

Abstract
Viruses take advantage of cellular machineries to facilitate their gene expression in the host. SR proteins, a superfamily of cellular precursor mRNA splicing factors, contain a domain consisting of repetitive arginine/serine dipeptides, termed the RS domain. The authentic RS domain or variants can also be found in some virus‐encoded proteins. Viral proteins may act through their own RS domain or through interaction with cellular SR proteins to facilitate viral gene expression. Numerous lines of evidence indicate that cellular SR proteins are important for regulation of viral RNA splicing and participate in other steps of post‐transcriptional viral gene expression control. Moreover, viral infection may alter the expression levels or modify the phosphorylation status of cellular SR proteins and thus perturb cellular precursor mRNA splicing. We review our current understanding of the interplay between virus and host in post‐transcriptional regulation of gene expression via RS domain‐containing proteins.
Keywords: Alternative splicing, kinases, phosphatases, phosphorylation, post‐transcriptional control, pre‐mRNA splicing, RS domain, SR proteins, viral problems, virus
Abbreviations
- CTE
constitutive transport element
- E4
early region 4
- EV
epidermodysplasia verruciformis
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- hnRNP
heterogeneous nuclear ribonucleoprotein
- HPV
human papillomavirus
- HSV
herpes simplex virus
- IRES
internal ribosome entry site
- N
nucleocapsid
- PP
protein phosphatase
- SARS‐CoV
severe acute respiratory syndrome coronavirus
- snRNP
small nuclear ribonucleoprotein
- SRPK
SR protein‐specific kinase
Introduction
Arginine/serine (RS) dipeptide repeats are present in a number of cellular proteins, termed SR proteins, that primarily participate in nuclear precursor (pre)‐mRNA splicing [1, 2, 3]. RS domain variants, such as serine and arginine‐rich motifs or arginine–aspartate or arginine–glutamate dipeptide‐rich domains, are also found in many nuclear proteins. In addition to the RS domains, SR splicing factors often contain one or more RNA recognition motifs. SR proteins function in both constitutive and regulated splicing via binding to cis‐elements of pre‐mRNA or interaction with other splicing factors. The RS domain interacts with both proteins and RNAs [1, 2, 3]. In particular, intermolecular interactions between SR proteins, which are important for spliceosome assembly and splice site determination during pre‐mRNA splicing, are mediated by their RS domains [3]. The RS domain also acts as a nuclear localization signal and targets SR proteins to nuclear speckled domains, where splicing factors are concentrated, for storage [1].
An important biochemical property of the RS domain is its differential phosphorylation at multiple serine and threonine residues. The RS domain is primarily phosphorylated by SR protein‐specific kinases (SRPKs), and the Clk/Sty family of kinases, and is probably dephosphorylated by protein phosphatase (PP)1/PP2A family phosphatases [2, 4]. RS domain phosphorylation can modulate protein–protein and protein–RNA interactions of SR proteins [1, 2, 3, 4, 5]. Reversible phosphorylation of SR proteins is important for assembly and function of the spliceosome and for proper regulation of alternative splicing, and also controls their subnuclear localization and nucleocytoplasmic transport [1, 2, 3, 4, 5, 6]. Moreover, environmental signals or viral infection can control the phosphorylation status of SR proteins, and subsequently affect mRNA splicing patterns [7, 8].
Authentic RS domain and its variants can also be found in some virus‐encoded proteins. For example, the human papillomavirus (HPV) E2 transcriptional regulator contains a prototypical RS domain [9]. Moreover, various lengths of the R/S‐rich motifs are found in some other viral proteins, such as the human hepatitis B virus (HBV) core protein and coronavirus nucleocapsid (N) protein [10, 11, 12]. SR protein kinases may phosphorylate these viral proteins and thus modulate viral activities in the infected host [12, 13]. Also, some viral proteins interact with cellular SR proteins, and thereby may influence host or viral gene expression at the post‐transcriptional level. In this review, we describe these viral SR proteins and also discuss the interplay between host and virus via their RS domain‐containing proteins.
Virus‐encoded SR proteins
HPV E2 protein
HPVs are a large family of small, double‐stranded DNA viruses. HPV infection causes a variety of cutaneous and mucosal lesions, ranging from warts to neoplasia and even cancer [14]. A subset of HPV types are associated with epidermodysplasia verruciformis (EV), a rare hereditary disease characterized by the development of multiple cutaneous warts [15]. Certain types of EV HPVs also have oncogenic potential. The E2 protein encoded by HPVs primarily regulates the transcription of early promoters by binding to a consensus element within the long control region of the viral genome, and also functions together with the E1 protein in viral DNA replication [16].
The E2 protein consists of the N‐terminal transactivation domain and the C‐terminal DNA‐binding domain. These two functional domains are linked by a hinge region that varies in length and sequence among HPV types. Notably, the relatively long hinge of EV HPV E2 proteins contains RS dipeptide repeats (Fig. 1), which suggests a function in pre‐mRNA splicing. Indeed, an EV HPV E2 protein interacts with cellular splicing factors, including prototypical SR proteins and RS domain‐containing small nuclear ribonucleoprotein (snRNP) components [9]. Functional investigation of this E2 protein has indicated that its RS‐rich hinge domain can facilitate splicing of the transcripts made via transactivation by E2 itself [9]. Therefore, the EV HPV E2 transactivator may recruit cellular splicing factors to cotranscriptionally facilitate pre‐mRNA splicing, and thus plays a dual role in gene expression.
Figure 1.
