

Abstract
Proteases are enzymes that hydrolyze the peptide bond of peptide substrates and proteins. Despite significant progress in recent years, one of the greatest challenges in the design and testing of substrates, inhibitors and activity‐based probes for proteolytic enzymes is achieving specificity toward only one enzyme. This specificity is particularly important if the enzyme is present with other enzymes with a similar catalytic mechanism and substrate specificity but completely different functionality. The cross‐reactivity of substrates, inhibitors and activity‐based probes with other enzymes can significantly impair or even prevent investigations of a target protease. In this review, we describe important concepts and the latest challenges, focusing mainly on peptide‐based substrate specificity techniques used to distinguish individual enzymes within major protease families.
Keywords: activity‐based probe, endopeptidase, exopeptidase, protease, substrate specificity
Proteases are enzymes that hydrolyze the peptide bond of peptide substrates and proteins. Despite significant progress in recent years, one of the greatest challenges in the design and testing of substrates, inhibitors and activity‐based probes for proteases is achieving specificity toward only one enzyme. In this review, we describe important concepts in substrate specificity determination techniques used to distinguish individual proteases.
Abbreviations
- AAT
α‐1‐antitrypsin
- Abu
aminobutyric acid
- ABP
activity‐based probe
- ABZ
aminobenzoic acid
- ACC
7‐amino‐4‐carbamoylmethylcoumarin
- ACT
antichymotrypsin
- AMC
7‐amino‐4‐methyl‐coumarin
- AOMK
acyloxymethylketone
- CatG
cathepsin G
- CLiPS
cellular libraries of peptide substrates
- CoSeSuL
counter selection substrate library
- DUB
deubiquitylating enzyme
- Gr
granzyme
- hK
human kallikrein
- hLeu
homoleucine
- HyCoSuL
hybrid combinatorial substrate library
- IQF
internally quenched fluorescent substrate
- LtA4h
leukotriene A4 hydrolase
- MALT1
mucosa‐associated lymphoid tissue lymphoma translocation protein 1
- Met(O)2
methionine sulfone
- MMP
matrix metalloproteinase
- NE
neutrophil elastase
- Nle(O‐Bzl)
2‐amino‐6‐benzyloxyhexanoic acid
- Nle
norleucine
- NSP4
neutrophil serine proteinase 4
- NSP
neutrophil serine protease
- Oic
octahydroindole‐2‐carboxylic acid
- Pal
pyridyl‐L‐alanine
- PR3
proteinase 3
- PSA
prostate‐specific antigen
- PS‐SCL
positional scanning synthetic combinatorial library
- SAR
structure activity relationship
- SAS
substrate activity screening
- SENP
human desumoylating enzyme
- Tic
1,2,3,4‐tetrahydroisoquinoline‐3‐carboxylic acid
- UCH
ubiquitin C‐terminal hydrolase
Introduction
Proteases are enzymes that hydrolyze peptide bonds within endogenous substrates and peptides. They play a key role in regulating many physiological conditions, and protease activity is dysregulated in many diseases, including cancer, diabetes and neurological disorders 1, 2. To date, five major families of proteases have been described: serine, cysteine, metallo‐, aspartyl and threonine proteases (Fig. 1). The most abundant and explored families are the serine and cysteine proteases, named after the reactive nucleophilic groups in their active sites – the hydroxyl group in serine and thiol group in cysteine 3. Mechanistically, proteases hydrolyze peptide bonds in the substrate (endopeptidases) or at the N or C termini (exopeptidases) 3. Proteinases belonging to the same family, such as caspases, neutrophil serine proteases, aminopeptidases, cathepsins or kallikreins, typically have similar functions and process the same naturally occurring substrates. However, little is known about the individual physiological and pathophysiological functions of some proteases 4, 5, 6, 7. To answer these questions, chemical tools including substrates, inhibitors and activity‐based probes (ABPs) have become essential for monitoring changes in the activity of proteolytic enzymes in cells or even in whole organisms (Fig. 2) 8, 9, 10.
Figure 1.
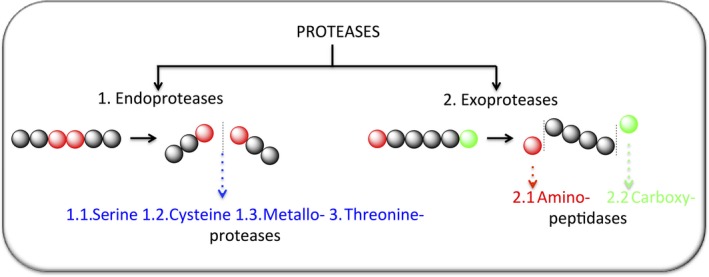
Schematic representation of the reviewed proteases and their catalytic properties.
Figure 2.
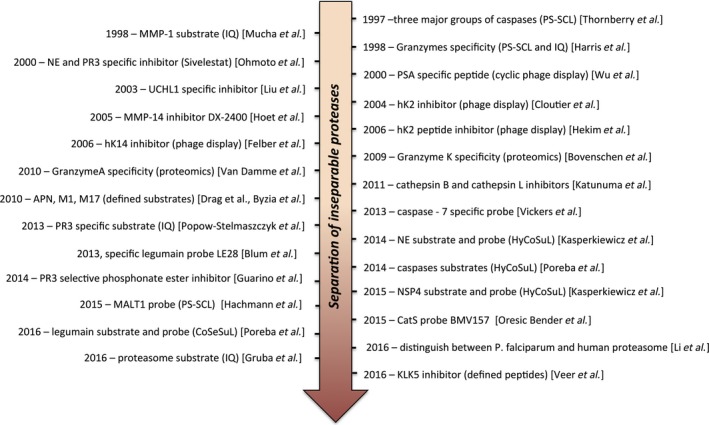
Notable achievements in the development of chemical tools for indistinguishable proteases. See text for details.
Despite the significant progress in recent years, one of the greatest challenges in the design and testing of substrates, inhibitors and ABPs is achieving specificity toward only one enzyme 1. This specificity is particularly important if the enzyme is in the presence of other enzymes with a similar catalytic mechanism but completely different functions. This cross‐reactivity of substrates, inhibitors and ABPs with other enzymes significantly impairs or even prevents investigations of a target protease 4. The specificity of the substrates, inhibitors and ABPs for proteolytic enzymes is optimized by selecting the appropriate amino acid sequences that interact with the binding pockets of the protease 8, 11.
Many different approaches have been proposed and used to investigate substrate specificity, including positional scanning synthetic combinatorial libraries (PS‐SCL), phage display, hybrid combinatorial substrate libraries (HyCoSuL), counter selection substrate libraries (CoSeSuL), internally quenched fluorescent substrate (IQF) libraries (also called fluorescence resonance energy transfer libraries) and proteomics, with varying results (Fig. 3) 8, 11, 12, 13, 14, 15.
Figure 3.
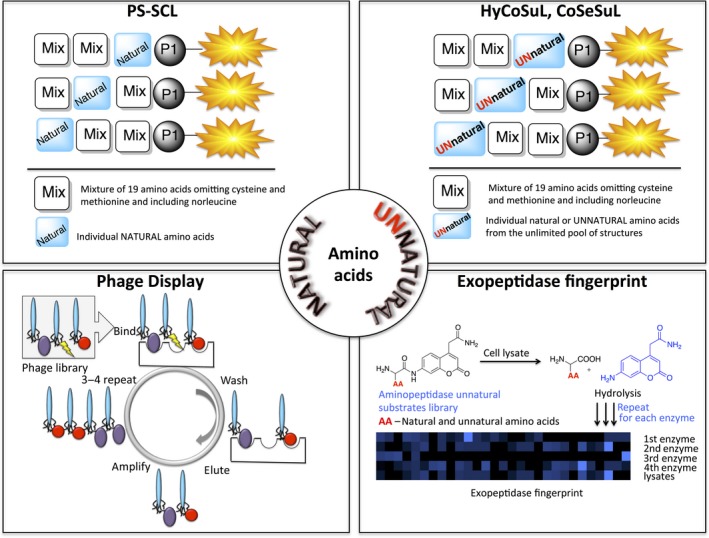
Scheme of different approaches that have been proposed and used to investigate substrate specificity, including PS‐SCL, HyCoSuL, CoSeSuL and exopeptidase fingerprint.
The first broad chemical approach to study protease substrate specificity was proposed by Thornberry and colleagues in 1997 16, 17. In this concept, the PS‐SCL (Fig. 3), the substrate library comprises three ‘sublibraries’ to study protease preferences at the P4, P3 and P2 positions. For instance, in the P4 sublibrary, this position was fixed with one of the natural amino acids (1–19), the P3–P2 positions were randomized with equimolar mixtures of natural amino acids (making this library useful to study various caspases), and the P1–P1′ positions were occupied by Asp–7‐amino‐4‐methyl‐coumarin (AMC). The caspase specificity preferences in the active site pockets were obtained by screening each position separately, making this approach ‘positional profiling’. Shortly after, the Ellman and Craik groups utilized a bifunctional 7‐amino‐4‐carbamoylmethylcoumarin (ACC) fluorophore as a reporter group in PS‐SCL, making introducing various amino acids at the P1 position much more effortless 18. To date, over 100 proteases have been profiled with the PS‐SCL approach, ranking this technology among the most useful tools in protease substrate specificity investigations 8. However, traditional PS‐SCL has been demonstrated to be insufficient when analyzing proteases with overlapping substrate specificity profiles (e.g. caspases, cysteine cathepsins, neutrophil proteases and deubiquitylating enzymes (DUBs)). The use of only natural amino acids in a PS‐SCL reduces the chance of finding a specific (and/or very active) peptide toward the protease of interest. This drawback was overcome by a novel approach developed in the Drag laboratory 19. This concept, the HyCoSuL, relies on the use of a broad panel of unnatural amino acids in the substrate sequences, which explore the protease active site much more accurately than a set of 20 natural analogues alone (Fig. 3). The use of unnatural amino acids in fluorogenic substrate libraries was also applied in the CoSeSuL 12. This approach is restricted to proteases that display very broad substrate specificity in one (or more) active site pockets. By comparing such a specificity matrix with other proteases displaying narrower specificity in corresponding pocket(s), it becomes possible to distinguish between enzymes. PS‐SCL, HyCoSuL and CoSeSuL are valuable tools to study protease substrate specificities; however, they are all limited to the non‐prime pockets of the enzyme active site. IQF substrates offer an excellent platform to study protease preferences in both prime and non‐prime regions (Fig. 4) 20. In brief, IQF substrates are peptides of various lengths flanked by a fluorescence group (donor) and a quencher group (acceptor). Such intact peptides are in a quenched state, as the fluorescence from the donor is quenched by an appropriate acceptor. Once the peptide is cleaved by the protease, the peptide fragment with the fluorophore is separated from the quencher, and the fluorescence is liberated. Currently, there is no single, unified technology using IQF substrates in protease screenings, and the architecture of an IQF substrate library is tailored to the protease being investigated. To date, various donor–acceptor pairs have been described in the literature and are reviewed elsewhere 8.
Figure 4.
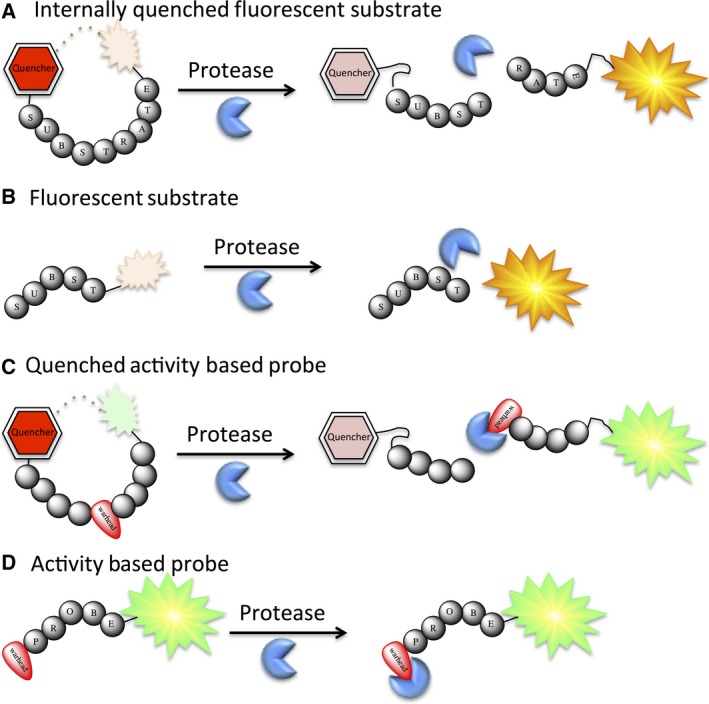
Schematic representation of the proteolysis mechanisms for commonly used types of substrates and probes for protease investigations: internally quenched fluorescent substrates (A), fluorescent substrates (B), quenched activity‐based probes (C), and activity‐based probes (D).
