

Abstract
Angiotensin‐converting enzyme‐2 (ACE2) may play an important role in cardiorenal disease and it has also been implicated as a cellular receptor for the severe acute respiratory syndrome (SARS) virus. The ACE2 active‐site model and its crystal structure, which was solved recently, highlighted key differences between ACE2 and its counterpart angiotensin‐converting enzyme (ACE), which are responsible for their differing substrate and inhibitor sensitivities. In this study the role of ACE2 active‐site residues was explored by site‐directed mutagenesis. Arg273 was found to be critical for substrate binding such that its replacement causes enzyme activity to be abolished. Although both His505 and His345 are involved in catalysis, it is His345 and not His505 that acts as the hydrogen bond donor/acceptor in the formation of the tetrahedral peptide intermediate. The difference in chloride sensitivity between ACE2 and ACE was investigated, and the absence of a second chloride‐binding site (CL2) in ACE2 confirmed. Thus ACE2 has only one chloride‐binding site (CL1) whereas ACE has two sites. This is the first study to address the differences that exist between ACE2 and ACE at the molecular level. The results can be applied to future studies aimed at unravelling the role of ACE2, relative to ACE, in vivo.
Keywords: angiotensin II, carboxypeptidase, chloride, metalloprotease, zinc
Abbreviations
- ACE
angiotensin‐converting enzyme
- Mca
(7‐methoxycoumarin‐4‐yl)acetyl
- tACE
testicular ACE
Angiotensin‐converting enzyme‐2 (ACE2) is a membrane protein with its active site exposed to the extracellular surface of endothelial cells, the renal tubular epithelium and also the epithelia of the lung and the small intestine [1, 2, 3]. Here ACE2 is poised to metabolize circulating peptides which may include angiotensin II, a potent vasoconstrictor and the product of angiotensin I cleavage by angiotensin‐converting enzyme (ACE; EC 3.4.15.1) [1, 4]. Indeed, ACE2 has been implicated in the regulation of heart and renal function where it is proposed to control the concentrations of angiotensin II relative to its hypotensive metabolite, angiotensin‐(1–7) [5, 6, 7, 8, 9, 10, 11, 12, 13]. Most recently, ACE2 has been identified as a functional receptor for the coronavirus which causes the severe acute respiratory syndrome (SARS) [14]. For recent reviews, see [15, 16].
ACE2 shares a number of characteristics with ACE, both being zinc‐containing enzymes which are sensitive to anion activation [4, 17, 18]. However, unlike ACE, ACE2 functions as a carboxypeptidase and is not susceptible to inhibition by the classical ACE inhibitors [1, 2]. After the elucidation of the crystal structure of testicular ACE (tACE), [19] a model of the active site of ACE2 was described which demonstrated the structural determinants underlying these differences in enzyme activity [17]. Critical residue substitutions were highlighted that gave rise to the elimination of the S2′ pocket found in ACE such that ACE2 is able to remove only a single amino acid from the C‐terminus of its substrates (whereas ACE is a peptidyl dipeptidase). Shortly after this, the structure of ACE2 was solved [20] which provided further insights into this enzyme in relation to its counterpart. However, it has become increasingly apparent that ACE2 is indeed both structurally and functionally distinct from ACE.
The extracellular domain structure of ACE2 was determined in the native and the inhibitor‐bound form [20]. The zinc protease domain is divided into two subdomains, which are defined by the movement of the subdomains relative to each other upon inhibitor binding. Subdomain I (N‐terminal) contains the zinc‐binding site, which faces into the deep cleft formed by the two subdomains connected at the base of the cleft. The hinge‐bending motion observed upon inhibitor binding occurs as subdomain I moves to close the gap, and in doing so brings critical residue groups around the substrate/inhibitor. This study provides the first investigation of the importance of key active‐site residues of ACE2 through site‐directed mutagenesis, with the aim of providing practical evidence for their role in substrate binding/hydrolysis. In addition, the effect of chloride activation is further addressed as the basis of the differing sensitivities of ACE2 and ACE to anions is not currently understood.
Results and Discussion
Binding of the C‐terminus of peptide substrates by ACE2
From the active‐site structure of ACE2 [17, 20], Arg273 is able to make a salt‐bridge with the C‐terminus of the ACE2 inhibitor, MLN‐4760 [21], and is hence proposed to be involved in binding of the C‐terminus of peptide substrates (Fig. 1A). To test this hypothesis, we used site‐directed mutagenesis to replace the arginine with a glutamine residue (R273Q), i.e. its counterpart in ACE. This represents a positive to neutral change in the side chain at this position while maintaining most of the hydrophobic surface area. For comparative purposes, the arginine residue was also replaced with a lysine in order to maintain the charge on the side chain (R273K). Stable expression of wild‐type soluble ACE2 and the R273Q/K mutants was established in HEK293 cells. The medium, containing the ACE2 protein, was collected, and total protein (30 µg) was separated by SDS/PAGE. Expression of the ACE2 mutant enzymes was successful, and the proteins migrated on SDS/PAGE with the same apparent M r as the wild‐type enzyme (Fig. 2A, top panel). ACE2 protein could not be detected in the medium collected from mock‐transfected cells. Incubation of the ACE2 wild‐type and mutant protein (30 µg total protein) with 25 μm (7‐methoxycoumarin‐4‐yl)acetyl (Mca)‐APK(Dnp) for 1 h revealed that the mutants, although expressed, were not active (Fig. 2A, bottom panel). Following from this, no enzyme activity was observed when R273Q/K protein (100 µg total protein) was incubated with the ACE2 substrate for 6 h. The positive side chain of Arg273 is therefore critical for binding of the substrate. Maintaining the positive charge at this position (R273K) is not sufficient for docking of the peptide into the ACE2 active site. In fact, the distance of this positive charge from the surface of the binding pocket is also crucial.
