

Abstract
The current opioid epidemic remains an ongoing challenge, exacerbated by the extreme potency of synthetic opioids (e.g., fentanyl and fentanyl analogues), leading to an increase in adulterated heroin-related deaths. The increasing prevalence of fentanyl and fentanyl analogues in mixtures with heroin and other adulterants, excipients, and bulking agents has placed an emphasis on trace analysis methods for their detection from complex drug mixtures. Here, gradient elution moving boundary electrophoresis (GEMBE), a robust and miniaturized electrophoretic separation technique, was employed for the separation and detection of fentanyl and nine (9) fentanyl analogues from mixtures. GEMBE incorporated a short capillary (5 cm × 15 μm i.d.) for the electrophoretic separation of analytes with an opposing bulk counterflow. As the velocity of the counterflow was varied, analytes with differing electrophoretic mobilities entered the separation channel at different times and were analyzed as moving boundaries by contactless conductivity detection. The continuous injection of sample, driven by a controlled and variable pressure, both provided selectivity of the analytes and prevented contaminants or particulate within the sample from entering the separation capillary. Fentanyl was successfully separated and detected down to 2.5 μmol/L, and demonstrated only 50 % to 60 % signal suppression in dilute binary mixtures with heroin and other common adulterants and excipients at 30:1 (compound:fentanyl) concentration ratios. In addition, GEMBE was successfully applied to a few adjudicated case samples of fentanyl-related mixtures exhibiting dyes and visible particulate. The short capillaries, contactless detection format utilized here, and continuous injection of sample allows for a small footprint platform that is easy-to-use for forensic analyses.
Graphical Abstract
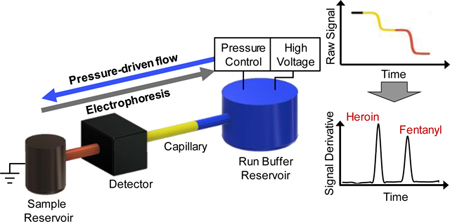
The ongoing opioid epidemic remains a unique challenge facing law enforcement, first responders, and medical personal. The 2017 World Drug Report by the United Nations Office on Drugs and Crime reported that the U.S. accounts for a quarter of the drug-related deaths worldwide, largely driven by opioids.1 A major contributor of this continuing epidemic has been correlated to the prevalence and potency of synthetic opioids. From 2015 to 2016, the number of deaths related to synthetic opioids doubled and the 2018 National Drug Threat Assessment stated that this class of drugs is currently involved in more deaths than any other illicit drug.2 In addition, the persistent and widespread adulteration of heroin with synthetic opioids, commonly fentanyl, has been cause for major concern due to the increased potency compared to heroin.3
Fentanyl is approximately 50 times more potent than heroin, with fentanyl and heroin at approximately 100 and 2.5 times more potent than morphine, respectively.3,4 The extreme potency of synthetic opioids can lead to potentially lethal effects at relatively low quantities of these fentanyl compounds. Fentanyl is oftentimes only a small component used as an adulterant in drug mixtures containing opioids, narcotics, other adulterants, or excipients, including bulking agents, fillers, and diluent compounds.2 The increasing prevalence of fentanyl and fentanyl analogues occurring as an adulterant in street samples, known or unknown by the user and other responding personnel, has caused an emphasis to be placed on trace analysis methods for the identification of fentanyl in complex drug mixtures.
Forensic laboratory analysis of seized street samples most commonly utilizes a colorimetric screen (i.e., the Marquis test) for heroin and fentanyl, followed by gas chromatography mass spectrometry (GC-MS) analysis.5 A targeted GC-MS method described by Ohta, et al. was able to identify fentanyl and 24 fentanyl analogues6 while Strano-Rossie et al. developed a rapid screening method for opiates, fentanyl, fentanyl analogues, and their respective metabolites in urine samples containing other narcotics for forensic toxicology applications7. Other common forensic laboratory techniques including Raman spectroscopy and ion mobility spectrometry (IMS) have also been applied for fentanyl analysis.8,9 Since fentanyl is commonly ≤10 % of an adulterated drug mixture,10 the weak intensity of Raman scattering makes it difficult to distinguish trace components in mixtures.7 Surface-enhanced Raman spectroscopy (SERS) has been applied to trace fentanyl detection as a more sensitive technique using analyte bonding to metal nanoparticles and novel substrates for increased sensitivity.10,11 Although street samples are rarely homogenous, minimal work has been explored using heterogenous mixtures. Alternatively, IMS can suffer from competitive ionization between fentanyl and excipients present in drug mixtures (e.g., procaine), as well as exhibit difficulties resolving heroin and fentanyl, creating challenges for analysis of complex drug mixtures.12,13
Extensive work has been reported for the detection of fentanyl using several liquid chromatography mass spectrometry (LC-MS) based techniques.14–20 Gergov et al. utilized LC-MS/MS for the detection of 16 opioids, fentanyl, and 8 fentanyl analogues in both blood and urine from ten autopsy samples16 whereas Elbardisy, et al. circumvented MS by applying high-performance liquid chromatography (HPLC) with diode array detection and HPLC with amperometry detection for analysis of heroin, fentanyl, and 10 fentanyl analogues21. LC-based instrumentation, however, is currently more readily available and utilized for forensic toxicology analyses in biological media.22 Equipping forensic laboratories separate from toxicology with LC-based techniques, i.e., LC-MS/MS, is challenging due to initial cost and requirement of expert users.