As an alternative to chemical‐based approaches, a phage display technology of biological origins has also been extensively developed and studied on proteolytic enzymes. This methodology was pioneered by Smith, who expressed a diverse pool of peptides on the surface of bacteriophage M13 21. This technique was initially oriented to studying protein–protein interactions; however, it was later modified and adapted for protease substrate specificity profiling (Fig. 3) 22. The main advantage of phage display is the generation of a very large and diverse pool of substrates (up to 1010 individual peptides), which would be very challenging through chemical synthesis. Another important feature of this technique is that these peptides are subjected to protease analysis after each round of expression/selection; thus, in subsequent cycles, peptides of increasing quality are generated. One of the main drawbacks of these methods is that the peptides are label‐free. Therefore, the kinetic analysis of the best substrates requires these peptides to be resynthesized with an appropriate reporter group. Since Matthews and Wells first described the use of phage display in protease screening, multiple enzymes have been profiled with this technique 23.
In this review, we describe important concepts and the latest challenges, focusing mainly on peptide‐based substrate specificity techniques used to distinguish individual enzymes within major protease families (Figs 3 and 4).
Endopeptidases
Serine proteases
Chymotrypsin‐like neutrophil serine proteases from primary granules
Neutrophil serine proteases (NSPs) of primary granules are some of the most abundant serine proteases present in almost all organisms. The NSP family includes neutrophil elastase (NE), cathepsin G (CatG), proteinase 3 (PR3) and neutrophil serine proteinase 4 (NSP4) 24, 25, 26. Among others, these enzymes are involved in killing bacteria via the formation of neutrophil extracellular traps 27, 28. NSPs evolved from a common ancestor by gene duplication, and they reveal a new branch of the chymotrypsinogen superfamily of serine proteases 29. NSPs consist of five exons and four introns. Along with the genes for NE and PR3, the NSP4 gene lies on chromosome 19p13.3 outside of the ZU1‐PRTN3‐ELANE‐CFD cluster 30, 31, 32, while CatG is located on chromosome 14q11.2 33. Regarding primary structure, the amino acid residues that determine the substrate recognition sites for NSPs are quite similar, and catalytic residues and substrate‐binding pockets are located in a crevice between two β‐barrels 32. Therefore, these enzymes share similar catalytic preferences and recognize the same endogenous inhibitors belonging to the serpin (serine protease inhibitors) family, such as antitrypsin or α‐1 proteinase inhibitor and human monocyte/neutrophil elastase inhibitor, which inhibit PR3, NE and CatG 34, 35.
Chemically, NSPs can be divided into groups: NE and PR3 recognize small aliphatic amino acid residues (Val and Ala) at the P1 position, CatG recognizes bulky, hydrophobic, aliphatic or aromatic amino acids (Phe, Leu and sometimes Arg) and NSP4 has restricted P1‐specificity for Arg 26, 36. NE and PR3 also have overlapping primary substrate specificity at P4–P2; therefore a specific substrate, inhibitor or activity‐based probe was not reported for these enzymes for many years. This specificity issue was confirmed in an elegant approach toward the global identification of neutrophil protease specificity that was recently described by O'Donoghue et al. 37. In this method, a rapid multiplex substrate profiling analysis using liquid chromatography–tandem mass spectrometry sequencing was applied to profile the substrates of neutrophil serine proteases in human neutrophil extracellular traps. The obtained data clearly showed that each enzyme had overlapping, yet distinct, endopeptidase activities, suggesting overlapping functions and substrate preferences between neutrophil serine proteases 24.
Recently, the Lesner group reported the design and synthesis of an internally quenched (IQF) PR3 substrate with the structure ABZ‐Tyr‐Tyr‐Abu‐Asn‐Glu‐Pro‐Tyr(3‐NO2)‐NH2 and an enzyme catalytic efficiency (k cat/K m) of 1534 × 103 m −1·s−1. The k cat/K m value for NE was not calculated using this substrate 38.
In 2014, Drag and colleagues enriched the classic PS‐SCL with a broad range of unnatural amino acids in an approach known as the HyCoSuL 19 (Fig. 3). This technology allows researchers to explore the P4–P2 binding sites in various structures. Surprisingly, in contrast to previously known NE substrates with sequence Ac‐AAPV‐AMC (Fig. 5), the HyCoSuL P4 screen identified the bulky 2‐amino‐6‐benzyloxyhexanoic acid (Nle(O‐Bzl)) residue. This result was confirmed later by Lechtenberg, who presented a crystal structure of an NE ABP containing Nle(O‐Bzl) at the P4 position 39 and demonstrated the fit of unnatural amino acids in the enzyme pockets in the active site. Moreover, methionine sulfone (Met(O)2) was identified at the S3 site, which suggested a potential post‐translational modification of natural NE substrates. Not surprisingly, both NE (octahydroindole‐2‐carboxylic acid, Oic) and PR3 prefer proline derivatives at the P2 position 19. Crucially, a comparison of the k cat/K m values for the synthesized substrate against both NE and PR3 showed that the Ac‐Nle(O‐Bzl)‐Met(O)2‐Oic‐Abu‐ACC (Fig. 5) substrate is approximately 900‐fold more specific for NE compared with PR3, exhibiting a relatively small K m of 0.28 μm for NE. An ABP for neutrophil elastase based on the optimal NE sequence was then used in in vitro studies, allowing researchers to discriminate between NE and PR3 and monitor the activity of neutrophil elastase in neutrophil traps for the first time 19.
Figure 5.
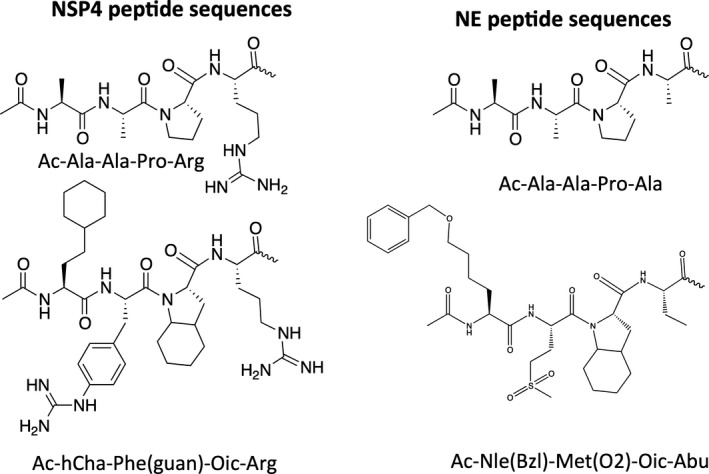
Examples of specific peptide sequences used in substrates and inhibitors for neutrophil serine proteases. hCha, homocyclohexylalanine; Phe(guan), 4‐guanidynophenylalanine.
CatG bears dual P1 chymotrypsin‐ and trypsin‐like substrate specificity 36. Due to the shape of the S1 pocket, CatG hydrolyzes the peptide bond after Phe and, with less potency, after Arg; therefore, this enzyme might cross‐react with NSP4 substrates. In 2015, Kasperkiewicz et al. used the HyCoSuL technology to search for NSP4‐specific molecules and investigate NSP4 primary specificity. Using this technology, specific NSP4 substrates and ABPs were synthesized and tested in in vitro assays. NSP4 recognized large hydrophobic residues at the P4 position (homocyclohexylalanine), large basic residues at the P3 (4‐guanidynophenylalanine) and the proline derivative Oic at the P2 position (Fig. 5). The most successful substrate was potent (k cat/K m 32 000 m −1·s−1) and approximately 70‐fold more specific for NSP4 compared with CatG. This discovery prompted the synthesis of ABPs, and resulted in the first covalently binding NSP4‐specific ABP, which exhibited approximately 1000‐fold more specific binding to NSP4 compared with CatG 40.
NSPs are involved in many diseases, particularly diseases associated with the lungs; thus, they are considered valuable drug targets. Moreover, a few drug trials have been conducted to discriminate serine proteases using inhibitors. Although a few of these inhibitors have been used in clinical trials 41, to date, only one compound, sivelestat (ONO‐5046, Ono Pharmaceuticals), was approved for use as a drug 42. This competitive inhibitor is specific for NE over trypsin, thrombin, chymotrypsin, CatG and plasmin with a relatively small half‐maximal inhibitory concentration (IC50) of 44 nm and K i of 0.2 μm toward NE.
The best‐known in vitro serine protease inhibitors are peptidyl derivatives of α‐aminoalkylphosphonate diphenyl esters, covalent inhibitors that bind to the hydroxyl group of serine in the active site 43. These compounds mimic natural amino acids, and they are some of the most frequently used NE inhibitors in in vitro studies. Oleksyszyn and colleagues modified the esters in this inhibitor class and subsequently found that an S‐methylsulfodiphenyl di‐ester was more potent than the unmodified ester 44, 45. Unfortunately, this approach was not sufficient to distinguish NE and PR3. Recently, Guarino et al. designed di(chlorophenyl) phosphonate esters characterized by an unexpected structure containing Asp at the P2 position, instead of Pro, which was described as the best‐fitting amino acid in this pocket. They used a probe with PEG66 [aminoomega‐carboxy poly(ethylene glycol)] as a linker and biotin as a tag characterized by a relatively small second‐order inhibition constant (k obs/[I]) against proteinase 3, which showed weak binding with NE, as a probe to detect PR3 in permeabilized neutrophils. The authors suggested that this probe may be used to monitor, detect and control PR3 activity in inflammatory diseases; however, no control was performed 46.
Granzymes
Human granzymes (Gr) were named from ‘granule enzymes’ in 1987 by Masson and Tschopp 47. They are closely related proteases with a nucleophilic serine residue stabilized by histidine and aspartic acid at the enzyme's active site and are expressed in cytotoxic T lymphocytes and natural killer (NK) cells. Those proteases are packed into cytoplasmic granules, and similarly to NSPs, they are secreted during stimulation. In humans, five Grs are distinguished (A, B, H, K and M) 48. However, granzyme genes were found on three chromosomes in three clusters (GrA, GrB and GrM clusters). The GrA cluster on chromosome 5 includes GrA 48, 49 and GrK; the GrB cluster on chromosome 14 includes GrB and GrH 50; and the GrM cluster on chromosome 19 includes GrM 51. Grs share distinctive structural features, such as the presence of a signal peptide and pro‐dipeptide, which are removed during activation. Although there are subtle differences in the crystal structures of Grs, their detection remains challenging because of the similarities between them. For instance, anti‐GrB binds both GrA and GrH. This problem is magnified since it was shown that Grs recognize and cleave the same natural substrates; for instance, 80 GrA substrates are reported to be GrB substrates as well, and among them, 22 have the same cleavage site 52. To date, no specific tool to distinguish between active and inactive Grs has been found; however, a few elegant approaches have been published and are discussed in this review.
GrA is the most abundant of all the granzymes 53. The effects of GrA and K on caspase‐independent cell death remain unclear 54; thus, it is necessary to distinguish between these enzymes to understand their individual roles. GrA and K primarily act in inflammation by promoting the secretion of cytokines, interleukins and tumor necrosis factor, and these enzymes recognize the same natural substrates 54, 55, 56. The extended substrate specificity for these enzymes is almost identical. These enzymes recognize Tyr, Asp and Val at the P3 position, Phe and Pro at the P2 position, and Asp and Met at the P4 position. GrK also recognizes Leu, Phe and Trp at the P4 position and prefers Glu and Gln at P3. These results reveal substantial differences between GrA and K, particularly at the S3 and S4 subsites. Few differences between the substrates cleaved by these enzymes in vitro have been identified, and GrA and K might have different physiological functions 56. Van Damme et al. 52 also explored and identified GrA substrate preferences in 2010 using a proteomic approach, with similar results. The utility of proteomics in granzyme specificity determination is reviewed elsewhere 15.
Granzyme B (GrB) is the only known serine protease with restricted specificity for Asp at the P1 position 57. This feature is common in caspases, which belong to the cysteine protease family. GrB and caspases have similar key functions, including the induction of apoptosis, although they belong to different protease families with different evolutionary origins 57. In 1998, Harris and colleagues analyzed the extended substrate specificity of GrB using filamentous (M13) phage display and explored the P3, P1′ and P2′ positions with an I‐P3‐P‐D‐P1′‐P2′ library with only one defined position; the other sites were fixed. Based on the results, GrB recognizes Glu at the P3 position and Gly at the P2′ position 58.
Unlike GrA and K, GrM and H have different preferences at the P1 position, thereby facilitating their differentiation. Several methods have been used to study GrM and H, including positional scanning libraries of coumarin substrates as well as screens of individual p‐nitroanilide and coumarin substrates 59. GrM hydrolyzes the peptide bond after the aliphatic amino acids Ile and Met and does not hydrolyze the bond after Phe. GrH also prefers hydrophobic amino acids at the P1 position, including Phe, but does not hydrolyze the peptide bond after Met 59, 60, 61. Powers and colleagues used a series of peptide thiobenzyl esters to determine the primary specificity of GrM, which exhibited a preference for Pro at P2 and Ala, Ser and Asp at the P3 position 62. Using P1‐Leu and P1‐Met PS‐SCL, Mahrus et al. 59 explored the P2–P4 subsites and showed that the substrate specificity profiles of the two libraries were similar at the P4 and P2 positions, but were slightly different at the P3 position, which might be an effect of subsite cooperativity. Further studies on the extended P4‐P4′ specificity were also conducted 63.