Figure 1.
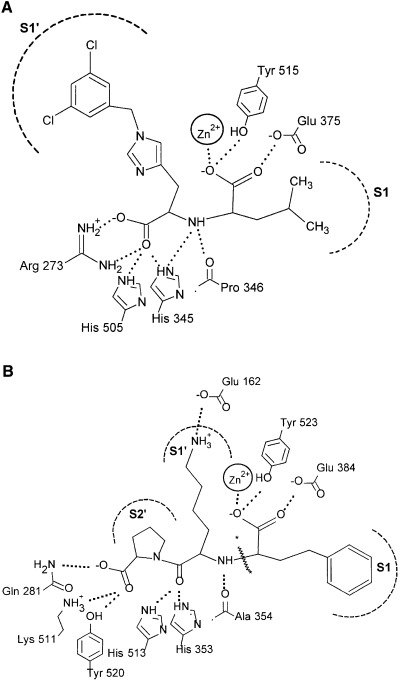
Schematic view of the active site of ACE2 and tACE. Binding interactions of the inhibitor (A) MLN‐4760 at the active site of ACE2 and (B) lisinopril at the active site of tACE. Hydrogen bonds to the ligand are shown (dotted lines). The different binding subsites are labelled. Adapted from [17].
Figure 2.
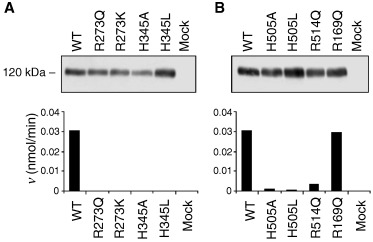
Expression of soluble ACE2 mutants. Medium, taken from mock‐transfected (empty vector) HEK293 cells and HEK293 cells transiently expressing soluble ACE2, was concentrated in a 10‐kDa cut‐off column. Aliquots, containing 30 µg total protein, were separated by SDS/PAGE (6% polyacrylamide gel) and then analysed by immunoelectrophoretic blotting using a human ACE2 polyclonal antibody (top panel). Total protein (30 µg) was incubated with the ACE2‐specific fluorogenic peptide, Mca‐APK(Dnp) (25 µm), as described in Experimental Procedures. Enzyme activity is expressed as mol product formed per min (bottom panel). Values are the mean of duplicate determinations.
Role of His505 and His345 in catalysis
Sequence alignment of ACE2 with ACE revealed that the ACE residue His1089, shown to be involved in the stabilization of the transition‐state intermediate [22], was conserved in ACE2 (His505). Indeed, the location (relative to the zinc‐binding motif) of His505 in the ACE2 sequence is very similar to the location of the catalytic histidines in both ACE and thermolysin, suggesting that His505 is the catalytic histidine of ACE2. On the basis of these features, we predicted and tested if His505 is the transition‐state stabilizing residue. In parallel, the role of His345, which can hydrogen‐bond with both the C‐terminus and the secondary amine group of the ACE2 inhibitor MLN‐4760 (Fig. 1A), was explored. To investigate the role of His505 and His345 in catalysis, the histidine residues were replaced with both alanine and leucine (H505A/L and H345A/L). Stable expression of the ACE2 mutants was established in HEK293 cells (Fig. 2, top panels). Upon incubation of the mutant protein (30 µg total protein) with the ACE2 fluorogenic substrate for 1 h, little or no enzyme activity was observed (Fig. 2, bottom panels). Subsequently, enzyme activity was examined under extensive hydrolytic conditions [100 µg total protein was incubated with 25 mm Mca‐APK(Dnp) for 6 h], and as a result the mutant enzymes were found to be considerably less active than the wild‐type enzyme (Table 1). With such little activity remaining (H505A ≈ 60‐fold, H505L ≈ 250‐fold, and H345A/L ≈ 300‐fold less active than the wild‐type enzyme), subsequent kinetic analysis of the ACE2 mutants was not feasible. These data establish an important role for both His505 and His345 as their replacement results in enzyme activity being dramatically reduced.
Table 1.
