

Abstract
The expression of p16/CDKN2A, the second most commonly inactivated tumour suppressor gene in cancer, is lost in the majority of chordomas. However, the mechanism(s) leading to its inactivation and contribution to disease progression have only been partially addressed using small patient cohorts. We studied 384 chordoma samples from 320 patients by immunohistochemistry and found that p16 protein was lost in 53% of chordomas and was heterogeneously expressed in these tumours. To determine if CDKN2A copy number loss could explain the absence of p16 protein expression we performed fluorescence in situ hybridisation (FISH) for CDKN2A on consecutive tissue sections. CDKN2A copy number status was altered in 168 of 274 (61%) of samples and copy number loss was the most frequent alteration acquired during clinical disease progression. CDKN2A homozygous deletion was always associated with p16 protein loss but only accounted for 33% of the p16‐negative cases. The remaining immunonegative cases were associated with disomy (27%), monosomy (12%), heterozygous loss (20%) and copy number gain (7%) of CDKN2A, supporting the hypothesis that loss of protein expression might be achieved via epigenetic or post‐transcriptional regulatory mechanisms. We identified that mRNA levels were comparable in tumours with and without p16 protein expression, but other events including DNA promoter hypermethylation, copy number neutral loss of heterozygosity and expression of candidate microRNAs previously implicated in the regulation of CDKN2A expression were not identified to explain the protein loss. The data argue that p16 loss in chordoma is commonly caused by a post‐transcriptional regulatory mechanism that is yet to be defined.
Keywords: chordoma, copy number, p16, CDKN2A, IHC, FISH, biomarker
Introduction
Chordoma is a rare primary malignant bone tumour showing notochordal differentiation and affected individuals have a median survival of 7 years from presentation 1, 2. Chordoma is characterised by expression of the embryonic transcription factor TBXT, also known as brachyury and T, which plays a critical role in the development of the disease 3, 4. Genomic studies have failed to identify recurrent genetic driver alterations other than copy number gain of TBXT in 27% of cases 5 in addition to occasional sporadic chromosomal rearrangements and alterations involving RB1, TP53 and cyclin dependent kinase inhibitor 2A (CDKN2A) 6.
The CDKN2A gene (chromosome 9p21) encodes the proteins p14ARF and p16INK4a, also referred to as p16, generated through alternative exon usage 7. p16 is transcribed using exons 1α, 2 and 3, whereas p14ARF is transcribed using exon 1β and exon 2. Both proteins are involved in cell cycle control via the Rb and p53 pathways which are critical for self‐renewal and ageing 8. p14ARF stabilises and activates the p53 pathway, whereas p16 blocks G1/S cell cycle progression by preventing phosphorylation of Rb: disruption of control of these pathways plays a pivotal role in the progression of a variety of cancers 9. CDKN2A is part of a locus that also contains CDKN2B, which encodes p15INK4b, a tumour suppressor that, like p16INK4a, inhibits CDK4/CDK6 10.
CDKN2A is the second most frequently inactivated tumour suppressor gene in cancer 9, 11 and its inactivation is achieved in the majority of cases via homozygous deletion or promoter hypermethylation 11. Germline mutations in CDKN2A confer susceptibility to melanoma and other tumours 12, 13, and haploinsufficiency of p14ARF has been implicated in genetic models of various cancers 12, 14.
The CDKN2A gene locus is deleted and p16 protein expression is lost in a number of chordoma cell lines 15, 16. Loss of p16 protein expression has also been reported in up to 80% of chordomas 6, 17, 18. The mechanism leading to its inactivation and the contribution of CDKN2A loss to disease progression have only been partially elucidated. Using small numbers of chordoma samples, it has previously been reported that 3–33% of chordoma cases harbour homozygous deletions of CDKN2A 6, 17, single nucleotide variations are rare 5, 18, and DNA promoter hypermethylation is an uncommon event 6. The absence of therapeutic options for patients with chordoma makes this observation clinically significant as p16 loss has been shown to sensitise to CDK4/6 inhibitors 15, 19, 20, making expression of p16 a potential biomarker for patient stratification and prognosis. This prompted us to interrogate a large number of chordoma samples with the aim of increasing our understanding of the role of CDKN2A inactivation in the pathogenesis of chordoma.
Materials and methods
Chordoma samples
Tumour diagnoses were made using the WHO classification 2. Frozen tumour material was available for 35 chordomas: 10 were analysed by whole genome sequencing and RNA sequencing and 26 by whole exome sequencing, the results of which have been reported previously 5. Formalin‐fixed paraffin‐embedded samples were obtained from the archive of the Royal National Orthopaedic Hospital and several other sites. The samples were used to construct tissue microarrays (TMAs), which were built as previously described 21.
Ethical approval for in‐house chordoma samples was obtained from the Cambridgeshire 2 Research Ethics Service (reference 09/H0308/165) (HTA Licence 12198). Samples were also obtained through the Brain UK Biobank (reference 14/006 – Large scale genetic and epigenetic screen of chordoma).
Chordoma cell lines
UCH‐1, UCH‐2, MUG‐Chor, UM‐Chor, UCH‐11, JHC7 (http://www.chordomafoundation.org/) and UCH‐7 16 are well characterised human chordoma cell lines; all derived from sacral tumours except UM‐Chor which was generated from a clival chordoma. U2OS (ATCC® HTB96™, ATCC, Manassas, VA, USA), an osteosarcoma cell line that lacks expression of TBXT, used as a control, was cultured according to ATCC guidelines. Cell lines were quality controlled by short‐tandem‐repeat analysis (DNA Diagnostic Centre, London, UK) and were regularly tested to ensure that they were mycoplasma‐free.