Whereas capillary electrophoresis (CE) is a well-established analysis technique in forensic DNA analysis, CE is less commonly used as an analytical tool for forensic analysis of seized drug material.23 Currently, CE is classified as a “Category B” technique by the Scientific Working Group for the Analysis of Seized Drugs (SWGDRUG) based on discriminating power. The forensic laboratory recommended use is in combination with another analysis technique of “Category A” discriminating power (e.g., mass spectrometry), to provide additional chemical structure or molecular weight information.24 However, CE is advantageous as a drug analysis technique due to the small sample volumes, high selectivity, and sensitivity for identification of a wide variety of analytes from mixtures, even enantiomers, by simply optimizing run buffer composition without the need for derivatization steps.25–28 The recommendation of orthogonal analytical techniques for complete forensic drug analysis of seized street samples necessitates the identification of techniques that are straightforward to optimize, allow for trace analysis of mixtures, and require minimal sample preparation.
Gradient elution moving boundary electrophoresis (GEMBE) was first described by Shackman, et al. in 2007 as an alternative CE technique.29 Conventional CE requires a precisely defined injection of a discrete zone of analytes into a capillary, followed by separation and electrokinetic migration of the analytes within that zone based on the differing electrophoretic mobilities. Alternatively, GEMBE utilizes continuous injection of analytes against a variable hydrodynamic counterflow into a short-length capillary (5 cm). In this format, the injected analytes are separated electrophoretically with an opposing pressure-controlled bulk counterflow. As the pressure-driven counterflow is ramped down, analytes sequentially enter the capillary as their electrophoretic velocity overcomes the bulk counterflow. This mechanism of separation is fundamentally different than conventional CE. With conventional CE, all analytes are injected and start migrating down the channel at the same time, and the separation of analytes is driven by the different amount of time each analyte takes to reach the detector. In contrast, with GEMBE, all analytes take the same amount of time to migrate from the capillary entrance to the detector. So, the separation of analytes is achieved because each analyte enters the capillary at a different time. Each separated analyte is detected within the capillary as a moving boundary using GEMBE, resulting in step-wise changes in conductivity, measured by contactless conductivity detection. The separation resolution is easily controlled by manipulating the pressure gradient and the applied electric field, without the need to change the capillary length or electroosmotic flow.
GEMBE has been previously applied to the analysis of inorganic salts30, organic acids31, enzymes32, amino acids33, and proteins34. The bulk counterflow utilized for GEMBE offers several advantages for the analysis of complex mixtures, which may contain environmental contaminants or interferents, including minimal or no additional sample preparation steps (e.g., filtration or centrifugation). The pressure-driven counterflow and judiciously chosen run buffer can be exploited to eliminate particulates, other sample components, or interfering analytes of opposite charge, from entering the separation capillary, thereby reducing fouling, capillary blockages, or other interferences.35,36 Strychalski et al. demonstrated the use of GEMBE with various complex samples including whole milk, dirt, leaves, ash, and blood serum with a single sample preparatory step of either dilution or suspension in run buffer.35
In this work, we describe the applicability of GEMBE for the separation and detection of fentanyl and fentanyl analogues within complex mixtures containing heroin and other common adulterants and excipients using capacitively coupled contactless conductivity detection (C4D). Fentanyl was also successfully detected from adjudicated case samples provided by the Maryland State Police, Forensic Science Division demonstrating no fouling or blockage of the separation capillary with particulate. The short capillaries, contactless detection format, and continuous injection of sample allowed for a small footprint platform that was easy-to-use for trace analysis of synthetic opioids from commonly encountered complex forensic mixtures.
MATERIALS AND METHODS
Chemicals.
The run buffer used for opioid and synthetic opioid separations was 12 mmol/L acetic acid, 3.3 mmol/L ammonium acetate pH 4.4 (Sigma-Aldrich, St. Louis, MO, USA) in ultrapure water (Millipore Milli-Q, 18.0 MΩ-cm). Acetaminophen, caffeine, mannitol, procaine, quinine, and methanol were all purchased from Sigma-Aldrich (St. Louis, MO, USA).
All fentanyl analogues (acetyl fentanyl, acryl fentanyl, benzyl fentanyl, butyryl fentanyl, furanyl fentanyl, methacryl fentanyl, methoxyacetyl fentanyl, phenyl fentanyl, valeryl fentanyl) were purchased as hydrochloride salts from Cayman Chemical (Ann Arbor, MI, USA) or, when possible, as 1 mg·mL−1 methanolic solutions. Powdered standards and samples were dissolved gravimetrically in methanol (Sigma-Aldrich, St. Louis, MO). The remaining opioids and narcotics (heroin, fentanyl, cocaine, U-47700) were purchased as 1 mg·mL−1 solutions in methanol or acetonitrile from Cerilliant (Round Rock, TX, USA). All sample dilutions were prepared in run buffer, and methanol was added as necessary to allow for a constant 4 % by volume organic solvent across all analyses. When increased analyte concentrations were desired, standards were concentrated to assure each sample contained a maximum of 4 % (v/v) solvent.