Activity of ACE2 mutants relative to wild‐type. Medium, taken from HEK293 cells stably expressing soluble ACE2, was concentrated in a 10‐kDa cut‐off column. The initial rate of ACE2 activity was determined by fluorimetric activity assay. Values are the mean of duplicate determinations ± SE.
| v (nmol·min−1·mg−1) | Relative activity (%) | |
|---|---|---|
| Wild‐type | 1.56 ± 0.018 | 100 |
| H345A | 0.005 ± 0.00002 | 0.3 |
| H345L | 0.005 ± 0.0006 | 0.3 |
| H505A | 0.023 ± 0.003 | 1.5 |
| H505L | 0.006 ± 0.0004 | 0.4 |
Modelling and structure determination, by Guy et al. [17] and Towler et al. [20], respectively, show that His505 and His345 (corresponding to His513 and His353 in tACE) play a key role in binding the substrate in ACE2. This structural information was used to probe further the role of these residues in catalysis. From the superposition of the active sites of ACE2 and tACE, several observations can be made. In both structures the histidine NE2 nitrogens (the protonated histidine side chain Nε nitrogen) of both residues are within hydrogen‐bonding distance of the carbonyl oxygen of the amide group of residue P1′ (tACE) (Fig. 1B) or equivalent terminal carboxylate oxygen (ACE2) (Fig. 1A) of the inhibitors. The first histidine (His353 in tACE and His345 in ACE2) is also within hydrogen‐bonding distance (3.2 Å in both ACE and ACE2) of the sp3 hybridized nitrogen of the inhibitors (which is the nitrogen involved in substrate peptide bond cleavage). It is therefore more likely to be this histidine that acts as a hydrogen bond donor/acceptor in the formation of the tetrahedral peptide intermediate in catalysis (Fig. 3) and not His505, which contradicts the role of His505 described by Towler et al. [20]. The closest potential nitrogen of His505(NE2) to the sp3 hybridized nitrogen is too far away (over 5 Å) for hydrogen‐bond formation. Instead, His505 may be important in hydrogen‐bonding to Tyr515(OH), which has been suggested to stabilize the carbonyl tetrahedral intermediate in ACE2 [20], and the equivalent Tyr in the Drosophila homologue, AnCE, [23] and Pyrococcus furiosus carboxypeptidase [24], all of which are zinc metalloproteases with similar overall tertiary structure. In the C‐domain of ACE, the equivalent residue to His505 in ACE2 has been suggested to be involved in stabilizing the carbonyl tetrahedral intermediate directly [22], with a hydrogen bond being formed between His1089(NE2) and the oxyanion formed during transition‐state binding. Certainly, a carbonyl tetrahedral intermediate modelled at the carbon C4 of MLN‐470 (the equivalent carbon of the carbonyl tetrahedral intermediate of the substrate) would support an identical role for His505 in ACE2 catalysis (Fig. 3). However, Towler et al. [20] suggest that His505 is too far from the zinc‐bound carboxylate to be directly involved in the stabilization of the carbonyl tetrahedral intermediate. Overall, the greater loss in activity for the H345A mutation (≈ 330‐fold decrease in activity) than the H505A mutation (≈ 60‐fold decrease) would support a more important role for His345 in catalysis rather than His505.
Figure 3.
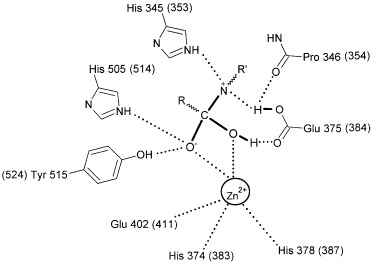
Role of His505 and His345 in catalysis. Schematic of the proposed reaction intermediate of ACE2, showing the importance of His345 and His505. Hydrogen bonds to the ligand are shown (dotted lines).The equivalent residues in tACE are given in parentheses.
Comparing the chloride‐binding sites of tACE and ACE2
The chloride dependence of ACE has long been recognized [25], and most recently mutagenesis studies have shown that it is in fact an arginine residue (Arg1098) that is essential for the chloride activation of ACE [18]. The structure of tACE revealed the location of two buried chloride ions [19]. The second chloride ion (CL2) was found to be bound to a water molecule and two amino‐acid residues, one being the equivalent residue to Arg1098 (Arg522). The presence of another chloride ion (CL1), located away from the active site, was unexpected. Again an arginine residue (Arg186) was found to play a key role in the positioning of the chloride ion at this site. Sequence alignment of ACE2 with ACE revealed that both the arginine residues at each chloride site, CL1 and CL2, were conserved in ACE2 (Arg169 and Arg514, respectively). The ACE2 mutants R169Q and R514Q were therefore created and were expressed in HEK293 cells (Fig. 2B, top panel). The effect of chloride ions on enzyme activity was investigated (Fig. 4). The hydrolysis of Mca‐APK(Dnp) by ACE2 is greatly enhanced in the presence of chloride ions [4, 17]. At high concentrations of NaCl (0.5 m), the activity of the wild‐type enzyme was increased ≈ 3.5‐fold (Fig. 5). The ACE2 mutant R514Q, however, did not show the expected loss of chloride activation. Instead, in the presence of high salt, enzyme activity was ≈ 11‐fold greater, and as such this mutant was much more sensitive to chloride ions than the wild‐type enzyme. Intriguingly, the ACE2 structure [20] revealed the absence of a bound chloride in the CL2 site. These data combined suggest that, unlike ACE, this second site in ACE2 does not exist and therefore does not contribute to the chloride effect. In the light of this finding, an assumption might be that it is actually the CL1 site that is responsible for chloride activation of ACE2. In contrast with the second site, a bound chloride in the ACE2 structure was reported in an identical position to the first binding site (CL1) of tACE. Yet, in the presence of NaCl, the R169Q mutant in which the CL1 chloride‐binding site has potentially been abolished, responds with an approximately fivefold increase in activation (Fig. 5), which is slightly greater than wild‐type. In fact, the activity profile for R169Q mirrors that obtained for the wild‐type enzyme (Fig. 4).