Fluorescence in situ hybridisation and immunohistochemistry
Fluorescence in situ hybridisation (FISH) was performed as described previously 22 using the p16/CDKN2A (9p21) (Vysis, Abbott Molecular, Abbott Park, IL, USA) and the TBXT (Custom probe to chr6:166526346–166 623 395; Agilent Technologies, Santa Clara, CA, USA) probes. Assessment of CDKN2A and TBXT FISH was undertaken as previously reported 22: for a probe signal to be counted as abnormal at least 15% of the nuclei analysed were required to reveal an aberrant signal on counting a minimum of 50 consecutive non‐overlapping nuclei. The following categories were determined as follows (1) monosomy (one p16/CDKN2A and one centromeric signal); (2) heterozygous deletion (loss of one copy of p16/CDKN2A in the presence of two centromeric signals); (3) homozygous deletion (loss of two copies of p16/CDKN2A in the presence of one or two centromeric signals) and (4) amplification (p16/CDKN2A centromeric ratio greater than 2).
Immunohistochemistry (IHC) was performed on a Leica Bond 3 as previously described 21. The p16 (JC8) antibody (Santa Cruz, USA, catalogue number SC‐56330) was used at a dilution of 1 of 200. This antibody was previously validated by knock‐down in vitro experiments 23. As TMAs are not fully representative of heterogenous tumours, IHC was repeated and validated on full sections in samples where there was loss of immunoreactivity: this provided a high concordance (88%, 5 false negatives/43). For those cases for which the results obtained using TMAs was inconclusive, the IHC and FISH were repeated on full tissue sections.
Real‐time quantitative PCR
Quantitative real‐time PCR (qPCR) for mRNA and miRNA expression was performed as previously reported 16 (see supplementary material, Supplementary Materials and Methods).
Genomic and transcriptomic analysis
The variant calling pipeline of the Cancer Genome Project at the Wellcome Trust Sanger Institute was used to call somatic mutations. The following algorithms, with standard settings, and no additional post‐processing were used on aligned DNA BAM files: CaVEMan (1.11.0) for substitutions 24; Pindel (2.1.0) for indels 25; BRASS (5.3.3; https://github.com/cancerit/BRASS) for rearrangements, and ASCAT NGS (4.0.0) for copy number aberrations 26. Sequenced RNA libraries were aligned with hisat2 (v2.1.0) to hg19 reference genome. EdgeR (v3.24.0) was used to count gene features.
DNA methylation analysis
DNA methylation array data (450K or EPIC array, Illumina, CA, USA) of 35 chordoma samples have been published previously 27 and EPIC array was performed on chordoma (UCH1, UM‐Chor, UCH7, MUG‐Chor) and U2OS cell lines. Nucleic acid was prepared as previously reported 5.
Statistical analysis
Continuous variables were compared via unpaired t‐test, whereas categorical variables were compared via Fisher's exact test and Chi Squared test using Wizard 1.9.21. Data were considered statistically significant when p < 0.05. Q values were calculated using the R package q value. In gene expression studies, data were judged to be statistically significant when P value was calculated as less than 0.05 by two‐tailed Student's t‐test. Statistical analysis was performed in GraphPad PRISM 5.0 (GraphPad Software, La Jolla, CA, USA).
Results
A total of 384 sporadic chordoma samples were collected from 320 patients from across Europe, including the UK, 286 of which were primary tumours, 86 local recurrences and 12 from metastatic disease. Samples (n = 2–6) from more than one time point were studied for 39 patients. The tumours were located in the skull base (n = 90), mobile spine (n = 48), sacrum/coccyx (n = 178) and extra‐axial sites (n = 4). The age at presentation of the patients ranged from 6 to 91 (median 60) years: those with tumours located at the skull base, mobile spine, sacro‐coccygeum and at extra‐axial sites presented between the ages of 6–68, 14–79, 14–76 and 24–28 years of age respectively. The female to male ratio was 1:1.53 (83 females, 127 males).
Genome sequencing data
Whole genome sequencing previously reported 5 revealed copy number alterations at the CDKN2A locus in 7 of 10 chordomas studied (see supplementary material, Table S1 and Figure S1). The patterns of CDKN2A deletion were variable: three cases showed complete loss of chromosome 9, two cases showed loss of several megabases and one case harboured a smaller deletion in the range of kilobases. One case revealed CDKN2A copy number gain; none of the samples showed single nucleotide variants (SNVs) or indels of CDKN2A. FISH revealed TBXT amplification in three cases: this did not correlate with a specific CDKN2A copy number status. Whole genomes and whole exomes (n = 35) were also analysed for the presence of copy number neutral loss of heterozygosity (LOH), which was identified in only one case where it was associated with retention of p16 expression in the absence of mutations.
p16/CDKN2A IHC and FISH
The result of p16 IHC performed on 303 informative samples demonstrated loss of expression in 53% (162/303) of cases (Figure 1A and Table 1), a finding not dissimilar to previous reports 6, 15. To determine if CDKN2A copy number loss could explain the absence of p16 protein expression we performed FISH on consecutive tissue sections to those used for IHC (Figure 1B and Table 1). Results from 274 of these samples were informative: the majority (167/274, 61%) of chordomas harboured CDKN2A copy number alterations, the most frequent event being copy number loss which was detected in 138/167 samples (83%) (Table 1).
