

Abstract
Objective
The objective of this study is to compare the risk of incident diabetes mellitus (DM) in patients with rheumatoid arthritis (RA) treated with biologic or targeted synthetic disease‐modifying antirheumatic drugs.
Methods
A new‐user observational cohort study was conducted using data from a US commercial (Truven MarketScan, 2005‐2016) claims database and a public insurance (Medicare, 2010‐2014) claims database. Patients with RA who did not have DM were selected into one of eight exposure groups (abatacept, infliximab, adalimumab, golimumab, certolizumab, etanercept, tocilizumab, or tofacitinib) and observed for the outcome of incident DM, defined as a combination of a diagnosis code and initiation of a hypoglycemic treatment. A stabilized inverse probability–weighted Cox proportional hazards model was used to account for 56 confounding variables and estimate hazard ratios (HRs) and 95% confidence intervals (CIs). All analyses were conducted separately in two databases, and estimates were combined using inverse variance meta‐analysis.
Results
Among a total of 50 505 patients with RA from Truven and 17 251 patients with RA from Medicare, incidence rates (95% CI) for DM were 6.8 (6.1‐7.6) and 6.6 (5.4‐7.9) per 1000 person‐years, respectively. After confounding adjustment, the pooled HRs (95% CI) indicated a significantly higher risk of DM among adalimumab (2.00 [1.11‐3.03]) and infliximab initiators (2.34 [1.38‐3.98]) compared with abatacept initiators. The pooled HR (95% CI) for the etanercept versus abatacept comparison was elevated but not statistically significant (1.65 [0.91‐2.98]). The effect estimates for certolizumab, golimumab, tocilizumab, and tofacitinib, compared with abatacept, were highly imprecise because of a limited sample size.
Conclusion
Initiation of abatacept was associated with a lower risk of incident DM in patients with RA compared with infliximab or adalimumab.
SIGNIFICANCE & INNOVATIONS.
Some preliminary evidence from observational studies has revealed a potentially lower risk of diabetes mellitus (DM) with tumor necrosis factor alpha inhibitors (TNF‐inhibitors), as well as with abatacept (a T‐cell co‐stimulation inhibitor), compared with nonbiologic disease‐modifying agents, which have general immunosuppressive properties. However, comparative risk of DM among patients with RA treated with different biologic and targeted synthetic disease‐modifying antirheumatic drugs is not well studied.
In this large cohort study that includes data from two nationwide data sources in the United States, we noted use of abatacept to be associated with a lower risk of incident DM, compared with TNF‐inhibitors, in patients with RA. Comparison of abatacept with other agents was inconclusive because of limited event counts available for valid treatment‐effect estimation.
INTRODUCTION
The contribution of inflammation in the pathogenesis of diabetes mellitus (DM) is now widely accepted, with studies unequivocally demonstrating an etiologic role of inflammation in the development of insulin resistance (1). Heightened systemic inflammatory activity in patients with rheumatoid arthritis (RA) contributes to a greater incidence of insulin resistance and DM. In a population‐based cohort study, a 50% higher risk of DM was observed among patients with RA compared with nonrheumatic controls (2). Comorbid DM in patients with RA increases the risk of a major cardiovascular adverse events by threefold (3).
Focusing on DM prevention efforts in patients with RA may be important to improve cardiovascular outcomes and reduce early mortality. Many biologic and targeted synthetic disease‐modifying antirheumatic drugs (DMARDs) directed toward specific components of the immune system, including tumor necrosis factor (TNF)–alpha, interleukins, Janus kinase enzyme, and T cells, have been successfully developed to target inflammation control in RA. Some preliminary evidence from observational studies has revealed a potentially lower risk of DM with TNF‐alpha inhibitors (TNF‐inhibitors) (4), as well as with abatacept (a T‐cell co‐stimulation inhibitor) (5), compared with nonbiologic disease‐modifying agents, which have general immunosuppressive properties.
There are 10 targeted disease‐modifying agents available for RA with potential differences in risks of various clinical outcomes, including infections and cardiovascular events (6, 7, 8). However, comparative risk of DM among patients with RA treated with different biologic and targeted synthetic DMARDs is not well studied. Abatacept, in particular, is of special interest with respect to DM risk because of prior observations of slowing the reduction in β‐cell functioning, compared with placebo treatment, in randomly assigned patients with type 1 diabetes (9) and association with delaying cardiovascular events in patients with existing DM, compared with TNF‐inhibitors, in a large nonrandomized study (8). A comparative evaluation of DM risk between various treatments of RA can provide insights regarding which treatment holds highest promise for modifying the risk of DM in RA. To that end, we used claims data from two large health care databases from the United States to report comparative risk estimates of developing incident DM in patients with RA treated with infliximab, etanercept, adalimumab, certolizumab, golimumab (all TNF‐inhibitors), tocilizumab (interleukin 6 inhibitor), abatacept (T‐cell co‐stimulation inhibitor), and tofacitinib (Janus kinase inhibitor). Rituximab (a CD20 activity blocker) and anakinra (interleukin 1 inhibitor) were excluded from consideration because they remain infrequently used as first‐line treatments in patients with RA and may represent a group of atypical patients with RA, with key differences in baseline comorbidities and RA disease activity (10, 11).
PATIENTS AND METHODS
Data source
An observational cohort study was designed using the Truven MarketScan (2005‐2016) administrative claims database and Medicare Fee‐for‐Service (parts A, B, and D; 2010‐2014). These databases contain longitudinal health care information for their enrollees, with Truven representing individuals enrolled in various employer‐sponsored commercial health plans and Medicare representing publicly insured individuals 65 years or older or with certain qualifying disabilities. Comprehensive information on hospital admissions, emergency department visits, outpatient visits, and outpatient surgical visits, as well as pharmacy dispensing, is available. Diagnoses are coded using the clinical modification of the International Classification of Diseases, Ninth Revision (ICD‐9) or International Classification of Diseases, 10th Revision (ICD‐10) system, and procedures are coded using the Current Procedural Terminology, Fourth Edition (CPT‐4). The Institutional Review Board (2017P001342) of Brigham and Women's Hospital approved the use of these databases for this study and approved the protocol for this study. Appropriate data use agreements were in place. Deidentified data are available from Truven and the Centers for Medicare and Medicare services through licensing. We did not involve patients or the public in our work.