Mahrus et al. 64 used the PS‐SCL approach to analyze GrM, K, H, B and A, and they identified differences in substrate preferences, allowing further investigations of specific inhibitors and ABPs.
In summary, a few peptide sequences for granzymes have been identified using PS‐SCL, phage display and proteomics, which will aid future research into discriminating their individual physiological functions. The Van Damme group performed hierarchical clustering of the surface surrounding the granzyme active sites using proteomics, which confirmed observations based on chemical methods, that the S1 pocket is the most restrictive and binds Asp for GrB, Arg for GrA and K, Met/Leu for GrM and Phe for GrH, while the S2–S4 pockets bind a broad range of amino acid residues 15, due to subtle differences in the primary specificity profiles. However, more research is necessary to distinguish among the granzymes, particularly GrA and K.
Kallikreins
The human tissue kallikrein family comprises at least 15 serine proteases (hK1–hK15), which are encoded by the largest protease gene cluster and are expressed in almost every tissue, including brain, breast and skin, among others 65. These enzymes possess at least 30% sequence identity, a similar molecular size (25–30 kDa) and (chymo)trypsin specificities 7.
Debela et al. determined the extended substrate specificity of seven human tissue kallikreins, hK3/prostate‐specific antigen (PSA), hK4, hK5, hK6, hK7, hK10 and hK11, using a combinatorial peptide library. Based on their data, hK3 and hK7 show chymotrypsin–trypsin‐like activity and hydrolyze peptide bonds after large hydrophobic (Tyr), aliphatic (Ala and Nle) or polar amino acids (Arg and Lys). The kallikreins hK4, hK5 and hK6 exhibit trypsin‐like specificity, with a strong preference for P1‐Arg, whereas hK10 and hK11 display dual specificity and hydrolyze P1 peptide bonds after both aliphatic (Nle, Met, and Ala) and basic amino acid residues (Lys and Arg). All investigated enzymes demonstrate broad specificity at the P4 and P3 subsites, but hK4 does not recognize Pro and hK5 does not recognize Asp at P3. All investigated kallikreins demonstrate subtle differences at the P2 and at the P1 positions, but hK3/hK7 and hK10/hK12 have similar substrate specificities 7. The PS‐SCL results agree with the structural data, and therefore might be helpful in future research. However, specific substrates for selected kallikreins have not been synthesized and characterized using these promising specificity profiles. Thus, subsite cooperativity has not been excluded and additional validation is necessary. Nevertheless, the presented data are valuable preliminary results.
PSA and hK2 display 79% sequence identity, which hinders the development of specific substrate, inhibitors and antibodies for these enzymes. In 2000, Wu et al. used a cyclic phage display peptide library to identify peptides specific for active PSA over other serine proteases, including chymotrypsin, CatG, kallikreins, trypsin and hK2 66. The identified sequence binds to the active site of PSA, allowing us to distinguish between active and inactive PSA, in contrast to antibody labeling, which binds both forms. Wu and colleagues used this discovery to developed new immunopeptidometric assays with unique specificities 67.
In addition, Hekim et al. used phage display to identify the specific sequence of a peptide inhibitor of hK2. In this approach, 10‐ and 11‐amino‐acid libraries were used and peptides binding to hK2 were isolated. A specific cyclic peptide substrate with no inhibitory activity and a linear peptide inhibitor (RFKXWW or ARRPXP) with no overlapping specificity toward PSA, chymotrypsin and various trypsin isoenzymes were obtained. These peptides differentiate between PSA and hK2, which was not previously possible 68.
In another approach, de Veer et al. 69 used a sparse matrix library consisting of 125 defined peptides to explore hK5 substrate specificity. GRSR, YRSR and GRNR were reported as the most active sequences, and an inhibitor was subsequently engineered by substituting the substrate sequences in the binding loop (P1, P2 and P4 residues) of sunflower trypsin inhibitor‐1. However, despite their high activity, the designed inhibitors lacked selectivity. The authors then substituted the P2′ residue, which generated a specific hK5 inhibitor with the kinetic constant K i = 4.2 ± 0.2 nm. The inhibitor displayed low activity toward hK7 and 12‐fold selectivity over hK14 69.
One of the most interesting and successful approaches for investigating specific molecules for kallikreins was described by Cloutier et al. 70 and Felber et al. 71. Both groups used the same strategy to modify serpins (endogenous inhibitors of serine proteases), the reactive center loop of which is crucial for inhibiting serine proteases. The amino acids near the scissile bond of the reactive center loop were modified in another approach by extracting structural fragments to obtain specific recombinant hK2 70 and hK14 71 inhibitors. In these approaches, phage display methodology was used to select peptide sequences for hK2 (six variants) and hK14 (two variants). hK14 has a dual (chymo)trypsin‐like substrate specificity, and Felber et al. described four variants of recombinant mutant serpins (two variants for serpin): α‐1‐antitrypsin (AAT) and antichymotrypsin (ACT). In both ACT and AAT, five residues surrounding the scissile bond were replaced with a pentapeptide sequence that was previously obtained using phage display 72. Specificity was tested against enzymes with a broad range of specificity, such as elastase, chymotrypsin, trypsin, plasma kallikreins and thrombin, and then tested against enzymes belonging to the same family, i.e. hK2, hK3, hK5, hK6, hK8 and hK13. One of the two variants of ACT and AAT displayed selectivity for hK14 (with the exception of chymotrypsin). All the variants specifically inhibited hK14, but two ACT‐derived inhibitors formed more stable complexes with hK14, in contrast to the other AAT inhibitors 71. For hK2, sequences were inserted into ACT and the resulting inhibitors were screened against human neutrophil elastase, as well as proteases with chymotrypsin‐like (chymotrypsin, PSA) and trypsin‐like (hK2, hK1, plasma kallikrein, urokinase‐type plasminogen activator) activity; two variants of modified ACT retained specific activity against hK2 70. Using this clever mixed strategy – modification of reactive center loop of serpins with different peptide sequences – the authors were able to distinguish between hK14 or hK2 and a broad range of serine proteases, with the exception of chymotrypsin.
Cysteine proteases
Caspases and paracaspases
Caspases are cysteine proteases that display very narrow preferences at the P1 position, primarily recognizing aspartic acid (with some exceptions) 73. Such primary specificity is quite rare among proteases. However, Wells and colleagues have recently shown that these enzymes can also cleave substrates after glutamic acid and phosphoserine residues, shedding new light on their physiological functions 74. Since the discovery and characterization of caspases 20 years ago, hundreds of papers have been published on their substrates and inhibitors 9, 75. Unfortunately, their exclusive specificity for Asp at the P1 position makes it very difficult to distinguish these enzymes 76. Using PS‐SCL as a tool, Thornberry and colleagues determined the substrate preferences of the S4–S2 pockets of nine human caspases 17. Based on these profiles, caspases were divided into three groups: caspase‐1, ‐4 and ‐5, which prefer the (W/L)EHD sequence, caspase‐3, ‐7 and ‐2, which recognize DE(V/H)D peptides, and caspase‐6, ‐8 and ‐9, which favor (V/L)EHD motifs. Nevertheless, the specificity matrix clearly shows that these proteases display overlapping substrate specificity, and the ‘optimal sequences’ proposed by the authors may not be specific for only one enzyme. Indeed, later work by other groups has shown that short inherent substrates containing tetrapeptide motifs display cross‐reactivity among the caspases, making it very difficult to use these substrates in cell extracts that contain different concentrations of several caspases 4, 77. In another approach to design caspase‐specific substrates, Stennicke and colleagues synthesized a panel of IQF substrates (Fig. 4) 78. This strategy also allowed the authors to screen caspase preferences in the prime region (i.e. to the right of the scissile bond). A kinetic analysis revealed that all tested caspases (1, 3, 6, 7 and 8) preferred small residues (Gly, Ser and Ala) at P1′, which could not be used as a discrimination factor to improve the specificity of the synthesized substrates. The inability to design caspase‐specific substrates using a PS‐SCL approach or by scanning internally quenched substrates could be due to their simple architecture. In these chemical approaches, the substrates are short linear peptides selected from a pool of peptide mixtures without an in‐depth caspase subsite cooperativity analysis. Biological approaches represent an alternative strategy for substrate identification. One such strategy tested on caspases was the application of cellular libraries of peptide substrates (CLiPS), developed by Boulware et al. 79, in which potential caspase substrates were randomly synthesized by bacteria and then subjected to fluorescence‐activated cell sorting analysis. This technique not only provided a general map of caspase preferences but also revealed detailed kinetic analyses of the substrates. However, the obtained results only confirmed that the preferred caspase‐3 cleavage motif is DXVD/G, and new specific sequences were not identified. The main limitation of the described methods (using both synthetic and natural substrates) is that all of these peptides contain only natural amino acids in their structure. A potential solution is an approach that has been recently proposed by Poreba et al. 80. In the HyCoSuL approach, P4–P2 peptide mixtures are enriched with over 100 unnatural amino acids, which provides a better exploration of the chemical space in the caspase active sites and attenuates the issue of overlapping specificity. The specificity of six human apoptotic caspases (3, 6, 7, 8, 9 and 10) toward the P4–P2 positions were screened using this approach. A detailed kinetic analysis of their preferences allowed the development of peptides containing unnatural amino acids that had much greater selectivity indices than the previously reported substrates (Fig. 6). One of these substrates was then used to specifically monitor caspase‐9 activation in HEK293F cytosolic extracts, in which apoptosis is triggered by cytochrome C 80.
Figure 6.
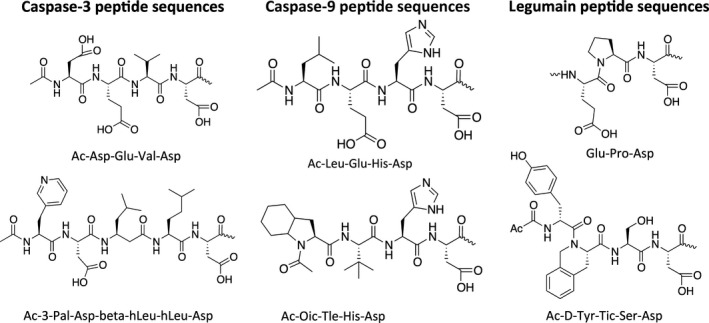
Examples of specific peptide sequences used in substrates and inhibitors for cysteine proteases.
Another challenging issue in distinguishing between caspases is the development of specific inhibitors. Because potent and selective caspase inhibitors appear as potential drug candidates, this area of research has been explored more rigorously than the identification of caspase substrates. To date, thousands of caspase inhibitors have been synthesized and evaluated 9. Some caspase inhibitors have been synthesized and used to study apoptosis in vitro and in vivo, whereas others are primarily used in pharmaceutical research 81, 82, 83. In general, almost all strategies for developing caspase‐specific inhibitors focus on (a) optimizing a peptide (or peptidomimetic) scaffold (non‐prime region), (b) exploring the S1′ pocket by introducing various warheads (cysteine traps) into PS‐SCL‐derived peptides (mostly DEVD, IETD and YVAD), or (c) optimizing lead non‐peptide scaffold structures, such as isatins, indolones and quinolines. In previous studies, synthetic caspase inhibitors were simple peptides equipped with an electrophilic warhead (e.g. acyloxymethylketone (AOMK), fluoromethyl ketone, or an aldehyde) 9. The peptide motifs were based on the PS‐SCL results reported by Thornberry et al. 17. However, these tools were subsequently shown to display only limited selectivity toward individual caspases 84, 85. As the inclusion of natural amino acids in the P4‐P1 positions has not produced specific inhibitors, unnatural amino acids have been introduced into peptide scaffolds. This work was pioneered by Bogyo and colleagues, who synthesized a P4–P2 combinatorial library of AOMK inhibitors containing 41 unnatural amino acids at each position 86. The same approach was further applied by Wolan and colleagues, who synthesized a P5–P2 peptide library containing an aldehyde as an electrophilic warhead 87, 88, 89. The introduction of unnatural amino acids into the peptide sequence resulted in increased inhibitor selectivity. Chemical approaches for developing caspase‐specific inhibitors and ABPs have recently been reviewed by Poreba et al. 9.
Mucosa‐associated lymphoid tissue lymphoma translocation protein 1 (MALT1) is a paracaspase and belongs to the cysteine protease family. MALT1 contains a domain homologous to caspases, and hence it was defined by Dixit et al. as a ‘paracaspase’ 90. MALT1, along with caspases, belongs to the CD clan and has the catalytic His–Cys diad characteristic of caspases. To date, only a few reports have described MALT1 substrates and probes. Probes were used on Jurkat cells, but extensive selectivity studies toward other enzymes have not been performed 91, 92, 93. Interestingly, the probes proposed by Xin et al. bind to MALT1, but a few other signals were also observed. Therefore, there is a need to distinguish between MALT1 and other proteases that share Arg P1 specificity. It is necessary to determine the precise substrate specificity to design optimal substrates and inhibitors. The Salvesen group used PS‐SCL to identify a few interesting features of MALT1. MALT1 has narrow specificity at the P4 and P1 positions, hydrolyzing Leu and Arg, respectively, but it has broad specificity at P3 and P2 91. This finding was helpful in the design of an ABP for this enzyme 92. However, this ABP only contains natural amino acid residues and its specificity toward other proteases remains unclear. It might be beneficial to use unnatural amino acids to further optimize this probe to reduce potential cross‐reactivity with other proteolytic enzymes.