Figure 1.

p16 IHC and CDKN2A FISH in chordoma samples. (A) Representative IHC images of chordoma cases negative (left) or positive (right) for p16. Objective magnification ×4. (B) Representative images of chordoma cases showing monosomic (left) or disomic (right) copy number status as assessed by FISH. Objective magnification ×100. (C) Sankey diagram of the IHC and FISH results. Results were available for both p16/CDKN2A IHC and FISH for at least one sample from 243 of 320 patients.
Table 1.
p16 IHC and CDKN2A FISH data for all informative samples from 243 patients
| p16 status (IHC) | CDKN2A copy number (FISH) | ||||
|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | |||
| Positive | 141 (47) | Normal | Disomy | 107 (39) | |
| Negative | 162 (53) | Copy number loss | 138 (50) | Monosomy | 46 (17) |
| Total | 303 | Deletion (hetero) | 44 (16) | ||
| Deletion (homo) | 48 (17) | ||||
| Copy number gain | 29 (11) | Amplification | 1 (1) | ||
| Polysomy | 28 (10) | ||||
| Total | 274 | ||||
Results were available for both p16/CDKN2A IHC and FISH for at least one sample from 243 of 320 patients. Copy number loss is represented by monosomy, heterozygous deletion, or homozygous deletion. Copy number gain is represented by amplification or polysomy.
On aligning the FISH with IHC data (Figure 1C), we found 100% correlation between homozygous loss of CDKN2A and p16 loss of expression. However, only 33% (48/147) of the p16 immunonegative samples revealed a CDKN2A homozygous deletion, leaving an explanation to be found for the loss of protein expression for the remaining 67% of samples. Notably, among p16 immunonegative cases, 27, 12 and 20% showed disomy, monosomy and heterozygous loss respectively (Figure 1C). This apparent discordance between the FISH and IHC results raises the possibility that protein loss might be achieved via either epigenetic or post‐transcriptional regulatory mechanisms.
DNA methylation
To determine if an epigenetic mechanism could explain the p16 protein loss in some chordomas, DNA promoter methylation status of CDKN2A was assessed in four chordoma cell lines (all p16‐negative) and 35 chordoma samples, 15 of which were negative for p16 immunoreactivity. We found low levels of methylation in all samples, with the exception of the single clival INI‐1‐negative poorly differentiated chordoma analysed and the clival‐derived INI‐1‐negative UM‐Chor cell line: both of these showed higher levels of promoter methylation compared to all other samples, including a second INI‐1 immunoreactive clival tumour and three INI‐1‐positive cell lines (UCH1, MUG‐Chor and UCH7) (see supplementary material, Figure S2A). This demonstrates, particularly when previous reports are considered 6, that DNA methylation rarely accounts for p16 protein loss in chordoma.
Polymorphism in CDKN2A
CDKN2A gene expression has been reported to be influenced by the SNP rs11515, which is the most frequent CDKN2A polymorphism located in the 3′UTR of the gene and has been associated with various cancers 28. Analysis of whole genomes and exomes revealed that, of 35 cases, 24 (68%) were homozygous (CC genotype) and 11 (32%) were heterozygous (CG genotype) for the major allele. This frequency is similar to that found in the general population 28 thereby excluding an association of the SNP rs11515 with chordoma. Moreover, this genotype did not correlate with p16 loss at the protein level (33% [3/9] CG cases, 60% [12/19] CC cases, p = 0.22), nor with CDKN2A genetic deletion (22% [4/14] CC cases, 30% [3/10] CG cases, p = 0.99). Analysis of RNA sequencing showed that transcriptional levels of p14ARF, p16INK4a and ANRIL (a long noncoding antisense transcript part of the CDKN2A locus which promotes CDKN2A transcriptional repression 29), were also not influenced by the SNP (see supplementary material, Figure S2B–D). Taken together these results argue against the SNP rs11515 influencing p16 expression in chordoma.
CDKN2A gene expression and IHC
To study further the loss of p16 protein expression in the absence of homozygous deletion we analysed the gene expression in 10 cases previously subjected to RNA sequencing 5 (see supplementary material, Table S1). Three of these cases, which showed loss of p16 protein expression, revealed monosomy in two of the cases and disomy in the third by FISH; however, the CDKN2A transcript levels were comparable to those that retained p16 immunoreactivity (Figure 2A). These data were supported by detectable levels of CDKN2A transcript associated with the loss of expression of p16 at the protein level in an additional 15 chordoma cases assessed by qPCR and in seven chordoma cell lines (three with homozygous deletions and four with monosomy) (Figure 2B‐C). This further supports the concept that loss of p16 protein expression is achieved at the post‐transcriptional level.
Figure 2.

CDKN2A transcript levels detected despite p16 immunonegativity in chordoma samples and cell lines and lack of correlation to miRNA expression. (A) Expression of CDKN2A transcript assessed in 10 chordoma samples by RNA‐sequencing. FPKM, fragments per kilobase of transcript per million mapped reads. (B) Expression of CDKN2A transcript assessed in 22 chordoma samples by qPCR. (C) Expression of CDKN2A transcript in chordoma cell lines by qPCR: p16‐negative with homozygous deletion (UCH1, UCH2, MUG‐Chor), p16 negative and monosomic (UCH7, UM‐Chor, UCH11), p16‐positive and monosomic (JHC7). (D–F) Expression of miRNA‐10, –24 and –125 in chordoma cases, as assessed by FISH, and showing positivity or negativity for p16 protein expression by IHC. Cases showing monosomic or disomic CDKN2A copy number were combined in this analysis.
Post‐transcriptional regulation of CDKN2A
miRNAs including miR‐24‐2 30, 31, miR‐10b‐5p 32 and miR‐125b 33 have been shown to regulate p16 expression at the post‐transcriptional level in various cancers. We therefore tested the expression of these candidate miRNAs in monoallelic (n = 16) and disomic (n = 18) chordoma cases that had retained or lost p16 immunoreactivity (Figure 2D–F), but we failed to identify a correlation with p16 expression by IHC, suggesting that other mechanisms yet to be identified control the expression of p16 in chordoma.
Patients with multiple samples
Next, we assessed tumour heterogeneity and evolution with respect to p16 IHC and CDKN2A copy number alteration from patients for whom there was more than one sample available including the primary tumour (Table 2). Informative samples from 36 patients revealed that 26 (72%) showed no change in the immunoreactivity status over time. Tumours from five (14%) patients showed loss of p16 over time and, when correlating these results with FISH on consecutive tissue sections, we observed a change in copy number in three cases (Table 2). This included a case of a p16‐positive primary tumour with CDKN2A disomy which revealed heterozygous loss in two local recurrences while retaining p16 positivity, and a homozygous deletion with p16 loss in a third local recurrence: this demonstrates a step‐wise acquisition of CDKN2A inactivation in chordoma evolution (case 212). However, in another four cases (5, 54, 213 and 214), despite the loss of p16 immunoreactivity over time, there was no change in the copy number, which was represented in all cases by either heterozygous loss or monosomy (Table 2).
Table 2.
p16 IHC and CDKN2A FISH of multiple samples from 39 patients
| Study unique ID | Anatomical site | Age at dx | Primary | LR1 | LR2 | LR3 | LR4 | LR5 | Met | Events per patient | IHC over time | FISH over time |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | Mobile spine | 53 | 2 | Pos > Neg | Mono > Mono | |||||||
| 20 | Mobile spine | 68 | 2 | Pos > Pos | Mono > Diso | |||||||
| 22 | Sacrum/coccyx | 63 | 2 | Neg > Neg | NI | |||||||
| 23 | Sacrum/coccyx | 72 | 2 | Pos > Pos | NI | |||||||
| 39 | Sacrum/coccyx | ‐ | 2 | Neg > Pos | Disomy > Disomy | |||||||
| 42 | Sacrum/coccyx | 58 | 2 | Neg > Neg | Del Homo > Del Homo | |||||||
| 54 | Sacrum/coccyx | 43 | 2 | Pos > Neg | Del Het > Del Het | |||||||
| 56 | Sacrum/coccyx | 61 | 2 | Neg > Neg | Del Homo > Del Homo | |||||||
| 61 | Sacrum/coccyx | 58 | 4 | Neg > Neg | NI | |||||||
| 67 | Sacrum/coccyx | 54 | 2 | Pos > Pos | Del Het > Del Het | |||||||
| 88 | Sacrum/coccyx | 67 | 2 | Neg > Pos | Mono > Mono | |||||||
| 90 | Sacrum/coccyx | 67 | 2 | Neg > Neg | Del Het > Del Het | |||||||
| 95 | Sacrum/coccyx | 63 | 3 | Neg > Neg | Mono > Mono | |||||||
| 131 | Sacrum/coccyx | 69 | 2 | Pos > Pos | NI | |||||||
| 134 | Sacrum/coccyx | 65 | 2 | NI | NI | |||||||
| 135 | Sacrum/coccyx | 65 | 2 | Pos > Pos | NI | |||||||
| 136 | Sacrum/coccyx | 73 | 2 | Neg > Neg | NI | |||||||
| 137 | Sacrum/coccyx | 69 | 2 | Neg > Neg | NI | |||||||
| 138 | Sacrum/coccyx | 61 | 3 | Neg > Neg | Del Het > Del Het | |||||||
| 143 | Sacrum/coccyx | ‐ | 6 | Pos > Pos | Poly > Poly | |||||||
| 153 | Sacrum/coccyx | 59 | 2 | Pos > Pos | Mono > Mono | |||||||
| 157 | Sacrum/coccyx | 53 | 2 | Pos > Pos | Mono > Poly | |||||||
| 162 | Skull base | 43 | 3 | Pos > Pos | Mono > Poly | |||||||
| 165 | Mobile spine | 49 | 2 | Pos > Pos | NI | |||||||
| 166 | Sacrum/coccyx | 63 | 2 | Neg > Neg | Mono > Mono | |||||||
| 168 | Skull base | 47 | 3 | NI | Diso > Mono | |||||||
| 173 | Skull base | 19 | 2 | NI | NI | |||||||
| 174 | Skull base | 35 | 2 | Pos > Pos | NI | |||||||
| 175 | Sacrum/coccyx | 35 | 3 | Neg > Neg | Del Het > Del Het | |||||||
| 177 | Skull base | 37 | 3 | Pos > Pos | Poly > Poly | |||||||
| 182 | Skull base | 65 | 2 | Pos > Pos | Diso > Diso | |||||||
| 209 | Skull base | 40 | 4 | Pos > Pos | Diso > Poly | |||||||
| 211 | Skull base | 63 | 3 | Neg > Pos | Del Het > Del Het | |||||||
| 212 | Skull base | 46 | 4 | Pos > Neg | Diso > Del het > Del homo | |||||||
| 213 | Skull base | 60 | 3 | Pos > Neg | Diso > Mono | |||||||
| 214 | Mobile spine | 57 | 2 | Pos > Neg | Diso > Del homo | |||||||
| 217 | Skull base | 31 | 3 | Neg > Pos | Mono > Mono | |||||||
| 218 | Vertebra | 53 | 5 | Neg > Neg | Del Homo > Del Homo | |||||||
| 219 | Mobile spine | 48 | 3 | Neg > Pos | Del Homo > Del Het |
Blue, samples analysed. p16 IHC was NI for three patients, leaving 36 patients available for analysis. Del Het, Heterozygous deletion; Del Mono, Homozygous deletion; Diso, disomy; Dx, diagnosis; LR1–LR5, first to fifth local recurrence; Met, metastasis; Mono, monosomy; Neg, p16‐negative; NI, non‐informative; Poly, polysomy; Pos, p16 positive.
Samples from another five patients (39, 88, 211, 217 and 219) revealed loss of p16 expression in the primary tumour but retention in one of the local recurrences (Table 2). We postulated that this could be explained by incomplete excision of an area of the primary tumour which was p16 immunoreactive. This was confirmed when we showed that p16 immunoreactivity was present focally in sections from one of two tissue blocks of the same specimen (case 39) (additional tissue blocks from the other four cases were not available for testing) (Figure 3 and see supplementary material, Table S2). We then tested for p16 expression in multiple blocks from another five cases (22, 56, 61, 90 and 95): three cases were consistently negative whereas two cases showed heterogeneous expression of p16 (see supplementary material, Figure S3 and Table S2). Tumour heterogeneity could therefore explain the finding of the absence of p16 in the primary tumours and the retention in local recurrences (cases 39, 88, 211, 217 and 219). This could also explain the finding of CDKN2A monosomy in a primary tumour and disomy (cases 20) or polyploidy (cases 157 and 162) in local recurrences. However, these FISH results could also be accounted for by complex structural alterations developing over time including whole chromosomal doubling, but a detailed analysis using whole genome sequencing would be required to confirm such a mechanism.
Figure 3.
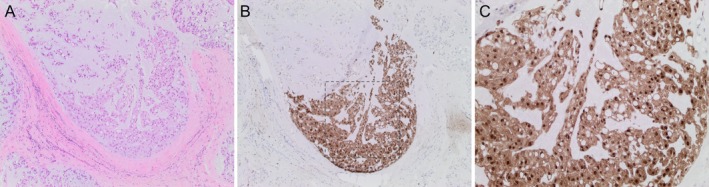
Heterogeneous expression of p16 in chordoma samples. (A–C) Representative IHC images of one chordoma case showing heterogeneous expression of p16: H&E (A, ×4 objective magnification), p16 (B and C, ×4 and ×10 objective magnification respectively). C shows approximately the region enclosed by the dotted lines in B.
p16/CDKN2A status and clinical data
Finally, we correlated CDKN2A FISH and IHC results with patients' clinical data (Tables 3 and 4). The anatomical location of the tumour did not influence CDKN2A copy number status or p16 protein expression. There was a trend towards CDKN2A copy number loss being more common in older patients (>40 years old, p = 0.020, q = 0.102) and in samples from local recurrences compared to primary tumours (p = 0.048, q = 0.154). Analysis of the 12 metastatic tumours available revealed that p16 protein expression was lost in all samples (p = 0.0005, q = 0.005, compared to primary tumours). CDKN2A copy number loss in metastatic disease was not significantly different form primary tumours (p = 0.225, q = 0.342).
Table 3.
p16 immunoreactivity and clinical information on samples from 243 patients with chordoma
| p16 (IHC) | Positive (%) | Negative (%) | P value | q value | |
|---|---|---|---|---|---|
| Age | <40 | 16/22 (72) | 6/22 (28) | ||
| >40 | 58/119 (49) | 61/119 (48) | 0.0617 | 0.1542 | |
| Stage of tumour | Primary | 103/211 (49) | 108/211 (51) | ||
| LR1–LR5 | 39/81 (48) | 42/81 (52) | 0.8960 | 0.8960 | |
| Metastasis | 0/12 (0) | 12/12 (100) | 0.0005 | 0.0050 | |
| Anatomical Site | Sacrum‐Coccygeal | 54/108 (50) | 54/108 (50) | ||
| Mobile Spine | 16/26 (62) | 10/26 (38) | 0.4829 | 0.5365 | |
| Skull Base | 20/57 (35) | 37/57 (65) | 0.1869 | 0.3417 | |
Results were available for both p16/CDKN2A IHC and FISH for at least one sample from 243 of 320 patients. Age relates to age at the time of diagnosis of the primary tumour. Anatomical site relates to the site at presentation.
Table 4.
CDKN2A copy number by FISH and clinical information for patients with chordoma
| CDKN2A (FISH) | Disomic (%) | Copy number loss (%) | Copy number gain (%) | P value | q value | |
|---|---|---|---|---|---|---|
| Age | <40 | 15/22 (68) | 6/22 (27) | 1/22 (5) | ||
| >40 | 44/113 (39) | 58/113 (51) | 11/113 (10) | 0.0204 | 0.1020 | |
| Stage of tumour | Primary | 86/193 (44) | 88/193 (46) | 19/193 (10) | ||
| LR1–LR5 | 19/70 (27) | 43/70 (61) | 8/70 (12) | 0.0480 | 0.1542 | |
| Anatomical site | Metastasis | 2/11 (18) | 7/11 (64) | 2/11 (18) | 0.2253 | 0.3417 |
| Sacrum‐coccyx | 36/93 (37) | 47/93(52) | 10/93 (11) | |||
| Mobile spine | 14/25 (56) | 10/25 (40) | 1/25 (4) | 0.2392 | 0.3417 | |
| Skull base | 28/56 (50) | 21/56 (37) | 7/56 (13) | 0.3342 | 0.4177 | |
Results were available for both p16/CDKN2A IHC and FISH for at least one sample from 243 of 320 patients.
Discussion
In this p16 IHC study of chordomas, the largest to date, we report that diffuse p16 loss is a frequent finding in this disease, occurring in at least 53% of cases, confirming the previously described frequent loss (66%) in 43 chordoma cases 15. Much of the reported evidence available to date implies that this loss is associated with either a heterozygous or homozygous deletion of the region covering the CDKN2A locus but the number of chordoma samples studied using both markers is limited. Specifically, the array CGH findings by Hallor and colleagues 17 showed that 70% of cases displayed genetic alterations but the protein expression was not studied. Le et al 6 performed immunohistochemical analysis on 18 cases and showed loss of expression in 83% of cases; they also observed loss of 9p, either through entire chromosome 9 loss or partial 9p loss alone, in 15/20 cases (75%) by array CGH. Our FISH results also corroborate the published array CGH findings, with 50% of chordomas revealing copy number loss represented by homozygous or heterozygous deletion or monosomy. As expected, homozygous deletion was always associated with loss of p16 expression but otherwise we found little correlation between copy number and p16 immunoreactivity. Notably, in our study 27% of chordomas with loss of p16 protein expression exhibited a normal diploid CDKN2A copy number status, a finding also reported by Le et al 6 in two of 18 cases. Furthermore, 30 of 147 (20%) cases with complete loss of p16 protein expression showed heterozygous loss and 18 of 147 (12%) showed monosomy. This raises the question of the mechanism by which the loss of protein occurs.
The frequent loss of p16 protein expression not associated with homozygous loss of CDKN2A could not be explained by a variety of mechanisms that we have investigated. Specifically, promoter DNA hypermethylation, one of the most common mechanisms implicated in the silencing of CDKN2A in tumours, was not found to account for this, confirming the work of others who reported that only one of 15 chordoma cases tested had definitive evidence of CDKN2A promoter methylation 6. Only one case, a clival chordoma, showed higher levels of DNA methylation for one of the probes at the promoter region. An interesting finding is that this case showed negativity for INI‐1, a feature that is found in poorly differentiated chordomas, which are known to exhibit a methylation profile different from conventional chordomas 34. Furthermore, with only one in 35 cases revealing copy number neutral LOH, a common copy number alteration caused by uniparental disomy and usually associated with homozygous mutations, homozygous deletions or alterations in cancer‐promoting imprinted genes 35, 36, this mechanism is unlikely to explain the loss of p16 protein in the absence of homozygous deletion. Indeed, the copy number neutral LOH identified in one chordoma was associated with p16 positivity in the absence of SNVs or indels, a finding rarely reported in other cancers 37.
We pursued two other major lines of enquiry in an attempt to explain the mechanism by which p16 protein could be lost. First, we analysed whether the SNP rs11515 was associated with CDKN2A expression because of the known association in melanoma, sporadic colorectal, skin, bladder, cervical, breast cancer and glioblastoma 28; however, we failed to detect any association. Second, we tested the expression of miRNAs which were previously shown to control p16 protein expression. MiR‐24‐2, a negative regulator of p16, blocks p16 translation in keratinocytes and chondrocytes 30, 31. Increased expression of miR‐10b‐5p correlates with reduced expression of its target genes including CDKN2A in renal papillary carcinoma and glioma 32. miR‐125b is a known tumour suppressor gene that blocks translation of a number of transcripts involved in the control of cell proliferation in various cancers 33, but a convincing association of miR‐125 and p16 expression has not been reported, with the exception of a study showing that hsa‐miR‐125b exhibited significant negative correlations with CDKN2A expression in glioblastoma multiforme 38. In this study we excluded an association between p16 and the expression of these miRNAs.
In view of the difficulty in explaining the loss of p16 protein by promoter hypermethylation and other selected epigenetic events, even though this has only been undertaken using a candidate approach, a number of reasons argue for a post transcriptional regulatory mechanism to explain the loss of protein in the presence of a retained allele as determined by FISH. It is otherwise difficult to explain how comparable levels of CDKN2A could be detected in chordomas with and without protein expression. Although CDKN2A mRNA would be expressed by non‐neoplastic cells in chordomas sample, this cannot be the case in the three chordoma cell lines in which mRNA was detected despite the loss of protein expression in the presence of monosomy for chromosome 9. Only one cell line (JHC7) was immunoreactive for p16 in the presence of monosomy, whereas the other three showed CDKN2A homozygous deletion and absence of protein expression.
The median life expectancy of patients with chordoma is 7 years 1. However, for individuals, it can range from months to more than 25 years (case 143), hence having a prognostic biomarker such as p16 IHC, which could be undertaken easily, would be valuable for patients and clinicians. However, retention of p16 expression cannot be reliably employed to predict less aggressive behaviour in chordoma, because it may be heterogeneously expressed, and detection is dependent on tumour sampling. Furthermore, as immunoreactivity is lost in 53% of our samples and others have reported an even greater percentage, we do not consider that it is useful to employ p16 IHC as a prognostic marker. However, p16 protein expression could be valuable for stratification of patients for the purposes of a clinical trial using a CDK4/6 inhibitor, in which it would be advisable to assess multiple areas 15. Finally, as much of the expression of p16 is controlled at a post transcriptional level, using FISH as a biomarker to determine protein expression provides limited information and is not recommended in a clinical setting.
Author contributions statement
AMF and LC conceived the study. AMF supervised the study. LC, NE, IU, LL and HY carried out the investigation. PL carried out bioinformatic analysis. RT, AMF and DL provided clinical data. AMF, SB, JP, NP, RT and FA curated samples. LC, AMF, IU and NP wrote the paper with input from other co‐authors.
Supporting information
Supplementary materials and methods
Figure S1. Patterns of CDKN2A copy number aberrations using whole genome sequencing data
Figure S2. Lack of CDKN2A promoter DNA methylation in chordoma samples and expression of p16, p14 and ANRIL in chordoma cases dependent on rs11515 SNP genotype
Figure S3. Heterogeneous expression of p16 in chordoma samples
Table S1. p16/CDKN2A assessed by IHC, genomic alterations and DNA methylation in 10 chordoma cases also analysed by whole genome sequencing
Table S2. p16 heterogeneity in chordoma
Acknowledgements
We would like to thank the UK Brain Bank for making samples available and the pathologists who provided the samples to the UK Brain Bank, including: Addenbrookes Hospital, Great Ormond Street Hospital, National Hospital for Neurology and Neurosurgery, Hull Royal Infirmary, Lancaster Teaching Hospital, Wellington, Plymouth Hospital. Funding for this project was received from Chordoma UK (a registered charity, RCN 1173201), Chordoma Foundation, and the Pathological Society of Great Britain and Ireland (Reference No: TSGS 2016 10 02). AMF is a NIHR senior investigator, and AMF, SB and NP are supported by the National Institute for Health Research, UCLH Biomedical Research Centre. AMF and NP are supported by the CRUK Experimental Cancer Centre. NP is a Cancer Research UK clinician scientist, grant number 18387. We would like to thank Dr Christopher Steele for critical input with the statistical analysis.
No conflicts of interest were declared.
Data availability
Infinium Methylation array data of chordoma and U2OS cell lines have been deposited in the National Center for Biotechnology Information GEO database under GEO accession number (GSE139410).
References
- 1. Stacchiotti S, Sommer J, Chordoma Global Consensus Group . Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol 2015; 16: e71–e83. [DOI] [PubMed] [Google Scholar]
- 2. Flanagan A, Yamaguchi T. Chordoma In: WHO Classification of Tumours of Soft Tissue and Bone. IARC Press: Lyon, 2013; 328–329. [Google Scholar]
- 3. Vujovic S, Henderson S, Presneau N, et al Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol 2006; 209: 157–165. [DOI] [PubMed] [Google Scholar]
- 4. Presneau N, Shalaby A, Ye H, et al Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional‐based study. J Pathol 2011; 223: 327–335. [DOI] [PubMed] [Google Scholar]
- 5. Tarpey PS, Behjati S, Young MD, et al The driver landscape of sporadic chordoma. Nat Commun 2017; 8: 890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le LP, Nielsen GP, Rosenberg AE, et al Recurrent chromosomal copy number alterations in sporadic chordomas. PLoS One 2011; 6: e18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16INK4A and their relevance to cancer. Biochemistry 2011; 50: 5566–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gil J, Peters G. Regulation of the INK4b‐ARF‐INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7: 667–677. [DOI] [PubMed] [Google Scholar]
- 9. Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res 2005; 576: 22–38. [DOI] [PubMed] [Google Scholar]
- 10. Krimpenfort P, IJpenberg A, Song JY, et al p15Ink4bis a critical tumour suppressor in the absence of p16Ink4a. Nature 2007; 448: 943–946. [DOI] [PubMed] [Google Scholar]
- 11. Zhao R, Choi BY, Lee MH, et al Implications of genetic and epigenetic alterations of CDKN2A (p16INK4a) in cancer. EBioMedicine 2016; 8: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krimpenfort P, Quon KC, Mooi WJ, et al Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001; 413: 83–86. [DOI] [PubMed] [Google Scholar]
- 13. Sharpless NE, Bardeesy N, Lee KH, et al Loss of p16Ink4a with retention of p19 predisposes mice to tumorigenesis. Nature 2001; 413: 86–91. [DOI] [PubMed] [Google Scholar]
- 14. Kamijo T, Bodner S, Van De Kamp E, et al Tumor spectrum in ARF‐deficient mice. Cancer Res 1999; 59: 2217–2222. [PubMed] [Google Scholar]
- 15. von Witzleben A, Goerttler LT, Marienfeld R, et al Preclinical characterization of novel chordoma cell systems and their targeting by pharmocological inhibitors of the CDK4/6 cell‐cycle pathway. Cancer Res 2015; 75: 3823–3831. [DOI] [PubMed] [Google Scholar]
- 16. Scheipl S, Barnard M, Cottone L, et al EGFR inhibitors identified as a potential treatment for chordoma in a focused compound screen. J Pathol 2016; 239: 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hallor KH, Staaf J, Jonsson G, et al Frequent deletion of the CDKN2A locus in chordoma: analysis of chromosomal imbalances using array comparative genomic hybridisation. Br J Cancer 2008; 98: 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choy E, MacConaill LE, Cote GM, et al Genotyping cancer‐associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PLoS One 2014; 9: e101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dickson MA. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin Cancer Res 2014; 20: 3379–3383. [DOI] [PubMed] [Google Scholar]
- 20. Schlenk R, Barth T, Groschel S, et al CDK4/6 inhibition in locally advanced/metastatic chordoma (NCT‐PMO‐1601). Ann Oncol 2017; 28 (Suppl 5): 538. [Google Scholar]
- 21. Shalaby AA, Presneau N, Idowu BD, et al Analysis of the fibroblastic growth factor receptor‐RAS/RAF/MEK/ERK‐ETS2/brachyury signalling pathway in chordomas. Mod Pathol 2009; 22: 996–1005. [DOI] [PubMed] [Google Scholar]
- 22. Amary MFC, Berisha F, Bernardi FDC, et al Detection of SS18‐SSX fusion transcripts in formalin‐fixed paraffin‐embedded neoplasms: analysis of conventional RT‐PCR, qRT‐PCR and dual color FISH as diagnostic tools for synovial sarcoma. Mod Pathol 2007; 20: 482–496. [DOI] [PubMed] [Google Scholar]
- 23. Ferdinando D, Wilson S, Wu H, et al The specificity and patterns of staining in human cells and tissues of p16INK4a antibodies demonstrate variant antigen binding. PLoS One 2013; 8: e53313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones D, Raine KM, Davies H, et al cgpCaVEManWrapper: simple execution of caveman in order to detect somatic single nucleotide variants in NGS data. Curr Protoc Bioinformatics 2016; 56: 15.10.1–15.10.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ye K, Schulz MH, Long Q, et al Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired‐end short reads. Bioinformatics 2009; 25: 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Loo P, Nordgard SH, Lingjaerde OC, et al Allele‐specific copy number analysis of tumors. Proc Natl Acad Sci U S A 2010; 107: 16910–11695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cottone L, Hookway ES, Cribbs A, et al Epigenetic inactivation of oncogenic brachyury (TBXT) by H3K27 histone demethylase controls chordoma cell survival. BioRxiv 2018; 44: 1–22. [Google Scholar]
- 28. Royds JA, Pilbrow AP, Ahn A, et al The rs11515 polymorphism is more frequent and associated with aggressive breast tumors with increased ANRIL and decreased p16INK4a expression. Front Oncol 2016; 5: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aguilo F, Zhou M, Walsh MJ. Long noncoding RNA, Polycomb, and the ghosts haunting INK4b‐ARF‐INK4a expression. Cancer Res 2011; 71: 5365–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lal A, Navarro F, Maher C, et al miR‐24 inhibits cell proliferation by suppressing expression of E2F2, MYC and other cell cycle regulatory genes by binding to “seedless” 3′UTR microRNA recognition elements. Mol Cell 2009; 35: 610–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Philipot D, Guérit D, Platano D, et al p16INK4a and its regulator miR‐24 link senescence and chondrocyte terminal differentiation‐associated matrix remodeling in osteoarthritis. Arthritis Res Ther 2014; 16: R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gabriely G, Yi M, Narayan RS, et al Human glioma growth is controlled by microRNA‐10b. Cancer Res 2011; 71: 3563–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yin H, Sun Y, Wang X, et al Progress on the relationship between miR‐125 family and tumorigenesis. Exp Cell Res 2015; 339: 252–260. [DOI] [PubMed] [Google Scholar]
- 34. Hasselblatt M, Thomas C, Hovestadt V, et al Poorly differentiated chordoma with SMARCB1/INI1 loss: a distinct molecular entity with dismal prognosis. Acta Neuropathol 2016; 132: 149–151. [DOI] [PubMed] [Google Scholar]
- 35. Teh MT, Blaydon D, Chaplin T, et al Genomewide single nucleotide polymorphism microarray mapping in basal cell carcinomas unveils uniparental disomy as a key somatic event. Cancer Res 2005; 65: 8597–8603. [DOI] [PubMed] [Google Scholar]
- 36. Fitzgibbon J, Smith LL, Raghavan M, et al Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res 2005; 65: 9152–9154. [DOI] [PubMed] [Google Scholar]
- 37. Sulong S, Moorman AV, Irving JA, et al A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood 2014; 113: 100–107. [DOI] [PubMed] [Google Scholar]
- 38. Feng J, Kim ST, Liu W, et al An integrated analysis of germline and somatic, genetic and epigenetic alterations at 9p21.3 in glioblastoma. Cancer 2012; 118: 232–240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Patterns of CDKN2A copy number aberrations using whole genome sequencing data
Figure S2. Lack of CDKN2A promoter DNA methylation in chordoma samples and expression of p16, p14 and ANRIL in chordoma cases dependent on rs11515 SNP genotype
Figure S3. Heterogeneous expression of p16 in chordoma samples
Table S1. p16/CDKN2A assessed by IHC, genomic alterations and DNA methylation in 10 chordoma cases also analysed by whole genome sequencing
Table S2. p16 heterogeneity in chordoma
Data Availability Statement
Infinium Methylation array data of chordoma and U2OS cell lines have been deposited in the National Center for Biotechnology Information GEO database under GEO accession number (GSE139410).