Figure 4.
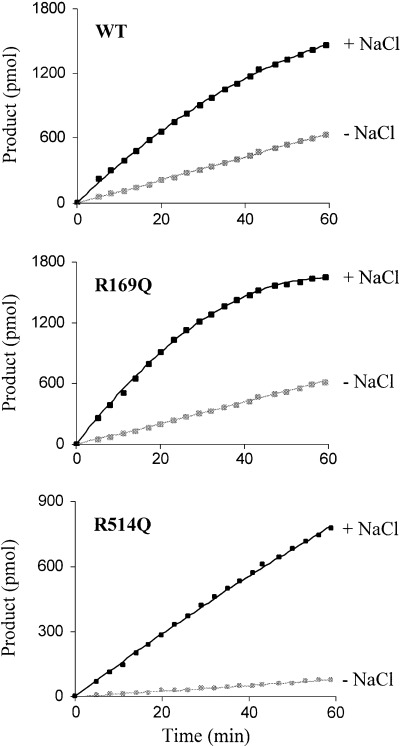
Effect of chloride ions on the activity of the ACE2 mutants (R169Q/R514Q). Medium, taken from HEK293 cells stably expressing soluble ACE2, was concentrated in a 10‐kDa cut‐off column and extensively dialysed against 50 mm Hepes/KOH buffer, pH 7.5, to remove chloride ions. Total protein (10 µg) was incubated with the ACE2‐specific fluorogenic peptide, Mca‐APK(Dnp) (25 µm), as described in Experimental Procedures in the absence (grey) or presence (black) of NaCl (500 mm). Enzyme activity is expressed as mol product formed over time. Product was quantified using pure standards. Values are the mean of four independent determinations.
Figure 5.
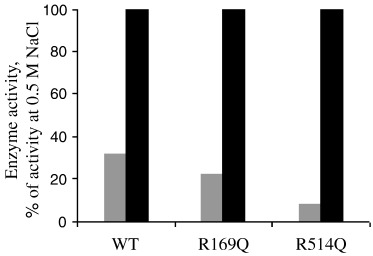
Activities of wild‐type and R169Q and R514Q ACE2 mutants in the absence (grey) and presence (black) of NaCl (500 mm). Medium, taken from HEK293 cells stably expressing soluble ACE2, was concentrated in a 10‐kDa cut‐off column and extensively dialysed against 50 mm Hepes/KOH buffer, pH 7.5, to remove chloride ions. Total protein (10 µg) was incubated with the ACE2‐specific fluorogenic peptide, Mca‐APK(Dnp) (25 µm), as described in Experimental Procedures in the absence (grey bars) or presence (black bars) of NaCl (500 mm). Enzyme activity (mol product formed·min−1) is expressed as the percentage of activity with 500 mm NaCl. Product was quantified using pure standards. Values are mean ± SE from four independent determinations.
Superimposing the structure of ACE2 on to tACE in inhibitor‐bound states revealed significant changes between the chloride ion‐binding sites of each enzyme. The designated CL2 site in tACE is absent from ACE2 [20], and this is due to the substitution of Pro407 and Pro519 in ACE for Glu398 and Ser511 in ACE2. The side chains of Glu398 and Ser511 project into the location of the chloride ion‐binding site and Ser511 hydrogen bonds with Arg514 [the equivalent Arg522 (NH1) coordinates the chloride ion in tACE]. Arg514 in ACE2 is displaced relative to Arg522 in ACE and is somewhat closer to the zinc‐binding site. The CL2 binding site is in close proximity to the catalytic site (10.4 Å away from the zinc) and is located at the interface between subdomains I and II (Table 2). Residue Pro407 in ACE (equivalent to Glu398 in ACE2) is in the hinge region between the two subdomains [20]. Hence, the binding of chloride to this site might be expected to affect zinc and ligand binding as well as the interactions between subdomain I and II. This effect would only be present in ACE as the site is absent from ACE2. Interestingly, the R514Q mutant at the CL2 site of ACE2 has limited effect on the activity of ACE2 in either the presence or absence of chloride ions. This is possibly a result of the fact Arg514 is not involved in chloride binding in ACE2 and its influence on activity is restricted to its effect on substrate binding, which would appear to be fairly small. However, site‐directed mutagenesis studies of the equivalent residue R1098Q in somatic ACE (sACE; equivalent to Arg522 in tACE) have a dramatic effect [18], with the mutation causing increased activity with the angiotensin I substrate relative to wild‐type in the absence of chloride. Presumably, this is the result of chloride binding at the CL2 site in ACE stabilizing complex formation. Therefore, removing this site (by mutation) stabilizes the complex formation and decreases the chloride dependency to that exerted by the occupancy of the CL1 site alone.