Experimental Apparatus.
The details of the GEMBE apparatus (Fig. 1) are described elsewhere.35,† Briefly, two reservoirs were separated by a 5 cm fused silica capillary (360 μm od, 15 μm id) that was threaded through a TraceDec C4D detector (Innovative Sensor Technologies GmbH, Strasshof, Austria), approximately 2 cm from the capillary inlet. Detector settings were the following: frequency, 2× high; voltage, 0 dB; gain, 200%. A Mensor 600 Series (San Marcos, TX, USA) pressure controller was used to drive the run buffer from a 2 mL background electrolyte (BGE) reservoir into a 0.2 mL sample reservoir. A high voltage dc power supply (PS350, Stanford Research Systems) was applied through platinum wires and used to induce electrophoretic migration between reservoirs. The acetate run buffer was replaced at least once per day. After each sample analysis, the sample reservoir was rinsed with 0.2 mL water or run buffer and a process blank of run buffer was analyzed to assess carryover (found to be below detectable limits).
Figure 1.

Schematic of a separation performed using GEMBE. (A) With a constant applied voltage, an initial high pressure is applied to the run buffer (BGE) reservoir so pressure-driven flow dominates and analytes remain in the sample reservoir prior to separation. (B) Pressure is then decreased, at a constant decreasing rate here, allowing analytes to enter the separation capillary connecting the sample and BGE reservoir when the electrophoretic mobility of an analyte (yellow) overcomes the counterflow. (C) As the pressure is further reduced over time, all analytes (yellow and red) have entered the capillary, separated based on the time each analyte entered the capillary and detected as a step-wise change in conductivity.
Experimental Procedure.
The following procedure and base parameters were used unless otherwise noted for specific tests. An initial prescan step was applied with the pressure constant at +30 kPa for 5 s (voltage off) to condition the capillary with run buffer and hold all sample solution/analytes in the sample reservoir until the start of the separation. The starting counterflow pressure was then set at a value between +12.5 and +14.5 kPa, depending on method optimization for shorter analysis times, and held constant for approximately 10 s while a −2800 V (−560 V/cm field strength) voltage was applied. This was followed by the initiation of a −25 Pa/s pressure ramp. The pressure ramp lasted approximately 360 s, until the final pressure reached +5500 Pa, and all analytes of interest had eluted. After which, the pressure was reset to +14.5 kPa and held for 10 s. A final postscan step was applied with the pressure held constant at +30 kPa for 30 s (voltage off) to flush and condition the capillary for subsequent runs. The run control and C4D data-logging were performed using a custom-written LabVIEW (National Instruments, TX) program. Unless specifically noted, total GEMBE analysis times were approximately 6 min for each sample, to achieve resolved steps for heroin, fentanyl, and fentanyl analogues.
Data Analysis.
The raw detector signal demonstrated steps in conductivity (Fig. 2A), the derivative of which yielded the more common peak-based representation (Fig. 2B). Peak-based electropherograms were processed in MATLAB (R2017a, Mathworks, Inc., Natick, MA, USA) with a Savitzky-Golay filter and used solely for visualization purposes. All quantitative data analyses (e.g., limits of detection and signal suppression studies) were derived from fitting the raw stepwise data to an error function and linear baseline offset in MATLAB. Similarly, the separation resolution of two adjacent steps was calculated by fitting the raw data with the sum of two error functions and a linear offset (Supplementary Equation 1).
Figure 2.

GEMBE separation of binary mixtures containing heroin and fentanyl at various concentration ratios. (A) Representative raw data showing stepwise decreases in the conductivity from the elution of heroin and fentanyl with binary mixtures of 10:1, 5:1, 3:1, 1:1, 0:1. Fentanyl concentration held constant at 10 μmol/L. (B) Derivative of raw stepwise data in (A) for representation as peaks. Separation conditions included a −560 V/cm electric field and pressure starting at 14.5 kPa and decreasing at −25 Pa/s. Data shifted vertically for visualization. (C) Fentanyl signal suppression for increasing amounts of heroin in a binary mixture for fentanyl at 5 μmol/L, 10 μmol/L, and 25 μmol/L. Average and standard deviations represented for n = 7 and dotted curve included as a guide.