Cysteine cathepsins
Similar to caspases, cathepsins are group of cysteine proteases that display overlapping substrate specificity. A broad study of the substrate preferences of these enzymes was first described by Choe et al. 7, who used a PS‐SCL approach to dissect the P4–P1 substrate specificity of six human cysteine cathepsins (L, V, K, S, F and B). The six cathepsins exhibited similar substrate preferences; the only significant difference was that cathepsin K recognized Pro at P2, whereas all other cathepsins did not. This distinguishing feature was sufficient to develop a cathepsin K‐specific tetrapeptide substrate that was not hydrolyzed by other family members. However, an AOMK inhibitor with this sequence displayed off‐target activity by also inhibiting cathepsin B. Another strategy for the rapid development of cathepsin substrates was substrate activity screening (SAS), proposed by Patterson et al. 94, 95, 96. In this method, a library of aminocoumarin derivatives with various N‐acyl groups was screened for activity against cathepsin S and the most active substrates were further converted into inhibitors. With the exception of PS‐SCL and SAS, no other rapid methods for the discovery of selective cathepsin substrates have been reported. Most substrates developed for cathepsins are the result of extensive SAR studies, in which individual fluorogenic peptide substrates with variations in only one position were screened against a panel of cathepsins and the best candidates were selected for further optimization 97, 98, 99.
Similar to other proteases, the development of cathepsin‐specific inhibitors mostly relies on studying a peptide or peptidomimetic backbone in the unprimed site or the investigation of electrophilic warheads at P1′; some of these examples have recently been reviewed 100. One classic example for distinguishing between cathepsins is the optimization of E‐64, a natural broad‐spectrum inhibitor. Katunuma and colleagues have developed several highly selective cathepsin B (CA‐030 and CA‐074) and cathepsin L (CLIK‐148 and CLIK‐195) inhibitors by replacing the substituents in the epoxide electrophilic warhead and changing the non‐prime recognition group 101. Other cathepsin S‐ (CLIK‐060) and cathepsin K‐specific inhibitors (CLIK‐163) were developed based on the structures of leupeptin and pyridoxal phosphate, respectively 101. These examples clearly show that optimization of lead structures (mostly broad spectrum inhibitors) is an excellent strategy for distinguishing closely related enzymes. Another strategy for the development of potent and selective cathepsin inhibitors is the previously mentioned SAS, in which the best substrates are converted into potent inhibitors by replacing the aminocoumarin reporter group with an electrophilic pharmacophore 95. One such seminal example is the cathepsin S probe (BMV157) developed by Oresic Bender et al. 102. BMV157 is a near‐infrared quenched‐activity‐based probe that becomes fluorescent only when bound to the cathepsin S active site. This molecule contains a Cy‐5 fluorophore attached to a P1‐Lys residue, a peptidomimetic fragment in the P3–P2 region and an acyloxymethylketone warhead equipped with QSY21, a bulky quenching group. The specificity of this probe toward cathepsin S was tested in both in vivo and in vitro models, demonstrating the wide range of its biological applications.
Legumain
Legumain (asparaginyl endopeptidase) is a cysteine protease that is up‐regulated in inflammatory diseases and multiple human cancers. Legumain is also very active in the tumor microenvironment; thus, it is thought to promote tumorigenesis. This protease shares some similarities with both previously described protease groups, caspases and cysteine cathepsins. An analysis of the tertiary structure of legumain assigned it to the CD clan (together with caspases, gingipains and separases). However, the localization of this enzyme is mainly lysosomal/endosomal, overlapping with cysteine cathepsins 103. Thus, to specifically target legumain, there is a need to distinguish this protease from caspases and cysteine cathepsins. However, this goal is very challenging as legumain displays narrow specificity at the P1 position, recognizing only Asn and Asp residues. The first (quenched) activity‐based probe that specifically targeted legumain in vivo (LE‐28) was developed by the Bogyo group 104. It contained a three‐peptide scaffold (Glu‐Pro‐Asp) equipped with an acyloxymethylketone warhead and flanked by two bulky groups (Cy‐5 as a fluorophore and QSY21 as a fluorescence quencher) (Fig. 4). As Glu‐Pro‐Asp is the preferred motif of caspases, the specificity of LE‐28 was obtained not by sequence itself, but by introducing bulky groups that drive LE‐28 cellular uptake into the lysosomes. Another approach to distinguish legumain from caspases was recently proposed by Poreba et al. 12. Using HyCoSuL, the authors demonstrated that the legumain specificity at the P4 position was very broad, tolerating even d‐amino acids (Fig. 6). Thus, it was possible to use counter selection to extract a P4–P1 tetrapeptide motif (with Asp at P1) that was not recognized by apoptotic caspases. This motif was then used to develop a legumain‐specific substrate, inhibitor, and activity‐based probe for tracking legumain activity in various biological models. This study provided proof of principle for the use of the CoSeSuL approach to develop specific tools for proteases that display broad substrate specificity.
Deubiquitylating and desumoylating enzymes
The human genome encodes approximately 100 DUBs, representing more than 15% of all human proteases. These enzymes also display some similarities in substrate preferences. High‐throughput screening has been one of the most successful strategies applied to differentiate these enzymes. The screening of over 40 000 compounds against the closely related ubiquitin C‐terminal hydrolase L1 (UCH‐L1) and L3 (UCH‐L3) enzymes allowed the development of a UCHL1‐specific inhibitor based on an isatin O‐acyl oxime scaffold 105. In another approach, Borodovsky and colleagues used a chemical ligation method (intein strategy), in which the C terminus of the ubiquitin protein was modified with different warheads 106, 107. Depending on the type of warhead, these probes exhibited some level of specificity toward different DUBs. In another approach, Drag et al. used PS‐SCL to identify the precise substrate specificity of three human DUBs, ovarian tumor 1, isopeptidase T and UCH‐L3, and one viral ubiquitin‐specific protease, the papain‐like protease from the severe acute respiratory syndrome coronavirus 108. A kinetic analysis of the P4–P2 positions revealed differences between these enzymes that can be further exploited in the design of specific substrate candidates. The PS‐SCL library approach was also used to analyze the substrate specificity of human desumoylating enzymes (sentrin‐specific proteases; SENPs). The results from the screen of this library clearly showed similar substrate specificities for natural amino acids in the P4, P3 and P2 positions between SENPs. The design of specific probes for DUBs and SENPs remains a substantial challenge.
Metalloproteinases
Matrix metalloproteinases
Matrix metalloproteinases (MMPs) include the 23 members of the zinc‐dependent endopeptidase family in the metzincin class of metalloendopeptidases that share a common domain structure 109. The most common classification of MMPs is based on the historical assessment of their catalytic preferences and their cellular localization. Therefore, one can distinguish four groups of MMPs: collagenases, gelatinases, stromelysins and membrane‐type MMPs. Nevertheless, there are several MMPs that are not classified into these traditional groups 110, 111.
MMPs were initially associated with cancer progression due to their ability to degrade connective tissues between the linings of blood vessels and between cells. These enzymes are involved in a wide variety of biological processes, such as tissue remodeling associated with angiogenesis, morphogenesis and tissue repair 112. Due to their important roles in humans, MMPs are subjected to very precise spatial and temporal control to prevent them from becoming destructive 109. Over the last 15 years, many studies have focused on identifying selective molecules to discriminate between MMPs. The application of the high‐throughput proteomic identification of protease cleavage site method, which investigates primary and extended substrate specificity, profiled MMPs 1, 2, 3, 7, 8, 9, 12, 13 and 14 using 4300 biologically diverse peptides. This study allowed the general specificity features shared by most MMPs to be determined and explored features characterizing particular MMPs 110, 111. However, the greatest challenge in distinguishing between MMPs in the above‐mentioned groups, as well as between these groups, is the ability of MMPs to hydrolyze similar peptide substrates. In 2003 MMP‐2 and MMP‐9 were shown by Chen et al. to exhibit overlapping substrate recognition, but they could be distinguished by their S2 pocket preferences 113. Precise kinetic studies of the synthetic peptides showed that replacement of one amino acid residue in the P2 position can convert a relatively selective MMP‐2 substrate into an MMP‐9 substrate. This finding underlined the significance of the P2 position and suggested that the S2 subsites of MMP‐2 and MMP‐9 interact with substrates differently 113.
In 2014, Ratnikov et al. compared the MMP cleavage yield of a selected and extended set of phage peptide substrates from approximately 64 million sequences and quantified the structure‐function dependence of eight different MMPs representing three phylogenetic subfamilies 114. The chosen proteases included MMP‐2 and ‐9 from the gelatinase subfamily, transmembrane MMPs 14, 15, 16 and 24; and MMP‐17 and ‐25 from the GPI‐MMP subfamily that contain a glycophosphatidyl inositol (GPI) cell surface anchor. The calculated second‐order rate constants (k obs) constitute a good distance measure of the functional similarities between the matrix metalloproteinase branches 114.
Based on the pathological role of MMPs in disease, attempts have been made to identify drugs that harness the therapeutic potential of MMP inhibition. More than 50 inhibitors targeting MMP activity have been developed, but all have failed during clinical trials due to a lack of specificity toward the targeted enzymes 1.
Preliminary trials to develop MMP inhibitors have focused on compounds that chelate the catalytic Zn2+ ion and possess backbones mimicking the natural peptide substrate for the individual MMPs. The first‐generation MMP inhibitors were composed of a collagen backbone and a hydroxamic acid (–C(O)NHOH) 115. Based on this scaffold, several collagen‐based peptidomimetic hydroxamates were developed, including ilomastat, marimastat and batimastat. Batimastat, the first MMP inhibitor to enter clinical trials 116, showed strong inhibition toward MMP‐1 (IC50 = 3 nm), ‐2 (IC50 = 4 nm), ‐7 (IC50 = 6 nm) and ‐9 (IC50 = 1 nm); however, the low specificity observed in the clinical trials was disappointing. A new generation of hydroxamate‐based MMP inhibitors consisting of a substituted aryl, a sulfonamide and a zinc‐binding group was proposed. MMI‐270 and MMI‐166 were identified as selective inhibitors of MMP‐2, ‐9 and ‐14 117.
Reverse hydroxamates and non‐hydroxamate inhibitors were developed to avoid the limitations associated with hydroxamate‐based inhibitors, such as metabolic inactivation and cross‐reactivity with other metalloproteases. As the structures of MMPs were revealed by crystallography, the next generation of MMP inhibitors was no longer restrained to substrate‐like compounds and new molecules with diverse peptidomimetic and non‐peptidomimetic scaffolds were designed 118. The investigation of numerous different structures led to the identification of selective MMP inhibitors. For example, the thiol‐based inhibitor SB‐3CT, which is composed of a deep‐pocket‐binding diphenylether scaffold, was specific for MMP‐2 and ‐9 with inhibition constants (K i) of 14 and 600 nm, respectively 119.
Due to the high structural homology between different members of the MMP family, it is difficult to distinguish the enzymes. Recent studies have turned to targeting less conserved sites of MMPs instead of the previously targeted catalytic sites. MMPs, like all proteolytic enzymes, possess prime and non‐prime subsites. However, the S1′ pocket was the most important site for substrate recognition 109, and thus exhibits the greatest degree of variability among MMPs in terms of the amino acid sequence and pocket shape. Based on this knowledge, a number of S1′ pocket‐specific inhibitors were designed that showed improved selectivity for MMP‐2 over MMP‐9 120.
Another trend in distinguishing between members of the MMP family is the application of antibody‐based therapeutics that utilize highly selective and potent function‐blocking antibodies. An extremely selective MMP‐14 inhibitor, DX‐2400, was identified using a human antibody phage display library and automated selection strategies 121. Further examples can be found in the literature 122, 123.
Almost all previous studies that focused on distinguishing MMPs constitute a useful starting point for future research. In the 1998 study by Mucha et al., the use of unusual amino acids improved MMP substrate specificity 124 and provided further perspectives for this type of study.
Exopeptidases
Metallo‐aminopeptidases
Aminopeptidases are exopeptidases that hydrolyze one or two amino acid residues from the N‐terminal fragment of the substrate 125, 126. Enzymes belonging to the aminopeptidase M1 family share several common features that are important for catalytic activation, such as the highly conserved HEXXH(X)18E zinc‐binding and GAMEN motifs 127. Until recently, aminopeptidase studies using selective substrates were generally neglected, mainly due to the lack of specific chemical tools, and only a few generic substrates were used in aminopeptidase studies. Classic examples include alanine and leucine substrates used for the M1 and M17 family, respectively 3. The aminopeptidase fingerprint approach published by Drag et al. overcame this limitation 128 and led to the first scrupulous analysis of aminopeptidase N specificity in different species. A defined fluorogenic library with the sequence NH2‐X‐ACC, where X represents amino acid residues enriched with 42 unnatural amino acid derivatives, was applied. Subsequently, the library was later expanded with the addition of several new derivatives and has been applied to several aminopeptidases from humans and other organisms (e.g. bacteria, animals and plants) 129, 130, 131. These studies have led to the general conclusion that unnatural amino acids are more preferred by aminopeptidases than natural amino acids (Fig. 7). Moreover, this approach was applied to distinguish aminopeptidases in cell lysates and provide clear information about the presence of individual aminopeptidases, which was not previously possible 5. To date, this aminopeptidase fingerprint approach using unnatural amino acids is the best‐known method to distinguish between aminopeptidases.