Study design and study population
We designed a new‐user observational cohort study (12). Patients aged 18 years or older entered the study cohort on the date when they filled a new prescription for a study medication, defined as the cohort entry date, after a 365‐day period of continuous insurance enrollment, defined as the baseline period. During the baseline period, we required patients to have one inpatient or two outpatient diagnosis codes for RA 7 to 365 days apart. This algorithm of identifying RA from administrative claims is reported to have an 87% positive predicted value (PPV) (13). Using all available information, we excluded patients if they had prevalent use of any study medication of interest, rituximab, or anakinra any time prior to the index date (14). In addition, patients with an existing diagnosis of DM (or use of antidiabetic medications) or a malignancy diagnosis during the baseline period were excluded. Patients were assigned into one of eight exposure groups at cohort entry: infliximab, etanercept, adalimumab, certolizumab, golimumab, tocilizumab, abatacept, or tofacitinib. We identified abatacept as the reference exposure a priori.
Follow‐up and outcome
The outcome of interest was incident diagnosis of DM, defined by an ICD‐9 or ICD‐10 code for DM and a prescription dispensing for an antidiabetic medication, with the date of medication initiation defined as the outcome date. Requiring medication use along with diagnosis codes to identify DM is reported to result in a PPV of 96.5% (15). An as‐treated follow‐up model was used with follow‐up beginning at treatment initiation and censoring on treatment discontinuation (defined as no refill of an existing prescription within 30 days of the end date of the most recent fill), switch to a different study medication, health plan disenrollment, or administrative end point.
Covariates
A total of 56 potential confounders were used for risk adjustment, including the following: demographics (age, sex, geographic region, and race [not available in Truven]); comorbid conditions, including alcoholism, heart failure, hyperlipidemia, hypertension, hypothyroidism, chronic liver disease, myocardial infarction, obesity, psychosis, pulmonary disease, chronic renal dysfunction, smoking, and stroke; comedications, including nonbiologic DMARDs (methotrexate, hydroxychloroquine, sulfasalazine, or other agents), steroids (indicators for any use in last 365 days, recent use in last 30 days, and cumulative dose in milligrams of prednisone equivalents), inhibitors of the renin‐angiotensin system, beta blockers, calcium‐channel blockers, nonsteroidal anti‐inflammatory drugs, statins, other lipid‐lowering agents, inhaled steroids, anticoagulants, antidepressants, antiplatelets, antipsychotics, benzodiazepines, diuretics, and opioids; and health care use characteristics, including counts of physician office visits, total number of prescription medications, indicators for any hospitalization or emergency department visit, and counts of laboratory test orders (acute‐phase reactants, cyclic citrullinated peptide, basic metabolic panels, comprehensive metabolic panels, and glycated hemoglobin). All characteristics were measured during the 365‐day baseline period.
Statistical analysis
Patient characteristics were presented descriptively, stratified by the exposure group. Incidence rates (IRs) for DM were calculated along with 95% confidence intervals (CIs) overall and by exposure groups. For confounding adjustment, all 56 baseline covariates were included in a multinomial logistic model to calculate a propensity score. Inverse probability treatment weighting with the predicted probability of receiving the observed treatment was conducted to achieve covariate balance. Inverse probability treatment weights (IPTWs) were stabilized by marginal probability of each treatment to avoid large weights and variance inflation. Weighted Cox proportional hazards models were used to derive hazard ratios (HRs), and 95% CIs were calculated using robust SEs to account for weighting (16). Kaplan‐Meier plots were reported for the weighted population. All analyses were conducted separately in the two data sources. Fixed‐effects meta‐analytic methods were used to combine results across two data sources.
Bias analysis
Obesity is strongly associated with the risk of developing DM and is incompletely captured in administrative claims. Therefore, we performed a post hoc bias analysis to evaluate the potential impact of confounding by obesity on our results. We used a multiplicative bias term to understand the magnitude of imbalance in the distribution of obesity across the exposure groups, which is required to fully explain the observed association. This was achieved by applying a correction factor (17) (the multiplicative bias term) for unmeasured confounding to the naïve or apparent relative risks (RRs) that did not account for unmeasured confounding:
A baseline prevalence of 30% for obesity in RA in the reference group (PC0) was used based on prior literature (18), and prevalence in the exposed group (PC1) was varied from 10% to 40% to calculate corrected RRs under varying degrees of imbalances. A risk ratio of 4.0 for the association between obesity and DM (RRCD) was used based on estimates from a previous study (19). Corrected RR estimates were calculated and plotted for point estimates of the observed estimates as well as for lower and upper confidence‐bound estimates to appropriately address uncertainty.
RESULTS
Study cohorts
A total of 50 505 patients with RA from Truven and 17 251 patients with RA from Medicare met all our inclusion criteria. Etanercept was the most commonly used drug in the Truven cohort (38.4%), followed by adalimumab (36.1%), infliximab (13.4%), and abatacept (5.1%). Infliximab was the most frequently initiated drug in the Medicare cohort (32.9%), followed by etanercept (17.8%), adalimumab (15.0%), and abatacept (14.9%). There were important differences in the baseline characteristics across initiators of different agents in both cohorts. Specifically, abatacept initiators were, on average, older and had a higher prevalence of certain cardiovascular comorbid conditions, including heart failure and myocardial infarction, compared with etanercept, adalimumab, or infliximab initiators in both cohorts (Table 1). After applying stabilized IPTWs, standardized differences in all covariates between each exposure group and the reference group (abatacept) moved considerably closer to 0 and were lower than the threshold of 10 for most covariates (Appendix Figures 1 and 2). Appendix Table 1 contains the distribution of patient characteristics by exposure groups in the weighted sample.
Table 1.