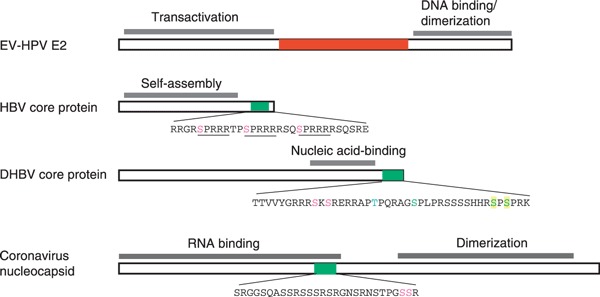
Viral SR proteins. The diagram shows domain structures of the representative viral proteins containing either a canonical RS domain (red) or an R/S‐rich motif (green). In the HBV core protein, three SPRRR motifs are underlined. Phosphorylation of the highlighted serine and threonine residues has been reported (see the text). Different highlights in the DHBV core protein represent different phosphorylation sites determined by three independent studies (see the text). The coronavirus (SARS‐CoV) nucleocapsid protein contains multiple phosphorylation sites (see the text); the two highlighted residues serve as the major phosphorylation sites of SRPK1 in vitro [12].
HBV core protein
HBV is a small dsDNA virus that replicates in hepatocytes. Chronic infection with HBV causes hepatocellular carcinoma. The HBV core protein plays several roles during the viral life cycle, including positive‐strand and minus‐strand DNA synthesis, pre‐genomic RNA packaging, and virion formation and release [17, 18]. This core protein is a phosphoprotein, and its function may be modulated by phosphorylation [10, 18, 19, 20]. The C‐terminal region of the core protein contains several nonconsecutive RS dipeptides as well as three SPRRR repeats (Fig. 1). Analysis of an HBV strain has revealed that phosphorylation mainly occurs at the SPRRR repeats [10]. Another report shows that the core protein C‐terminal domain may be phosphorylated by SRPK1/2 in host cells [13]. However, a more recent study revealed that although SRPK1/2 could suppress viral replication by interfering with pre‐genomic RNA packaging, the kinase activity appeared to be dispensable [20]. Therefore, the role of SRPK1/2 in HBV core protein phosphorylation, if any, remains to be investigated.
The core protein of duck HBV is not well conserved with its human counterpart, but still contains several RS repeats (Fig. 1). Phosphorylation of multiple serine residues within this region is required for first‐strand DNA synthesis during reverse transcription [18]. Analogously, a hyperphosphorylation‐mimetic mutant of the core protein fails to accumulate dsDNA, indicating that reversible phosphorylation of the core protein is critical for completion of viral reverse transcription [19]. However, the determination of which cellular kinases and phosphatases are responsible for such functionally related phosphorylation/dephosphorylation still requires further investigation.
Coronavirus nucleocapsid protein
The coronavirus genome is a positive‐sense, ssRNA. Infection with coronavirus primarily causes respiratory and enteric syndromes in a wide range of animals [21]. The nucleocapsid (N) protein is the most abundant viral protein produced throughout viral infection, and plays multiple roles in the viral life cycle, including in viral encapsidation, replication and transcription [21]. Both the N‐terminal and C‐terminal domains of the N protein contribute to nucleic acid binding, and the latter is additionally involved in oligomerization [22, 23, 24]. Coronavirus N proteins of different species share limited similarity with each other, but all contain an R/S‐rich segment in the central region (Fig. 1). Phosphorylation may occur at multiple sites within this R/S‐rich motif [12, 25, 26]. Experimental analyses have indicated that various cellular kinases, including cyclin‐dependent kinases, GSK, casein kinase II, mitogen‐activated protein kinases, and SRPKs, may phosphorylate coronavirus N proteins [11, 12]. We have recently observed that the severe acute respiratory syndrome coronavirus (SARS‐CoV) N protein redistributes to cytoplasmic stress granules in response to environmental stress [12]. However, such redistribution can be prevented by overexpression of SRPK1, which suggests that SRPK1 targets the N protein in cells [12]. Nevertheless, heterogeneous phosphorylation of the N protein may indicate its dynamic phosphorylation status and perhaps multiple functions during viral infection [27].
Phosphorylation of the RS‐rich motif may influence the biochemical activities of the N protein. Recent evidence suggests that phosphorylated infectious bronchitis virus N protein preferentially recognizes viral RNA over nonviral RNA [22]. We recently reported that phosphorylation of the SARS‐CoV N protein within the RS motif moderately impairs its multimerization [12]. Therefore, it is possible that the phosphorylation status of coronavirus N protein determines its activity in viral RNA transcription and packaging. Moreover, coronavirus N protein may also influence various cellular processes. Coronavirus infection causes cellular translation shutoff in the host, probably via the activity of the N protein [28]. Our recent report shows that the SARS‐CoV N protein can suppress translation, and that this activity depends on the RS motif of SARS‐CoV N protein, but is attenuated by its phosphorylation [12]. Therefore, we speculate that coronavirus N protein contributes to viral infection‐induced translation inhibition, which can be governed by the level of N protein phosphorylation.
Interactions between viral proteins and cellular SR proteins
Several viral proteins interact with SR splicing factors
HPV E2
As described above, the E2 protein of EV‐associated HPVs interacts with RS domain‐containing splicing factors via its RS dipeptide‐rich hinge. The interaction between an EV HPV E2 protein and a set of canonical SR proteins, including SRp20, ASF/SF2, SC35, SRp40, SRp55 and SRp75, was detected by a protein‐blotting analysis [9]. We also detected the interaction of this E2 protein with two SR family snRNP components, U1‐70K and U5‐100kD. Therefore, the RS‐rich hinge of EV HPV E2 functions to recruit splicing factors to facilitate cotranscriptional splicing [9].