Table 2.
Subdomain boundaries of tACE and ACE2. The zinc protease domain of both tACE and ACE2 is divided into two subdomains [20]. Subdomain I contains the zinc ion and the N‐terminus. The C‐terminus is found in subdomain II.
| tACE | ACE2 | |
|---|---|---|
| Subdomain I | 40–121 | 19–102 |
| 299–406 | 290–397 | |
| 426–434 | 417–430 | |
| Subdomain II | 122–298 | 103–289 |
| 407–425 a | 398–416 | |
| 439 a −617 | 431–615 |
Residues 426–438 not present in tACE structure.
The designated CL1 is in subdomain II (Table 2). Residues coordinating the chloride ligand in tACE, Trp485, Arg186 and Arg489, are largely conserved in ACE2, as Trp477, Arg169 and Lys481. However, the Arg to Lys change may have some effect on chloride affinity. The Lys(NZ)–CL1 (5.2 Å) interaction in ACE2 is less intimate than that of Arg489(NH1)–CL1 (3.2 Å) in ACE (Fig. 6). Furthermore, the large difference in solvation energy between Arg and Lys might be expected to weaken chloride affinity in ACE2 relative to ACE. Presumably, the role of the CL1‐binding site is to stabilize the native state of subdomain II when in complex with the ligand. Although this is unlikely to have a direct influence on zinc binding (the zinc ion is separated from CL1 by 20.7 Å in tACE and is coordinated by residues in subdomain I), it may be important for stabilizing complex formation for those residues in subdomain II involved directly in substrate binding (Arg273 in ACE2 and Gln281, Lys511, Tyr520 in tACE, which bind the C‐terminal carboxy group of the respective inhibitors) and catalysis (His505 in ACE2 and the equivalent His511 in tACE). A similar hypothesis has been proposed for the role of the CL1 site in both domains of ACE [26]. It is surprising that the mutation R169Q has very little effect on activity in either the presence or absence of chloride. This may be the result of the fact that the mutant enzyme is still able to bind chloride at this site and retain its activating effect. Other residues within the site may be more important for chloride binding and their substitution may give rise to a more dramatic effect on chloride activation. For example, Asp499 found in the coordination shell of the chloride ion is in close proximity to some of the catalytic machinery and therefore may affect stabilization of the active enzyme complex. Trp478 hydrogen‐bonds with Asp499, and so, indirectly, its replacement may also elicit an effect on chloride activation. Less obviously, Trp271 lies two residues upstream of Arg273 (critical for substrate binding) and so it might ‘transmit’ effects on binding/catalysis of the substrate. The replacement of Trp477, which hydrogen‐bonds with the chloride ion, may cause a loss in chloride‐binding affinity. However, like Arg169, this may not cause underlying changes in chloride activation.
Figure 6.
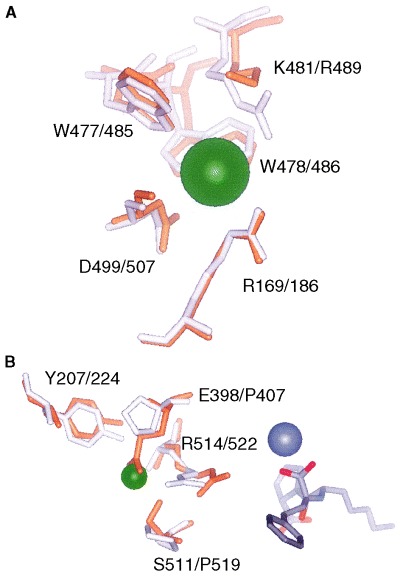
Chloride binding to ACE2 (yellow) and tACE (white). (A) Binding site of CL1 in ACE2 and tACE; (B) binding site of CL2 in ACE2 and tACE. Residue numbering for ACE2 is first. The chloride ion is green and the zinc ion is grey (both in spacefill). (B) The lisinopril ligand is coloured according to atom type (CPK) and the chloride ion is shown with a reduced radius to demonstrate its overlap with Glu398 in ACE2 more clearly.
The difference in chloride sensitivity between ACE2 and ACE makes sense in view of the fact that ACE2 has only one chloride‐binding site (CL1) whereas ACE has two sites (one in each subdomain). In addition to this, the CL1 site is found in subdomain II, some distance from the active site, and, with a likely difference in affinity for chloride at this site being suggested for ACE2 (see above), further explains this phenomenon. Liu et al. [18] showed a dramatic loss of chloride activation in the C‐domain of ACE where the CL2 site has been abolished. However, this effect is not all or nothing in that some enzyme activation is still observed. In this case, it is likely that ACE behaves more like ACE2, with only a single occupied site (CL1) contributing to the activation of substrate hydrolysis by chloride. Interestingly, Natesh et al. [27] suggest that, compared with ACE, the CL1 binding site in AnCE may also be altered and the CL2 site may be absent.