Figure 7.
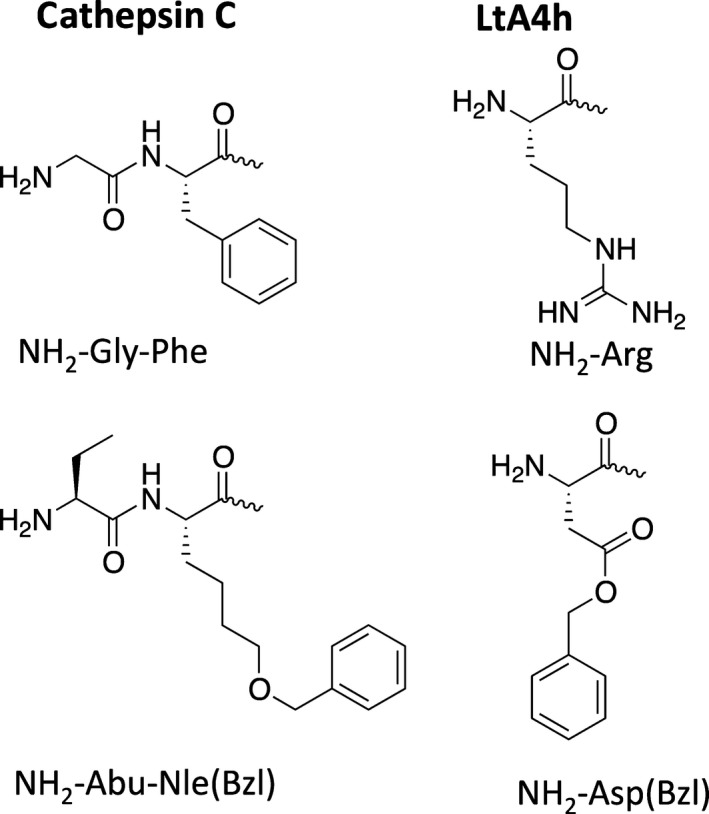
Examples of specific peptide sequences used in substrates for aminopeptidases. LtA4h, leukotriene A4 hydrolase.
Carboxypeptidases
Carboxypeptidases are enzymes responsible for the hydrolysis of C‐terminal amino acids of their peptide substrates, and this attribute underlies their participation in various essential biological processes. To date, there are 30 known representatives of human carboxypeptidases or peptidases with established carboxypeptidase activity (reported in the MEROPS database) 132, 133. They are classified by the chemical nature of their active site. Therefore, one can distinguish metallo‐carboxypeptidases (families M2, M14, M20 and M28), serine carboxypeptidases (families S10 and S28) and cysteine carboxypeptidases (family C1). The substrate specificity of numerous carboxypeptidases has been determined using various techniques, primarily kinetic analyses of synthetic substrates and biologically relevant peptides 134, as well as proteome‐derived peptide libraries 135. In 2002, Barinka et al. used a novel assay to determine the hydrolytic activity of recombinant human glutamate carboxypeptidase II, which was based on the fluorimetric detection of released α‐amino groups, and adopted obtained results for enzyme characterization 136. Dipeptide libraries were synthesized via solid‐phase peptide synthesis, and the action of pure recombinant enzymes on a panel of dipeptides was tested. The release of a C‐terminal amino acid residue was determined fluorometrically utilizing OPA derivatization 136. The substrate specificity of carboxypeptidase M was determined in similar way by Deiteren et al. in 2007, the difference being the utilization of a series of benzoyl‐Xaa‐Arg substrates 137. Not long after, Fricker and colleagues defined the enzymatic preferences of carboxypeptidase A4 and A6 for the first time by using the overlapping strength of three interdependent approaches – a kinetic analysis of chromogenic substrates, the individual cleavage of synthetic peptides and a quantitative peptidomics mass spectrometry‐based approach 138, 139. To date, there have been many further studies of carboxypeptidases and their substrate specificities, which can be found in the literature 140.
One enzyme, few catalytic activities
Proteasome
The ubiquitin–proteasome system plays a pivotal role in the controlled degradation of proteins in eukaryotic cells. Its proteolytic action on regulatory proteins makes it one of the most important enzyme systems, and it is involved in almost all biological processes, including cell development and differentiation. Proteolysis of the substrates occurs within the 20S proteasome core particle, which is a barrel‐shaped protein complex containing four stacked rings that are composed of seven α subunits and seven β subunits, all of which are unique. Only three of these subunits possess the catalytic threonine residue required for proteolytic cleavage: the β1, β2 and β5 subunits 141.
To date, studies conducted by Harris et al. have been the most important because they provided primary knowledge of the substrate specificity of individual proteasome subunits. The primary N‐terminal and extended substrate specificity of the human 20S proteasome were defined in the presence of 11S proteasome activators using a comprehensive, composite set of fluorogenic substrates. The data promoted the full characterization of proteasome activity and the dissection of the proteolytic effects of the individual proteasome subunits, information that is essential for the design of more specific substrates and inhibitors 141. This approach constituted a powerful tool to determine the non‐prime residues of a peptide substrate. Other methods were employed to examine prime‐side substrate specificity. For example, Lesner et al. employed combinatorial chemistry with internally quenched fluorescent peptide substrates to synthesize peptide substrates optimized at both the prime and non‐prime regions of the peptide chain. This approach was used to determine the trypsin‐like subunit substrate specificity and select the best substrates (ABZ‐Val‐Val‐Ser‐Arg‐Ser‐Leu‐Gly‐Tyr(3NO2)‐NH2, k cat/K m = 934 000 m −1·s−1; and ABZ‐Val‐Val‐Ser‐GNF‐Ala‐Met‐Gly‐Tyr(3NO2)‐NH2, k cat/K m = 1 980 000 m −1·s−1), which showed significant selectivity over different proteasome units 142. Later, the Lesner group investigated the chymotrypsin‐like activity of the 20S proteasome subunit (β5) and identified the selective substrate ABZ‐Val‐Val‐Ser‐Tyr‐Ala‐Met‐Gly‐Tyr(3NO2)‐NH2 and determined its kinetic parameters (k cat/K m = 9.7 × 105 m −1·s−1, k cat = 8 s−1). These were the first studies on the chymotrypsin‐like activity alone, leading to the optimization of the prime area of the substrate and avoiding non‐selective processing by other proteases with overlapping specificity 143.
Many natural or synthetic compounds can inhibit protein degradation by the proteasome. Natural inhibitors include lactacystin, epoxyketones and the TMC‐95 cyclic peptides. The first synthetic compounds reported to inhibit proteasome activity were peptide aldehydes such as Z‐Leu‐Leu‐Leu‐al (MG132), Z‐Ile‐Glu(OBut)‐Ala‐Leu‐al (PSI), or peptide vinyl sulfones. These compounds bind to the active sites within the 20S core particle. However, these reactive compounds have many drawbacks, including a lack of specificity toward individual proteasome active subunits. Additionally, most of these compounds are also known to inhibit serine and cysteine proteases (e.g. calpains and cathepsins); therefore, it would be inadvisable to use these compounds as drugs 144.
To date, the three most important proteasome inhibitors that have been approved as drugs are bortezomib 145, carfilzomib 146 and ixazomib 147. All three preferentially bind to the β5 subunit, which is responsible for the chymotrypsin‐like activity of the proteasome, but they also inhibit the caspase‐ and trypsin‐like proteolytic sites at higher doses. G. de Bruin and colleagues reported the most significant attempt to distinguish particular proteasome subunits. Based on a previous study, this group presented a set of ABPs that promoted the gel‐based detection of all catalytic human proteasome subunits and also developed specific inhibitors for individual proteasome activities 148.
Recently, the Bogyo group designed inhibitors based on amino acid preferences specific to Plasmodium falciparum and not the human proteasome 149. This elegant work showed that proteasome orthologs can be distinguished using natural amino acids; in this case, the bulky tryptophan was a key amino acid. The human proteasome showed chymotrypsin‐like (bulky amino acids), trypsin‐like (basic amino acids) and caspase‐like (acidic, Asp) activity, whereas the P. falciparum proteasome preferred aromatic Trp at the S2 and S3 pockets 149.
Summary
Our knowledge about proteases has increased dramatically over the years (Table 1). However, studies were typically restricted to one particular group of enzymes, not individual enzymes, due to the lack of specific substrates, inhibitors and ABPs. For several years, many studies have attempted to identify specific molecules that act on different enzymes with similar substrate preferences (Fig. 2). A number of modern design techniques have been applied to identify protease‐specific sequences, including phage display, PS‐SCL, CLiPS and proteomics. However, these methods are limited to natural amino acids, which are not always sufficient to distinguish between closely related enzymes such as caspases, neutrophil serine proteases, aminopeptidases or cathepsins. The recently described HyCoSuL and CoSeSuL techniques incorporate a broad range of unnatural structures and are currently the methods of choice to distinguish between enzymes with similar substrate specificities that cannot be successfully targeted with chemical tools possessing only natural amino acids.
Table 1.
Examples of strategies used in substrates, inhibitors and activity‐based probes design for indistinguishable proteases
| Enzyme | Strategy | Specific tool | Specificity | Reference |
|---|---|---|---|---|
| Neutrophil serine proteases | IQF | ± | Within the enzyme family; in biological extracts | 38, 150, 151 |
| HyCoSuL | ✓ | Within the enzyme family; activity‐based probe can be used in cellular assays | 19, 40 | |
| Multiple non‐peptide inhibitors | ± | Within the enzyme family, in vitro assays | 42 | |
| Peptide inhibitors | ± | Within the enzyme family | 43, 44 | |
| Granzymes | Phage display | ± | Within the enzyme family | 58 |
| Proteomics | ± | Within the enzyme family | 52 | |
| PS‐SCL | ± | Within the enzyme family | 64 | |
| Kallikreins | PS‐SCL | ± | Within the enzyme family | 7 |
| Phage display | ✓ | Within the enzyme family | 66, 67, 68, 152 | |
| Sparse matrix library and modification of sunflower trypsin inhibitor‐1 | ✓ | Within the enzyme family | 69 | |
| Serpin modifications | ✓ | Within the enzyme family | 70, 71 | |
| Caspases | PS‐SCL | ± | Within the enzyme family | 17 |
| IQF substrates | ± | Within the enzyme family | 78 | |
| CLiPS | − | — | 79 | |
| HyCoSuL | ✓ | Within the enzyme family; starting point for selective probe design, in vitro assay (cell lysates) | 80 | |
| PS‐SCL‐based inhibitors | ± | Within the enzyme family | 84, 85 | |
| Peptide inhibitors with unnatural amino acids | ± | Within the enzyme family | 86, 87, 88, 89 | |
| P1′ (warhead) SAR studies (peptide and peptidomimetic) | ± | Within the enzyme family | Reviewed in 9 | |
| Non‐peptide, SAR studies on lead structures | ± | Within the enzyme family | ||
| Cathepsins | PS‐SCL | ± | Within the enzyme family | 7 |
| SAS | ± | Within the enzyme family | 94, 95 | |
| IQF and peptide–fluorophore substrates | ± | Within the enzyme family | Multiple examples | |
| Optimization of broad‐spectrum lead structure | ✓ | Within the enzyme family, in vitro assays, imaging of cathepsin S | Reviewed in 101, 102 | |
| Case‐by‐case optimization of electrophilic warhead | ± | Within the enzyme family | Reviewed in 100, 153 | |
| Proteomics | ± | Within the enzyme family | 154, 155 | |
| DUBs | PS‐SCL screening | ± | Within the enzyme family | 108 |
| High‐throughput screening | ✓ | Within the enzyme family | 105 | |
| Metallo‐aminopeptidases | PS‐SCL enriched with unnatural amino acids | ✓ | Within the enzyme family; in cell lysates | 5, 129, 130 |
| MMPs | Phage display | − | Within the enzyme family | 114 |
| Substrate profiling | ± | Within the enzyme family | 113 | |
| Proteomic identification of protease cleavage site | − | Within the enzyme family | 110, 111 | |
| Synthetic substrates with unnatural amino acids | − | Within the enzyme family | 124 | |
| Proteasome 20S | PS‐SCL | ± | Within the enzyme family | 141 |
| IQF | ✓ | Within the enzyme family | 142, 143 | |
| Natural inhibitor modifications | ± | Within the enzyme family | 144, 145, 146 | |
| Unnatural amino acids in peptide scaffold | ✓ | Within the enzyme family; cellular assays (P. falciparum schizonts) | 147 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Author contributions
PK, MP, KG and MD wrote and reviewed the manuscript.