RESULTS AND DISCUSSION
A schematic representation of the GEMBE setup and typical separation is described in Figure 1. Initially, a constant separation voltage and a high positive pressure (greater than the critical starting pressure37) are applied to the sample reservoir. Under these conditions, the pressure-driven flow dominates, allowing the analytes to remain in the sample reservoir prior to separation (Fig. 1A). Then, as a negative pressure gradient is applied, shown as a constant decreasing rate in Figure 1, the electrophoretic mobilities begin to offset the counterflow pressure allowing each analyte to enter the separation capillary connecting the sample and background electrolyte (BGE) reservoir. Each analyte enters the capillary sequentially at the time when the electrophoretic velocities exceed the pressure-driven counterflow (Fig. 1B). The analytes then migrate down the capillary as a moving boundary at constant acceleration equal to the counterflow acceleration, due to the continuous injection format. The separation of analytes is achieved due to the different time each entered the capillary, which is governed by the differing electrophoretic mobilities. For analysis of various opioids, synthetic opioids, excipients, and other narcotics with GEMBE, the signal at the detector will decrease for each analyte over time, creating a series of steps (Fig. 1C). The step spacing for the analyte boundaries is equal to the different times the analytes initially entered the capillary, allowing the pressure gradient to be optimized for a known sample to decrease overall analysis time.
The buffer composition was initially optimized for the separation and detection of heroin and fentanyl, resulting in acetic acid-ammonium acetate buffer at pH 4.4. Based on the respective pKa’s of heroin, fentanyl, and the fentanyl analogues, all target species were protonated in the chosen buffer. In addition, due to safety considerations, the standard samples were purchased as solutions in organic solvent (see Materials and Methods section), a small fraction of which remained in analyzed sample solution. Therefore, the impact of the solvent concentration on the step height and resolution was also evaluated. Solvent concentrations ≤ 20 % by volume, methanol or acetonitrile, had little to no impact on the step heights or resolution, however, for consistency, the amount of organic solvent in each sample was held constant at 4 % (v/v).
Similarly, for a specified electric field, the resolution was easily manipulated by the pressure ramp rate (Supplementary Figure S2). Decreasing the ramp rate of the pressure-controlled bulk counterflow directly enhanced the resolution of the heroin and fentanyl steps. However, manipulating the electric field strength or pressure gradient to increase the resolution had opposing effects on the separation time. Unlike increasing the electric field, as the pressure ramp rate was decreased, the run time for the separation increased (Supplementary Figure S2). This analysis identified optimal ranges for base parameters to achieve separation of heroin and fentanyl (as specified in the Materials and Methods section), which were applied to the remaining investigations. In addition, parameter limits, beyond which heroin and fentanyl were not fully resolved, were identified (Figures S1 and S2). Cursory sensitivity measurements of fentanyl and heroin were also evaluated using the optimized parameters. Empirical limits of detection (LOD) were determined by serially diluting neat 100 μmol/L samples of fentanyl and heroin, ultimately yielding LODs of 2.5 μmol/L and 1.0 μmol/L, respectively (Supplementary Figure S3).
A preliminary optimization of the electric field strength and applied pressure gradient, the main factors in the trade-off between resolution and the separation time, was performed. Both GEMBE parameters were easily controlled by the instrument software allowing for relatively quick method development. Contrastingly, conventional CE requires consideration of the electric field, capillary length, and the apparent mobility in the separation medium as pressure gradient is not available for manipulation. Since fentanyl is most commonly found in mixtures with heroin, their step resolution was characterized as a function of these system parameters. Similar to conventional CE, increasing the electric field strength in GEMBE resulted in improved resolution and reduced separation times (Supplementary Figure S1). The improvement in resolution was limited by Joule heating and Taylor-Aris dispersion at elevated electric fields, where the step width began increasing linearly with electric field strength.37 The optimal electric field was identified just before this transition.
Fentanyl analysis in binary mixtures.
Although trace analysis capabilities and low detection limits are imperative for the identification of opioids, fentanyl is oftentimes encountered in mixtures that can include a range of compounds such as heroin and common adulterants (e.g., acetaminophen, quinine, and procaine). Previous work has highlighted some of the capabilities and hurdles of analytical techniques such as IMS. Specifically, IMS faces challenges due to the overlapping retention times and signal suppression of fentanyl within mixtures.7 Sisco, et al. determined that certain compounds present in drug mixtures can create significant matrix effects, notably fentanyl ion suppression due to competitive ionization with increasing amounts of competing compound.12 Although competitive ionization, as demonstrated with IMS, is not a concern for GEMBE, the overall effects of increasing amounts of excipient (i.e., heroin, cocaine, procaine, and quinine) relative to fentanyl in binary mixtures were evaluated.
Figure 2 demonstrates the GEMBE response for a constant 10 μmol/L concentration of fentanyl in increasing amounts of heroin. The binary mixture was evaluated at concentration ratios of heroin to fentanyl ranging from pure fentanyl (0:1) to 10× as much heroin as fentanyl (10:1). Figure 2A shows the representative raw data with stepwise decreases in conductivity for the elution of heroin and fentanyl at each ratio. Similarly, the derivative plots of the raw stepwise data are shown in Figure 2B, resembling a more traditional electropherogram. As the ratio and concentration of heroin in the binary mixture increased, the fentanyl response (i.e., step height and associated derivative peak height) decreased relative to a sample with no heroin (0:1 ratio). The resulting fentanyl signal suppression (relative to a neat 10 μmol/L fentanyl sample) was quantified across the concentration ratio range (0:1, 1:1, 3:1, 5:1, and 10:1) and is displayed in Figure 2C. Figure 2C demonstrates an approximately 50 % suppression of the fentanyl signal (at 10 μmol/L loading) in the presence of 10× as much heroin (100 μmol/L). Further extending the ratio out to 30:1 (300 μmol/L heroin) only increased the signal suppression, yielding a fentanyl signal of about 40 % relative to a neat sample (Figure 3A). These results demonstrated that GEMBE detected fentanyl from mixtures with heroin at typically observed ratios.