Baseline patient characteristics in patients with RA initiating various targeted disease‐modifying antirheumatic drugs prior to inverse probability treatment weighting
| Truven MarketScan | Medicare | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ABT | ADL | CZP | ETN | GOL | INF | TCZ | TOF | ABT | ADL | CZP | ETN | GOL | INF | TCZ | TOF | |
| Total, n | 2595 | 18 237 | 1069 | 19 408 | 1332 | 6765 | 452 | 647 | 2578 | 2597 | 1634 | 3080 | 801 | 5668 | 438 | 455 |
| Age, mean (SD), y | 54.2 (13.1) | 49.0 (11.8) | 53.2 (13.7) | 49.6 (12.2) | 50.5 (13) | 53.5 (13.4) | 51.7 (14.0) | 55 (12.1) | 73.0 (6.2) | 71.6 (6.0) | 72.4 (5.9) | 71.4 (5.8) | 71.9 (5.7) | 72.1 (5.7) | 72.3 (6.2) | 72.5 (6.3) |
| Female sex, % | 84.2 | 73 | 77 | 74 | 76.1 | 74.4 | 81 | 81.8 | 86.8 | 80.6 | 79.6 | 80.4 | 81.3 | 79.3 | 86.1 | 86.2 |
| White race, % | … | … | … | … | … | … | … | … | 90.2 | 81 | 92 | 84.9 | 87.3 | 91.9 | 89.3 | 82.9 |
| RA‐related drug use | ||||||||||||||||
| HCQ, % | 32.3 | 26.1 | 25.4 | 29.2 | 26.3 | 19.1 | 20.4 | 32.8 | 30.2 | 26.1 | 26.6 | 29.9 | 31.2 | 23.7 | 27.9 | 32.1 |
| MTX, % | 55.8 | 68.4 | 65.7 | 68.6 | 69 | 64.6 | 45.6 | 61.1 | 61.9 | 66.7 | 63.1 | 65.6 | 66.4 | 71.5 | 54.8 | 55.4 |
| LEF, % | 18.2 | 12.3 | 12.8 | 12.9 | 13.7 | 10.3 | 14.4 | 22.9 | 20.9 | 16.2 | 17.1 | 17.7 | 18.9 | 16.7 | 18.9 | 23.3 |
| SSZ, % | 10.8 | 13.2 | 11.9 | 13.8 | 10.6 | 9.8 | 5.5 | 14.2 | 12.1 | 12.8 | 11.9 | 13.1 | 11.9 | 10.9 | 11.9 | 13.2 |
| Other nonbiologics, % | 8.2 | 4.6 | 5.6 | 4.4 | 4.2 | 5.8 | 4 | 6.6 | 7.8 | 7.5 | 6.5 | 6.4 | 7 | 5.6 | 9.1 | 10.3 |
| Steroid injections, % | 16.8 | 13.4 | 16.1 | 13.4 | 13.1 | 15 | 15.7 | 15.1 | 22.4 | 19.4 | 25.8 | 19.8 | 21.2 | 20.9 | 23.3 | 16 |
| Last‐30‐d oral steroid use, % | 40 | 40.8 | 38.8 | 41.6 | 40.6 | 36.9 | 42.7 | 41.3 | 49.8 | 47.7 | 45.8 | 51.4 | 47.8 | 45.5 | 48.4 | 47.9 |
| Any oral steroid use in baseline period, % | 65.5 | 69.2 | 68.9 | 70.1 | 67.9 | 62.9 | 61.5 | 70.8 | 73.2 | 71 | 70.6 | 73.7 | 73.3 | 70.8 | 71.7 | 70.3 |
| Cumulative oral steroid dose, mean (SD) in prednisone equivalents (mg) | 1212.8 (3482.6) | 1216.9 (13 861.8) | 1103.2 (2567.8) | 1158 (5057.8) | 1166 (3646) | 1196.3 (9394.1) | 3123.9 (37 713.3) | 1541 (6858.1) | 1092.4 (1274.9) | 1132.2 (1506.9) | 1025.6 (1258.2) | 1164 (1347.7) | 1088.4 (1274.2) | 1039.1 (1281.5) | 1282.3 (1587.9) | 1184.4 (1381.8) |
| Comorbid conditions | ||||||||||||||||
| Alcoholism, % | 0.8 | 0.8 | 0.7 | 0.9 | 0.5 | 0.9 | 0.9 | 0.3 | 0.3 | 0.6 | 0.7 | 0.2 | 0.7 | 0.3 | 0.2 | 0.9 |
| Heart failure, % | 4 | 1.1 | 2.2 | 1.1 | 0.9 | 2.2 | 3.3 | 2.5 | 10.7 | 6.9 | 7.3 | 8.1 | 7.7 | 6.1 | 11.6 | 9.2 |
| Hyperlipidemia, % | 27.6 | 25.1 | 31.1 | 25.3 | 27.9 | 26.8 | 29 | 32.3 | 59.2 | 53.5 | 60.2 | 54.4 | 62.8 | 58.2 | 62.1 | 57.1 |
| Hypertension, % | 39.4 | 31.1 | 38 | 29.6 | 34 | 36.7 | 36.7 | 43 | 71.6 | 70 | 69.8 | 66.8 | 74.5 | 68.2 | 68.3 | 67.5 |
| Hypothyroidism, % | 15.7 | 13.5 | 14.7 | 13.5 | 13.7 | 13.1 | 15.5 | 20.2 | 29.7 | 25 | 25.6 | 24.4 | 28.1 | 26.1 | 24.9 | 25.1 |
| Chronic liver disease, % | 5.5 | 4.1 | 5 | 4.3 | 3.5 | 4.1 | 6.6 | 3.9 | 6.7 | 6.2 | 7.5 | 6.7 | 5.7 | 6.1 | 6.2 | 5.1 |
| Myocardial infarction, % | 1.7 | 0.7 | 1.3 | 0.7 | 0.8 | 1 | 2.4 | 1.1 | 4.3 | 3.5 | 2.6 | 3.4 | 3.7 | 2.9 | 2.5 | 4.6 |
| Obesity, % | 8.9 | 8.1 | 8.1 | 7.2 | 10.2 | 6.9 | 9.7 | 11.3 | 6.7 | 8.4 | 10.6 | 7.6 | 11.1 | 7.6 | 10.7 | 10.5 |
| Psychosis, % | 20.3 | 19.6 | 20.1 | 19.4 | 20.3 | 17.5 | 23.2 | 21.3 | 24.9 | 26.1 | 24.3 | 25.6 | 28.2 | 22.4 | 28.1 | 30.1 |
| Pulmonary disease, % | 19.9 | 13 | 15.2 | 14 | 14.6 | 16.8 | 17.3 | 17.6 | 28.1 | 27.1 | 26.9 | 27.5 | 28 | 25.4 | 30.6 | 25.1 |
| Chronic renal dysfunction, % | 3.5 | 1.5 | 2.9 | 1.8 | 2.2 | 2.2 | 3.5 | 3.7 | 9.9 | 8.1 | 8.8 | 7.8 | 10.5 | 7.4 | 10 | 9.5 |