Acknowledgements
This project received funding from the Polish National Center of Science under grant agreements 2014/13/B/ST5/00240 (to MD), 2014/14/M/ST5/00619 (to MD), and 2013/11/N/ST5/01995 (to PK) as well as the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska‐Curie grant agreement No 661187 (to MP). PK is the beneficiary of a START scholarship from the Foundation for Polish Science. KG is the beneficiary of a Diamentowy Grant in Poland.
Contributor Information
Paulina Kasperkiewicz, Email: paulina.kasperkiewicz@pwr.edu.pl.
Marcin Drag, Email: marcin.drag@pwr.edu.pl.
References
- 1. Drag M & Salvesen GS (2010) Emerging principles in protease‐based drug discovery. Nat Rev Drug Discov 9, 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Turk B (2006) Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov 5, 785–799. [DOI] [PubMed] [Google Scholar]
- 3. Rawlings ND, Tolle DP & Barrett AJ (2004) MEROPS: the peptidase database. Nucleic Acids Res 32, D160–D164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McStay GP, Salvesen GS & Green DR (2008) Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ 15, 322–331. [DOI] [PubMed] [Google Scholar]
- 5. Byzia A, Szeffler A, Kalinowski L & Drag M (2016) Activity profiling of aminopeptidases in cell lysates using a fluorogenic substrate library. Biochimie 122, 31–37. [DOI] [PubMed] [Google Scholar]
- 6. Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA & Craik CS (2006) Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem 281, 12824–12832. [DOI] [PubMed] [Google Scholar]
- 7. Debela M, Magdolen V, Schechter N, Valachova M, Lottspeich F, Craik CS, Choe Y, Bode W & Goettig P (2006) Specificity profiling of seven human tissue kallikreins reveals individual subsite preferences. J Biol Chem 281, 25678–25688. [DOI] [PubMed] [Google Scholar]
- 8. Poreba M & Drag M (2010) Current strategies for probing substrate specificity of proteases. Curr Med Chem 17, 3968–3995. [DOI] [PubMed] [Google Scholar]
- 9. Poreba M, Szalek A, Kasperkiewicz P, Rut W, Salvesen GS & Drag M (2015) Small molecule active site directed tools for studying human caspases. Chem Rev 115, 12546–12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deu E, Verdoes M & Bogyo M (2012) New approaches for dissecting protease functions to improve probe development and drug discovery. Nat Struct Mol Biol 19, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rut W, Kasperkiewicz P, Byzia A, Poreba M, Groborz K & Drag M (2015) Recent advances and concepts in substrate specificity determination of proteases using tailored libraries of fluorogenic substrates with unnatural amino acids. Biol Chem 396, 329–337. [DOI] [PubMed] [Google Scholar]
- 12. Poreba M, Solberg R, Rut W, Lunde NN, Kasperkiewicz P, Snipas SJ, Mihelic M, Turk D, Turk B, Salvesen GS et al (2016) Counter selection substrate library strategy for developing specific protease substrates and probes. Cell Chem Biol 23, 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vizovišek M, Vidmar R, Fonović M & Turk B (2016) Current trends and challenges in proteomic identification of protease substrates. Biochimie 122, 77–87. [DOI] [PubMed] [Google Scholar]
- 14. Sanman LE & Bogyo M (2014) Activity‐based profiling of proteases. Annu Rev Biochem 83, 249–273. [DOI] [PubMed] [Google Scholar]
- 15. Plasman K, Maurer‐Stroh S, Gevaert K & Van Damme P (2014) Holistic view on the extended substrate specificities of orthologous granzymes. J Proteome Res 13, 1785–1793. [DOI] [PubMed] [Google Scholar]
- 16. Rano TA, Timkey T, Peterson EP, Rotonda J, Nicholson DW, Becker JW, Chapman KT & Thornberry NA (1997) A combinatorial approach for determining protease specificities: application to interleukin‐1beta converting enzyme (ICE). Chem Biol 4, 149–155. [DOI] [PubMed] [Google Scholar]
- 17. Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia‐Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP et al (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 272, 17907–17911. [DOI] [PubMed] [Google Scholar]
- 18. Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA & Craik CS (2000) Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci USA 97, 7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kasperkiewicz P, Poreba M, Snipas SJ, Parker H, Winterbourn CC, Salvesen GS & Drag M (2014) Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc Natl Acad Sci USA 111, 2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yaron A, Carmel A & Katchalski‐Katzir E (1979) Intramolecularly quenched fluorogenic substrates for hydrolytic enzymes. Anal Biochem 95, 228–235. [DOI] [PubMed] [Google Scholar]
- 21. Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317. [DOI] [PubMed] [Google Scholar]
- 22. Matthews DJ & Wells JA (1993) Substrate phage: selection of protease substrates by monovalent phage display. Science 260, 1113–1117. [DOI] [PubMed] [Google Scholar]
- 23. Deperthes D (2002) Phage display substrate: a blind method for determining protease specificity. Biol Chem 383, 1107–1112. [DOI] [PubMed] [Google Scholar]
- 24. O'Donoghue AJ, Jin Y, Knudsen GM, Perera NC, Jenne DE, Murphy JE, Craik CS & Hermiston TW (2013) Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PLoS ONE 8, e75141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kettritz R (2016) Neutral serine proteases of neutrophils. Immunol Rev 273, 232–248. [DOI] [PubMed] [Google Scholar]
- 26. Perera NC, Wiesmuller KH, Larsen MT, Schacher B, Eickholz P, Borregaard N & Jenne DE (2013) NSP4 is stored in azurophil granules and released by activated neutrophils as active endoprotease with restricted specificity. J Immunol 191, 2700–2707. [DOI] [PubMed] [Google Scholar]
- 27. Stapels DA, Geisbrecht BV & Rooijakkers SH (2015) Neutrophil serine proteases in antibacterial defense. Curr Opin Microbiol 23, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y & Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. [DOI] [PubMed] [Google Scholar]
- 29. Salvesen G & Enghild JJ (1991) Zymogen activation specificity and genomic structures of human neutrophil elastase and cathepsin G reveal a new branch of the chymotrypsinogen superfamily of serine proteinases. Biomed Biochim Acta 50, 665–671. [PubMed] [Google Scholar]
- 30. Zimmer M, Medcalf RL, Fink TM, Mattmann C, Lichter P & Jenne DE (1992) Three human elastase‐like genes coordinately expressed in the myelomonocyte lineage are organized as a single genetic locus on 19pter. Proc Natl Acad Sci USA 89, 8215–8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sturrock AB, Espinosa R 3rd, Hoidal JR & Le Beau MM (1993) Localization of the gene encoding proteinase‐3 (the Wegener's granulomatosis autoantigen) to human chromosome band 19p13.3. Cytogenet Cell Genet 64, 33–34. [DOI] [PubMed] [Google Scholar]
- 32. Perera NC, Schilling O, Kittel H, Back W, Kremmer E & Jenne DE (2012) NSP4, an elastase‐related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci USA 109, 6229–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Caughey GH, Schaumberg TH, Zerweck EH, Butterfield JH, Hanson RD, Silverman GA & Ley TJ (1993) The human mast cell chymase gene (CMA1): mapping to the cathepsin G/granzyme gene cluster and lineage‐restricted expression. Genomics 15, 614–620. [DOI] [PubMed] [Google Scholar]
- 34. Remold‐O'Donnell E, Chin J & Alberts M (1992) Sequence and molecular characterization of human monocyte/neutrophil elastase inhibitor. Proc Natl Acad Sci USA 89, 5635–5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Travis J & Salvesen GS (1983) Human plasma proteinase inhibitors. Annu Rev Biochem 52, 655–709. [DOI] [PubMed] [Google Scholar]
- 36. Polanowska J, Krokoszynska I, Czapinska H, Watorek W, Dadlez M & Otlewski J (1998) Specificity of human cathepsin G. Biochim Biophys Acta 1386, 189–198. [DOI] [PubMed] [Google Scholar]
- 37. O'Donoghue AJ, Eroy‐Reveles AA, Knudsen GM, Ingram J, Zhou M, Statnekov JB, Greninger AL, Hostetter DR, Qu G, Maltby DA et al (2012) Global identification of peptidase specificity by multiplex substrate profiling. Nat Methods 9, 1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Popow‐Stellmaszyk J, Wysocka M, Lesner A, Korkmaz B & Rolka K (2013) A new proteinase 3 substrate with improved selectivity over human neutrophil elastase. Anal Biochem 442, 75–82. [DOI] [PubMed] [Google Scholar]
- 39. Lechtenberg BC, Kasperkiewicz P, Robinson H, Drag M & Riedl SJ (2015) The elastase‐PK101 structure: mechanism of an ultrasensitive activity‐based probe revealed. ACS Chem Biol 10, 945–951. [DOI] [PubMed] [Google Scholar]
- 40. Kasperkiewicz P, Poreba M, Snipas SJ, Lin SJ, Kirchhofer D, Salvesen GS & Drag M (2015) Design of a selective substrate and activity based probe for human neutrophil serine protease 4. PLoS ONE 10, e0132818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abbenante G & Fairlie DP (2005) Protease inhibitors in the clinic. Med Chem 1, 71–104. [DOI] [PubMed] [Google Scholar]
- 42. Ohmoto K, Yamamoto T, Horiuchi T, Imanishi H, Odagaki Y, Kawabata K, Sekioka T, Hirota Y, Matsuoka S, Nakai H et al (2000) Design and synthesis of new orally active nonpeptidic inhibitors of human neutrophil elastase. J Med Chem 43, 4927–4929. [DOI] [PubMed] [Google Scholar]
- 43. Oleksyszyn J & Powers JC (1989) Irreversible inhibition of serine proteases by peptidyl derivatives of alpha‐aminoalkylphosphonate diphenyl esters. Biochem Biophys Res Commun 161, 143–149. [DOI] [PubMed] [Google Scholar]
- 44. Sienczyk M, Winiarski L, Kasperkiewicz P, Psurski M, Wietrzyk J & Oleksyszyn J (2011) Simple phosphonic inhibitors of human neutrophil elastase. Bioorg Med Chem Lett 21, 1310–1314. [DOI] [PubMed] [Google Scholar]
- 45. Winiarski L, Oleksyszyn J & Sienczyk M (2012) Human neutrophil elastase phosphonic inhibitors with improved potency of action. J Med Chem 55, 6541–6553. [DOI] [PubMed] [Google Scholar]
- 46. Guarino C, Legowska M, Epinette C, Kellenberger C, Dallet‐Choisy S, Sienczyk M, Gabant G, Cadene M, Zoidakis J, Vlahou A et al (2014) New selective peptidyl di(chlorophenyl) phosphonate esters for visualizing and blocking neutrophil proteinase 3 in human diseases. J Biol Chem 289, 31777–31791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Masson D & Tschopp J (1987) A family of serine esterases in lytic granules of cytolytic T lymphocytes. Cell 49, 679–685. [DOI] [PubMed] [Google Scholar]
- 48. Masson D, Nabholz M, Estrade C & Tschopp J (1986) Granules of cytolytic T‐lymphocytes contain two serine esterases. EMBO J 5, 1595–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wilharm E, Parry MA, Friebel R, Tschesche H, Matschiner G, Sommerhoff CP & Jenne DE (1999) Generation of catalytically active granzyme K from Escherichia coli inclusion bodies and identification of efficient granzyme K inhibitors in human plasma. J Biol Chem 274, 27331–27337. [DOI] [PubMed] [Google Scholar]
- 50. Odake S, Kam CM, Narasimhan L, Poe M, Blake JT, Krahenbuhl O, Tschopp J & Powers JC (1991) Human and murine cytotoxic T lymphocyte serine proteases: subsite mapping with peptide thioester substrates and inhibition of enzyme activity and cytolysis by isocoumarins. Biochemistry 30, 2217–2227. [DOI] [PubMed] [Google Scholar]
- 51. Smyth MJ, Sayers TJ, Wiltrout T, Powers JC & Trapani JA (1993) Met‐ase: cloning and distinct chromosomal location of a serine protease preferentially expressed in human natural killer cells. J Immunol 151, 6195–6205. [PubMed] [Google Scholar]
- 52. Van Damme P, Maurer‐Stroh S, Hao H, Colaert N, Timmerman E, Eisenhaber F, Vandekerckhove J & Gevaert K (2010) The substrate specificity profile of human granzyme A. Biol Chem 391, 983–997. [DOI] [PubMed] [Google Scholar]
- 53. Lieberman J (2010) Granzyme A activates another way to die. Immunol Rev 235, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, Kim S, Raja SM, Shi L, Simon MM et al (2008) Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity 29, 720–733. [DOI] [PubMed] [Google Scholar]