Herpes simplex virus (HSV)‐1 ICP27
The HSV‐1 immediate‐early protein ICP27 plays multiple roles in post‐transcriptional regulation, and is essential for expression of viral late genes. ICP27 interacts with SR proteins such as SRp20 and U1‐70K [29, 30]. Moreover, ICP27 modulates the kinase activity and cellular localization of SRPK1, which results in hypophosphorylation of SR proteins and, consequently, downregulation of cellular pre‐mRNA splicing [29, 30]. ICP27 acts through the nuclear export receptor TAP/NXF1 of host cells to facilitate viral intronless mRNA export, and also participates in translation of viral mRNAs [31]. Perhaps ICP27 takes advantage of its interacting SR proteins to recruit TAP/NXF1 to viral RNAs for nuclear export and even for translation activation.
Adenovirus E4‐ORF4
Adenovirus produces a complex set of alternatively spliced viral mRNAs during replication. The early region 4 (E4)‐ORF4 protein plays an important role in regulation of the IIIa pre‐mRNA splicing at the late phase of the infectious cycle [32]. Cellular SR proteins bind to an intronic element of the IIIa pre‐mRNA to inhibit exon IIIa inclusion. E4‐ORF4 interacts directly with the SR proteins ASF/SF2 and SRp30c. Meanwhile, E4‐ORF4 recruits PP2A to dephosphorylate these SR proteins, which leads to derepression of IIIa pre‐mRNA splicing [33, 34]. Moreover, adenovirus infection alters cellular localization of SR proteins [35]. At the intermediate stages of viral infection, SR proteins and snRNPs are recruited to particular nuclear locations where viral pre‐mRNAs are transcribed and processed [36]. Therefore, adenovirus makes efficient use of the cellular splicing machinery to facilitate its own gene expression and, in addition, adenovirus infection may profoundly alter the cellular mRNA splicing pattern.
The hepatitis C virus (HCV) core protein interacts with RNA helicase DDX3
HCV is a major cause of chronic liver diseases, and its core protein plays an important role in hepatitis and hepatocarcinogenesis [37]. Translation of the viral polyprotein occurs through an internal ribosome entry site (IRES) located in the 5′‐nontranslated region [38]. This IRES‐mediated translation can be stimulated by an optimal dose of the core protein [39, 40]. The core protein interacts directly with a cellular RS domain‐containing protein, DDX3 [41, 42, 43]. DDX3 is a phylogenetically conserved member of the DEAD box RNA helicase family, and is involved in various mRNA metabolic events, including pre‐mRNA splicing, mRNA export and mRNA translation [44, 45, 46, 47, 48]. Coincident with the HCV core–DDX3 interaction, a mild activation of HCV IRES‐mediated translation has been observed after overexpression of DDX3 [48]. Moreover, it has also been shown that depletion of DDX3 from human hepatoma HuH‐7 cells decreases HCV RNA expression [49]. Therefore, DDX3 not only interacts with an HCV protein but may also modulate viral activity. Notably, upregulation of DDX3 has been observed in hepatocellular carcinoma [50]. Therefore, whether DDX3 exerts any cooperative effect with the HCV core protein on viral translation and replication control or modifies the activity of the core protein in hepatoma remains an interesting question.
HPV E1^E4 protein interacts with SRPK1
The HPV E1^E4 protein is highly expressed in epithelial cells during the viral productive stage, and perhaps functions throughout the early and late stages of the virus life cycle [51]. E1^E4 protein interacts with SRPK1 through an arginine‐rich domain and an oligomerization domain, and impairs autophosphorylation of SRPK1 [52]. In terminally differentiated cells, E1^E4 protein also recruits SRPK1 to inclusion bodies to colocalize with HPV E4 proteins. E4 proteins exist in several different proteolytic forms, and may have multiple biological activities [52]. E4 proteins function to promote viral DNA synthesis in suprabasal keratinocytes, where their phosphorylation occurs [51]. Interestingly, SRPK1 can phosphorylate E4 proteins, and may modulate their function in the host [52]. Moreover, by sequestration of SRPK1, E1^E4 protein may perturb cellular mRNA processing and thus alter the gene expression pattern of virus‐infected cells.
Cellular SR proteins modulate viral gene expression or function
SR splicing factors modulate viral RNA splicing
Retroviruses and DNA viruses produce complicated mRNA patterns via splicing. For example, more than 40 mRNA species of HIV are generated by alternative splicing of the single primary transcript [53]. Alternative splicing is regulated by cellular splicing factors, including SR proteins and heterogeneous nuclear ribonucleoprotein (hnRNP) proteins, which bind to the regulatory cis‐elements of viral mRNAs. In general, SR proteins bind to exonic splicing enhancers to facilitate the use of proximal splice sites, whereas hnRNPs inhibit splice site usage by binding to exonic or intronic splicing silencers [54]. Extensive reviews regarding the regulation of viral RNA splicing exist elsewhere [53, 55, 56]; therefore, we will describe only a few representative examples in this review.