| Smoking, % | 11.5 | 12.3 | 11.1 | 12.2 | 11.7 | 11.6 | 13.1 | 13 | 12.1 | 13.3 | 14.5 | 14.8 | 19.5 | 12.2 | 15.8 | 18 |
| Stroke, % | 2.6 | 1.1 | 2.2 | 1.3 | 1.4 | 1.8 | 2.4 | 1.7 | 7.3 | 6.2 | 6.5 | 6.9 | 7.1 | 6 | 5.5 | 6.6 |
| Charlson comorbidity index, mean (SD) | 1.4 (0.8) | 1.2 (0.6) | 1.3 (0.7) | 1.2 (0.7) | 1.2 (0.7) | 1.2 (0.7) | 1.4 (0.8) | 1.4 (0.8) | 1.9 (1.2) | 1.8 (1.1) | 1.8 (1.1) | 1.8 (1.2) | 1.9 (1.1) | 1.7 (1.1) | 1.9 (1.2) | 1.9 (1.2) |
| Comedication use | ||||||||||||||||
| Renin‐angiotensin system blockers, % | 25.4 | 20.8 | 24 | 20.3 | 22.4 | 22.6 | 21.9 | 29.1 | 43.4 | 41.5 | 43 | 42.1 | 46.8 | 42 | 40.9 | 40 |
| Beta blockers, % | 18.4 | 13.5 | 14.2 | 13.3 | 12 | 15 | 15 | 15.6 | 33.7 | 32.3 | 31.5 | 30.4 | 35.3 | 30.8 | 33.8 | 31 |
| Calcium‐channel blockers, % | 13.9 | 9.9 | 13.4 | 10.1 | 10.4 | 11.9 | 11.7 | 13.6 | 26.8 | 25.2 | 23.3 | 25.7 | 25.6 | 25 | 28.8 | 26.4 |
| NSAIDs, % | 44.2 | 57.1 | 52 | 56.5 | 53.6 | 47.7 | 38.1 | 50.5 | 39.8 | 43.5 | 41.7 | 42.4 | 41.3 | 42.9 | 38.1 | 39.1 |
| Other lipid‐lowering agents, % | 4.8 | 4.4 | 4.3 | 4.3 | 4.1 | 5.4 | 3.5 | 2.8 | 7.5 | 7.5 | 6.5 | 6.1 | 6.7 | 6.9 | 7.5 | 5.1 |
| Inhaled steroids, % | 17.8 | 17.1 | 18.1 | 17.8 | 17.4 | 15.6 | 17 | 21.6 | 20.6 | 21.9 | 22.2 | 22.9 | 24.5 | 20.3 | 23.5 | 24.6 |
| Anticoagulants, % | 5.7 | 2.9 | 4.5 | 2.9 | 2.9 | 3.7 | 6.6 | 4.3 | 10.1 | 7.5 | 9 | 8.2 | 9 | 9.3 | 9.1 | 8.1 |
| Antidepressants, % | 34.3 | 33.8 | 34.3 | 34 | 32 | 32.2 | 31.4 | 30.3 | 38 | 35.3 | 33.9 | 36.4 | 38.8 | 35.1 | 39 | 35.6 |
| Antiplatelets, % | 3.6 | 2.7 | 3.1 | 2.2 | 2.3 | 3.4 | 3.1 | 4 | 7.9 | 6.4 | 6.6 | 7.5 | 6.2 | 6.2 | 5.7 | 7.3 |
| Antipsychotics, % | 2.6 | 2.5 | 2.4 | 2.4 | 1.7 | 2.5 | 1.3 | 1.7 | 2 | 3.1 | 2.3 | 2.5 | 1.4 | 1.9 | 0.9 | 2.2 |
| Benzodiazepines, % | 21.2 | 19.1 | 19.3 | 19.4 | 17 | 19.8 | 16.6 | 17.2 | 10.2 | 9.2 | 14.4 | 9.5 | 16 | 7.6 | 17.6 | 19.1 |
| Diuretics, % | 25.8 | 21.5 | 22 | 21.3 | 22.7 | 24.7 | 21.9 | 24.9 | 43.3 | 42.6 | 40.3 | 39.4 | 41.8 | 41.3 | 41.1 | 38.7 |
| Opioids, % | 62.4 | 61 | 58.7 | 61.1 | 55.9 | 60.1 | 56.2 | 58.4 | 66.8 | 67.4 | 63.4 | 67.6 | 63.9 | 64.1 | 64.8 | 65.7 |
| Statins, % | 18.3 | 16.1 | 20.8 | 16.5 | 16.6 | 18.1 | 18.4 | 18.5 | 38.6 | 37.3 | 38.2 | 36.8 | 43.4 | 37.5 | 41.3 | 37.6 |
| No. of prescription drugs, mean (SD) | 12.3 (7.7) | 12.5(6.7) | 12(6.7) | 12.4 (6.6) | 11.6 (6.5) | 11.2 (7.2) | 10.2(7.6) | 12.5 (6.5) | 13.1 (6.2) | 13.8 (6.4) | 12.3 (5.8) | 13.9 (6.3) | 13(6.2) | 12.3 (5.9) | 12.8 (6.3) | 13.6 (6.4) |
| Health care use | ||||||||||||||||
| No. of office visits, mean (SD) | 13.5 (7.9) | 11.7(6.7) | 12.5 (8.0) | 11.9 (7.0) | 11.9 (6.9) | 12.7 (7.5) | 12.1 (7.6) | 11.9 (7.7) | 17.8 (10.1) | 16.2 (9.6) | 17.1 (9.6) | 16.3 (9.9) | 15.9 (8.4) | 16.6 (9.2) | 16.5 (9.4) | 15.1 (9.2) |
| ≥1 ED visit, % | 29.2 | 24.8 | 26.5 | 24.4 | 23.3 | 28.4 | 30.1 | 24.1 | 35.3 | 36.9 | 31.8 | 35.3 | 32.2 | 31.3 | 33.8 | 34.3 |
| ≥1 hospitalization, % | 15.7 | 10.2 | 13.2 | 10.3 | 9.9 | 13.7 | 17.7 | 11.9 | 22.7 | 20 | 18.5 | 20.5 | 18.5 | 19.8 | 20.5 | 21.1 |
| Laboratory test orders, mean (SD) | ||||||||||||||||
| No. of acute‐phase reactant tests ordered | 2.6 (2.4) | 2.6 (2.1) | 2.5 (2.2) | 2.6 (2.2) | 2.6 (2.1) | 2.4 (2.4) | 2.4 (2.5) | 2.4 (2.3) | 3.6 (2.6) | 3.1 (2.4) | 3.4 (2.3) | 3.3 (2.6) | 3.4 (2.3) | 3.6 (2.5) | 3.7 (2.7) | 3.1 (2.6) |
| No. of HbA1c tests ordered | 0.4 (0.7) | 0.6 (0.8) | 0.5 (0.7) | 0.6 (0.8) | 0.5 (0.7) | 0.5 (0.8) | 0.3 (0.6) | 0.4 (0.7) | 0.5 (0.8) | 0.5 (0.8) | 0.5 (0.8) | 0.5 (0.8) | 0.5 (0.8) | 0.5 (0.8) | 0.4 (0.7) | 0.3 (0.6) |