Figure 3.

Fentanyl signal suppression as a function of competing compound concentration in binary mixtures of heroin, cocaine, quinine, and procaine across mixture ratios of 0:1, 1:1, 3:1, 5:1, 10:1, 20:1, and 30:1 (competing compound:fentanyl) with fentanyl at constant 10 μmol/L. Average and standard deviations represented for 7 to 10 replicates.
Given the conductivity-based detection scheme employed here, not only was the ratio of species in a binary mixture of interest, but also the overall sample concentrations (and therefore conductivity). In addition to the 10 μmol/L fentanyl case described above, constant fentanyl concentrations of 5 μmol/L and 25 μmol/L were also investigated across the same ratio range (0:1, 1:1, 3:1, 5:1, and 10:1). Figure 2C demonstrates not only the increased fentanyl signal suppression for increasing amounts of heroin, but also an increased suppression for overall increases in sample concentration (fentanyl and heroin). This was not completely unexpected given the mode of GEMBE separation and conductivity detection scheme. Optimal detection was exhibited for low analyte concentrations relative to the buffer, such that the background conductivity was much greater than the change in conductivity due to the presence or absence of an analyte. As a result, the greatest fentanyl signal suppression was observed for the binary mixtures with 25 μmol/L fentanyl. At the 10:1 ratio (250 μmol/L heroin: 25 μmol/L fentanyl), fentanyl demonstrated an average signal of only approximately 30 % that of the neat sample.
An identical approach was used to evaluate the fentanyl signal suppression in binary mixtures with other interfering compounds relative to the neat 10 μmol/L fentanyl response (0:1 ratio). In addition to heroin, signal suppression from binary mixtures of fentanyl with frequently encountered adulterants and excipients was evaluated, including cocaine, quinine, procaine, mannitol, acetaminophen, and caffeine.38 Mannitol, acetaminophen, and caffeine all eluted well after fentanyl and the pressure gradient switch where the bulk pressure-driven counterflow dominated the injection of analytes into the separation capillary. Consequently, these analytes did not interfere with the detection, nor suppress the signal, of fentanyl and were not considered further (Supplementary Figure S4). Cocaine, quinine, and procaine all eluted with step times before heroin and fentanyl (Supplementary Figures S5 and S6). Figure 3 shows the relative fentanyl signal suppression induced by each competing species (i.e., heroin, cocaine, quinine, and procaine) out to concentration ratios of 30:1. The competing compound that resulted in the greatest signal suppression of fentanyl was heroin, and the competing compound with the least amount of signal suppression observed was quinine. The overall sensitivity for the detection of fentanyl decreased as the concentration of the competing compound increased, which resulted in increased step heights, due to the increased change in conductivity from the background conductivity (Supplementary Figure S7). Of the competing compounds evaluated, the migration order was as follows: quinine, procaine, cocaine, heroin, and fentanyl.
A subset of the ever-increasing array of fentanyl analogues was also investigated to determine relative elution times and signal suppression. Since heroin resulted in the greatest impact on fentanyl signal suppression, and is a common constituent found in street samples containing fentanyl, binary mixtures containing fentanyl analogues with heroin were evaluated. Common fentanyl analogues, including acryl, butyryl, furany1, valeryl, acetyl, and benzyl fentanyl, were analyzed at a concentration ratio of 20:1 with heroin (heroin:analogue). Acetyl and benzyl fentanyl exhibited step times close to that of heroin in pure samples and were unresolvable from heroin in binary mixtures for the chosen separation parameters. Further optimization of the counterflow pressure gradient or alternative buffers would need to be assessed to resolve these co-migrating analytes. The remaining fentanyl analogues were resolved from heroin in binary mixtures as displayed in Figure 4A. The fentanyl analogues investigated here all exhibited step times around, and just following, heroin. The fentanyl analogue signal suppression (relative to a neat 10 μmol/L sample) in a 20:1 binary mixture with heroin was quantified as shown in Figure 4B. A similar trend was observed between the fentanyl analogues and fentanyl, where the signal was suppressed approximately 50 % to 60 % in the presence of heroin.
Figure 4.

(A) GEMBE separations of binary mixtures containing heroin and fentanyl analogues at 20:1 concentration ratios (200 μmol/L heroin : 10 μmol/L analogue), including (top to bottom) acryl, butyryl, furanyl, and valeryl fentanyl. Separation conditions included a −560 V/cm electric field and pressure starting at 12.5 kPa and decreasing at −25 Pa/s. Data shifted vertically for visualization. (B) Fentanyl analogue signal suppression for binary mixtures with heroin at 20:1 concentration ratios. Dashed red line denotes the average fentanyl signal suppression in a binary mixture with heroin at 20:1 for comparison. Average and standard deviations represented for 7 replicates.