In HIV‐1 pre‐mRNA, the SR proteins ASF/SF2 and SC35 bind exonic enhancer elements to activate tat exon 3 utilization [54]. On the other hand, the negative regulator hnRNP A1 initially binds a high‐affinity exonic element, and subsequently occupies the upstream region of this binding site to preclude splicing activators and hence inhibit splicing. Overexpression of ASF/SF2 can antagonize the negative effect of hnRNP A1 on splicing of the HIV‐1 tat RNA [54]. Moreover, ASF/SF2 promotes proximal or weak 5′‐splice site utilization of the adenovirus E1A and influenza virus M1 mRNAs [57, 58]. ASF/SF2 also activates the use of the proximal 3′‐splice site of bovine papillomavirus type 1 late pre‐mRNA by binding to the enhancer elements between two alternative 3′‐splice sites [59]. Notably, this splicing regulation occurs in a differentiation‐specific manner in keratinocytes, and can be controlled by the phosphatidylinositol 3‐kinase/Akt signaling pathway [59]. Therefore, viral RNA splicing in host is controlled in a cell type‐dependent or time‐dependent manner, and is determined by the relative expression levels between different splicing factors.
SR proteins stimulate polyadenylation in Rous sarcoma virus
SR proteins also participate in regulation of mRNA polyadenylation. The simple avian retrovirus Rous sarcoma virus produces unspliced RNAs for replication. The negative regulator of splicing element within the gag gene acts as a decoy 5′‐splice site to be recognized by SR proteins and U1/U11 snRNPs; this, however, results in splicing inhibition [60]. Via binding to this negative regulator of splicing element, SR proteins also recruit the 3′‐polyadenylation machinery to promote polyadenylation of unspliced RNAs [60]. This stimulatory activity of SR proteins in polyadenylation is counteracted by hnRNP H [61]. Therefore, SR proteins, together with other RNA‐binding proteins, coordinate the coupling of splicing and polyadenylation.
SR proteins participate in viral protein translation
SR proteins are primarily localized in the nucleus; however, a subset of SR proteins, including SRp20, 9G8 and ASF/SF2, shuttle continuously between the nucleus and the cytoplasm [6]. The shuttling SR proteins may participate in mRNA export and exert translation control. ASF/SF2 activates cap‐dependent translation via its binding to the exonic splicing enhancers of mRNAs [62]. In addition, SR proteins can facilitate IRES‐mediated translation [63]. Translation of the poliovirus genome is mediated through an IRES within the 5′‐noncoding region. SRp20 probably cooperates with the poly(rC) binding protein 2, which directly binds to the poliovirus IRES, to facilitate viral IRES‐mediated translation [63].
Simple retroviruses mediate the export of unspliced viral mRNAs and genomic RNA through the constitutive transport element (CTE) within the retained introns [64]. In host cells, the nuclear export factor TAP/NXF1 directly binds the Mason–Pfizer monkey virus CTE to facilitate unspliced viral RNA export [64]. However, TAP/NXF1‐mediated mRNA export in general involves adaptors such as shuttling SR proteins, which may subsequently promote translation [62, 65]. Coincidently, a recent report shows that the TAP‐interacting and shuttling SR protein 9G8 can enhance translation of the CTE‐containing viral RNAs by promoting their association with polysomes [66]. Thus, shuttling SR proteins could provide links between mRNA export and translation control for both cellular and viral mRNAs.
SR proteins affect viral production
SR proteins have a broad range of effects on viral gene expression. However, it is still not well understood how SR proteins affect various viral activities in the host. An in vivo analysis has revealed that overexpression of SR proteins reduces the yield of HIV genomic RNA and structural proteins, perhaps through their general activity in splicing promotion, and thereby downregulates virion production [67]. However, different SR proteins give rise to different viral RNA splicing patterns [68]. For example, ASF/SF2 overexpression increases the vpr mRNA expression level, whereas SC35 and 9G8 overexpression promotes production of tat mRNA. This is probably because each SR protein prefers its own specific binding elements on viral RNA. Moreover, phosphorylation of SRp75 can significantly enhance HIV expression [69], suggesting that modulation of SR protein phosphorylation levels may also have an effect on viral production.
Viral infection affects cellular SR proteins
Changes in SR protein expression level
To optimize the cellular environment for viral life cycle progression, viruses may profoundly alter the proteomes of the infected cells through various mechanisms, such as modification of host cell gene expression patterns, microRNA levels, or cellular signaling pathways. Conceivably, viruses also modify cellular SR proteins in order to take control of the host cell RNA splicing machinery.
It has been observed that, during persistent infection by HIV‐1 in macrophages, SC35 expression level initially increases and then declines [70]. Overexpression of SC35 can specifically increase tat mRNA expression [68]. Perhaps, to facilitate viral activity, HIV induces SC35 expression in the host during a specific time window. HPV‐16 infection upregulates the expression of both ASF/SF2 and its antagonistic factor hnRNP A1 in differentiated epithelial cells [71]. Therefore, HPV may utilize these cellular factors to coordinate appropriate alternative splicing control of viral late transcripts.
Changes in SR protein phosphorylation
Viruses modulate cellular SR protein phosphorylation levels and thereby affect viral and cellular pre‐mRNA splicing by several distinct mechanisms. As described above, the adenovirus E4‐ORF4 protein recruits PP2A to dephosphorylate SR proteins, and thereby activates IIIa pre‐mRNA splicing [35]. The HSV ICP27 protein instead interacts with and inactivates SRPK1, which also results in hypophosphorylation of SR proteins and hence pre‐mRNA splicing inhibition [30]. Moreover, adenovirus infection induces de novo synthesis of ceramide followed by nonapoptotic cell death, and adenovirus E4‐ORF4 can act through this ceramide signaling pathway to modulate SR protein phosphorylation levels [72]. This is reminiscent of the scenario of FAS ligand‐induced ceramide accumulation, which results in dephosphorylation of SR proteins and, hence, changes in alternative splicing patterns [73]. Similarly, infection of vaccinia virus induces dephosphorylation and inactivation of SR proteins [74]. Vaccinia virus encodes its own dual‐specificity PP, VH1, and thus its infection causes more severe dephosphorylation of SR proteins than adenovirus and HSV [74].
Manipulating SR protein phosphorylation levels in the host may be an antiviral strategy. It has been shown that reduced activity of SR proteins resulting from viral infection can be recovered by overexpression of SR proteins or by their rephosphorylation in the host cells [74], and that HIV expression can be greatly increased when SRp75 is phosphorylated by SRPK2 [69]. Therefore, SR protein phosphorylation inhibitors can be used as antiviral agents [75].
Conclusion and perspectives
In this review, we discuss the interplay between viral and cellular SR proteins in the regulation of both viral and host gene expression (Table 1). Through the RS domain or R/S‐rich motifs present in viral proteins, a virus may efficiently make use of the cellular splicing machinery to benefit its own gene expression. Phosphorylation of the RS domain can modulate the biological function of viral SR proteins, which may in turn impact on viral gene expression or other activities (Fig. 2). Moreover, through interactions with cellular SR proteins or by modifying their phosphorylation status, several non‐SR viral proteins may interfere with cellular gene expression at the post‐transcriptional level (Fig. 2). Cellular SR proteins and their cooperative or antagonistic factors may play a critical role in the life cycle control of viruses, which involves a series of alternative splicing events for expression of viral genome or proteins. However, if the expression of these host factors is spatially or temporally controlled, viral RNA splicing patterns may differ between cell types and differentiation stages. Nevertheless, there is still much to learn about how viruses undergo alternative splicing in various host cell environments.
Table 1.
Functional interplay between viral proteins and cellular SR proteins as well as SR kinases/phosphatases.
| Viral protein | Cellular proteins | Function | Reference |
|---|---|---|---|
| SR proteins | |||
| EV HPV E2 | SR | Splicing activation | [9] |
| HBV core | SRPK1/2 | Viral replication | [13] |
| SARS‐CoV N | SRPK1 (?) | Translation inhibition | [12] |
| Non‐SR proteins | |||
| HSV ICP27 | SR and SRPK1 | Splicing inhibition | [29, 30] |
| Adenovirus E4‐ORF4 | SR and PP2A | Splicing regulation | [33, 34] |
| HCV core | DDX3 | Viral translation and replication | [41, 42, 43, 49] |
| HPV E1^E4 | SRPK1 | Cellular RNA processing (?) | [52] |
Figure 2.
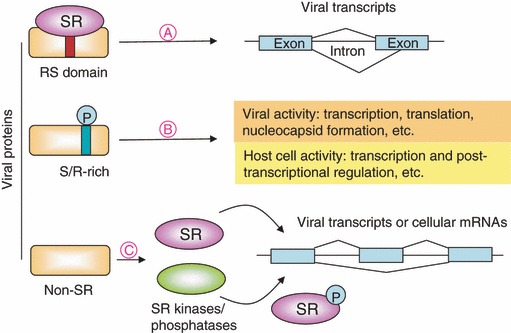
Viral and host gene expression is modulated through the interplay between viral proteins and cellular SR proteins. (A) Viral SR proteins (rectangle) recruit cellular SR proteins (oval) to promote splicing efficiency and/or modulate alternative splicing of viral transcripts. (B) Phosphorylation of viral proteins in the S/R‐rich motif may modulate their function and thereby influence viral and cellular activities. (C) Non‐SR viral proteins (rectangle) may interact directly with cellular SR proteins (purple oval) or modulate their phosphorylation status via SR protein kinases or phosphatases (green oval) and thereby determine the splicing patterns of both viral and cellular RNAs. In general, viral infection may influence the cellular splicing machinery, particularly SR proteins, thereby altering viral and host cell gene expression at the post‐transcriptional level.
The interplay between viral and cellular SR proteins certainly has a substantial effect on post‐transcriptional control of both viral and host gene expression. Although this process is not yet well understood, cellular SR proteins have been implicated as antiviral targets or therapeutic agents. When tethered to a pre‐mRNA, a synthetic RS repeat‐containing peptide is able to rescue defective splicing caused by mutations in the cis‐regulatory element [76]. Indole derivatives have been used to reduce HIV‐1 RNA synthesis and viral particle assembly by specifically interfering with the splicing activity of ASF/SF2, which is involved in the expression of HIV‐1 viral proteins [77]. To modulate the phosphorylation level of SR proteins, SR protein kinase inhibitors have recently been developed [75]. An inhibitor specific to SRPK1/2 can suppress HIV expression, perhaps by inactivating SRp75 [69]. Moreover, downregulation of a specific SR protein using RNA interference may be useful in manipulating viral activity. Certainly, a more comprehensive understanding of post‐transcriptional regulation governed by viruses will benefit the future development of antiviral strategies.
Acknowledgements
We acknowledge support from the Academia Sinica Investigator Award to W.‐Y. Tarn. We thank Drs Chiaho Shih and Steve S.‐L. Chen for comments on the manuscript.
References
- 1. Hertel KJ & Graveley BR (2005) RS domains contact the pre‐mRNA throughout spliceosome assembly. Trends Biochem Sci 30, 115–118. [DOI] [PubMed] [Google Scholar]
- 2. Stamm S (2008) Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem 283, 1223–1227. [DOI] [PubMed] [Google Scholar]
- 3. Lin S & Fu XD (2007) SR proteins and related factors in alternative splicing. Adv Exp Med Biol 623, 107–122. [DOI] [PubMed] [Google Scholar]
- 4. Shi Y, Reddy B & Manley JL (2006) PP1/PP2A phosphatases are required for the second step of pre‐mRNA splicing and target specific snRNP proteins. Mol Cell 23, 819–829. [DOI] [PubMed] [Google Scholar]
- 5. Fluhr R (2008) Regulation of splicing by protein phosphorylation. Curr Top Microbiol Immunol 326, 119–138. [DOI] [PubMed] [Google Scholar]
- 6. Huang Y & Steitz JA (2005) SRprises along a messenger’s journey. Mol Cell 17, 613–615. [DOI] [PubMed] [Google Scholar]
- 7. Guil S & Caceres JF (2007) Stressful splicing. Mol Cell 28, 180–181. [DOI] [PubMed] [Google Scholar]
- 8. Tarn WY (2007) Cellular signals modulate alternative splicing. J Biomed Sci 14, 517–522. [DOI] [PubMed] [Google Scholar]
- 9. Lai MC, Teh BH & Tarn WY (1999) A human papillomavirus E2 transcriptional activator. The interactions with cellular splicing factors and potential function in pre‐mRNA processing. J Biol Chem 274, 11832–11841. [DOI] [PubMed] [Google Scholar]
- 10. Liao W & Ou JH (1995) Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol 69, 1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Surjit M, Kumar R, Mishra RN, Reddy MK, Chow VT & Lal SK (2005) The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14‐3‐3‐mediated translocation. J Virol 79, 11476–11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peng TY, Lee KR & Tarn WT (2008) Phosphorylation of the arginine/serine dipeptides‐rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J 275, 4152–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Daub H, Blencke S, Habenberger P, Kurtenbach A, Dennenmoser J, Wissing J, Ullrich A & Cotten M (2002) Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J Virol 76, 8124–8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Longworth MS & Laimins LA (2004) Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev 68, 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pfister H (1992) Human papillomaviruses and skin cancer. Semin Cancer Biol 3, 263–271. [PubMed] [Google Scholar]
- 16. Ham J, Dostatni N, Gauthier JM & Yaniv M (1991) The papillomavirus E2 protein: a factor with many talents. Trends Biochem Sci 16, 440–444. [DOI] [PubMed] [Google Scholar]
- 17. Perri S & Ganem D (1997) Effects of mutations within and adjacent to the terminal repeats of hepatitis B virus pregenomic RNA on viral DNA synthesis. J Virol 71, 8448–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gazina E, Fielding JE, Lin B & Anderson DA (2000) Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J Virol 74, 4721–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lan YT, Li J, Liao WY & Ou JH (1999) Role of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259, 342–348. [DOI] [PubMed] [Google Scholar]
- 20. Zheng Y, Fu XD & Ou JH (2005) Suppression of hepatitis B virus replication by SRPK1 and SRPK2 via a pathway independent of the phosphorylation of the viral core protein. Virology 342, 150–158. [DOI] [PubMed] [Google Scholar]
- 21. Lai MM & Cavanagh D (1997) The molecular biology of coronaviruses. Adv Virus Res 48, 1–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan H, Ooi A, Tan YW, Wang S, Fang S, Liu DX & Lescar J (2005) The nucleocapsid protein of coronavirus infectious bronchitis virus: crystal structure of its N‐terminal domain and multimerization properties. Structure 13, 1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saikatenudu KS, Joseph JS, Subramanian V, Neuman BW, Buchmeier MJ, Stevens RC & Kuhn P (2007) Ribonucleocapsid formation of SARS‐CoV through molecular action of the N‐terminal domain of N protein. J Virol 81, 3913–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen CY, Chang CK, Chang YW, Sue SC, Bai HI, Riang L, Hsiao CD & Huang TH (2007) Structure of the SARS coronavirus nucleocapsid protein RNA‐binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J Mol Biol 368, 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen H, Gill A, Dove BK, Emmett SR, Kemp FC, Ritchie MA, Dee M & Hiscox JA (2005) Mass spectroscopic characterization of the coronavirus infectious bronchitis virus nucleoprotein and elucidation of the role of phosphorylation in RNA binding by using surface plasmon resonance. J Virol 79, 1164–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calvo E, Escors D, Lopez JA, Gonzalez JM, Alvarez A, Arza E & Enjuanes L (2005) Phosphorylation and subcellular localization of transmissible gastroenteritis virus nucleocapsid protein in infected cells. J Gen Virol 86, 2255–2267. [DOI] [PubMed] [Google Scholar]
- 27. Dove B, Brooks G, Bicknell K, Wurm T & Hiscox JA (2006) Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J Virol 80, 4147–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zuniga S, Sola I, Moreno JL, Sabella P, Plana‐Duran J & Enjuanes L (2007) Coronavirus nucleocapsid protein is an RNA chaperone. Virology 357, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bryant HE, Wadd S, Lamond AI, Silverstein SJ & Clements JB (2001) Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome‐associated protein 145 and inhibits splicing prior to the first catalytic step. J Virol 75, 4376–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sciabica KS, Dai QJ & Sandri‐Goldin RM (2003) ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBO J 22, 1608–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen IH, Sciabica KS & Sandri‐Goldin RM (2002) ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J Virol 76, 12877–12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanopka A, Muhlemann O & Akusjarvi G (1996) Inhibition by SR proteins of splicing of a regulated adenovirus pre‐mRNA. Nature 381, 535–538. [DOI] [PubMed] [Google Scholar]
- 33. Estmer Nillsson C, Petersen‐Mahrt S, Durot C, Shtrichman R, Krainer AR, Kleinberger T & Akusjarvi G (2001) The adenovirus E4‐ORF4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J 20, 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shtrichman R, Sharf R & Kleinberger T (2000) Adenovirus E4orf4 protein interacts with both Bα and B′ subunits of protein phosphatase 2A, but E4orf4‐induced apoptosis is mediated only by Bα. Oncogene 19, 3757–3765. [DOI] [PubMed] [Google Scholar]
- 35. Kanopka A, Muhlemann O, Petersen‐Mahrt S, Estmer C, Ohrmalm C & Akusjarvi G (1998) Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature 393, 185–187. [DOI] [PubMed] [Google Scholar]
- 36. Lindberg A, Gama‐Carvalho M, Carmo‐Fonseca M & Kreivi JP (2004) A single RNA recognition motif in splicing factor ASF/SF2 directs it to nuclear sites of adenovirus transcription. J Gen Virol 85, 603–608. [DOI] [PubMed] [Google Scholar]
- 37. Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T & Koike K (1998) The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 4, 1065–1067. [DOI] [PubMed] [Google Scholar]
- 38. Honda M, Ping LH, Rijnbrand RC, Amphlett E, Clarke B, Rowlands D & Lemon SM (1996) Structural requirements for initiation of translation by internal ribosome entry within genome‐length hepatitis C virus RNA. Virology 222, 31–42. [DOI] [PubMed] [Google Scholar]
- 39. Boni S, Lavergne JP, Boulant S & Cahour A (2005) Hepatitis C virus core protein acts as a trans‐modulating factor on internal translation initiation of the viral RNA. J Biol Chem 280, 17737–17748. [DOI] [PubMed] [Google Scholar]
- 40. Lourenco S, Costa F, Debarges B, Andrieu T & Cahour A (2008) Hepatitis C virus internal ribosome entry site‐mediated translation is stimulated by cis‐acting RNA elements and trans‐acting viral factors. FEBS J 275, 4179–4197. [DOI] [PubMed] [Google Scholar]
- 41. Mamiya N & Worman HJ (1999) Hepatitis C virus core protein binds to a DEAD box RNA helicase. J Biol Chem 274, 15751–15756. [DOI] [PubMed] [Google Scholar]
- 42. Owsianka AM & Patel AH (1999) Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology 257, 330–340. [DOI] [PubMed] [Google Scholar]
- 43. You LR, Chen CM, Yeh TS, Tsai TY, Mai RT, Lin CH & Lee YH (1999) Hepatitis C virus core protein interacts with cellular putative RNA helicase. J Virol 73, 2841–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yedavalli VS, Neuveut C, Chi YH, Kleiman L & Jeang KT (2004) Requirement of DDX3 DEAD box RNA helicase for HIV‐1 Rev‐RRE export function. Cell 119, 381–392. [DOI] [PubMed] [Google Scholar]
- 45. Chuang RY, Weaver PL, Liu Z & Chang TH (1997) Requirement of the DEAD‐box protein Ded1p for messenger RNA translation. Science 275, 1468–1471. [DOI] [PubMed] [Google Scholar]
- 46. Lai MC, Lee YH & Tarn WY (2008) The DEAD‐box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip‐associated protein and participates in translational control. Mol Biol Cell 19, 3847–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee CS, Dias AP, Jedrychowski M, Patel AH, Hsu JL & Reed R (2008) Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res 36, 4708–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shih JW, Tsai TY, Chao CH & Wu Lee YH (2008) Candidate tumor suppressor DDX3 RNA helicase specifically represses cap‐dependent translation by acting as an eIF4E inhibitory protein. Oncogene 27, 700–714. [DOI] [PubMed] [Google Scholar]
- 49. Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T & Kato N (2007) DDX3 DEAD‐box RNA helicase is required for hepatitis C virus RNA replication. J Virol 81, 13922–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang JS, Chao CC, Su TL, Yeh SH, Chen DS, Chen CT, Chen PJ & Jou YS (2004) Diverse cellular transformation capability of overexpressed genes in human hepatocellular carcinoma. Biochem Biophys Res Commun 315, 950–958. [DOI] [PubMed] [Google Scholar]
- 51. Nakahara T, Peh WL, Doorbar J, Lee D & Lambert PF (2005) Human papillomavirus type 16 E1circumflexE4 contributes to multiple facets of the papillomavirus life cycle. J Virol 79, 13150–13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bell I, Martin A & Roberts S (2007) The E1^E4 protein of human papillomavirus interacts with the serine‐arginine‐specific protein kinase SRPK1. J Virol 81, 5437–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stoltzfus CM & Madsen JM (2006) Role of viral splicing elements and cellular RNA binding proteins in regulation of HIV‐1 alternative RNA splicing. Curr HIV Res 4, 43–55. [DOI] [PubMed] [Google Scholar]
- 54. Black DL (2003) Mechanisms of alternative pre‐messenger RNA splicing. Annu Rev Biochem 72, 291–336. [DOI] [PubMed] [Google Scholar]
- 55. Schwartz S (2008) HPV‐16 RNA processing. Front Biosci 13, 5880–5891. [DOI] [PubMed] [Google Scholar]
- 56. Akusjarvi G (2008) Temporal regulation of adenovirus major late alternative RNA splicing. Front Biosci 13, 5006–5015. [DOI] [PubMed] [Google Scholar]
- 57. Caceres JF, Stamm S, Helfman DM & Krainer AR (1994) Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science 265, 1706–1709. [DOI] [PubMed] [Google Scholar]
- 58. Shih SR & Krug RM (1996) Novel exploitation of a nuclear function by influenza virus: the cellular SF2/ASF splicing factor controls the amount of the essential viral M2 ion channel protein in infected cells. EMBO J 15, 5415–5427. [PMC free article] [PubMed] [Google Scholar]
- 59. Liu X, Mayeda A, Tao M & Zheng ZM (2003) Exonic splicing enhancer‐dependent selection of the bovine papillomavirus type 1 nucleotide 3225 3′ splice site can be rescued in a cell lacking splicing factor ASF/SF2 through activation of the phosphatidylinositol 3‐kinase/Akt pathway. J Virol 77, 2105–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maciolek NL & McNally MT (2007) Serine/arginine‐rich proteins contribute to negative regulator of splicing element‐stimulated polyadenylation in Rous sarcoma virus. J Virol 81, 11208–11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wilusz JE & Beemon KL (2006) The negative regulator of splicing element of Rous sarcoma virus promotes polyadenylation. J Virol 80, 9634–9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sanford JR, Gray NK, Beckmann K & Caceres JF (2004) A novel role for shuttling SR proteins in mRNA translation. Genes Dev 18, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bedard KM, Daijogo S & Semler BL (2007) A nucleo‐cytoplasmic SR protein functions in viral IRES‐mediated translation initiation. EMBO J 26, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gruter P, Tabernero C, von Kobbe C, Schmitt C, Saavedra C, Bachi A, Wilm M, Felber BK & Izaurralde E (1998) TAP, the human homolog of Mex67p, mediates CTE‐dependent RNA export from the nucleus. Mol Cell 1, 649–659. [DOI] [PubMed] [Google Scholar]
- 65. Dreyfuss G, Kim VN & Kataoka N (2002) Messenger‐RNA‐binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3, 195–205. [DOI] [PubMed] [Google Scholar]
- 66. Swartz JE, Bor YC, Misawa Y, Rekosh D & Hammarskjold ML (2007) The shuttling SR protein 9G8 plays a role in translation of unspliced mRNA containing a constitutive transport element. J Biol Chem 282, 19844–19853. [DOI] [PubMed] [Google Scholar]
- 67. Jacquenet S, Decimo D, Muriaux D & Darlix JL (2005) Dual effect of the SR proteins ASF/SF2, SC35 and 9G8 on HIV‐1 RNA splicing and virion production. Retrovirology 2, 33, doi: DOI: 10.1186/1742-4690-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ropers D, Ayadi L, Gattoni R, Jacquenet S, Damier L, Branlant C & Stevenin J (2004) Differential effects of the SR proteins 9G8, SC35, ASF/SF2, and SRp40 on the utilization of the A1 to A5 splicing sites of HIV‐1 RNA. J Biol Chem 279, 29963–29973. [DOI] [PubMed] [Google Scholar]
- 69. Fukuhara T, Hosoya T, Shimizu S, Sumi K, Oshiro T, Yoshinaka Y, Suzuki M, Yamamoto N, Herzenberg LA, Herzenberg LA et al. (2006) Utilization of host SR protein kinases and RNA‐splicing machinery during viral replication. Proc Natl Acad Sci USA 103, 11329–11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dowling D, Nasr‐Esfahani S, Tan CH, O’Brien K, Howard JL, Jans DA, Purcell DF, Stoltzfus CM & Sonza S (2008) HIV‐1 infection induces changes in expression of cellular splicing factors that regulate alternative viral splcing and virus production in macrophages. Retrovirology 5, 18, doi: DOI: 10.1186/1742-4690-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cheunim T, Zhang J, Milligan SG, McPhillips MG & Graham SV (2008) The alternative splicing factor hnRNP A1 is up‐regulated during virus‐infected epithelial cell differentiation and binds the human papillomavirus type 16 late regulatory element. Virus Res 131, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kanj SS, Dandashi N, El‐Hed A, Harik H, Maalouf M, Kozhaya L, Mousallem T, Tollefson AE, Wold WS, Chalfant CE et al. (2006) Ceramide regulates SR protein phosphorylation during adenoviral infection. Virology 345, 280–289. [DOI] [PubMed] [Google Scholar]
- 73. Chalfant CE, Ogretmen B, Galadari S, Kroesen BJ, Pettus BJ & Hannun YA (2001) FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J Biol Chem 276, 44848–44855. [DOI] [PubMed] [Google Scholar]
- 74. Huang TS, Nisson CE, Punga T & Akusjarvi G (2002) Functional inactivation of the SR family of splicing factors during a vaccinia virus infection. EMBO Rep 3, 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hagiwara M (2005) Alternative splicing: a new drug target of the post‐genome era. Biochim Biophys Acta 1754, 324–331. [DOI] [PubMed] [Google Scholar]
- 76. Cartegni L & Krainer AR (2003) Correction of disease‐associated exon skipping by synthetic exon‐specific activators. Nat Struct Biol 10, 120–125. [DOI] [PubMed] [Google Scholar]
- 77. Bakkour N, Lin YL, Maire S, Ayadi L, Mahuteau‐Betzer F, Nguyen CH, Mettling C, Portales P, Grierson D, Chabot B et al. (2007) Small‐molecule inhibition of HIV pre‐mRNA splicing as a novel antiretroviral therapy to overcome drug resistance. PLoS Pathog 3, 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]