Conclusion
This study has highlighted the importance of Arg273 in binding of the C‐terminus of peptide substrates by ACE2. The complete loss of activity observed as a result of replacing this single residue could not have been predicted simply by analysis of the active‐site structure. Further to this, the role of His505 and His345 in catalysis has been probed which has provided new insight into transition‐state stabilization by ACE2 and other zinc proteases, e.g. ACE. Our mutagenesis data therefore provide additional critical information over that obtained from X‐ray data alone. The knowledge gained from these mutagenesis data will be valuable in directing the design of modulators of ACE2 activity. At present, very few studies have been carried out to develop inhibitors of ACE2 [24, 28, 29] despite the emerging importance of this enzyme in both cardiovascular homoeostasis and viral entry mechanisms. Finally, a comprehensive explanation for the differing sensitivity of ACE2 and ACE to chloride ions has been suggested, but how this relates to the physiological significance of chloride activation remains to be explored.
Experimental procedures
Materials
The peptide Mca‐APK(Dnp) was synthesized by G. Knight (University of Cambridge, UK).
Site‐directed mutagenesis
Mutagenic PCRs were carried out in 0.2 mL Eppendorf tubes with 50 µL reaction volumes. A typical reaction would contain: 1 µL 10 mm dNTP‐Mix; 5 µL 10 × reaction buffer; 0.5 µL 50 µm forward primer; 0.5 µL 50 µm reverse primer; 0.5 µL denatured template DNA (0.5 µg); 2.5 U PfuTurbo DNA polymerase (Stratagene, La Jolla, CA, USA) made up to 50 µL with sterile deionized water. The following PCR profile was used: one cycle (95 °C for 5 min); 16 cycles (95 °C for 30 s, 60 °C for 1 min, 68 °C for 12 min); one cycle (0 °C for 1 h). The amplification reaction was treated with 2 µL of the DpnI restriction enzyme (20 U·µL−1). After this addition, the reaction mixtures were thoroughly mixed by pipetting the solution up and down several times. The reaction mixture was centrifuged in a microcentrifuge for 1 min and incubated for 5 h at 37 °C. An aliquot (1 µL) of the DpnI‐treated DNA was used to transform competent Escherichia coli cells. Plasmid DNA was prepared from a single colony and fully sequenced to ensure the presence of the desired point mutations and the absence of unintended mutations.
Expression of ACE2 in HEK293 cells
Before transfection (24 h), cells were grown to 50–60% confluence in a Petri dish. For transient transfection, the monolayer was washed twice with Dulbecco's modifed Eagle's medium (DMEM) before transfection with 5 µg plasmid DNA (pCI‐neo containing nucleotides 104–2323 of ACE2 cDNA encoding a truncated protein lacking the transmembrane and cytosolic domains in‐frame with the FLAG peptide) per dish. GeneJuice transfection reagent was used at a ratio of DNA to reagent of 1 : 3 (w/v). This was added to the Petri dish in 2.5 mL DMEM and incubated for 16 h before the addition of supplemented DMEM. The medium was removed 24 h after the start of transfection, the monolayer rinsed twice with OptiMem, and then 5 mL of was added to each flask. This was incubated for a further 16 h before harvesting of the medium, containing soluble secreted ACE2 protein. The media samples containing protein were concentrated using Centricon (Millipore, Billerica, MA, USA) 10 kDa cut‐off filter units. For the chloride activation assays, the medium was harvested and exchanged into 50 mm Hepes/KOH, pH 7.5, using Centricon 10 kDa cut‐off filter units.
To obtain a stable cell line expressing soluble ACE2, the transfected cells were incubated in supplemented medium from 16 h after the start of transfection. At 72 h the cells were passaged and allowed to grow in supplemented medium containing the antibiotic G418 (1 mg·mL−1). The cells were subjected to repeated rounds of selection with G418 until they reached 80% confluence when they were passaged and allowed to continue to grow in selection medium.
One‐step RT‐PCR
RNA preparations from cells stably expressing soluble ACE2 were performed using a Qiagen (Valencia, CA, USA) RNeasy Mini Kit according to the manufacturer's guidelines. Reverse transcriptase (RT)‐PCR was carried out using the TITANIUM One‐Step RT‐PCR Kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer's guidelines with ACE2‐specific primers. The following PCR profile was used: one cycle (50 °C for 1 h); 1 cycle (94 °C for 3 min); 30 cycles (94 °C for 1 min, 55 °C for 1 min, 68 °C for 1 min); one cycle (68 °C for 10 min). Amplicons were sequenced to confirm the integrity of the ACE2 product, and this process was carried out for each of the mutants made for this study.