| No. of basic metabolic panels, BUN tests, or serum creatinine tests ordered | 1.2 (2.1) | 1.1 (1.8) | 0.9 (1.8) | 1.2 (2) | 1 (1.7) | 1.1 (1.9) | 1.1 (2) | 0.9 (1.9) | 2.2 (2.8) | 1.8 (2.4) | 1.8 (2.3) | 1.9 (2.4) | 1.8 (2.3) | 2.1 (2.6) | 1.9 (2.6) | 1.7 (2.4) |
| No. of comprehensive metabolic panels ordered | 2.2 (2.4) | 2.2 (2.3) | 2.1 (2.2) | 2.1 (2.3) | 2.3 (2.6) | 2 (2.3) | 2.1 (2.6) | 2.2 (2.2) | 3.7 (2.8) | 3.3 (2.6) | 3.5 (2.5) | 3.4 (2.7) | 3.7 (2.7) | 3.6 (2.8) | 3.8 (3) | 3.6 (2.7) |
| No. of rheumatoid factor tests ordered | 0.1 (0.4) | 0.1 (0.4) | 0.1 (0.4) | 0.1 (0.4) | 0.1 (0.4) | 0.1 (0.3) | 0.2 (0.4) | 0.2 (0.4) | 0.2 (0.5) | 0.2 (0.5) | 0.2 (0.5) | 0.2 (0.5) | 0.2 (0.5) | 0.2 (0.5) | 0.2 (0.5) | 0.2 (0.5) |
| No. of anti‐CCP tests ordered | 0.4 (0.6) | 0.5 (0.6) | 0.4 (0.7) | 0.5 (0.6) | 0.4 (0.6) | 0.4 (0.6) | 0.3 (0.6) | 0.4 (0.7) | 0.5 (0.6) | 0.4 (0.6) | 0.5 (0.6) | 0.4 (0.6) | 0.5 (0.7) | 0.5 (0.6) | 0.4 (0.6) | 0.3 (0.6) |
Abbreviation: ABT, abatacept; ADL, adalimumab; BUN, blood urea nitrogen; CCP, cyclic citrullinated peptide; CZP, certolizumab; ED, emergency department; ETN, etanercept; GOL, golimumab; HbA1c, hemoglobin A1c; HCQ, hydroxychloroquine; INF, infliximab; LEF, leflunomide; MTX, methotrexate; NSAID, nonsteroidal anti‐inflammatory drug; RA, rheumatoid arthritis; SSZ, sulfasalazine; TCZ, tocilizumab; TOF, tofacitinib.
Risk of incident DM
A total of 313 events in the Truven cohort and 114 events in the Medicare cohort were observed over an average follow‐up time of 368 days and 332 days, respectively, corresponding to IRs (95% CI) of 6.8 (6.1‐7.6) and 6.6 (5.4‐7.9) per 1000 person‐years (Table 2). Event counts were low in certolizumab, golimumab, tocilizumab, and tofacitinib groups because of a relatively small sample size and limited follow‐up time. Among individual exposure groups with at least 1000 person‐years of follow‐up in each data source, IRs (95% CI) per 1,000 person‐years ranged from 4.1 (2.0‐7.3) in the abatacept group to 7.6 (5.7‐9.7) in the infliximab group in the Truven cohort and from 3.7 (1.6‐7.2) in the abatacept group to 9.6 (7.6‐12.0) in the infliximab group in the Medicare cohort.
Table 2.
Crude incidence rates of diabetes mellitus in patients with rheumatoid arthritis initiating various targeted disease‐modifying antirheumatic drugs
| Exposure | Sample Size | Average Follow‐up, d | Crude Event Count | Person‐years of Follow‐up | Incidence Rates per 1000 Person‐years | 95% Lower Confidence Limit | 95% Upper Confidence Limit |
|---|---|---|---|---|---|---|---|
| Truven | |||||||
| Abatacept | 2595 | 306.8 | 8 | 2180.9 | 3.7 | 1.6 | 7.2 |
| Adalimumab | 18 237 | 312.8 | 104 | 15 627.6 | 6.7 | 5.4 | 8.1 |
| Certolizumab | 1069 | 233.3 | 2 | 683.2 | 2.9 | 0.4 | 10.6 |
| Etanercept | 19 408 | 331.7 | 115 | 17 639.5 | 6.5 | 5.4 | 7.8 |
| Golimumab | 1332 | 290.2 | 3 | 1059.0 | 2.8 | 0.6 | 8.3 |
| Infliximab | 6765 | 436.9 | 78 | 8097.3 | 9.6 | 7.6 | 12.0 |
| Tocilizumab | 452 | 251.9 | 0 | 311.9 | 0.0 | … | 11.8 |
| Tofacitinib | 647 | 202.2 | 3 | 358.5 | 8.4 | 1.7 | 24.5 |
| Total | 50 505 | 332.1 | 313 | 45 958.0 | 6.8 | 6.1 | 7.6 |
| Medicare | |||||||
| Abatacept | 2578 | 380.5 | 11 | 2687.6 | 4.1 | 2.0 | 7.3 |
| Adalimumab | 2597 | 287.3 | 15 | 2044.1 | 7.3 | 4.1 | 12.1 |
| Certolizumab | 1634 | 253.3 | <11a | 1133.8 | 5.3 | 1.9 | 11.5 |
| Etanercept | 3080 | 310.0 | 14 | 2615.6 | 5.4 | 2.9 | 9.0 |
| Golimumab | 801 | 228.7 | <11a | 502.0 | 12.0 | 4.4 | 26.0 |
| Infliximab | 5668 | 503.2 | 59 | 7814.2 | 7.6 | 5.7 | 9.7 |
| Tocilizumab | 438 | 281.1 | <11a | 337.3 | 5.9 | 0.7 | 21.4 |
| Tofacitinib | 455 | 196.2 | <11a | 244.6 | 4.1 | 0.1 | 22.8 |
| Total | 17 251 | 367.7 | 114 | 17 379.2 | 6.6 | 5.4 | 7.9 |
Counts <11 are suppressed to comply with the data use agreement from the Centers for Medicare and Medicaid Services.