Fentanyl analysis in complex mixtures.
With presumptive knowledge of the constituents of a mixture or targeted analysis (e.g., screening for fentanyl-related species), the GEMBE separation method can be easily optimized for reducing the overall analysis time. The simplest way to reduce the separation time is to implement a multistep pressure gradient with different ramp rates. Figure 5 shows exemplary derivative plots for a separation of quinine, heroin, and fentanyl with the respective pressure gradients in blue. In the first separation (Fig. 5A), a single constant decreasing pressure rate for the counterflow was implemented. A slower constant pressure gradient would be ideal for a completely unknown sample to be sure that any possible analyte constituent was detected. However, if a presumptive test was performed or the analysis was targeting fentanyl-related species, Figure 5B separation plot demonstrates an example of multiple different pressure rates used to shorten the analysis time. The previous analysis demonstrated that the fentanyl analogues all eluted in the same region of the electropherogram, providing a basis for targeted separations. In the shortest separation time (Fig 5B), two different decreasing pressure rates were chosen. A faster (4×) initial pressure rate was utilized to decrease the time spent early in the separation, before the opioids were known to elute and where precise resolution was not required. The slower pressure rate was then applied for the elution of heroin and fentanyl based on previous optimization studies to assure that the two steps (peaks in the derivative plots) were fully resolved.
Figure 5.

Representative derivative plots using multiple pressure gradients for decreased run time with a mixture of fentanyl, heroin, and quinine at concentrations of 20 μmol/L, 200 μmol/L, and 200 μmol/L, respectively. (A) Derivative plot with a constant pressure gradient of −25 Pa/s (1) and longest run time. (B) Two pressure gradients of −100 Pa/s (1) and −25 Pa/s (2), respectively, were utilized to decrease the overall run time by ~3.25 min compared to a single constant pressure ramp. Separation conditions included start pressure of 14.5 kPa and −560 V/cm. Data shifted vertically for visualization.
However, if the sample to be analyzed was completely unknown, the separation time may need to be extended by implementing a decreased counterflow pressure gradient to ensure resolution of all analyte steps. Adjudicated case samples that contained fentanyl and several additional compounds were provided by the Maryland State Police, Forensic Science Division to demonstrate the capabilities of GEMBE. The derivative plots of two different case samples are shown in Figure 6 with images of each sample in methanolic solutions (~1 mg/mL to 2 mg/mL) before ~25× dilution in run buffer for a total of 4 % by volume methanol. Figure 6A demonstrates the GEMBE separation from a blue-colored suspension with undissolved particulate. The sample composition was verified using GC-MS and contained the synthetic opioid U-47700, fentanyl, and an unknown cutting agent (Supplementary Figure S8). Standard solutions were used to confirm analyte identification with GEMBE. The second case sample was a clear solution with nonuniform, undissolved white particulate as shown in Figure 6B. Six different components were identified and confirmed with standard solutions as lidocaine, heroin, 6-monoacetylmorphine, methoxyacetyl fentanyl, phenyl fentanyl, and methacryl fentanyl (listed in migration order). The contents of the sample were verified using GC-MS, however, caffeine and trace levels of codeine were also identified. As discussed previously, caffeine did not interfere with the identification of fentanyl using the developed GEMBE method due to the elution after the pressure gradient switch (where the bulk pressure-driven counterflow dominates). In addition, the concentration of codeine in the sample was believed to be below the limits of detection for the GEMBE system. A slower negative pressure gradient was implemented at 5 Pa/s to observe increased resolution between steps #2 and #3, heroin and 6-monoacetylmorphine, respectively, to aid in analyte verification (Fig. 6B inset).
Figure 6.

Representative GEMBE separations for complex drug mixtures containing fentanyl related species. (A-B) Adjudicated case samples that contained fentanyl and several additional compounds provided by the Maryland State Police, Forensic Science Division. (A) Labeled derivative plot from a blue-colored suspension with undissolved particulate (photo) that contained the synthetic opioid U-47700, fentanyl, and an unidentified cutting agent. (B) Labeled derivative plot from a clear solution with nonuniform, undissolved white particulate (photo) containing lidocaine (1), heroin (2), 6-monoacetylmorphine (3), methoxyacetyl fentanyl (4), phenyl fentanyl (5), methacryl fentanyl (6), caffeine, and codeine with inset from a slowed pressure gradient (−5 Pa/s). Separation conditions included a −560 V/cm electric field and pressure starting at 14.5 kPa and decreasing at −25 Pa/s (unless noted otherwise)
For both case samples there was no additional sample preparatory steps required to remove the undissolved particulate before performing the analyses using GEMBE. Simply, the unknown adjudicated samples were suspended in methanol (for both GC-MS and GEMBE) and then further diluted in run buffer, resulting in minimal consumption of the sample solution for analysis with GEMBE (~10 μL of the methanolic suspension). In addition to minimal sample preparation and consumption, the overall run time required for fentanyl identification was ~ 6 min using a −25 Pa/s pressure gradient and overall run times of ~10 – 12 min was used for GC-MS verification.
