it is rather difficult to establish a structure–activity relationship (SAR). Thus, at this point of time, we are not able to conjecture the interaction of different moieties of the compound with specific amino acid residues of nsP1. Chemical modification by addition or removal of chemical moiety/moieties may result in enhanced potency of 5‐IT in terms of increased efficacy and improved pharmacokinetic properties. With the availability of the structure of nsP1 from the alphavirus genus, an extensive structure–activity study of 5‐IT will be required to use it as a reference compound for rational drug designing and identification/development of more potent and specific therapeutic agents targeting nsP1 protein.
In conclusion, a simple, robust, and versatile CE‐based assay for measuring MTase activity of VEEV and CHIKV nsP1 was developed. Favorable assay parameters for MTase activity of VEEV and CHIKV nsP1 viz pH, temperature, salt concentration, and metal ion (Mg2+) concentration were optimized. For the mechanistic study of nsP1, m7GTP formation by H37A mutant of nsP1 was analyzed with the help of a PVA capillary using a separate CE method. The assay was further used to evaluate the IC50 values of known nsP1 inhibitors, sinefungin, and ATA. Evaluation of enzymatic activity in the presence of potential inhibitors led to the identification of adenosine analog 5‐IT as the inhibitor of MTase activity of the enzyme. The potent hit compound was further evaluated in a secondary orthogonal ELISA assay that quantitates m7GMP‐nsP1 adduct formed after nsP1 GT activity. 5‐IT showed a marked reduction in CHIKV replication as evident by the results of the plaque reduction assay, viral‐induced CPE reduction, qRT‐PCR, and IFA studies. The developed CE‐based assay can further accelerate the screening of inhibitors against nsP1 of alphaviruses and identification or development of new antiviral drugs against alphaviruses. With the effective inhibition of CHIKV replication in cell culture by 5‐IT, the possible new approach of targeting alphaviral nsP1 with adenosine analogs can be further investigated for identification and development of pan‐alphavirus antiviral agents.
Author contribution
RM and SM conducted the experiments and analyzed the data. RM and ST planned experiments, analyzed the data, and wrote the manuscript.
Supporting information
Fig. S1. Inhibition of CPE in CHIKV‐infected cells by 5‐IT.
Acknowledgements
This work was supported by Science and Engineering Research Board, Department of Science and Technology, Government of India (EMR/2016/004938). RM is thankful to CSIR, and SM is thankful to MHRD for financial support. Authors are indebted to Andrew S Miller at Purdue University, US, for his help in MALDI‐TOF experiments. Authors would also like to acknowledge Mr. Ravi Kumar Marikanty, Mr. Jagmohan Singh, and Mr. Nitin Vig from Agilent Technologies, India, for technical assistance in CE analysis. The funders played no role in study design, data collection and analysis, the decision to publish, or the preparation of the manuscript.
Edited by Urs Greber
References
- 1. Strauss JH and Strauss EG (1994) The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58, 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jose J, Snyder JE and Kuhn RJ (2009) A structural and functional perspective of alphavirus replication and assembly. Future Microbiol 4, 837–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paessler S and Weaver SC (2009) Vaccines for Venezuelan equine encephalitis. Vaccine 27, D80–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ehrenkranz NJ and Ventura AK (1974) Venezuelan equine encephalitis virus infection in man. Ann Rev Med 25, 9–14. [DOI] [PubMed] [Google Scholar]
- 5. Powers AM and Logue CH (2007) Changing patterns of Chikungunya virus: re‐emergence of a zoonotic arbovirus. J Gen Virol 88, 2363–2377. [DOI] [PubMed] [Google Scholar]