- 55. Hua G, Wang S, Zhong C, Xue P & Fan Z (2009) Ignition of p53 bomb sensitizes tumor cells to granzyme K‐mediated cytolysis. J Immunol 182, 2152–2159. [DOI] [PubMed] [Google Scholar]
- 56. Bovenschen N, Quadir R, van den Berg AL, Brenkman AB, Vandenberghe I, Devreese B, Joore J & Kummer JA (2009) Granzyme K displays highly restricted substrate specificity that only partially overlaps with granzyme A. J Biol Chem 284, 3504–3512. [DOI] [PubMed] [Google Scholar]
- 57. Ewen CL, Kane KP & Bleackley RC (2012) A quarter century of granzymes. Cell Death Differ 19, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Harris JL, Peterson EP, Hudig D, Thornberry NA & Craik CS (1998) Definition and redesign of the extended substrate specificity of granzyme B. J Biol Chem 273, 27364–27373. [DOI] [PubMed] [Google Scholar]
- 59. Mahrus S, Kisiel W & Craik CS (2004) Granzyme M is a regulatory protease that inactivates proteinase inhibitor 9, an endogenous inhibitor of granzyme B. J Biol Chem 279, 54275–54282. [DOI] [PubMed] [Google Scholar]
- 60. Edwards KM, Kam CM, Powers JC & Trapani JA (1999) The human cytotoxic T cell granule serine protease granzyme H has chymotrypsin‐like (chymase) activity and is taken up into cytoplasmic vesicles reminiscent of granzyme B‐containing endosomes. J Biol Chem 274, 30468–30473. [DOI] [PubMed] [Google Scholar]
- 61. Smyth MJ, O'Connor MD, Trapani JA, Kershaw MH & Brinkworth RI (1996) A novel substrate‐binding pocket interaction restricts the specificity of the human NK cell‐specific serine protease, Met‐ase‐1. J Immunol 156, 4174–4181. [PubMed] [Google Scholar]
- 62. Rukamp BJ, Kam CM, Natarajan S, Bolton BW, Smyth MJ, Kelly JM & Powers JC (2004) Subsite specificities of granzyme M: a study of inhibitors and newly synthesized thiobenzyl ester substrates. Arch Biochem Biophys 422, 9–22. [DOI] [PubMed] [Google Scholar]
- 63. de Poot SA, Westgeest M, Hostetter DR, Van Damme P, Plasman K, Demeyer K, Broekhuizen R, Gevaert K, Craik CS & Bovenschen N (2011) Human and mouse granzyme M display divergent and species‐specific substrate specificities. Biochem J 437, 431–442. [DOI] [PubMed] [Google Scholar]
- 64. Mahrus S & Craik CS (2005) Selective chemical functional probes of granzymes A and B reveal granzyme B is a major effector of natural killer cell‐mediated lysis of target cells. Chem Biol 12, 567–577. [DOI] [PubMed] [Google Scholar]
- 65. Prassas I, Eissa A, Poda G & Diamandis EP (2015) Unleashing the therapeutic potential of human kallikrein‐related serine proteases. Nat Rev Drug Discov 14, 183–202. [DOI] [PubMed] [Google Scholar]
- 66. Wu P, Leinonen J, Koivunen E, Lankinen H & Stenman UH (2000) Identification of novel prostate‐specific antigen‐binding peptides modulating its enzyme activity. Eur J Biochem 267, 6212–6220. [DOI] [PubMed] [Google Scholar]
- 67. Wu P, Zhu L, Stenman UH & Leinonen J (2004) Immunopeptidometric assay for enzymatically active prostate‐specific antigen. Clin Chem 50, 125–129. [DOI] [PubMed] [Google Scholar]
- 68. Hekim C, Leinonen J, Narvanen A, Koistinen H, Zhu L, Koivunen E, Vaisanen V & Stenman UH (2006) Novel peptide inhibitors of human kallikrein 2. J Biol Chem 281, 12555–12560. [DOI] [PubMed] [Google Scholar]
- 69. de Veer SJ, Swedberg JE, Brattsand M, Clements JA & Harris JM (2016) Exploring the active site binding specificity of kallikrein‐related peptidase 5 (KLK5) guides the design of new peptide substrates and inhibitors. Biol Chem 397, 1237–1249. [DOI] [PubMed] [Google Scholar]
- 70. Cloutier SM, Kundig C, Felber LM, Fattah OM, Chagas JR, Gygi CM, Jichlinski P, Leisinger HJ & Deperthes D (2004) Development of recombinant inhibitors specific to human kallikrein 2 using phage‐display selected substrates. Eur J Biochem 271, 607–613. [DOI] [PubMed] [Google Scholar]
- 71. Felber LM, Kundig C, Borgono CA, Chagas JR, Tasinato A, Jichlinski P, Gygi CM, Leisinger HJ, Diamandis EP, Deperthes D et al (2006) Mutant recombinant serpins as highly specific inhibitors of human kallikrein 14. FEBS J 273, 2505–2514. [DOI] [PubMed] [Google Scholar]
- 72. Felber LM, Borgono CA, Cloutier SM, Kundig C, Kishi T, Ribeiro Chagas J, Jichlinski P, Gygi CM, Leisinger HJ, Diamandis EP et al (2005) Enzymatic profiling of human kallikrein 14 using phage‐display substrate technology. Biol Chem 386, 291–298. [DOI] [PubMed] [Google Scholar]
- 73. Denault JB & Salvesen GS (2002) Caspases: keys in the ignition of cell death. Chem Rev 102, 4489–4500. [DOI] [PubMed] [Google Scholar]
- 74. Seaman JE, Julien O, Lee PS, Rettenmaier TJ, Thomsen ND & Wells JA (2016) Cacidases: caspases can cleave after aspartate, glutamate and phosphoserine residues. Cell Death Differ 23, 1717–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pop C & Salvesen GS (2009) Human caspases: activation, specificity, and regulation. J Biol Chem 284, 21777–21781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Poreba M, Strozyk A, Salvesen GS & Drag M (2013) Caspase substrates and inhibitors. Cold Spring Harb Perspect Biol 5, a008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Benkova B, Lozanov V, Ivanov IP & Mitev V (2009) Evaluation of recombinant caspase specificity by competitive substrates. Anal Biochem 394, 68–74. [DOI] [PubMed] [Google Scholar]
- 78. Stennicke HR, Renatus M, Meldal M & Salvesen GS (2000) Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem J 350, 563–568. [PMC free article] [PubMed] [Google Scholar]
- 79. Boulware KT & Daugherty PS (2006) Protease specificity determination by using cellular libraries of peptide substrates (CLiPS). Proc Natl Acad Sci USA 103, 7583–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Poreba M, Kasperkiewicz P, Snipas SJ, Fasci D, Salvesen GS & Drag M (2014) Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ 21, 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kudelova J, Fleischmannova J, Adamova E & Matalova E (2015) Pharmacological caspase inhibitors: research towards therapeutic perspectives. J Physiol Pharmacol 66, 473–482. [PubMed] [Google Scholar]
- 82. Callus BA & Vaux DL (2007) Caspase inhibitors: viral, cellular and chemical. Cell Death Differ 14, 73–78. [DOI] [PubMed] [Google Scholar]
- 83. Linton SD (2005) Caspase inhibitors: a pharmaceutical industry perspective. Curr Top Med Chem 5, 1697–1717. [DOI] [PubMed] [Google Scholar]
- 84. Pereira NA & Song Z (2008) Some commonly used caspase substrates and inhibitors lack the specificity required to monitor individual caspase activity. Biochem Biophys Res Commun 377, 873–877. [DOI] [PubMed] [Google Scholar]
- 85. Berger AB, Sexton KB & Bogyo M (2006) Commonly used caspase inhibitors designed based on substrate specificity profiles lack selectivity. Cell Res 16, 961–963. [DOI] [PubMed] [Google Scholar]
- 86. Berger AB, Witte MD, Denault JB, Sadaghiani AM, Sexton KM, Salvesen GS & Bogyo M (2006) Identification of early intermediates of caspase activation using selective inhibitors and activity‐based probes. Mol Cell 23, 509–521. [DOI] [PubMed] [Google Scholar]
- 87. Vickers CJ, Gonzalez‐Paez GE, Litwin KM, Umotoy JC, Coutsias EA & Wolan DW (2014) Selective inhibition of initiator versus executioner caspases using small peptides containing unnatural amino acids. ACS Chem Biol 9, 2194–2198. [DOI] [PubMed] [Google Scholar]
- 88. Vickers CJ, Gonzalez‐Paez GE & Wolan DW (2013) Selective detection and inhibition of active caspase‐3 in cells with optimized peptides. J Am Chem Soc 135, 12869–12876. [DOI] [PubMed] [Google Scholar]
- 89. Vickers CJ, Gonzalez‐Paez GE & Wolan DW (2013) Selective detection of caspase‐3 versus caspase‐7 using activity‐based probes with key unnatural amino acids. ASC Chem Biol 8, 1558–1566. [DOI] [PubMed] [Google Scholar]
- 90. Uren AG, O'Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV & Dixit VM (2000) Identification of paracaspases and metacaspases: two ancient families of caspase‐like proteins, one of which plays a key role in MALT lymphoma. Mol Cell 6, 961–967. [DOI] [PubMed] [Google Scholar]
- 91. Hachmann J, Snipas SJ, van Raam BJ, Cancino EM, Houlihan EJ, Poreba M, Kasperkiewicz P, Drag M & Salvesen GS (2012) Mechanism and specificity of the human paracaspase MALT1. Biochem J 443, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hachmann J, Edgington‐Mitchell LE, Poreba M, Sanman LE, Drag M, Bogyo M & Salvesen GS (2015) Probes to monitor activity of the paracaspase MALT1. Chem Biol 22, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Xin BT, Schimmack G, Du Y, Florea BI, van der Marel GA, Driessen C, Krappmann D & Overkleeft HS (2016) Development of new Malt1 inhibitors and probes. Bioorg Med Chem 24, 3312–3329. [DOI] [PubMed] [Google Scholar]
- 94. Patterson AW, Wood WJ & Ellman JA (2007) Substrate activity screening (SAS): a general procedure for the preparation and screening of a fragment‐based non‐peptidic protease substrate library for inhibitor discovery. Nat Protoc 2, 424–433. [DOI] [PubMed] [Google Scholar]
- 95. Patterson AW, Wood WJ, Hornsby M, Lesley S, Spraggon G & Ellman JA (2006) Identification of selective, nonpeptidic nitrile inhibitors of cathepsin s using the substrate activity screening method. J Med Chem 49, 6298–6307. [DOI] [PubMed] [Google Scholar]
- 96. Wood WJ, Patterson AW, Tsuruoka H, Jain RK & Ellman JA (2005) Substrate activity screening: a fragment‐based method for the rapid identification of nonpeptidic protease inhibitors. J Am Chem Soc 127, 15521–15527. [DOI] [PubMed] [Google Scholar]
- 97. Tchoupe JR, Moreau T, Gauthier F & Bieth JG (1991) Photometric or fluorometric assay of cathepsin B, L and H and papain using substrates with an aminotrifluoromethylcoumarin leaving group. Biochim Biophys Acta 1076, 149–151. [DOI] [PubMed] [Google Scholar]
- 98. Puzer L, Cotrin SS, Alves MF, Egborge T, Araujo MS, Juliano MA, Juliano L, Bromme D & Carmona AK (2004) Comparative substrate specificity analysis of recombinant human cathepsin V and cathepsin L. Arch Biochem Biophys 430, 274–283. [DOI] [PubMed] [Google Scholar]
- 99. Alves MF, Puzer L, Cotrin SS, Juliano MA, Juliano L, Bromme D & Carmona AK (2003) S3 to S3’ subsite specificity of recombinant human cathepsin K and development of selective internally quenched fluorescent substrates. Biochem J 373, 981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Siklos M, BenAissa M & Thatcher GR (2015) Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors Acta Pharm Sin B 5, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Katunuma N (2011) Structure‐based development of specific inhibitors for individual cathepsins and their medical applications. Proc Jpn Acad Ser B Phys Biol Sci 87, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Oresic Bender K, Ofori L, van der Linden WA, Mock ED, Datta GK, Chowdhury S, Li H, Segal E, Sanchez Lopez M, Ellman JA et al (2015) Design of a highly selective quenched activity‐based probe and its application in dual color imaging studies of cathepsin S activity localization. J Am Chem Soc 137, 4771–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dall E & Brandstetter H (2016) Structure and function of legumain in health and disease. Biochimie 122, 126–150. [DOI] [PubMed] [Google Scholar]
- 104. Edgington LE, Verdoes M, Ortega A, Withana NP, Lee J, Syed S, Bachmann MH, Blum G & Bogyo M (2013) Functional imaging of legumain in cancer using a new quenched activity‐based probe. J Am Chem Soc 135, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liu Y, Lashuel HA, Choi S, Xing X, Case A, Ni J, Yeh LA, Cuny GD, Stein RL & Lansbury PT Jr (2003) Discovery of inhibitors that elucidate the role of UCH‐L1 activity in the H1299 lung cancer cell line. Chem Biol 10, 837–846. [DOI] [PubMed] [Google Scholar]
- 106. Borodovsky A, Ovaa H, Kolli N, Gan‐Erdene T, Wilkinson KD, Ploegh HL & Kessler BM (2002) Chemistry‐based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol 9, 1149–1159. [DOI] [PubMed] [Google Scholar]