CONCLUSIONS
The separation and sensitive detection of fentanyl and fentanyl analogues from complex mixtures was demonstrated using GEMBE with contactless conductivity detection. Due to the persistent and widespread adulteration of heroin with synthetic opioids, binary mixtures with fentanyl and fentanyl analogues were analyzed, exhibiting clear separation between heroin and fentanyl, as well as the analogues, acryl, butyryl, furanyl, valeryl, methacryl, methoxyacetyl, and phenyl fentanyl. However, for the GEMBE parameters and buffer employed here, acetyl and benzyl fentanyl were unresolvable from heroin. In addition to separation and detection from heroin, fentanyl was resolvable from a range of tested adulterants and excipients, including, U-47700, cocaine, phencyclidine (PCP), amphetamine, methamphetamine, 6-monoacetylmorphine, procaine, quinine, lidocaine, acetaminophen, mannitol, and caffeine. Signal suppression of fentanyl in binary mixtures with select competing compounds (i.e., heroin, cocaine, procaine, and quinine) was quantified, Fentanyl was detected in the presence of competing compound at 30× the fentanyl concentration, demonstrating around 50 % to 60 % signal suppression relative to a neat sample.
Fentanyl was also successfully identified in adjudicated case samples provided by the Maryland State Police, Forensic Science Division, without the need for additional sample preparation. The counterflow nature of the GEMBE platform prohibited the visible particulate from entering the capillary, eliminating the potential for blockages or capillary fouling.
The short capillaries, contactless detection, minimal sample preparatory steps, and continuous injection format of GEMBE allows for a platform that is easy-to-use and optimize for trace analysis of synthetic opioids from complex forensic drug mixtures. GEMBE also allows for multiplexed sample analysis for high-throughput screening from microfluidic chip-based devices of less than 1 in2 in area. Shackman et al. demonstrated the ability to perform eight simultaneous separations with fluorescence detection, requiring only n+1 reservoirs (eight sample reservoirs and one buffer reservoir).29 Similarly, Ross et al. demonstrated a multiplexed GEMBE system with a 16-capillary array utilizing 3 mm capillaries to connect individual sample reservoirs and a common buffer reservoir with current detection.32 As demonstrated here, fentanyl and fentanyl analogues were detected from complex mixtures in as short as 2 minutes, significantly shorter than traditional GC-MS (~10 – 20 min) and LC-based (~10 – 30 min) methods.21 A highly multiplexed GEMBE platform would enable parallel high-throughput screening of backlogged forensic samples.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank Amber Burns at the Maryland State Police, Forensic Science Division for providing the adjudicated case samples, as well as Ed Sisco at the National Institute of Standards and Technology for providing confirmatory GC-MS analysis. This research was performed while S.T.K. held a National Institute of Standards and Technology (NIST) National Research Council (NRC) Research Postdoctoral Associateship Award.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Additional experimental methods and figures as noted in the text (PDF)
Certain commercial equipment, instruments, or materials are identified in this article in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
Official contribution of the National Institute of Standards and Technology; not subject to copyright in the United States.
The authors have declared the following potential conflict of interest: D.R. is an inventor on two patents (describing GEMBE) assigned to the United States of America as represented by the Secretary of Commerce, The National Institute of Standards and Technology. T.P.F. and S.T.K. declare no competing financial interests.
REFERENCES
- (1).United Nations Office on Drugs and Crime, World Drug Report Vienne, Austria: UNODC: 2017. [Google Scholar]
- (2).Drug Enforcement Administration, 2018. National Drug Threat Assessment. https://www.dea.gov/sites/default/files/2018-11/DIR-032-182018NDTAfinallowresolution.pdf Accessed April 4, 2019.