After confounding adjustment with stabilized IPTWs, the pooled HRs (95% CI) across the two data sources indicated a significantly higher risk of DM among adalimumab (2.00 [1.11‐3.03]) and infliximab initiators (2.34 [1.38‐3.98]) compared with abatacept initiators (Table 3). The pooled HR (95% CI) for the etanercept versus abatacept comparison was numerically elevated but not statistically significant (1.65 [0.91‐2.98]). The effect estimates for certolizumab, golimumab, tocilizumab, and tofacitinib, compared with abatacept, were highly imprecise because of a limited sample size. Inspection of the Kaplan‐Meier plots in the weighted sample suggested separation of survival curves for infliximab, adalimumab, and etanercept from that for abatacept in the Truven cohort (Figure 1). The survival curves in the Medicare cohort had higher variability and inconsistent patterns because of low event counts.
Table 3.
HRs and 95% CIs for diabetes mellitus in patients with rheumatoid arthritis initiating various targeted disease‐modifying antirheumatic drugs
| Exposure | Truven, HR (95% CI) | Medicare, HR (95% CI) | Combined, HR (95% CI) |
|---|---|---|---|
| Crude | |||
| Abatacept | Reference | Reference | Reference |
| Adalimumab | 1.82 (0.89‐3.73) | 1.75 (0.8‐3.8) | 1.78 (1.05‐3.03) |
| Certolizumab | 0.81 (0.17‐3.8) | 1.21 (0.45‐3.28) | 1.08 (0.47‐2.49) |
| Etanercept | 1.78 (0.87‐3.65) | 1.28 (0.58‐2.83) | 1.54 (0.9‐2.61) |
| Golimumab | 0.78 (0.21‐2.95) | 2.64 (0.97‐7.16) | 1.70 (0.77‐3.77) |
| Infliximab | 2.61 (1.26‐5.4) | 1.92 (1.01‐3.66) | 2.2 (1.36‐3.56) |
| Tocilizumab | … | 1.38 (0.31‐6.21) | … |
| Tofacitinib | 2.32 (0.62‐8.77) | 0.9 (0.12‐7.01) | 1.76 (0.58‐5.35) |
| Inverse probability weighted | |||
| Abatacept | Reference | Reference | Reference |
| Adalimumab | 2.34 (1‐5.45) | 1.72 (0.75‐3.97) | 2.00 (1.11‐3.63) |
| Certolizumab | 0.89 (0.18‐4.47) | 0.96 (0.34‐2.74) | 0.94 (0.39‐2.26) |
| Etanercept | 2.54 (1.09‐5.92) | 1.09 (0.47‐2.49) | 1.65 (0.91‐2.98) |
| Golimumab | 0.63 (0.15‐2.61) | 2.14 (0.69‐6.65) | 1.33 (0.55‐3.23) |
| Infliximab | 3.53 (1.48‐8.41) | 1.84 (0.94‐3.59) | 2.34 (1.38‐3.98) |
| Tocilizumab | … | 1.23 (0.26‐5.83) | … |
| Tofacitinib | 1.71 (0.42‐6.97) | 0.34 (0.04‐2.68) | 1.02 (0.32‐3.27) |
Abbreviation: CI, confidence interval; HR, hazard ratio.
Figure 1.
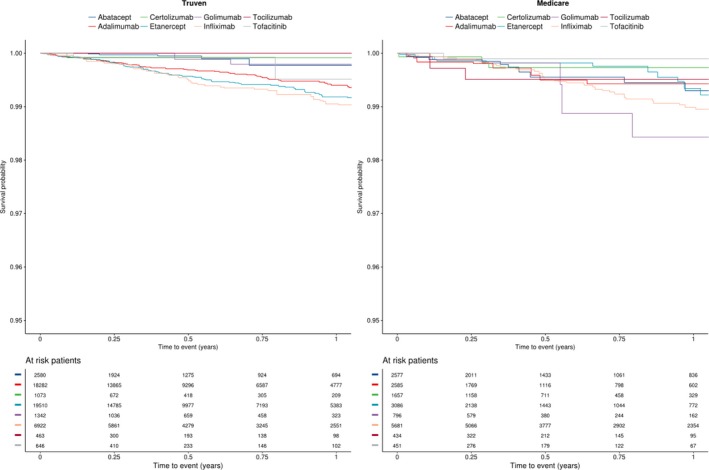
Kaplan‐Meier plots in the weighted population.
Bias analysis
The bias analysis (Figure 2) suggested that for the adalimumab versus abatacept comparison, an obesity prevalence of 38% or higher in the adalimumab group, versus 30% in the abatacept group, could bring the corrected RR corresponding to the lower 95% confidence bound of the observed RR (1.11) below the null value. For the infliximab versus abatacept comparison, an obesity prevalence of more than 50% in the infliximab group, versus 30% in the abatacept group, was required to bring the corrected RR corresponding to the lower 95% confidence bound of the observed RR (1.38) below the null value.
Figure 2.