- 6. Filipowicz W, Furuichi Y, Sierra JM, Muthukrishnan S, Shatkin AJ and Ochoa S (1976) A protein binding the methylated 5’‐terminal sequence, m7G pppN, of eukaryotic messenger RNA. Proc Nat Acad Sci USA 73, 1559–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hyde JL and Diamond MS (2015) Innate immune restriction and antagonism of viral RNA lacking 2′‐O methylation. Virology 479–480, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahola T and Kääriäinen L (1995) Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7‐methyl‐GMP. Proc Nat Acad Sci USA 92, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vasiljieva L, Merits A, Auvinen P and Kaariainen L (2000) Identification of a novel function of the alphavirus capping apparatus‐RNA 5’triphosphatase activity of Nsp2. J Biol Chem 275, 17281–17287. [DOI] [PubMed] [Google Scholar]
- 10. Wang H‐L, O'rear J and Stollar V (1996) Mutagenesis of the Sindbis virus nsP1 protein: effects on methyltransferase activity and viral infectivity. Virology 217, 527–531. [DOI] [PubMed] [Google Scholar]
- 11. Ahola T, Lampio A, Auvinen P and Kääriäinen L (1999) Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J 18, 3164–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laakkonen P, Ahola T and Kääriäinen L (1996) The effects of palmitoylation on membrane association of Semliki forest virus RNA capping enzyme. J Biol Chem 271, 28567–28571. [DOI] [PubMed] [Google Scholar]
- 13. Spuul P, Salonen A, Merits A, Jokitalo E, Kääriäinen L and Ahola T (2007) Role of the amphipathic peptide of Semliki forest virus replicase protein nsP1 in membrane association and virus replication. J Virol 81, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bullard‐Feibelman KM, Fuller BP and Geiss BJ (2016) A sensitive and robust high‐throughput screening assay for inhibitors of the Chikungunya virus nsP1 capping enzyme. PLoS ONE 11, e0158923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delang L, Li C, Tas A, Quérat G, Albulescu IC, De Burghgraeve T, Guerrero NS, Gigante A, Piorkowski G and Decroly E (2016) The viral capping enzyme nsP1: a novel target for the inhibition of Chikungunya virus infection. Sci Rep 6, 31819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaur R, Mudgal R, Narwal M and Tomar S (2018) Development of an ELISA assay for screening inhibitors against divalent metal ion dependent alphavirus capping enzyme. Virus Res 256, 209–221. [DOI] [PubMed] [Google Scholar]
- 17. Feibelman KM, Fuller BP, Li L, LaBarbera DV and Geiss BJ (2018) Identification of small molecule inhibitors of the Chikungunya virus nsP1 RNA capping enzyme. Antiviral Res 154, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Clercq E and Li G (2016) Approved antiviral drugs over the past 50 years. Clin Microbiol Rev 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Clercq E (2011) A 40‐year journey in search of selective antiviral chemotherapy. Annu Rev Pharmacol Toxicol 51, 1–24. [DOI] [PubMed] [Google Scholar]
- 20. De Clercq E (2010) In search of a selective therapy of viral infections. Antiviral Res 85, 19–24. [DOI] [PubMed] [Google Scholar]
- 21. Brecher M, Chen H, Li Z, Banavali NK, Jones SA, Zhang J, Kramer LD and Li H (2015) Identification and characterization of novel broad‐spectrum inhibitors of the flavivirus methyltransferase. ACS Infect Dis 1, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen H, Liu L, Jones SA, Banavali N, Kass J, Li Z, Zhang J, Kramer LD, Ghosh AK and Li H (2013) Selective inhibition of the West Nile virus methyltransferase by nucleoside analogs. Antiviral Res 97, 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jain R, Butler KV, Coloma J, Jin J and Aggarwal AK (2017) Development of a S‐adenosylmethionine analog that intrudes the RNA‐cap binding site of Zika methyltransferase. Sci Rep 7, 1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vernekar SKV, Qiu L, Zhang J, Kankanala J, Li H, Geraghty RJ and Wang Z (2015) 5′‐silylated 3′‐1,2,3‐triazolyl thymidine analogues as inhibitors of West Nile Virus and Dengue Virus. J Med Chem 58, 4016–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delang L, Segura Guerrero N, Tas A, Quérat G, Pastorino B, Froeyen M, Dallmeier K, Jochmans D, Herdewijn P, Bello F et al (2014) Mutations in the Chikungunya virus non‐structural proteins cause resistance to favipiravir (T‐705), a broad‐spectrum antiviral. J Antimicrob Chemother 69, 2770–2784. [DOI] [PubMed] [Google Scholar]
- 26. Markland W, McQuaid TJ, Jain J and Kwong AD (2000) Broad‐spectrum antiviral activity of the IMP dehydrogenase inhibitor VX‐497: a comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob Agents Chemother 44, 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Julander JG, Bowen RA, Rao JR, Day C, Shafer K, Smee DF, Morrey JD and Chu CK (2008) Treatment of Venezuelan equine encephalitis virus infection with (‐)‐carbodine. Antiviral Res 80, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stuyver LJ, Whitaker T, McBrayer TR, Hernandez‐Santiago BI, Lostia S, Tharnish PM, Ramesh M, Chu CK, Jordan R, Shi J et al (2003) Ribonucleoside analogue that blocks replication of bovine viral diarrhea and hepatitis C viruses in culture. Antimicrob Agents Chemother 47, 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pyrc K, Bosch BJ, Berkhout B, Jebbink MF, Dijkman R, Rottier P and van der Hoek L (2006) Inhibition of human coronavirus NL63 infection at early stages of the replication cycle. Antimicrob Agents Chemother 50, 2000–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ehteshami M, Tao S, Zandi K, Hsiao H‐M, Jiang Y, Hammond E, Amblard F, Russell OO, Merits A and Schinazi RF (2017) Characterization of β‐d‐N4‐hydroxycytidine as a novel inhibitor of Chikungunya virus. Antimicrob Agents Chemother 61, e02395‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Urakova N, Kuznetsova V, Crossman DK, Sokratian A, Guthrie DB, Kolykhalov AA, Lockwood MA, Natchus MG, Crowley MR, Painter GR et al (2018) β‐d‐N4‐hydroxycytidine is a potent anti‐alphavirus compound that induces a high level of mutations in the viral genome. J Virol 92, e01965‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaur R, Neetu, Mudgal R, Jose J, Kumar P and Tomar S (2019) Glycan‐dependent Chikungunya viral infection divulged by antiviral activity of NAG specific chi‐like lectin. Virology 526, 91–98. [DOI] [PubMed] [Google Scholar]
- 33. Singh H, Mudgal R, Narwal M, Kaur R, Singh VA, Malik A, Chaudhary M and Tomar S (2018) Chikungunya virus inhibition by peptidomimetic inhibitors targeting virus‐specific cysteine protease. Biochimie 149, 51–61. [DOI] [PubMed] [Google Scholar]
- 34. Li C, Guillén J, Rabah N, Blanjoie A, Debart F, Vasseur J‐J, Canard B, Decroly E and Coutard B (2015) mRNA capping by Venezuelan equine encephalitis virus nsP1: functional characterization and implication for Antiviral Res. J Virol 89, 8292–8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aggarwal M, Kaur R, Saha A, Mudgal R, Yadav R, Dash PK, Parida M, Kumar P and Tomar S (2017) Evaluation of antiviral activity of piperazine against Chikungunya virus targeting hydrophobic pocket of alphavirus capsid protein. Antiviral Res 146, 102–111. [DOI] [PubMed] [Google Scholar]
- 36. Sharma R, Fatma B, Saha A, Bajpai S, Sistla S, Dash PK, Parida M, Kumar P and Tomar S (2016) Inhibition of Chikungunya virus by picolinate that targets viral capsid protein. Virology 498, 265–276. [DOI] [PubMed] [Google Scholar]
- 37. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J et al (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gouet P, Robert X and Courcelle E (2003) ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31, 3320–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ahola T and Karlin DG (2015) Sequence analysis reveals a conserved extension in the capping enzyme of the alphavirus supergroup, and a homologous domain in nodaviruses. Biol Direct 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rozanov MN, Koonin EV and Gorbalenya AE (1992) Conservation of the putative methyltransferase domain: a hallmark of the ‘Sindbis‐like’ supergroup of positive‐strand RNA viruses. J Gen Virol 73, 2129–2134. [DOI] [PubMed] [Google Scholar]
- 41. Ahola T, Laakkonen P, Vihinen H and Kääriäinen L (1997) Critical residues of Semliki Forest virus RNA capping enzyme involved in methyltransferase and guanylyltransferase‐like activities. J Virol 71, 392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Friedberg MA, Hinsdale M and Shihabi ZK (1997) Effect of pH and ions in the sample on stacking in capillary electrophoresis. J Chromatogra A 781, 35–42. [Google Scholar]
- 43. Shihabi ZK (1995) Sample stacking by acetonitrile‐salt mixtures. J Capillary Electropho 2, 267–271. [PubMed] [Google Scholar]
- 44. Laakkonen P, Hyvönen M, Peränen J and Kääriäinen L (1994) Expression of Semliki Forest virus nsP1‐specific methyltransferase in insect cells and in Escherichia coli. J Virol 68, 7418–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tomar S, Narwal M, Harms E, Smith JL and Kuhn RJ (2011) Heterologous production, purification and characterization of enzymatically active Sindbis virus nonstructural protein nsP1. Protein Expr Purif 79, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dorgan KM, Wooderchak WL, Wynn DP, Karschner EL, Alfaro JF, Cui Y, Zhou ZS and Hevel JM (2006) An enzyme‐coupled continuous spectrophotometric assay for S‐adenosylmethionine‐dependent methyltransferases. Anal Biochem 350, 249–255. [DOI] [PubMed] [Google Scholar]
- 47. Duchin S, Vershinin Z, Levy D and Aharoni A (2015) A continuous kinetic assay for protein and DNA methyltransferase enzymatic activities. Epigenetics Chromatin 8, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ibáñez G, McBean JL, Astudillo YM and Luo M (2010) An enzyme‐coupled ultrasensitive luminescence assay for protein methyltransferases. Anal Biochem 401, 203–210. [DOI] [PubMed] [Google Scholar]
- 49. Schäberle TF, Siba C, Höver T and König GM (2013) An easy‐to‐perform photometric assay for methyltransferase activity measurements. Anal Biochem 432, 38–40. [DOI] [PubMed] [Google Scholar]
- 50. Drake KM, Watson VG, Kisielewski A, Glynn R and Napper AD (2014) A sensitive luminescent assay for the histone methyltransferase NSD1 and other SAM‐dependent enzymes. Assay Drug Dev Technol 12, 258–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Decroly E, Ferron F, Lescar J and Canard B (2012) Conventional and unconventional mechanisms for capping viral mRNA. Nat Rev Microbiol 10, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kong F, Sivakumaran K and Kao C (1999) The N‐terminal half of the brome mosaic virus 1a protein has RNA capping‐associated activities: specificity for GTP and S‐adenosylmethionine. Virology 259, 200–210. [DOI] [PubMed] [Google Scholar]
- 53. Li YI, Chen YJ, Hsu YH and Meng M (2001) Characterization of the AdoMet‐dependent guanylyltransferase activity that is associated with the N terminus of bamboo mosaic virus replicase. J Virol 75, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Magden J, Takeda N, Li T, Auvinen P, Ahola T, Miyamura T, Merits A and Kääriäinen L (2001) Virus‐specific mRNA capping enzyme encoded by hepatitis E virus. J Virol 75, 6249–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Merits A, Kettunen R, Mäkinen K, Lampio A, Auvinen P, Kääriäinen L and Ahola T (1999) Virus‐specific capping of tobacco mosaic virus RNA: methylation of GTP prior to formation of covalent complex p126–m7GMP. FEBS Lett 455, 45–48. [DOI] [PubMed] [Google Scholar]
- 56. Vlot AC, Menard A and Bol JF (2002) Role of the alfalfa mosaic virus methyltransferase‐like domain in negative‐strand RNA synthesis. J Virol 76, 11321–11328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parkinson FE and Geiger JD (1996) Effects of iodotubercidin on adenosine kinase activity and nucleoside transport in DDT1 MF‐2 smooth muscle cells. J Pharmacol Exp Ther 277, 1397–1401. [PubMed] [Google Scholar]
- 58. Zhang X, Jia D, Liu H, Zhu N, Zhang W, Feng J, Yin J, Hao B, Cui D, Deng Y et al (2013) Identification of 5‐Iodotubercidin as a genotoxic drug with anti‐cancer potential. PLoS ONE 8, e62527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fedele DE, Gouder N, Güttinger M, Gabernet L, Scheurer L, Rülicke T, Crestani F and Boison D (2005) Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain 128, 2383–2395. [DOI] [PubMed] [Google Scholar]
- 60. Boison D (2008) The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol 84, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gouder N, Scheurer L, Fritschy J‐M and Boison D (2004) Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci 24, 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Broeckel R, Sarkar S, May NA, Totonchy J, Kreklywich CN, Smith P, Graves L, DeFilippis VR, Heise MT, Morrison TE et al (2019) Src family kinase inhibitors block translation of alphavirus subgenomic mRNAs. Antimicrob Agents Chemother 63, e02325‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Inhibition of CPE in CHIKV‐infected cells by 5‐IT.