- (3).Ciccarone D. Int. J. Drug Policy 2017, 46, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Suzuki J; El-Haddad S Drug Alcohol Depen. 2017, 171, 107–116. [DOI] [PubMed] [Google Scholar]
- (5).Marinetti LJ; Ehlers BJ J. Anal. Toxicol. 2014, 38, 592–598. [DOI] [PubMed] [Google Scholar]
- (6).Ohta H; Suzuki S; Ogasawara KJ Anal. Toxicol. 1999, 23, 280–285. [DOI] [PubMed] [Google Scholar]
- (7).Strano-Rossi S; Bermejo AM; de la Torre X; Botre F Anal. Bioanal. Chem. 2011, 399, 1623–1630. [DOI] [PubMed] [Google Scholar]
- (8).Verkouteren JR; Staymates JL Forensic Sci. Int. 2011, 206, 190–196. [DOI] [PubMed] [Google Scholar]
- (9).Leonard J; Haddad A; Green O; Birke RL; Kubic T; Kocak A; Lombardi JR J. Raman Spectrosc. 2017, 48, 1323–1329. [Google Scholar]
- (10).Haddad A; Comanescu MA; Green O; Kubic TA; Lombardi JR Anal. Chem. 2018, 90, 12678–12685. [DOI] [PubMed] [Google Scholar]
- (11).Salemmilani R; Moskovits M; Meinhart CD Analyst 2019, 144, 3080–3087 [DOI] [PubMed] [Google Scholar]
- (12).Sisco E; Verkouteren J; Staymates J; Lawrence J Forensic Chem. 2017, 4, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zaknoun H; Binette MJ; Tam M Int. J. Ion Mobil. Spectrom. 2019, 22, 1–10. [Google Scholar]
- (14).Huynh NH; Tyrefors N; Ekman L; Johansson MJ Pharmaceut. Biomed. 2005, 37, 1095–1100. [DOI] [PubMed] [Google Scholar]
- (15).Lurie IS; Iio RJ Chromatogr. A 2009, 1216, 1515–1519. [DOI] [PubMed] [Google Scholar]
- (16).Gergov M; Nokua P; Vuori E; Qjanpera I Forensic Sci. Int. 2009, 186, 36–43. [DOI] [PubMed] [Google Scholar]
- (17).Ghassabian S; Moosavi SM; Valero YG; Shekar K; Fraser JF; Smith MT J. Chromatogr. B 2012, 903, 126–133. [DOI] [PubMed] [Google Scholar]
- (18).Lurie IS; Berrier AL; Casale JF; Iio R; Bozenko JS Forensic Sci. Int. 2012, 220, 191–196. [DOI] [PubMed] [Google Scholar]
- (19).Wu AH; Gerona R; Armenian P; French D; Petrie M; Lynch KL Clin. Toxicol. 2012, 50, 733–742. [DOI] [PubMed] [Google Scholar]
- (20).Nan Q; Ping X; Shen BH; Zhuo XY; Yan S; Song FY J. Chromatogr. B 2019, 1124, 82–99. [DOI] [PubMed] [Google Scholar]
- (21).Elbardisy HM; Foster CW; Cumba L; Antonides LH; Gilbert N; Schofield CJ; Belal TS; Talaat W; Sutcliffe OB; Daabees HG; Banks CE Anal. Methods 2019, 11, 1053–1063. [Google Scholar]
- (22).Armenian P; Vo KT; Barr-Walker J; Lynch KL Neuropharmacology 2018, 134, 121–132. [DOI] [PubMed] [Google Scholar]
- (23).Tagliaro F; Pascali J; Fanigliulo A; Bortolotti F Electrophoresis 2010, 31, 251–259. [DOI] [PubMed] [Google Scholar]
- (24).SWGDRUG, Scientific Working Group for the Analysis of Seized Drugs Recommendations, Version 7.1; 2016. http://www.swgdrug.org/Documents/SWGDRUGRecommenda-tionsVersion7-1.pdf
- (25).Lurie IS Forensic Sci. Int. 1998, 92, 125–136. [Google Scholar]
- (26).Lurie IS; Klein RFX; Dalcason TA; Lebelle MJ; Brenneisen R; Weinberger RE Anal. Chem. 1994, 66, 4019–4026. [DOI] [PubMed] [Google Scholar]
- (27).Koppenhoefer B; Zhu XF; Jakob A; Wuerthner S; Lin BC J. Chromatogr. A 2000, 875, 135–161. [DOI] [PubMed] [Google Scholar]
- (28).Millot MC J. Chromatogr. B 2003, 797, 131–159. [DOI] [PubMed] [Google Scholar]
- (29).Shackman JG; Munson MS; Ross D Anal. Chem. 2007, 79, 565–571. [DOI] [PubMed] [Google Scholar]
- (30).Flanigan PM; Ross D; Shackman JG Electrophoresis 2010, 31, 3466–3474. [DOI] [PubMed] [Google Scholar]
- (31).Ross D; Romantseva EF Anal. Chem. 2009, 81, 7326–7335. [DOI] [PubMed] [Google Scholar]
- (32).Ross D; Kralj JG Anal. Chem. 2008, 80, 9467–9474. [DOI] [PubMed] [Google Scholar]
- (33).Ross D; Shackman JG; Kralj JG; Atencia J Lab Chip 2010, 10, 3139–3148. [DOI] [PubMed] [Google Scholar]
- (34).Kralj JG; Munson MS; Ross D Electrophoresis 2014, 35, 1887–1892. [DOI] [PubMed] [Google Scholar]
- (35).Strychalski EA; Henry AC; Ross D Anal. Chem. 2009, 81, 10201–10207. [DOI] [PubMed] [Google Scholar]
- (36).Strychalski EA; Henry AC; Ross D Anal. Chem. 2011, 83, 6316–6322. [DOI] [PubMed] [Google Scholar]
- (37).Ross D Electrophoresis 2010, 31, 3658–3664. [DOI] [PubMed] [Google Scholar]
- (38).Cole C; Jones L; McVeigh J; Kicman A; Syed Q; Bellis MA, CUT: A Guide to Adulterants, Bulking Agents and Other Contaminants Found in Illicit Drugs; 2010. https://phi.ljmu.ac.uk/wp-content/uploads/2012/08/cut-a-guide-tothe-adulterants-bulking-agents-and-other-contaminants-found-in-illicit-drugs.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.