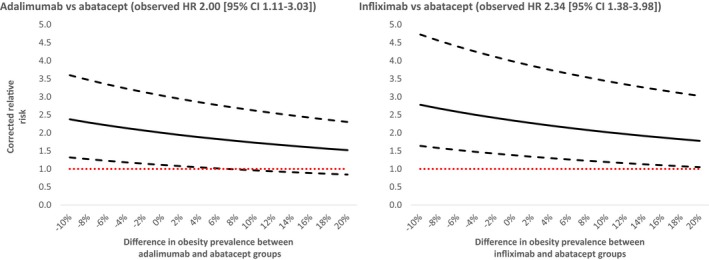
Bias analysis to investigate the potential impact of unmeasured obesity on the observed associations. Solid lines indicate corrected relative risks corresponding to the point estimates, and dashed lines indicate corrected relative risks corresponding to upper and lower bounds of the 95% confidence intervals (CIs) at various levels of differences in obesity prevalence between the abatacept (assumed 30% baseline obesity) (18) and adalimumab or infliximab groups. Negative numbers indicate higher prevalence of obesity in the abatacept group and positive numbers indicate higher prevalence in adalimumab or infliximab group. The dotted lines indicate corrected relative risk of null. HR, hazard ratio.
DISCUSSION
In this large cohort study that includes data from two nationwide data sources in the United States, we noted use of abatacept to be associated with a lower risk of incident DM, compared with use of two TNF‐inhibitors (infliximab and adalimumab), in patients with RA. Comparison of abatacept with other agents was inconclusive because of limited event counts available for valid treatment‐effect estimation.
Results from this study provide a novel contribution to the literature by quantifying the RRs for development of incident DM in patients with RA exposed to different biologics. The finding of potentially lower risk of DM with abatacept is in line with a previous investigation by Ozen et al (5), who reported an HR of 0.52 (95% CI 0.31‐0.89) for developing DM in patients with RA receiving abatacept versus methotrexate monotherapy. More aggressive inflammation control, compared with methotrexate monotherapy, may be offered as a potential explanation for the risk reduction in DM conferred by abatacept in the investigation by Ozen et al (5). However, aggressive inflammation control alone may be an insufficient explanation for the risk reduction observed in this study because we compared abatacept with individual TNF‐inhibitors and other targeted immunomodulators, which are expected to be equivalent, with respect to inflammation control, to abatacept. Indeed, Solomon et al (4) reported an HR of 0.62 (95% CI 0.42‐0.91) for incident DM in patients treated with TNF‐inhibitors compared with patients treated with nonbiologic DMARDs. We posit that direct effects of abatacept on glucose metabolism due to inhibition of T‐cell co‐stimulation may play a key role in explaining our results. In a recent prospective study of 15 patients with RA treated with abatacept, Ursini et al (20) reported an improvement in the insulin sensitivity index as well as a reduction in glycated hemoglobin values after 6 months of treatment. In a randomized controlled trial, abatacept was also shown to be associated with slowing the reduction in β‐cell functioning, compared with placebo treatment, in patients with type 1 diabetes (9). If confirmed in future randomized controlled studies, the observation that abatacept is potentially associated with a lower risk of developing DM, compared with TNF‐inhibitors, may have important clinical implications because it may allow physicians to select a treatment that alters the risk of DM in patients with RA with a higher risk of developing DM, such as those with a family history of DM or other metabolic disturbances.
As our study is observational in nature, residual confounding due to important unmeasured confounding factors could threaten the validity of the observed results. One of the key risk factors for DM development is obesity, which is imperfectly captured in claims data. In our post hoc bias analysis, we noted that prevalence difference in obesity at baseline of more than 8% and 20% in the adalimumab and infliximab groups, respectively, compared with the abatacept group, could explain the observed effect estimates. In recent years, disease activity has been recognized as a potentially important risk factors for DM development in patients with RA (21). Because our data sources lacked measurement of disease activity, our results may partially be explained by residual confounding if abatacept is preferentially used in patients with lower disease activity. However, previous investigations of patients with RA initiating abatacept and TNF‐inhibitors as first‐line therapies have reported no such preference and similar disease activities at baseline (22, 23).
There are some additional limitations of this analysis that deserve mention. First, we did not have availability of laboratory test results and relied on diagnosis codes and prescription claims to identify the outcome of interest. Although this approach is known to have good specificity in identifying DM (15), we may have missed some DM events (ie, diet‐controlled DM) because of a low sensitivity of this approach. Next, we restricted this analysis to new initiators of biologic or targeted synthetic DMARDs to avoid known biases, including confounding by treatment duration and confounding by unmeasured RA duration. Non‐TNF biologics are frequently initiated as second‐line treatments after insufficient response to TNF‐inhibitors (10). Thus, our analysis may have underrepresented patients with RA at later stages of the disease. Because of underlying heterogeneity in coverage of biologics across health plans, it is possible that certain agents are more frequently used as first‐line agent in certain health plans. Because we did not have access to plan formulary structures, we were unable to account for this variation, which may introduce bias. Finally, there were few DM events in our study among initiators of newer agents, including certolizumab, golimumab, tocilizumab, and tofacitinib which limited our ability to draw conclusions regarding the impact of these treatments on DM risk. Even for abatacept, the total number of outcomes we observed were only 19, which suggests that our results should be considered preliminary and should be replicated in other sources. We also did not consider patients solely treated with nonbiologic DMARDs as a comparison group because of concerns related to confounding by RA severity. Biologics and tofacitinib are generally reserved for patients who are not adequately responding to nonbiologics and who are, hence, likely to be inherently different with respect to RA severity compared with nonbiologic initiators.
In conclusion, we observed a lower risk of incident DM in patients with RA initiating abatacept compared with patients with RA initiating infliximab or adalimumab. A limited number of DM events and incomplete capture of important risk factors for DM development, including obesity and RA disease activity, in administrative claims used to conduct this study precludes a causal conclusion. Future randomized prospective studies are necessary to determine the causality of this association.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Desai, Kim.
Acquisition of data
Desai, Dejene, Jin, Liu, Kim.
Analysis and interpretation of data
Desai, Dejene, Jin, Liu, Kim.
ROLE OF THE STUDY SPONSOR
Bristol‐Myers Squibb was given the opportunity to make nonbinding comments on a draft of the manuscript. The authors independently designed the study, collected and analyzed the data, interpreted the results, and had the final decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Bristol‐Myers Squibb.
Supporting information
Appendix
Supported by an investigator‐sponsored grant from Bristol‐Myers Squibb (IM101‐699).
Dr. Desai has received research grants to Brigham and Women's Hospital from Bayer, Novartis, and Vertex for unrelated studies. Dr. Kim has received research grants to Brigham and Women's Hospital from Roche, Pfizer, and AbbVie for unrelated studies. No other disclosures relevant to this article were reported.