Protein determination
Protein concentrations were determined using the bicinchoninic acid assay with BSA as standard [30].
PNGase F treatment
PNGase F treatment (New England Biolabs, Beverly, MA, USA) was performed according to the manufacturer's instructions.
SDS/PAGE
Proteins were separated by SDS/PAGE by the method of Laemmli [31]. Samples were prepared in gel loading buffer [0.313 m Tris/HCl, pH 6.8, 10% (w/v) SDS, 20% (v/v) glycerol, 20% (v/v) 2‐mercaptoethanol, 0.02% (w/v) bromophenol blue] and heated to 100 °C for 5 min. Broad‐range prestained protein standards were run alongside the samples.
Immunoelectrophoretic analysis
The proteins were electrophoretically transferred from a polyacrylamide gel to a poly(vinylidene difluoride) membrane using a semidry blotter (Bio‐Rad, Hercules, CA, USA). The membrane was incubated overnight in TBS (10 mm Tris/HCl, pH 7.4, 150 mm NaCl) containing 5% (w/v) nonfat dry milk (TBSM) at 4 °C. After a quick rinse with TBS containing 0.1% (v/v) Tween 20 (TBST) the membrane was incubated for 2–3 h at room temperature in the presence of primary antibody. The following primary antibody was diluted as specified in TBSM: human ACE2 polyclonal antibody (1 : 500) was obtained from R & D Systems Europe Ltd (Abingdon, Oxon, UK). After a quick rinse in TBST the membrane was washed twice for 15 min in TBST at room temperature. The membrane was then incubated for 1 h at room temperature in the appropriate secondary antibody, diluted as specified in TBSM: horseradish peroxidase‐conjugated anti‐goat IgG (1 : 10 000) was obtained from Sigma. The TBST washes were repeated before visualization of the immunoreactive proteins by chemiluminescence using an ECL kit. For densitometric analysis, data were captured using a Fuji LAS‐1000 Imaging System CCD camera (aida 2.11 software for analysis).
ACE2/ACE activity assays
Fluorogenic assays using the synthetic ACE2 substrate, Mca‐APK(Dnp) [4] (final concentration 25 µm) were carried out at room temperature. The assay was monitored continuously by measuring the increase in fluorescence (excitation = 340 nm, emission = 430 nm) upon substrate hydrolysis using a Wallac Victor2 fluorescence plate reader (Turku, Finland). Initial velocities were determined from the linear rate of fluorescence increase over the 0–60 min time course. The reaction product was quantified by using standard solutions of Mca.
Acknowledgements
We thank the Medical Research Council of Great Britain (MRC) and the National Heart Research Fund (NHRF) for financial support.
References
- 1. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G & Turner AJ (2000) A human homolog of angiotensin‐converting enzyme: cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem 275, 33238–33243. [DOI] [PubMed] [Google Scholar]
- 2. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R et al. (2000) A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87, E1–E9. [DOI] [PubMed] [Google Scholar]
- 3. Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ & van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS cornavirus. A first step in understanding SARS pathogenesis. J Pathol 203, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F et al. (2002) Hydrolysis of biological peptides by human angiotensin‐converting enzyme‐related carboxypeptidase. J Biol Chem 277, 14838–14843. [DOI] [PubMed] [Google Scholar]
- 5. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira‐dos‐Santos AJ, da Costa J, Zhang L, Pei Y et al. (2002) Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature 417, 822–828. [DOI] [PubMed] [Google Scholar]
- 6. Allred AJ, Donoghue M, Acton S & Coffman TM (2002) Regulation of blood pressure by the angiotensin converting enzyme homologue ACE2. Am J Nephrol 13, 52A. [Google Scholar]
- 7. Donoghue M, Wakimoto H, Maguire CT, Acton S, Hales P, Stagliano N, Fairchild‐Huntress V, Xu J, Lorenz JN, Kadambi V et al. (2003) Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins, J Mol Cell Cardiol 35, 1043–1053. [DOI] [PubMed] [Google Scholar]
- 8. Chappell MC, Jung F, Gallagher PE, Averill DB, Crackower MA, Penninger JM & Ferrario CM (2002) Omapatrilat treatment is associated with increased ACE2 and angiotensin (1–7) in spontaneously hypertensive rats. Hypertension 40, 409. [Google Scholar]
- 9. Neves LA, Williams AF, Averill DB, Ferrario CM, Walkup MP & Brosnihan KB (2003) Pregnancy enhances the angiotensin (Ang)‐(1–7) vasodilator response in mesenteric arteries and increases the renal concentration and urinary excretion of Ang‐(1–7). Endocrinology 144, 3338–3343. [DOI] [PubMed] [Google Scholar]
- 10. Brosnihan KB, Neves LAA, Joyner J, Averill DB, Chappell MC, Sarao R, Penninger J & Ferrario CM (2003) Enhanced renal immunocytochemical expression of Ang‐(1–7) and ACE2 during pregnancy. Hypertension 42, 749–753. [DOI] [PubMed] [Google Scholar]
- 11. Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR & Canver CC (2003) Increased angiotensin‐(1–7)‐forming activity in failing human heart ventricles. Evidence for upregulation of the angiotensin‐converting enzyme homologue ACE2. Circulation 108, 1707–1712. [DOI] [PubMed] [Google Scholar]
- 12. Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J & Cooper ME (2003) Characterization of renal angiotensin‐converting enzyme 2 in diabetic nephropathy. Hypertension 41, 392–397. [DOI] [PubMed] [Google Scholar]
- 13. Ye, M , Wysocki J, Naaz P, Salabat MR, LaPointe MS & Batlle D (2004) Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice. A renoprotective combination? Hypertension 43, 1120–1125. [DOI] [PubMed] [Google Scholar]
- 14. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, et al. (2003) Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guy JL, Lambert DW, Warner FJ, Hooper NM & Turner AJ (2005) Membrane‐associated zinc peptidase families: comparing ACE and ACE2. Biochim Biophys Acta in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Turner AJ, Hiscox JA & Hooper NM (2004) ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci 25, 291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guy JL, Jackson RM, Acharya KR, Sturrock ED, Hooper NM & Turner AJ (2003) Angiotensin‐converting enzyme‐2 (ACE2): Comparative modelling of the active site, substrate specificity and chloride sensitivity. Biochemistry 42, 13185–13192. [DOI] [PubMed] [Google Scholar]
- 18. Liu X, Fernandez M, Wouters MA, Heyberger S & Husain A (2001) Arg (1098) is critical for the chloride dependence of human angiotensin I‐converting enzyme C‐domain catalytic activity, J Biol Chem 276, 33518–33525. [DOI] [PubMed] [Google Scholar]
- 19. Natesh R, Schwager SL, Sturrock ED & Acharya KR (2003) Crystal structure of the human angiotensin‐converting enzyme‐lisinopril complex. Nature 421, 551–554. [DOI] [PubMed] [Google Scholar]
- 20. Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA et al. (2004) ACE2 X‐ray structures reveal a large hinge‐bending motion important for inhibitor binding and catalysis. J Biol Chem 279, 17996–18007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dales NA, Gould AE, Brown JA, Calderwood EF, Guan B, Minor CA, Gavin JM, Hales P, Kaushik VK, Stewart M, et al. (2002) Substrate‐based design of the first class of angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) inhibitors. J Am Chem Soc 124, 11852–11853. [DOI] [PubMed] [Google Scholar]
- 22. Fernandez M, Liu X, Wouters MA, Heyberger S & Husain A (2001) Angiotensin I‐converting enzyme transition state stabilization by HIS1089: evidence for a catalytic mechanism distinct from other gluzincin metalloproteinases. J Biol Chem 276, 4998–5004. [DOI] [PubMed] [Google Scholar]
- 23. Kim HM, Shin DR, Yoo OJ, Lee H & Lee JO (2003) Crystal structure of Drosophila angiotensin I‐converting enzyme bound to captopril and lisinopril. FEBS Lett 538, 65–70. [DOI] [PubMed] [Google Scholar]
- 24. Arndt JW, Hao B, Ramakrishnan V, Cheng T, Chan SI & Chan MK (2002) Crystal structure of a novel carboxypeptidase from the hyperthermophilic archaeon Pyrococcus furiosus . Structure (Camb) 10, 215–224. [DOI] [PubMed] [Google Scholar]
- 25. Bunning P & Riordan JF (1983) Activation of angiotensin converting enzyme by monovalent anions. Biochemistry 22, 110–116. [DOI] [PubMed] [Google Scholar]
- 26. Tzakos AG, Galanis AS, Spyroulias GA, Cordopatis P, Manessi‐Zoupa E & Gerothanassis IP (2003) Structure‐function discrimination of the N‐ and C‐catalytic domains of human angiotensin‐converting enzyme: implications for Cl‐activation and peptide hydrolysis mechanisms. Protein Eng 16, 993–1003. [DOI] [PubMed] [Google Scholar]
- 27. Natesh R, Schwager SLU, Evans HR, Sturrock ED & Acharya KR (2004) Structural details on the binding of the antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I‐converting enzyme. Biochemistry 43, 8718–8724. [DOI] [PubMed] [Google Scholar]
- 28. Huang L, Sexton DJ, Skogerson K, Devlin M, Smith R, Sanyal I, Parry T, Kent R, Enright J, Wu Q et al. (2003) Novel peptide inhibitors of angiotensin‐converting enzyme 2. J Biol Chem 278, 15532–15540. [DOI] [PubMed] [Google Scholar]
- 29. Huentelman MJ, Zubcevic J, Hernandez Prada JA, Xiao X, Dimitrov DS, Raizada MK & Ostrov DA (2004) Structure‐based discovery of a novel angiotensin‐converting enzyme 2 inhibitor. Hypertension 44, 903–906. [DOI] [PubMed] [Google Scholar]
- 30. Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ & Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150, 76–85. [DOI] [PubMed] [Google Scholar]
- 31. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]