- 107. Hemelaar J, Borodovsky A, Kessler BM, Reverter D, Cook J, Kolli N, Gan‐Erdene T, Wilkinson KD, Gill G, Lima CD et al (2004) Specific and covalent targeting of conjugating and deconjugating enzymes of ubiquitin‐like proteins. Mol Cell Biol 24, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Drag M, Mikolajczyk J, Bekes M, Reyes‐Turcu FE, Ellman JA, Wilkinson KD & Salvesen GS (2008) Positional‐scanning fluorigenic substrate libraries reveal unexpected specificity determinants of DUBs (deubiquitinating enzymes). Biochem J 415, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Vandenbroucke RE & Libert C (2014) Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov 13, 904–927. [DOI] [PubMed] [Google Scholar]
- 110. Eckhard U, Huesgen PF, Schilling O, Bellac CL, Butler GS, Cox JH, Dufour A, Goebeler V, Kappelhoff R, Auf dem Keller U et al (2016) Active site specificity profiling datasets of matrix metalloproteinases (MMPs) 1, 2, 3, 7, 8, 9, 12, 13 and 14. Data Brief 7, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Eckhard U, Huesgen PF, Schilling O, Bellac CL, Butler GS, Cox JH, Dufour A, Goebeler V, Kappelhoff R, Keller UAD et al (2016) Active site specificity profiling of the matrix metalloproteinase family: proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses. Matrix Biol 49, 37–60. [DOI] [PubMed] [Google Scholar]
- 112. Overall CM & Lopez‐Otin C (2002) Strategies for MMP inhibition in cancer: innovations for the post‐trial era. Nat Rev Cancer 2, 657–672. [DOI] [PubMed] [Google Scholar]
- 113. Chen EI, Li W, Godzik A, Howard EW & Smith JW (2003) A residue in the S2 subsite controls substrate selectivity of matrix metalloproteinase‐2 and matrix metalloproteinase‐9. J Biol Chem 278, 17158–17163. [DOI] [PubMed] [Google Scholar]
- 114. Ratnikov BI, Cieplak P, Gramatikoff K, Pierce J, Eroshkin A, Igarashi Y, Kazanov M, Sun Q, Godzik A, Osterman A et al (2014) Basis for substrate recognition and distinction by matrix metalloproteinases. Proc Natl Acad Sci USA 111, E4148–E4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Whittaker M, Floyd CD, Brown P & Gearing AJH (1999) Design and therapeutic application of matrix metalloproteinase inhibitors. Chem Rev 99, 2735–2776. [DOI] [PubMed] [Google Scholar]
- 116. Macaulay VM, O'Byrne KJ, Saunders MP, Braybrooke JP, Long L, Gleeson F, Mason CS, Harris AL, Brown P & Talbot DC (1999) Phase I study of intrapleural batimastat (BB‐94), a matrix metalloproteinase inhibitor, in the treatment of malignant pleural effusions. Clin Cancer Res 5, 513–520. [PubMed] [Google Scholar]
- 117. Iwasaki M, Nishikawa A, Fujimoto T, Akutagawa N, Manase K, Endo T, Yoshida K, Maekawa R, Yoshioka T & Kudo R (2002) Anti‐invasive effect of MMI‐166, a new selective matrix metalloproteinase inhibitor, in cervical carcinoma cell lines. Gynecol Oncol 85, 103–107. [DOI] [PubMed] [Google Scholar]
- 118. Jacobsen JA, Jourden JLM, Miller MT & Cohen SM (2010) To bind zinc or not to bind zinc: an examination of innovative approaches to improved metalloproteinase inhibition. Biochim Biophys Acta 1803, 72–94. [DOI] [PubMed] [Google Scholar]
- 119. Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang ML, Fridman R & Mobashery S (2005) Potent mechanism‐based inhibitors for matrix metalloproteinases. J Biol Chem 280, 33992–34002. [DOI] [PubMed] [Google Scholar]
- 120. Fabre B, Filipiak K, Diaz N, Zapico JM, Suarez D, Ramos A & de Pascual‐Teresa B (2014) An integrated computational and experimental approach to gaining selectivity for MMP‐2 within the gelatinase subfamily. ChemBioChem 15, 399–412. [DOI] [PubMed] [Google Scholar]
- 121. Hoet RM, Cohen EH, Kent RB, Rookey K, Schoonbroodt S, Hogan S, Rem L, Frans N, Daukandt M, Pieters H et al (2005) Generation of high‐affinity human antibodies by combining donor‐derived and synthetic complementarity‐determining‐region diversity. Nat Biotechnol 23, 344–348. [DOI] [PubMed] [Google Scholar]
- 122. Bu ZB, Pan YY, Shang BX, Cao ZF, Zhou QS & Ruan CG (2012) SZ‐117, a monoclonal antibody against matrix metalloproteinase‐2 inhibits tumor cell‐mediated angiogenesis. Hybridoma 31, 63–67. [DOI] [PubMed] [Google Scholar]
- 123. Hu JL, Van den Steen PE, Houde M, Ilenchuk TT & Opdenakker G (2004) Inhibitors of gelatinase B/matrix metalloproteinase‐9 activity comparison of a peptidomimetic and polyhistidine with single‐chain derivatives of a neutralizing monoclonal antibody. Biochem Pharmacol 67, 1001–1009. [DOI] [PubMed] [Google Scholar]
- 124. Mucha A, Cuniasse P, Kannan R, Beau F, Yiotakis A, Basset P & Dive V (1998) Membrane type‐1 matrix metalloprotease and stromelysin‐3 cleave more efficiently synthetic substrates containing unusual amino acids in their P1’ positions. J Biol Chem 273, 2763–2768. [DOI] [PubMed] [Google Scholar]
- 125. Taylor A (1993) Aminopeptidases: structure and function. FASEB J 7, 290–298. [DOI] [PubMed] [Google Scholar]
- 126. Taylor A (1993) Aminopeptidases: towards a mechanism of action. Trends Biochem Sci 18, 167–171. [PubMed] [Google Scholar]
- 127. Luan Y, Ma C, Wang Y, Fang H & Xu W (2012) The characteristics, functions and inhibitors of three aminopeptidases belonging to the m1 family. Curr Protein Pept Sci 13, 490–500. [DOI] [PubMed] [Google Scholar]
- 128. Drag M, Bogyo M, Ellman JA & Salvesen GS (2010) Aminopeptidase fingerprints, an integrated approach for identification of good substrates and optimal inhibitors. J Biol Chem 285, 3310–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Poreba M, Gajda A, Picha J, Jiracek J, Marschner A, Klein CD, Salvesen GS & Drag M (2012) S1 pocket fingerprints of human and bacterial methionine aminopeptidases determined using fluorogenic libraries of substrates and phosphorus based inhibitors. Biochimie 94, 704–710. [DOI] [PubMed] [Google Scholar]
- 130. Poreba M, McGowan S, Skinner‐Adams TS, Trenholme KR, Gardiner DL, Whisstock JC, To J, Salvesen GS, Dalton JP & Drag M (2012) Fingerprinting the substrate specificity of M1 and M17 aminopeptidases of human malaria, Plasmodium falciparum. PLoS ONE 7, e31938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gras S, Byzia A, Gilbert FB, McGowan S, Drag M, Silvestre A, Niepceron A, Lecaille F, Lalmanach G & Brossier F (2014) Aminopeptidase N1 (EtAPN1), an M1 metalloprotease of the apicomplexan parasite Eimeria tenella, participates in parasite development. Eukaryot Cell 13, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Reznik SE & Fricker LD (2001) Carboxypeptidases from A to z: implications in embryonic development and Wnt binding. Cell Mol Life Sci 58, 1790–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Petrera A, Lai ZW & Schilling O (2014) Carboxyterminal protein processing in health and disease: key actors and emerging technologies. J Proteome Res 13, 4497–4504. [DOI] [PubMed] [Google Scholar]
- 134. Gardell SJ, Craik CS, Clauser E, Goldsmith EJ, Stewart CB, Graf M & Rutter WJ (1988) A novel rat carboxypeptidase, CPA2: characterization, molecular cloning, and evolutionary implications on substrate specificity in the carboxypeptidase gene family. J Biol Chem 263, 17828–17836. [PubMed] [Google Scholar]
- 135. Tanco S, Lorenzo J, Garcia‐Pardo J, Degroeve S, Martens L, Aviles FX, Gevaert K & Van Damme P (2013) Proteome‐derived peptide libraries to study the substrate specificity profiles of carboxypeptidases. Mol Cell Proteomics 12, 2096–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Barinka C, Rinnova M, Sacha P, Rojas C, Majer P, Slusher BS & Konvalinka J (2002) Substrate specificity, inhibition and enzymological analysis of recombinant human glutamate carboxypeptidase II. J Neurochem 80, 477–487. [DOI] [PubMed] [Google Scholar]
- 137. Deiteren K, Surpateanu G, Gilany K, Willemse JL, Hendriks DF, Augustyns K, Laroche Y, Scharpe S & Lambeir AM (2007) The role of the S1 binding site of carboxypeptidase M in substrate specificity and turn‐over. Biochim Biophys Acta 1774, 267–277. [DOI] [PubMed] [Google Scholar]
- 138. Lyons PJ & Fricker LD (2010) Substrate specificity of human carboxypeptidase A6. J Biol Chem 285, 38234–38242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Tanco S, Zhang X, Morano C, Aviles FX, Lorenzo J & Fricker LD (2010) Characterization of the substrate specificity of human carboxypeptidase A4 and implications for a role in extracellular peptide processing. J Biol Chem 285, 18385–18396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Willemse JL, Polla M, Olsson T & Hendriks DF (2008) Comparative substrate specificity study of carboxypeptidase U (TAFIa) and carboxypeptidase N: development of highly selective CPU substrates as useful tools for assay development. Clin Chim Acta 387, 158–160. [DOI] [PubMed] [Google Scholar]
- 141. Harris JL, Alper PB, Li J, Rechsteiner M & Backes BJ (2001) Substrate specificity of the human proteasome. Chem Biol 8, 1131–1141. [DOI] [PubMed] [Google Scholar]
- 142. Gruba N, Wysocka M, Brzezinska M, Debowski D, Rolka K, Martin NI & Lesner A (2016) Novel internally quenched substrate of the trypsin‐like subunit of 20S eukaryotic proteasome. Anal Biochem 508, 38–45. [DOI] [PubMed] [Google Scholar]
- 143. Gruba N, Wysocka M, Brzezinska M, Debowski D, Sienczyk M, Gorodkiewicz E, Guszcz T, Czaplewski C, Rolka K & Lesner A (2016) Bladder cancer detection using a peptide substrate of the 20S proteasome. FEBS J 283, 2929–2948. [DOI] [PubMed] [Google Scholar]
- 144. Kisselev AF & Goldberg AL (2001) Proteasome inhibitors: from research tools to drug candidates. Chem Biol 8, 739–758. [DOI] [PubMed] [Google Scholar]
- 145. Richardson PG, Hideshima T & Anderson KC (2003) Bortezomib (PS‐341): a novel, first‐in‐class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control 10, 361–369. [DOI] [PubMed] [Google Scholar]
- 146. Khan ML & Stewart AK (2011) Carfilzomib: a novel second‐generation proteasome inhibitor. Future Oncol 7, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F et al (2010) Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res 70, 1970–1980. [DOI] [PubMed] [Google Scholar]
- 148. de Bruin G, Xin BT, Kraus M, van der Stelt M, van der Marel GA, Kisselev AF, Driessen C, Florea BI & Overkleeft HS (2015) A set of activity‐based probes to visualize human (immuno) proteasome activities. Angew Chem 55, 4199–4203. [DOI] [PubMed] [Google Scholar]
- 149. Li H, O'Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PC & Bogyo M (2016) Structure‐ and function‐based design of Plasmodium‐selective proteasome inhibitors. Nature 530, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Korkmaz B, Attucci S, Hazouard E, Ferrandiere M, Jourdan ML, Brillard‐Bourdet M, Juliano L & Gauthier F (2002) Discriminating between the activities of human neutrophil elastase and proteinase 3 using serpin‐derived fluorogenic substrates. J Biol Chem 277, 39074–39081. [DOI] [PubMed] [Google Scholar]
- 151. Korkmaz B, Attucci S, Moreau T, Godat E, Juliano L & Gauthier F (2004) Design and use of highly specific substrates of neutrophil elastase and proteinase 3. Am J Respir Cell Mol Biol 30, 801–807. [DOI] [PubMed] [Google Scholar]
- 152. Wu P, Stenman UH, Pakkala M, Narvanen A & Leinonen J (2004) Separation of enzymatically active and inactive prostate‐specific antigen (PSA) by peptide affinity chromatography. Prostate 58, 345–353. [DOI] [PubMed] [Google Scholar]
- 153. Powers JC, Asgian JL, Ekici OD & James KE (2002) Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev 102, 4639–4750. [DOI] [PubMed] [Google Scholar]
- 154. Vizovisek M, Vidmar R, Van Quickelberghe E, Impens F, Andjelkovic U, Sobotic B, Stoka V, Gevaert K, Turk B & Fonovic M (2015) Fast profiling of protease specificity reveals similar substrate specificities for cathepsins K, L and S. Proteomics 15, 2479–2490. [DOI] [PubMed] [Google Scholar]
- 155. Biniossek ML, Nagler DK, Becker‐Pauly C & Schilling O (2011) Proteomic identification of protease cleavage sites characterizes prime and non‐prime specificity of cysteine cathepsins B, L, and S. J Proteome Res 10, 5363–5373. [DOI] [PubMed] [Google Scholar]