REFERENCES
- 1. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov 2014;13:465–76. [DOI] [PubMed] [Google Scholar]
- 2. Solomon DH, Love TJ, Canning C, Schneeweiss S. Risk of diabetes among patients with rheumatoid arthritis, psoriatic arthritis and psoriasis. Ann Rheum Dis 2010;69:2114–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Innala L, Möller B, Ljung L, Magnusson S, Smedby T, Södergren A, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011;13:R131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Solomon DH, Massarotti E, Garg R, Liu J, Canning C, Schneeweiss S. Association between disease‐modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA 2011;305:2525–31. [DOI] [PubMed] [Google Scholar]
- 5. Ozen G, Pedro S, Holmqvist ME, Avery M, Wolfe F, Michaud K. Risk of diabetes mellitus associated with disease‐modifying antirheumatic drugs and statins in rheumatoid arthritis. Ann Rheum Dis 2017;76:848–54. [DOI] [PubMed] [Google Scholar]
- 6. Desai RJ, Thaler KJ, Mahlknecht P, Gartlehner G, McDonagh MS, Mesgarpour B, et al. Comparative risk of harm associated with the use of targeted immunomodulators: a systematic review. Arthritis Care Res (Hoboken) 2016;68:1078–88. [DOI] [PubMed] [Google Scholar]
- 7. Jin Y, Kang EH, Brill G, Desai RJ, Kim SC. Cardiovascular (CV) risk after initiation of abatacept versus TNF inhibitors in rheumatoid arthritis patients with and without baseline CV disease. J Rheumatol 2018;45:1240–8. [DOI] [PubMed] [Google Scholar]
- 8. Kang EH, Jin Y, Brill G, et al. Comparative cardiovascular risk of abatacept and tumor necrosis factor inhibitors in patients with rheumatoid arthritis with and without diabetes mellitus: a multidatabase cohort study. J Am Heart Assoc 2018;7:e007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co‐stimulation modulation with abatacept in patients with recent‐onset type 1 diabetes: a randomised, double‐blind, placebo‐controlled trial. Lancet 2011;378:412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desai RJ, Solomon DH, Jin Y, Liu J, Kim SC. Temporal trends in use of biologic DMARDs for rheumatoid arthritis in the United States: a cohort study of publicly and privately insured patients. J Manag Care Spec Pharm 2017;23:809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim SC, Jin Y, Brill G, Lewey J, Choi NK, Patorno E, et al. Diabetes and other comorbidities in rheumatoid arthritis patients starting a biologic DMARD: a multi‐database cohort study [abstract]. American College of Rheumatology: Abstracts from the 2016 ACR/ARHP Annual Meeting, Washington, DC, November 11‐16, 2016. URL: https://acrabstracts.org/abstract/diabetes-and-other-comorbidities-in-rheumatoid-arthritis-patients-starting-a-biologic-dmard-a-multi-database-cohort-study/. [Google Scholar]
- 12. Ray WA. Evaluating medication effects outside of clinical trials: new‐user designs. Am J Epidemiol 2003;158:915–20. [DOI] [PubMed] [Google Scholar]
- 13. Kim SY, Servi A, Polinski JM, Mogun H, Weinblatt ME, Katz JN, et al. Validation of rheumatoid arthritis diagnoses in health care utilization data. Arthritis Res Ther 2011;13:R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brunelli SM, Gagne JJ, Huybrechts KF, Wang SV, Patrick AR, Rothman KJ, et al. Estimation using all available covariate information versus a fixed look‐back window for dichotomous covariates. Pharmacoepidemiol Drug Saf 2013;22:542–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Solberg LI, Engebretson KI, Sperl‐Hillen JM, Hroscikoski MC, O'Connor PJ. Are claims data accurate enough to identify patients for performance measures or quality improvement? The case of diabetes, heart disease, and depression. Am J Med Qual 2006;21:238–45. [DOI] [PubMed] [Google Scholar]
- 16. Desai RJ, Franklin JM. Alternative approaches for confounding adjustment in observational studies using weighting based on the propensity score: a primer for practitioners. BMJ 2019;367:l5657. [DOI] [PubMed] [Google Scholar]
- 17. Schneeweiss S. Sensitivity analysis and external adjustment for unmeasured confounders in epidemiologic database studies of therapeutics. Pharmacoepidemiol Drug Saf 2006;15:291–303. [DOI] [PubMed] [Google Scholar]
- 18. Crowson CS, Matteson EL, Davis JM III, Gabriel SE. Contribution of obesity to the rise in incidence of rheumatoid arthritis. Arthritis Care Res (Hoboken) 2013;65:71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Twig G, Afek A, Derazne E, Tzur D, Cukierman‐Yaffe T, Gerstein HC, et al. Diabetes risk among overweight and obese metabolically healthy young adults. Diabetes Care 2014;37:2989–95. [DOI] [PubMed] [Google Scholar]
- 20. Ursini F, Russo E, Letizia Hribal M, Mauro D, Savarino F, Bruno C, et al. Abatacept improves whole‐body insulin sensitivity in rheumatoid arthritis: an observational study. Medicine (Baltimore) 2015;94:e888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baker J, England B, George M, Cannon G, Mikuls T. Disease activity, cytokine profiles, and the risk of incident diabetes in rheumatoid arthritis [abstract]. American College of Rheumatology: Abstracts from the 2019 ACR/ARP Annual Meeting, Atlanta, GA, Nobember 8‐13, 2019. URL: https://acrabstracts.org/abstract/disease-activity-cytokine-profiles-and-the-risk-of-incident-diabetes-in-rheumatoid-arthritis/. [Google Scholar]
- 22. Klink AJ, Curtice TG, Gupta K, Tuell KW, Szymialis AR, Nero D, et al. Real‐world outcomes among patients with early rapidly progressive rheumatoid arthritis. Am J Manag Care 2019;25:e288–95. [PubMed] [Google Scholar]
- 23. Ramiro S, Landewé R, van der Heijde D, Harrison D, Collier D, Michaud K. Discontinuation rates of biologics in patients with rheumatoid arthritis: are TNF inhibitors different from non‐TNF inhibitors? [Extended report]. RMD Open 2015;1:e000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix