

Abstract
The fabrication of electrospun composite polyurethane fibers capable of dual-action antibacterial dendrimer release is reported. Generation 4 (G4) poly(amidoamine) dendrimers were functionalized with octyl alkyl chain or quaternary ammonium (QA) moieties followed by modification of the resulting secondary amines with N-diazeniumdiolate nitric oxide (NO) donors to produce dual-action antibacterial dendrimers. Control and NO-releasing dendrimers were doped into polyurethane solutions prior to electrospinning of the polymer to yield well-defined dendrimer-doped composite polyurethane fibers. The fiber mats were semi-porous (≥30% porosity) and exhibited high water uptake (>100% relative to fiber mass). Dendrimer- and NO-release characteristics (rates and totals) were dependent on the dendrimer modification and polyurethane composition, with total dendrimer- and NO-release amounts ranging from 10 – 80 μg/mg and 0.027 – 0.072 μmol NO/mg, respectively. The antibacterial action of the fibers was evaluated against Gram-negative and Gram-positive bacterial strains. Nitric oxide-releasing fibers demonstrated broad-spectrum bactericidal action at short (2 h) and long (24 h) timescales.
Keywords: electrospinning, antibacterial, wound dressing, poly(amidoamine) dendrimer, N-diazeniumdiolate, nitric oxide
Graphical Abstract
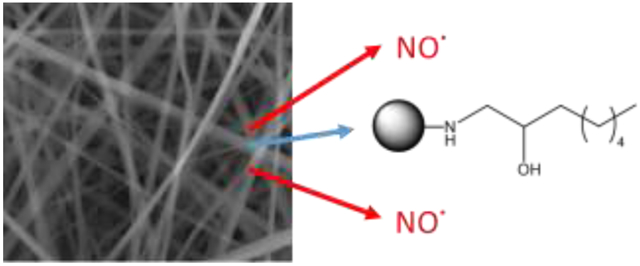
Introduction
The successful treatment of chronic wounds such as diabetic foot ulcers, pressure ulcers, and venous leg ulcers is often hindered by the inefficient eradication of opportunistic pathogens such as Pseudomonas aeruginosa and Staphyloccocus aureus.1-3 Left untreated, the proliferation of bacteria in chronic wounds often leads to significant morbidity (e.g., limb amputation).3 While the use of wound dressings may facilitate some wound healing, the presence of bacteria hinders complete wound closure. An ideal wound dressing would provide facile gaseous and fluid exchange, absorb excess wound exudates, and act as a physical barrier to infectious microorganisms while also reducing the bacterial burden in the wound site.4
Polyurethane materials are often used as wound dressings because they exhibit adequate barrier properties, oxygen permeability, and tissue compatibility.5-8 Commercially-available wound dressings such as semi-permeable polyurethane films and foams have demonstrated utility as physical barriers, inhibiting the migration of bacteria to wounds while promoting wound closure.4, 9-10 Incorporating silver ions into these polyurethane wound dressings as an active release antimicrobial agent has further improved wound healing and reduced pain-related symptoms.11-13 However, significant accumulation of wound fluid beneath polyurethane bandages continues to be a disadvantage of these dressings, requiring frequent wound aspiration to prevent leakage and infection.5, 14-15 The fabrication of porous wound dressing materials via electrospinning represents a recent strategy to further improve the wound healing capabilities of polyurethane dressings. The electrospinning technique, consisting of the application of a high potential difference between the head of a syringe needle containing a polymer solution and a grounded collector, produces nonwoven nanofibrous polymer webs.4, 16-17 As a result of their inherent porosity and large effective surface area, these mats have properties essential for wound healing. Indeed, the high surface area to volume ratio and porous architecture of such materials allows for enhanced water absorption, cell respiration, and protection of the wound from bacterial infection.4 In adult male guinea pigs, Khil et al. reported improved wound healing for wounds treated with electrospun polyurethane fibers versus conventional Tegaderm©, a commercially-available polyurethane wound dressing.5 The electrospun dressings both increased the rate of epithelialization and prevented the permeation of infectious microorganisms with little observed fluid accumulation.
Electrospun fibers have been modified to exhibit antibacterial action via the incorporation of therapeutic compounds blended into an all-in-one dressing.4, 18-19 Further, the use of a core-sheath structure when fabricating electrospun fibers allows for greater control over therapeutic release from the fibers.17, 20-21 Initial studies in the development of antibacterial electrospun wound dressings demonstrated the controlled release of antibiotics18-19, 22 and silver ions23-24 from electrospun fibers. However, bacterial resistance to antibiotics and silver25-27 warrants the evaluation of dressings doped with next generation antibacterial agents that are unlikely to foster resistance. Our study thus aimed to develop electrospun polyurethane fibers capable of releasing novel antibacterial scaffolds to enhance bactericidal activity.
Nitric oxide (NO) is a promising antibacterial agent that exhibits broad-spectrum antibacterial and wound-healing actions.28-30 Macromolecular scaffolds capable of controllable NO storage and release represent a viable strategy for delivering bactericidal NO doses.31-32 Further, the use of macromolecular scaffolds allows for the combination of antibacterial NO release with a non-depleting, contact-based biocide (e.g., quaternary ammonium moieties, alkyl chains), which has proven especially effective at producing broad-spectrum dual-action antibacterial agents.33-36 The co-administration of two mechanistically different biocides in this manner reduces the likelihood of resistance and improves the bactericidal efficacy of the scaffold, thereby lowering the required therapeutic dose.37-38 We have previously reported on the eradication of Gram-negative and Gram-positive biofilms with NO-releasing alkyl chain-modified dendrimers as a function of dendrimer generation (i.e., size) and modification.34-35 Herein, we report the 1) fabrication of electrospun composite polyurethane fibers doped with NO-releasing alkyl chain- or quaternary ammonium (QA)-modified poly(amidoamine) dendrimers, and 2) evaluation of their antibacterial action against Gram-negative and Gram-positive bacteria. This study aims to determine the utility of dual-action dendrimer biocide release from electrospun polyurethane fibers for potential antibacterial wound dressing applications.
Experimental Section
Materials.
Triethylamine (TEA), rhodamine B isothiocyanate (RITC), dimethyloctylamine, epichlorohydrin, phenazine methosulfate (PMS), fetal bovine serum (FBS), trypsin, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS), penicillin streptomycin (PS), and propidium iodide (PI) were purchased from Sigma-Aldrich (St. Louis, MO). Methyl acrylate, 1,2-epoxyoctane, and ethylenediamine (EDA) were purchased from the Aldrich Chemical Company (Milwaukee, WI). Sodium methoxide (5.4 M solution in methanol) was purchased from Acros Organics (Geel, Belgium). Colloidal silica in methanol was obtained from Nissan Chemical America Corporation (Houston, TX). Cellulose ester dialysis membranes (500-1000 MWCO) were purchased from Spectrum Laboratories, Inc. (Rancho Dominguez, CA). Tecophilic (HP-93A-100) and Tecoflex (SG-80A) polyurethanes were obtained from Thermedics (Woburn, MA). Tecoplast (TP-470) polyurethane was a gift from Lubrizol (Cleveland, OH). Dulbecco's modified Eagle's medium (DMEM) and Dulbecco’s phosphate buffered saline (PBS) were obtained from Lonza Group (Basel, Switzerland). 4,5-Diaminofluorescein diacetate (DAF-2 DA) was purchased from Calbiochem (San Diego, CA). Tryptic soy broth (TSB), tryptic soy agar (TSA), Luria-Bertani (LB) broth, LB, and Mueller Hinton agar were obtained from Becton, Dickinson and Company (Franklin Lakes, NJ). Pseudomonas aeruginosa (P. aeruginosa; ATCC #19143), Eschericia coli (E. coli; ATCC #35150), Staphylococcus aureus (S. aureus; ATCC #29213), and methicillin-resistant Staphylococcus aureus (MRSA; ATCC #33591) were obtained from American Type Tissue Culture Collection (Manassas, VA). L929 mouse fibroblasts were obtained from the UNC Tissue Culture Facility (Chapel Hill, NC). Nitrogen (N2), argon (Ar), carbon dioxide (CO2), and nitric oxide (NO) calibration (25.87 PPM, balance N2) gases were purchased from National Welders (Raleigh, NC). Pure nitric oxide (NO) gas (99.5%) was purchased from Praxair (Sanford, NC). Common laboratory salts and solvents were purchased from Fisher Scientific (Fair Lawn, NJ). Distilled water was purified using a Millipore Milli-Q UV Gradient A-10 system (Bedford, MA), resulting in a total organic content of ≤6 ppb and a final resistivity of 18.2 mΩ·cm. Unless noted otherwise, all materials were analytical-reagent grade and used as received without further purification.
Synthesis of QA- and Alkyl Chain-Modified PAMAM Dendrimers.
Poly(amidoamine) (PAMAM) scaffolds were synthesized as described previously,34, 39-40 by repeated alkylation/amidation steps using methyl acrylate and EDA from an EDA core. G4 PAMAM dendrimers were then modified with either octyl or octylQA moieties. To form octyl-modified dendrimers,35 G4 PAMAM (100.0 mg) was dissolved in 5 mL methanol, and one equivalent of triethylamine (with respect to the molar amount of primary amines) and one molar equivalent of epoxyoctane were then added to the vial. This solution was stirred at room temperature for 3 d. After reaction completion, excess epoxide was removed in vacuo. To ensure removal of any unreacted epoxide, dendrimers were re-dissolved in 5 mL methanol and kept under vacuum overnight. Complete removal of the epoxide was verified via 1H NMR spectroscopy (Bruker 400 MHz spectrometer; Billerica, MA).
To form octyl QA-modified dendrimers, quaternary ammonium epoxides (octylQA-epoxide) were first synthesized as described previously.33, 36 Briefly, 0.04 mmol epichlorohydrin was reacted with 0.01 mmol N,N-dimethyloctylamine at room temperature overnight (~18 h). The mixture was then added dropwise to cold ether while sonicating, and the solid/viscous liquid octylQA-epoxide was collected via centrifugation (810×g, 5 min). The supernatant was decanted, and the octylQA-epoxide was washed with 50 mL of cold ether and sonicated extensively. This washing procedure was repeated three times before drying the product in vacuo. A ring-opening reaction was then carried out between the octylQA-epoxide and the terminal primary amines of the PAMAM dendrimers. G4 PAMAM (100.0 mg) was dissolved in 5 mL of methanol. One equivalent of triethylamine (with respect to the molar amount of primary amines) and 2.5 molar equivalents of octylQA-epoxide were then added to the vial. The solution was stirred at room temperature for 4 d. Solvent was then removed in vacuo. The dendrimers were subsequently dissolved in water, followed by dialysis against water overnight and lyophilization.
Representative 1H NMR data of modified G4 PAMAM included the following peaks. G4 octyl: 1H NMR (400 MHz, MeOD, δ) 2.29 (s, NCH2CH2C(O)NH), 1.35–1.23 (m, NHCH2CH(OH)(CH2)5CH3), 0.83–0.80 (t, NHCH2CH(OH)C(H2)5CH3). G4 octylQA: 1H NMR (400 MHz, CD3OD, δ) 2.31 (s, NCH2CH2C(O)NH), 1.80 (s, CH2N+(CH3)2CH2CH2(CH2)5CH3), 1.31–1.23 (m, CH2N+(CH3)2CH2CH2(CH2)5CH3), 0.83 (t, CH2N+(CH3)2CH2CH2(CH2)5CH3).
N-Diazeniumdiolation of QA- and Alkyl Chain-Modified PAMAM Dendrimers.
To form N-diazeniumdiolate NO donors on the modified dendrimer scaffold, single-action G4 PAMAM (30 mg) were added to 1 mL solutions of 1:1 MeOH:THF. One molar equivalent (e.g., with respect to the molar amount of primary amines) of sodium methoxide was added after mixing (vortexing) the solutions. The resulting dendrimer solutions were placed in a stainless steel reactor with continuous magnetic stirring and connected to an in-house NO reactor. The vessel was flushed with Ar three times to a pressure of 7 bar, followed by three longer Ar purges (10 min) to remove trace oxygen from the solutions. Following deoxygenation, the reactor was pressurized to 10 bar with NO gas pre-scrubbed with KOH. The pressure was maintained at 10 bar for 4 d, after which the solutions were again purged with Ar three times for short durations followed by extended purges (3 × 10 min) to remove unreacted NO. Solvent was removed in vacuo, and the NO-releasing dendrimers were dissolved in anhydrous methanol in a 1 dram glass vial, capped and parafilmed, and stored at −20 °C.
Fabrication of Electrospun Polyurethane Fibers.
All polyurethane solutions used were prepared at a concentration of 10 wt% (100 mg/mL) in 3:1:1 THF:DMF:MeOH. Polyurethane solutions containing control and NO-releasing dendrimers (5, 15, or 25 mg/mL) were prepared by first dissolving the polyurethane in THF and DMF, followed by the addition of dopant solution in the remaining equivalent of methanol.
Electrospun fibrous mats were fabricated using a custom electrospinning apparatus41-42 consisting of a ES20P-20W High Voltage power supply (Gamma High Voltage Research, Ormond Beach, FL), two Kent Scientific Genie Plus syringe pumps (Torrington, CT), and a grounded steel collector plate covered in aluminum foil placed at a 45° angle to the needle. All fibers were fabricated using an applied voltage of 15 kV and 15 cm tip-to-collector distance. Composite polyurethane fibers were formed using a co-axial needle (Ramè-Hart; Succasunna, NY) composed of two concentric needles (inner gauge: 22, outer gauge: 13). The co-axial needle was supplied by both individual core and sheath solutions, each connected to a separate syringe pump (Figure S1). Core solutions were composed of either 10 wt% Tecophilic (HP 93A) or Tecoflex (SG 80A) polyurethanes doped with control or NO-releasing dendrimer (blank fibers contained no dendrimer dopant in the core solution). Sheath solutions were composed of 10 wt% Tecoplast (TP 470) or Tecoflex (SG 80A) polyurethanes with no dendrimer dopant. Fibers prepared with a SG 80A sheath and HP 93A core were doped with either 5, 15, or 25 mg/mL dendrimer, while the core solutions for all fibers prepared using a TP 470 sheath were doped with 25 mg/mL dendrimer. To form fibrous mats, 0.5 mL core solution (10 μL/min flow rate) and 1.5 mL sheath solutions (30 μL/min flow rate) were electrospun and collected on aluminum foil. After collection, fibers were cut using a 1.27 cm-diameter hole-punch to yield individual “coupon” samples with a resultant surface area of 1.267 cm2. Each fiber sample was weighed, and any sample with a mass below 1 mg or above 3.5 mg was excluded.
Characterization of NO Storage and Release.
Nitric oxide release was evaluated in real-time in deoxygenated PBS (pH 7.4, 37 °C) using a Sievers NOA 280i chemiluminescence NO analyzer (NOA, Boulder, CO).43 Prior to analysis, the NO analyzer was calibrated with air passed through a NO zero filter (0 ppm NO) and a 25.87 ppm NO standard gas (balance N2). Fibrous mat coupons (surface area: 1.267 cm2) or 0.5 mg aliquots of N-diazeniumdiolate-functionalized PAMAM in methanol were added to 30 mL deoxygenated PBS to initiate NO release. Nitrogen was flowed through the solution at a flow rate of 80 mL/min to carry the liberated NO to the analyzer. Additional nitrogen flow was supplied to the flask to match the collection rate of the instrument at 200 mL/min. Nitric oxide analysis was terminated when NO levels decreased to below 1 pmol NO/cm2 for fibers (9.7 ppb) or 10 ppb NO/mg dendrimer.
Nitric oxide release was also measured in 50 mM HCl at 37 °C to calculate the mass of dendrimer encapsulated within the fiber (μg dendrimer/mg fiber). The total amount of dendrimer dopant incorporated into the fibers (t[dendrimer]) was determined by the following equation, where t[NO]dendrimer is the total NO payload released per milligram of dendrimer scaffold and t[NO]HCl is the total NO payload released per milligram of fiber in 50 mM HCl:
| Eq. 1 |
Characterization of Electrospun Polyurethane Fibers.
Fiber diameter and morphology were assessed using a FEI Helios 600 Nanolab Dual Beam System (Hillsboro, OR) without additional metal coating. Fiber diameters were determined using NIH ImageJ software (Bethesda, MD) and averaged for at least 300 measurements over three separate fiber samples.
Water absorption capabilities of the electrospun fibrous mats were assessed by comparing weights of the dry and hydrated samples. Dry electrospun fibers were weighed before soaking in Milli-Q water overnight at room temperature. The hydrated samples were removed from water, and the excess surface water was removed by dabbing with a Kimwipe before weighing the samples again. Water absorption was calculated by the following equation, where WH is the weight of the hydrated sample and WD is the initial weight of the dry fiber mat:
| Eq. 2 |
The porosity of the electrospun fibrous mats was determined using the liquid intrusion method.44-45 Fiber mats were weighed prior to immersion in 100% ethanol at room temperature overnight to allow diffusion of ethanol into the void volume. After this incubation, fibers were removed from ethanol, dabbed with a Kimwipe, and weighed again. Porosity was calculated by dividing the volume of intruded ethanol (determined by the change in mass and the density of ethanol, 0.789 g/mL) by the total volume after intrusion (volume of ethanol and fibers, determined by initial fiber mass and polyurethane density, 1.1 g/mL).
Leaching assays for determining dendrimer delivery were performed by doping RITC-tagged G4 PAMAM dendrimers into the fibers. Fluorescently-labeled G4 PAMAM dendrimers were synthesized as described previously.35-36, 46 Briefly, 100 mg G4 PAMAM was added to a vial containing one molar equivalent of RITC per mole dendrimer (3.8 mg) in 2 mL methanol. One equivalent of triethylamine (with respect to the molar amount of primary amines) was then added to the vial. The solution was stirred for 24 h in the dark, followed by solvent removal in vacuo. Dendrimers were dissolved in water, dialyzed against water (3 d), and then lyophilized. The above procedures for dendrimer modification and fiber mat fabrication were performed in the dark to yield RITC-tagged electrospun fibrous mats. Individual fiber mats were incubated in 500 μL PBS (pH 7.4, 37 °C) for 30 min, 2 h, 6 h, and 24 h. After incubation, 100 μL of each solution was transferred to a black 96-well plate in triplicate. The fluorescence intensity was measured using a BMG PolarStar Omega fluorescence plate reader (Ortenberg, Germany). Calibration standards were prepared at concentrations ranging from 0 – 200 μg/mL in triplicate.
Zone of Inhibition Assays.
Lyophilized P. aeruginosa, E. coli, S. aureus, and MRSA were reconstituted in tryptic soy broth (TSB) or Luria-Bertani (LB) broth (E. coli) and cultured overnight at 37 °C. A 0.5 mL aliquot of culture was grown in 50 mL TSB or LB broth to a concentration of 108 colony forming units per mL (cfu/mL), collected by centrifugation (2355×g), resuspended in 15% glycerol (v/v in PBS), and stored at −80 °C in 1 mL aliquots. For daily experiments, colonies of bacteria culture were inoculated in 2 mL TSB or LB broth overnight at 37 °C and recultured in fresh TSB or LB broth (50 mL) the next day.
The antibacterial action of the electrospun fibers was evaluated using a corrected zone of inhibition test.47 Briefly, P. aeruginosa, E. coli, S. aureus, and MRSA were cultured in TSB or LB broth (E. coli) to a concentration of 108 colony forming units per mL (cfu/mL), and 50 μL of the bacterial suspension in broth was spared over Mueller Hinton agar plates. Individual fiber mat samples (blank, control, and NO-releasing) were placed on the bacteria-containing agar. The plates were then incubated at 37 °C overnight. Zones of growth inhibition (ZOI) surrounding the fibers were measured (in mm) from the edge of the sample using calipers. Of note, no plate dehydration was observed. All zone of inhibition tests were repeated in triplicate.
Bacterial Log Reduction Assays.
An adapted time-kill kinetic log reduction assay was also employed to further assess the antibacterial action of the electrospun fibrous mats.47 Briefly, P. aeruginosa, E. coli, S. aureus, and MRSA were cultured in TSB or LB broth (E. coli) to a concentration of 108 cfu/mL. Individual fiber mat samples (blank, control, and NO-releasing) were added to 1 dram glass vials and sterilized under UV light for 2 h prior to the bacteria assays. Fibers were exposed to 200 μL 108 cfu/mL bacteria in broth for either 2 or 24 h at 37 °C with light agitation. Untreated controls (blanks) were included in each experiment to ensure the bacteria remained viable over the 2 or 24 h assays. After 2 h exposure, the bacteria solutions were vortexed and a 10 μL aliquot was removed for dilution and plating on tryptic soy agar (TSA) or LB agar (E. coli) plates. The bacteria and fiber solutions were then returned to 37 °C with light agitation for the remainder of the 24 h incubation. After exposure, the bacteria solutions were vortexed and then spiral-plated at 100-, 1000-, and 10,000-fold dilutions on TSA or LB agar plates using an Eddy Jet spiral plater (IUL; Farmingdale, NY). Bacterial viability was assessed by counting the number of colonies formed on the agar plate using a Flash & Go colony counter (IUL; Farmingdale, NY). Log reductions in bacterial viability were determined by the following equation, where Blank Viability is the viability of the unexposed bacterial blank solution and Exposed Viability is the resulting viability of bacteria solutions exposed to either blank, control, or NO-releasing electrospun fibers:
| Eq. 3 |
Fluorescence Microscopy for Detection of Intracellular NO and Cell Death.
P. aeruginosa was cultured in TSB to a concentration of 108 cfu/mL, collected by centrifugation (2355×g), and resuspended in PBS containing 30 μM PI and 20 μM DAF-2 DA at 108 cfu/mL. Nitric oxide-releasing fibers were exposed to 200 μL P. aeruginosa in PBS/PI/DAF-2 DA for 30 min. Every 5 min, a 10 μL aliquot of the bacteria solution was deposited on a glass slide with a coverslip for wide-field fluorescence imaging. An Olympus iX80 inverted microscope with an Olympus light source (Chroma) and Hamamatsu ORCA detector were used to image bacteria on slides. Fluorescent PI images (red) were obtained using a BP 542 – 582 nm excitation and BP 604 – 644 nm emission filters. Green DAF-2 images were obtained using BP 464 – 500 nm emission and BP 516 – 556 nm excitation filters. Images were acquired using Metamorph software and a 0.45 NA lens with a 20× objective.
In Vitro Cytotoxicity.
L929 mouse fibroblasts were grown in DMEM supplemented with 10 vol% FBS and 1 wt% PS and incubated in 5 vol% CO2 under humidified conditions at 37 °C. After reaching 80% confluency, cells were trypsinized and seeded onto tissue culture-treated polystyrene. To assess fiber cytotoxicity, L929 cells were seeded onto tissue culture-treated polystyrene 24-well plates at a density of 105 cells/mL, and incubated at 37 °C for 72 h. The supernatant was then aspirated and replaced with 1 mL of fresh growth medium and the electrospun fiber mats. Dimethyl sulfoxide (10%) was used as a positive control. After incubation for 2 or 24 h at 37 °C, the fiber mats were removed from the wells and the supernatant was aspirated. Next, 500 μL of a mixture of DMEM/MTS/PMS (105/20/1, v/v/v) was added to each well. After 1.5 h incubation at 37 °C, 100 μL of the colored solution was transferred to a 96-well plate in triplicate. The absorbance was quantified at 490 nm using a Thermoscientific Multiskan EX plate reader (Waltham, MA), with the DMEM/MTS/PMS mixture and untreated cells used as blanks and controls, respectively. Results were expressed as percentage of relative cell viability as follows:
| Eq. 4 |
To determine dendrimer toxicity, L929 cells were seeded onto tissue culture-treated polystyrene 96-well plates at a density of 2 × 104 cells/mL, and incubated at 37 °C for 72 h. The supernatant was then aspirated and replaced with 200 μL of fresh growth medium and 50 μL of varying concentrations of dendrimer in PBS. Dimethyl sulfoxide (10%) and 50 μL PBS were used as positive and negative controls, respectively. After 2 or 24 h incubation at 37 °C, the supernatant was aspirated and 120 μL of a mixture of DMEM/MTS/PMS (105/20/1, v/v/v) was added to each well. After 1.5 h incubation at 37 °C, the absorbance of the colored solutions was quantified at 490 nm with percent cell viability determined using Eq. 4. A killing curve was constructed for each dendrimer modification by plotting % cell viability versus dendrimer concentration. IC50 values, defined as the dendrimer concentration that corresponded to a 50% reduction in cell viability, were determined from each plot.
Results and Discussion
Generation 4 (G4) poly(amidoamine) (PAMAM) dendrimers were modified with octyl alkyl chains or octylQA moieties through a ring-opening reaction at the peripheral primary amines (Scheme 1).35-36 Covalent chemical modifications were confirmed using 1H NMR spectroscopy. On average, 40 – 50 functional groups were added to the G4 scaffold, resulting in ~70% functionalization of the terminal primary amines (Table S1).
Scheme 1.

Reaction of G4 PAMAM scaffold with either (A) octyl alkyl chain or (B) octylQA epoxides to yield G4 octyl and G4 octylQA dendrimers, respectively, followed by reaction with high pressures of NO to generate NO-releasing dendrimers.
The resulting secondary amines were converted to N-diazeniumdiolate NO donors under high pressures of NO. Nitric oxide storage was tuned by adjusting the ratio of THF:methanol, resulting in similar NO payloads of ~1 μmol/mg for each scaffold (Table S1). The NO-releasing octyl-modified dendrimers released NO for ~9 h with a half-life (t1/2) of ~25 min. G4 octylQA/NO dendrimers were characterized by slower NO-release kinetics, with an initial max flux about half that of the G4 octyl/NO dendrimers and extended NO-release (duration exceeding 16 h and t1/2 of 115 min). This longer NO release is the result of the permanent positive charge of the QA moiety stabilizing the N-diazeniumdiolate group. Using dendrimer scaffolds with distinct NO-release kinetics, we sought to evaluate the ability to tune NO-release characteristics via different dendrimer dopants.
Fabrication and Characterization of Electrospun Polyurethane Fibers.
Electrospun fibers were formed using Tecoplast, Tecoflex, and Tecophilic medical grade thermoplastic polyurethanes. Tecoplast (TP 470) was the most hydrophobic system evaluated due to its rigid, aromatic structure. Alternately, Tecoflex (SG 80A) and Tecophilic (HP 93A) are hydrophilic aliphatic polyurethanes. HP 93A is a polyether-based polyurethane with the greatest water uptake capability. The dendrimer-containing core solution was composed of either SG 80A or HP 93A aliphatic polyurethanes, while the outer (non-dendrimer-containing) sheath polymer was either SG 80A or TP 470. For clarity, the polymer compositions are denoted Sheath/Core-Dopant; for example, a G4 octyl-doped HP 93A core combined with a TP 470 sheath is referred to as TP 470/HP 93A-G4 octyl. Fibers were fabricated using a custom electrospinning apparatus with a co-axial setup using concentric needles to introduce core and sheath polymer solutions (Figure S1).
Blank (no dendrimer dopant), control (non-NO-releasing), and NO-releasing composite electrospun polyurethane fibers were prepared using three polymer compositions (i.e., SG 80A/HP 93A, TP 470/SG 80A, and TP 470/HP 93A) and two dendrimer modifications (i.e., G4 octyl and G4 octylQA). The resulting semi-porous electrospun fibrous mats exhibited smooth morphology with little to no bead formation (Figure 1). Altering the sheath and/or core polyurethane composition greatly influenced the macroscopic physical characteristics of the electrospun fiber mats (Figure 2). Fiber mats fabricated using SG 80A as either the core or sheath polyurethane were elastic and adhered to each other. Alternately, the TP 470/HP 93A electrospun fibers were easier to handle and maintained good shape over time. Fiber diameter distributions were more dependent on the identity of the sheath polyurethane versus the core (Figure 3, Figure S2). The SG 80A/HP 93A fiber diameters skewed larger, averaging around 600 nm compared to 400 – 450 nm for both TP 470 sheath formulations. These differences in fiber diameter were attributed to greater kinematic viscosity of the Tecoflex polyurethane solutions.42
Figure 1.

Scanning electron micrographs of blank, control, and NO-releasing G4 octyl-doped electrospun SG 80A/HP 93A, TP 470/SG 80A, and TP 470/HP 93A fibers.
Figure 2.

Image of electrospun fiber substrates (1.267 cm2). From left to right: SG 80A/HP 93A, TP 470/SG 80A, and TP 470/HP 93A.
Figure 3.

Histograms depicting fiber diameter distribution in nm for blank, control, and NO-releasing G4 octyl-doped electrospun SG 80A/HP 93A, TP 470/SG 80A, and TP 470/HP 93A fibers.
For potential wound dressings, a porous nanofiber structure would allow for adequate wound respiration and help maintain an appropriately moist environment.4 The porosity of dendrimer-doped electrospun fibrous mats was thus evaluated via liquid intrusion by measuring the ability of a non-wetting liquid to permeate the pores.44-45 The SG 80A/HP 93A fibers exhibited greater mat porosity compared to fibers containing a TP 470 sheath (Table 1), most likely the result of the larger fiber diameter distributions of the SG 80A/HP 93A fibers. Further, composite fibers containing TP 470 exhibited similar porosity regardless of core polyurethane. While adding dendrimers to the TP 470/SG 80A fibers increased the fiber mat porosity by ~10%, control and NO-releasing TP 470/HP 93A fibers exhibited porosities ~10% lower than blank fibers. Overall, the electrospun fiber characteristics were more influenced by the composition of the sheath polymer than either the core polyurethane or dendrimer dopant.
Table 1.
Characterization of electrospun polyurethane fibersa
| Substrate Mass (mg) |
Fiber Diameter (nm) |
Porosity (%) |
Water Absorption (%) |
|
|---|---|---|---|---|
| SG 80A/HP 93A | 1.41 ± 0.40 | 645 ± 292 | 71.4 ± 8.3 | 128 ± 41 |
| SG 80A/HP 93A-G4 octyl | 1.58 ± 0.34 | 585 ± 228 | 78.0 ± 4.0 | 145 ± 26 |
| SG 80A/HP 93A-G4 octyl/NO | 1.63 ± 0.60 | 590 ± 217 | 70.1 ± 7.4 | 120 ± 42 |
| TP 470/SG 80A | 1.41 ± 0.25 | 394 ± 156 | 29.4 ± 5.0 | 306 ± 40 |
| TP 470/SG 80A-G4 octyl | 1.74 ± 0.52 | 412 ± 127 | 40.3 ± 3.8 | 397 ± 44 |
| TP 470/SG 80A-G4 octyl/NO | 1.36 ± 0.35 | 393 ± 157 | 42.6 ± 4.0 | 210 ± 48 |
| TP 470/HP 93A | 1.77 ± 0.43 | 443 ± 152 | 51.3 ± 4.0 | 156 ± 70 |
| TP 470/HP 93A-G4 octyl | 1.40 ± 0.28 | 409 ± 176 | 37.1 ± 5.0 | 333 ± 107 |
| TP 470/HP 93A-G4 octyl/NO | 1.99 ± 0.78 | 433 ± 165 | 37.4 ± 8.3 | 379 ± 80 |
| TP 470/HP 93A-G4 octylQA | 1.68 ± 0.36 | 440 ± 182 | 45.9 ± 5.3 | 435 ± 68 |
| TP 470/HP 93A-G4 octylQA/NO | 1.82 ± 0.55 | 418 ± 155 | 39.0 ± 10.5 | 335 ± 90 |
For all measurements, n ≥ 3 pooled experiments.
Water absorption by the electrospun fibers is an attractive dressing characteristic for treating highly exuding wounds, preventing wound stagnation.4, 24 Although all of the fiber samples demonstrated sufficient water uptake capabilities (>100%), SG 80A/HP 93A fibers absorbed roughly half the water as fibers prepared using a TP 470 sheath (Table 1). This result was initially surprising due to the greater hydrophobicity of the TP 470 polyurethane versus SG 80A. However, the water absorption capabilities of the electrospun polyurethane fibers correlated directly with the porosity of the fibrous mat. Electrospun SG 80A/HP 93A fiber mats were characterized as having the largest porosity (~70%) and modest water absorption (~130%), while fibers prepared using a TP 470 sheath had lower porosities (30 – 50%) and absorbed more water (>300%). Control and NO-releasing TP 470/HP 93A fibers exhibited ~10% lower porosity than the blank fibers with enhanced water absorption (150 to ~350%). The increased water uptake with lower fiber mat porosity is attributed to the greater surface area available per volume allowing for more efficient water absorption into the individual fibers.
Total NO and dendrimer payloads were determined by measuring the NO release under acidic conditions (50 mM HCl). Despite doping the core polyurethane solution with the same dendrimer concentration (25 mg/mL), total dendrimer incorporation varied dramatically between polyurethane compositions (Table 2). SG 80A/HP 93A-G4 octyl/NO fibers demonstrated the largest dendrimer storage (~110 μg/mg), followed by the TP 470/SG 80A-G4 octyl/NO (~80 μg/mg) and the TP 470/HP 93A-G4 octyl/NO (~60 μg/mg) fibers. Additionally, the total dendrimer incorporation into the fibers varied between dendrimer modifications, with the TP 470/HP 93A-G4 octylQA/NO fibers exhibiting the second largest storage (~95 μg/mg). The identity of the core or sheath polyurethane had no clear effect on the amount of dendrimer incorporated into the fibers, suggesting the dendrimer loading cannot be projected prior to fiber fabrication due to potential dendrimer loss during the electrospinning process. However, the total dendrimer incorporation could be tuned by varying the concentration of dendrimer in the initial core polyurethane solution. Doping G4 octyl/NO dendrimers into SG 80A/HP 93A fibers at initial concentrations of 5, 15, and 25 mg/mL led to greater total dendrimer incorporation with increasing initial dendrimer concentration. Due to the low dendrimer loading within the 5 mg/mL SG 80A/HP 93A-G4 octyl/NO fibers (~5 μg/mg), only the 15 and 25 mg/mL compositions were evaluated further.
Table 2.
Total nitric oxide storage and dendrimer encapsulation by fiber massa
| [Dendrimer]b (mg/mL) |
t[NO]c (nmol/mg) |
t[NO]HCld (nmol/mg) |
t[dendrimer]e (μg/mg) |
|
|---|---|---|---|---|
| SG 80A/HP 93A-G4 octyl/NO | 5 | 1.8 ± 0.8 | 3.8 ± 0.4 | 5 ± 1 |
| SG 80A/HP 93A-G4 octyl/NO | 15 | 26.0 ± 3.6 | 35.0 ± 5.2 | 42 ± 6 |
| SG 80A/HP 93A-G4 octyl/NO | 25 | 72.3 ± 9.0 | 93.8 ± 7.4 | 111 ± 10 |
| TP 470/SG 80A-G4 octyl/NO | 25 | 42.4 ± 4.4 | 71.1 ± 8.3 | 81 ± 6 |
| TP 470/HP 93A-G4 octyl/NO | 25 | 27.3 ± 4.1 | 51.0 ± 12.7 | 63 ± 13 |
| TP 470/HP 93A-G4 octylQA/NO | 25 | 44.7 ± 11.0 | 88.8 ± 18.9 | 94 ± 23 |
For all measurements, n ≥ 3 pooled experiments.
Dendrimer concentration in the original polymer solution.
Total NO payload released per mg of fiber in PBS (pH 7.4).
Total NO payload released per mg of fiber in 50 mM HCl.
Total amount of dendrimer encapsulated per mg of fiber (determined by total NO release in 50 mM HCl).
To study the dendrimer delivery from composite polyurethane fibers, control and NO-releasing dendrimers were modified with a fluorescent RITC tag prior to electrospinning. The resulting RITC-containing electrospun fiber mats (Figure S3) were soaked in PBS to quantify the amount of dendrimer released over 24 h. Impact of the fluorescent label on the scaffold was minimized by limiting the amount of RITC modification to one molecule per dendrimer scaffold (~3% of G4 PAMAM mass). For all of the composite polyurethane fibers, most of the dendrimer delivery occurred over the first 2 h, with only a slight increase in dendrimer release over the next 22 h (Figure 4). Of note, an insignificant increase in dendrimer delivery was observed after 7 d, indicating the majority of dendrimer release is achieved during the first 24 h (data not shown).
Figure 4.

(A) Delivery of control (solid line) and NO-releasing (dashed line) G4 octyl dendrimers from electrospun SG 80A/HP 93A fibers at 15 (blue) and 25 (purple) mg/mL initial dendrimer concentration. (B) Delivery of control (solid line) and NO-releasing (dashed line) G4 octyl dendrimers from electrospun SG 80A/HP 93A (blue), TP 470/SG 80A (red), TP 470/HP 93A (black) fibers. (C) Delivery of control (solid line) and NO-releasing (dashed line) G4 octyl (square) and G4 octylQA (triangle) dendrimers from electrospun TP 470/HP 93A fibers. For all measurements, n ≥ 3 pooled experiments with error bars representing standard deviation of the mean.
The polyurethane composition dramatically influenced the rate of control (i.e., non-NO-releasing) dendrimer delivery from the composite electrospun fibers. Fibers containing a SG 80A sheath delivered more dendrimer than fibers comprised of TP 470 as the sheath polyurethane (Figure 4A-B). Both the 15 and 25 mg/mL SG 80A/HP 93A-G4 octyl fibers exhibited similar dendrimer release (~30 μg/mL), corresponding to 65 and 30% of the total incorporated dendrimer, respectively. TP 470/HP 93A and TP 470/SG 80A fibers released dendrimer doses corresponding to ~10 and ~20% of the total incorporated dendrimer, respectively. Clearly, the identify of the core and sheath polyurethanes influenced the rate of dendrimer delivery, with the hydrophobic TP 470 polyurethane acting as a more effective barrier layer compared to the aliphatic SG 80A. The enhanced release of G4 octyl dendrimers from TP 470/SG 80A fibers compared to TP 470/HP 93A fibers is attributed to greater charge stabilization of the dendrimers within the more hydrophilic, polyether-based HP 93A polyurethane.
The release of the NO-releasing dendrimers was dependent on the amount of dendrimer incorporated and sheath polyurethane, with the fibers containing an SG 80A polyurethane sheath resulting in greater overall dendrimer release. Indeed, the 25 mg/mL SG 80A/HP 93A-G4 octyl/NO fibers delivered the largest total dendrimer dose (~60 μg/mg), representing ~50% of the total dendrimer storage. The remaining G4 octyl/NO-doped fibers exhibited similar dendrimer release (~40 μg/mg) regardless of sheath or core polyurethane composition. In contrast to the TP 470 sheath fibers that released roughly 55% of the total incorporated dendrimer, ~100% of the dendrimer was delivered when using 15 mg/mL SG 80A/HP 93A-G4 octyl/NO fibers. The greater percent dendrimer delivery for the SG 80A sheath fibers again demonstrates that the aliphatic polyurethane provides a less effective barrier than hydrophobic TP 470.
Larger doses of NO-releasing dendrimer were delivered relative to the control dendrimers for all of the polyurethane compositions. The N-diazeniumdiolate NO donor is zwitterionic and likely destabilizes the dendrimer scaffold within the polyurethane fibers due to the inherent electrostatic charge.48 In a similar manner, both control and NO-releasing TP 470/HP 93A-G4 octylQA fibers delivered greater dendrimer doses relative to the G4 octyl system (Figure 4C), which lacks a cationic QA moiety. Dendrimer release rates were generally dependent on the resulting charge of the dendrimer scaffold after modification (e.g., QA moiety or N-diazeniumdiolate), with increased charge resulting in decreased dendrimer stability within the polyurethane fibers and faster delivery.
Similar to dendrimer delivery, the NO-release kinetics of the composite electrospun fibers were dependent on the polyurethane identity and dendrimer modification. To evaluate NO-release kinetics from composite electrospun fibers under physiological conditions (pH 7.4, 37 °C), fiber mats were cut into circular coupons with a standard surface area (1.27 cm2). Regardless of polyurethane composition, all of the NO-releasing fibers demonstrated an initial maximum burst of NO followed by a steady decline in release (Figure S4). The initial NO burst for both of the SG 80A/HP 93A-G4 octyl/NO fiber compositions was greater than that for either of the fibers containing a TP 470 sheath (Table 3). This difference in initial NO flux was attributed to the aliphatic SG 80A polyurethane being a less effective barrier layer to water and the dendrimers, leading to a faster release of dendrimers to solution and greater water access for N-diazeniumdiolate breakdown. While the 15 mg/mL SG 80A/HP 93A-G4 octyl/NO fibers exhibited similar NO totals as the G4 octyl/NO-doped fibers containing a TP 470 sheath (~45 nmol/cm2), the NO release was more rapid. Indeed, the SG 80A/HP 93A-G4 octyl/NO fibers had a shorter NO release duration (~2 h) than either of the fibers fabricated using a TP 470 sheath (duration ~3 h), indicating the sheath polyurethane plays a greater role on NO release than the core polyurethane composition.
Table 3.
Nitric oxide-release properties for NO-releasing electrospun fibers in PBS (pH 7.4, 37 °C)a
| [Dendrimer]b (mg/mL) |
[NO]maxc (pmol/cm2) |
tmaxd (min) |
t[NO]e (nmol/cm2) |
t[NO]2hf (nmol/cm2) |
t1/2g (min) |
tdh (h) |
|
|---|---|---|---|---|---|---|---|
| SG 80A/HP 93A-G4 octyl/NO | 15 | 21.8 ± 5.7 | 6.1 ± 2.9 | 44.6 ± 11.1 | 43.7 ± 10.2 | 22.4 ± 3.3 | 2.2 ± 0.3 |
| SG 80A/HP 93A-G4 octyl/NO | 25 | 27.6 ± 11.7 | 6.2 ± 2.2 | 87.2 ± 19.1 | 71.2 ± 17.3 | 35.7 ± 3.9 | 4.2 ± 0.8 |
| TP 470/SG 80A-G4 octyl/NO | 25 | 14.0 ± 2.1 | 3.3 ± 0.6 | 46.7 ± 9.5 | 38.3 ± 7.2 | 47.8 ± 6.9 | 3.6 ± 0.5 |
| TP 470/HP 93A-G4 octyl/NO | 25 | 17.2 ± 6.2 | 4.0 ± 1.6 | 44.3 ± 10.1 | 39.9 ± 9.1 | 39.0 ± 4.5 | 2.9 ± 0.4 |
| TP 470/HP 93A-G4 octylQA/NO | 25 | 10.6 ± 1.5 | 4.3 ± 0.6 | 66.5 ± 16.5 | 36.2 ± 5.6 | 102.4 ± 24.6 | 6.8 ± 1.5 |
For all measurements, n ≥ 3 pooled experiments.
Dendrimer concentration in the original polymer solution.
Maximum flux of NO release.
Time required to reach maximum flux.
Total NO payload released per surface area.
NO payload released after 2 h.
NO release half-life.
Duration of NO release.
Between the two SG 80A/HP 93A-G4 octyl/NO formulations, NO-release totals and kinetics were dependent on total dendrimer incorporation. The 25 mg/mL composition exhibited larger total NO storage (87 versus 45 nmol/cm2) and longer NO release duration (4.2 versus 2.2 h) than the 15 mg/mL fibers. Similarly, the TP 470/HP 93A-G4 octylQA/NO fibers exhibited a prolonged NO-release duration (~7 h) relative to the G4 octyl/NO-containing fibers. These results were expected due to the extended NO-release properties characteristic of the G4 octylQA/NO scaffold (NO-release half-life ~115 min), indicating that fiber NO-release kinetics are highly dependent on the dendrimer modification.
Zone of Inhibition.
For antibacterial evaluation, electrospun fibers doped with 25 mg/mL control and NO-releasing dendrimers were cut into circular coupons with a standard surface area (1.27 cm2). P. aeruginosa, E. coli, and S. aureus (including methicillin-resistant strains) are among the most commonly isolated species in chronic wounds.2, 49-50 These pathogens were selected to evaluate the potential antibacterial action of control and NO-releasing electrospun fibers. The use of two Gram-negative (P. aeruginosa, E. coli) and two Gram-positive (S. aureus, MRSA) bacterial strains allowed for an initial evaluation of the effect of Gram designation on antibacterial action.
As expected, electrospun composite polyurethane fibers prepared without dendrimers (i.e., blanks) were void of antibacterial action. Nearly every control and NO-releasing polyurethane composition exhibited low antibacterial activity (ZOI <0.5 mm) against each of the Gram-negative strains, with only the TP 470/SG 80A-G4 octyl/NO fibers displaying an inhibition zone of 1.7 mm (Table 4). Moderate bactericidal action (ZOI >0.5mm) was observed against the Gram-positive bacteria. Control and NO-releasing SG 80A/HP 93A-G4 octyl fibers resulted in inhibition zones of ~1 mm, while the TP 470/SG 80A-G4 octyl/NO fibers exhibited inhibition zones of 2.0 and 1.6 mm against S. aureus and MRSA, respectively. The similar antibacterial action for the control and NO-releasing SG 80A/HP 93A-G4 octyl fibers is attributed to the large dendrimer doses delivered by these fibers. While NO-releasing TP 470/HP 93A fibers (i.e., G4 octyl and G4 octylQA) resulted in low to moderate antibacterial action against both Gram-positive strains (ZOI 0.4 – 0.7 mm), the control TP 470/HP 93A fibers exhibited little to no bactericidal activity against either Gram-negative or Gram-positive bacteria (ZOI ≤0.2 mm). Despite small inhibition zones, bacterial growth was not observed beneath any of the control or NO-releasing fiber samples (Figure S5). The larger zones of inhibition observed for these fibers (ZOI >0.6 mm) agree with results published by Vogt et al. who demonstrated the antibacterial efficacy of NO-releasing nanofibers against S. aureus (ZOI 0.75 – 1.2 mm).51 However, as NO diffuses in all directions, not just along the agar surface,51 we evaluated the reduction of bacterial growth in solution in conjunction with the zone of inhibition assay to fully assess the antibacterial action of NO-releasing electrospun polyurethane fiber mats.
Table 4.
Average zone of inhibition against planktonic bacteriaa
|
P. aeruginosa ZOI (mm) |
E. coli ZOI (mm) |
S. aureus ZOI (mm) |
MRSA ZOI (mm) |
|
|---|---|---|---|---|
| SG 80A/HP 93A-G4 octyl | 0.4 ± 0.3 | 0.5 ± 0.1 | 1.0 ± 0.1 | 1.2 ± 0.3 |
| SG 80A/HP 93A-G4 octyl/NO | 0.5 ± 0.3 | 0.4 ± 0.1 | 0.9 ± 0.3 | 0.6 ± 0.1 |
| TP 470/SG 80A-G4 octyl | 0.4 ± 0.5 | 0.4 ± 0.3 | 0.2 ± 0.3 | 0.0 ± 0.0 |
| TP 470/SG 80A-G4 octyl/NO | 1.7 ± 0.3 | 1.8 ± 0.3 | 2.0 ± 0.1 | 1.6 ± 0.2 |
| TP 470/HP 93A-G4 octyl | 0.1 ± 0.3 | 0.1 ± 0.2 | 0.2 ± 0.3 | 0.2 ± 0.3 |
| TP 470/HP 93A-G4 octyl/NO | 0.4 ± 0.5 | 0.1 ± 0.2 | 0.7 ± 0.2 | 0.6 ± 0.1 |
| TP 470/HP 93A-G4 octylQA | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| TP 470/HP 93A-G4 octylQA/NO | 0.3 ± 0.5 | 0.6 ± 0.4 | 0.6 ± 0.2 | 0.4 ± 0.2 |
For all measurements, n ≥ 3 pooled experiments.
Bacterial Log Reduction.
For log reduction tests, a compound is considered bactericidal if it reduces bacterial viability by at least three orders of magnitude.47, 52 Using this assay, we evaluated the ability of blank, control, and NO-releasing TP 470/HP 93A fibers to reduce the viability of P. aeruginosa, E. coli, S. aureus, or MRSA planktonic cultures as a function of NO-release kinetics and dendrimer modification. These studies were performed under growth conditions (i.e., in nutrient broth) over 2 and 24 h to determine the short- and long-term bactericidal action of control and NO-releasing polyurethane fibers. Fiber mats displayed high antibacterial activity if they exhibited at least a 3-log reduction in bacterial viability, moderate activity for a 1- to 3-log reduction, and low activity for <1-log reduction.47
As expected, blank TP 470/HP 93A fibers did not eradicate bacteria (Figure S6, Figure S7), while the antibacterial activity of control (i.e., non-NO-releasing) fibers was dependent on the dendrimer dopant. Similar to the zone of inhibition studies, TP 470/HP 93A-G4 octylQA fibers demonstrated little to no antibacterial action against the pathogens at either timepoint (Table 5). In contrast, TP 470/HP 93A-G4 octyl fibers were moderately antibacterial against the majority of bacterial strains tested (≥1-log reduction), only exhibiting low antibacterial action against S. aureus (<1-log reduction). The increased bactericidal action of the G4 octyl-doped fibers over their G4 octylQA counterparts was surprising due to the lower corresponding dendrimer dose. Nevertheless, these results corroborate our previous observations that alkyl chains are more potent biocides than QA moieties.35-36 The G4 octyl-doped fibers were generally more effective at shorter timescales, with an average 2-log bacterial reduction after 2 h and only a 1-log reduction after 24 h. This behavior was expected, however, as the majority of the low dendrimer dose is delivered over the first 2 h.
Table 5.
Average log reduction against planktonic bacteriaa
| P. aeruginosa | E. coli | S. aureus | MRSA | |||||
|---|---|---|---|---|---|---|---|---|
| 2 h | 24 h | 2 h | 24 h | 2 h | 24 h | 2 h | 24 h | |
| TP 470/HP 93A | 0.4 ± 0.8 | 0.6 ± 0.7 | 0.0 ± 0.2 | 0.2 ± 0.2 | 0.0 ± 0.1 | −0.2 ± 0.2 | 0.0 ± 0.1 | 0.7 ± 0.8 |
| G4 octyl | 2.0 ± 0.6 | 1.2 ± 1.4 | 2.5 ± 0.5 | 1.1 ± 1.0 | 0.7 ± 1.0 | 0.4 ± 0.9 | 1.5 ± 0.8 | 2.4 ± 1.5 |
| G4 octyl/NO | 4.0 ± 0.8 | 3.7 ± 0.4 | 3.0 ± 0.6 | 4.2 ± 0.8 | 2.7 ± 0.8 | 4.3 ± 1.4 | 2.6 ± 0.6 | 4.4 ± 1.0 |
| G4 octylQA | 0.5 ± 0.9 | 0.1 ± 0.5 | 0.0 ± 0.3 | 0.2 ± 0.3 | 0.2 ± 0.4 | −0.2 ± 0.1 | 0.5 ± 0.5 | 0.4 ± 0.8 |
| G4 octylQA/NO | 4.1 ± 0.9 | 3.5 ± 1.3 | 2.3 ± 0.5 | 2.6 ± 0.8 | 3.4 ± 0.7 | 4.7 ± 1.2 | 3.2 ± 1.0 | 4.7 ± 0.9 |
For all measurements, n ≥ 3 pooled experiments.
Nitric oxide-releasing electrospun fibers proved to have greater antibacterial action than either blank or control fibers. Moderate to high antibacterial activity was consistently observed against the bacterial strains evaluated. The TP 470/HP 93A-G4 octyl/NO fibers demonstrated greater bactericidal action against Gram-negative pathogens while TP 470/HP 93A-G4 octylQA/NO fibers were more effective against the Gram-positive strains, although the differences in efficacy were modest (~1-log difference between the two dendrimer modifications). After 2 h exposure, the NO-releasing fibers exhibited an average 3-log reduction in bacterial viability, an unequivocal improvement over previously reported NO-releasing nanofibers.53 In contrast to control fibers, the NO-releasing TP 470/HP 93A fibers were more effective at reducing bacterial viability at extended time periods (24 h), suggesting the greater doses provided by the NO-releasing polyurethane fibers allows for continued bactericidal action of the dendrimer scaffold in solution. At least one of the NO-releasing fiber formulations displayed high antibacterial activity against each bacterial strain, with an average 4-log reduction in bacterial viability observed at 24 h. While TP 470/HP 93A-G4 octylQA/NO fibers deliver almost twice as much dendrimer per milligram of fiber (~35 and 80 μg/mg for G4 octyl/NO and G4 octylQA/NO fibers, respectively), the TP 470/HP 93A-G4 octyl/NO fibers exhibited greater broad-spectrum bactericidal activity with less antibacterial dendrimer delivery. The broad-spectrum antibacterial action demonstrated by the NO-releasing electrospun fibers is noteworthy, particularly in comparison to certain commercially available silver-releasing dressings that do not exhibit adequate bactericidal action against Gram-positive bacteria.47
To elucidate the mechanism of antibacterial action for the NO-releasing electrospun fibers, DAF-2 DA and PI fluorescent probes were used to visualize intracellular NO and cell membrane damage, respectively. After 5 min exposure to TP 470/HP 93A-G4 octyl/NO fibers, planktonic P. aeruginosa bacteria showed evidence of intracellular NO and substantial membrane damage (Figure 5). In previous studies, observation of NO-mediated cell death with fluorescence microscopy yielded a bright green fluorescent signal from DAF-2 (indicative of intracellular NO buildup) prior to the detection of red fluorescence (PI) signifying membrane disruption and subsequent cell death.28, 36 The lack of overlap between the bacterial cells with compromised membranes and the localization of intracellular NO herein indicates the biocidal actions of G4 octyl-modified dendrimers and NO are exerted independently from one another. The diminished intracellular NO levels after 15 min and undetectable DAF-2 fluorescence at later periods (i.e., 30 min, 2 h) suggests that any antibacterial activity derived from NO release is only observed at finite periods. As such, the majority of bactericidal action exhibited by the NO-releasing fibers is attributed to dendrimer-induced bacterial killing, corroborating our observation that greater dendrimer delivery from NO-releasing fibers increases bactericidal action compared to control fibers.
Figure 5.

Fluorescence microscopy images of P. aeruginosa exposed to TP 470/HP 93A-G4 octyl/NO fibers for 5 and 15 minutes. DAF-2 green fluorescence depicts intracellular NO, while PI red fluorescence indicates compromised membranes. Threshold inverted for clarity.
In Vitro Cytotoxicity.
Toxicity of the electrospun fibers was assessed using L929 mouse fibroblast cells at exposure periods of 2 and 24 h. For comparison, the inhibitory concentrations at 50% cell viability (IC50) against L929 mouse fibroblasts were evaluated for each of the control and NO-releasing dendrimers at both time points. As expected, both control and NO-releasing dendrimers were significantly more toxic at extended exposure periods, with IC50 values ranging from 200 – 360 and 40 – 100 μg/mL after 2 and 24 h, respectively (Table S2). Due to the observed toxicity of octyl- and octylQA-modified dendrimers alone at relatively low concentrations, we hypothesized that the NO-releasing polyurethane fibers would exhibit substantial toxicity to L929 cells after 24 h exposure. Yet, all of the fiber formulations were relatively non-toxic (≥ 80% cell viability) regardless of time (Figure 6, Figure S8). The lowest cell viabilities were observed for the blank and control SG 80A/HP 93A fibers after 24 h (86 and 82% cell viability, respectively), while the SG 80A/HP 93A-G4 octyl/NO fibers exhibited no reduction in viability. The reduction in cell viability observed for the blank fibers may be the result of cell adhesion to the fiber mat and subsequent removal from the culture plate. All of NO-releasing fibers demonstrated at least ~95% cell viability or greater, suggesting that releasing antibacterial dendrimers from electrospun fibers over time reduces the overall stress to mammalian cells compared to direct exposure.
Figure 6.

Viability (%) of L929 mouse fibroblast cells following (A) 2 h or (B) 24 h exposure to blank, control, and NO-releasing electrospun SG 80A/HP 93A (blue), TP 470/SG 80A (red), and TP 470/HP 93A (gray) fibers. For all measurements, n ≥ 3 pooled experiments with error bars representing standard deviation of the mean.
Conclusions
The utility of dendrimer- and NO-releasing electrospun polyurethane fibers as potential antibacterial wound dressings was evaluated as a function of dendrimer and polyurethane compositions. Dendrimer-doped electrospun fibers exhibited adequate porosity and water absorption properties that should enable dynamic gas and fluid exchange in a chronic wound setting. The release of antibacterial dendrimers and NO were tunable by adjusting the sheath polyurethane hydrophobicity, dendrimer modification, and amount of dendrimer incorporated into the fibers. Fibers fabricated using a less hydrophobic sheath polyurethane were characterized by faster dendrimer and NO release, while the charged dendrimer modifications (e.g., N-diazeniumdiolate or QA moieties) resulting in greater dendrimer delivery. Similarly, NO-release kinetics were dependent on the dendrimer modification, reflecting the relative kinetics of the NO-releasing dendrimers in solution. The NO-releasing TP 470/HP 93A fibers demonstrated moderate to high antibacterial activity against several Gram-negative and Gram-positive species at short and long timescales, averaging a 4-log reduction in bacterial viability after 24 h exposure. These materials represent highly promising candidates for wound healing applications due to their established antibacterial activity and negligible toxicity to mammalian cells.
Supplementary Material
Acknowledgements
Financial support was provided by NIH DE025207. BVW gratefully acknowledges a graduate research fellowship from Eastman Chemical Company (Kingsport, TN). The authors thank Wallace Ambrose for assistance with fluorescence microscopy (Chapel Hill Analytical and Nanofabrication Laboratory).
Footnotes
Supporting Information
Characterization, NO-release properties, and cytotoxicity of the dendrimer scaffolds. Scanning electron micrographs, fiber diameter histograms, and cytotoxicity of TP 470/HP 93A-G4 octylQA fibers. Zone of inhibition images and viability of planktonic bacteria exposed to electrospun fibers.
Disclosure
The corresponding author declares competing financial interest. Mark Schoenfisch is a co-founder and a member of the board of directors, and maintains a financial interest in Novan Therapeutics, Inc. Novan Therapeutics is commercializing macromolecular nitric oxide storage and release vehicles for dermatological clinical indications.
REFERENCES
- 1.James GA; Swogger E; Wolcott R; Secor P; Sestrich J; Costerton JW; Stewart PS, Biofilms in chronic wounds. Wound Repair Regen 2008, 16, 37–44, DOI: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 2.Bjarnsholt T; Kirketerp-Møller K; Jensen PØ; Madsen KG; Phipps R; Krogfelt K; Høiby N; Givskov M, Why chronic wounds will not heal: a novel hypothesis. Wound Repair Regen 2008, 16, 2–10, DOI: 10.1111/j.1524-475X.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 3.Blakytny R; Jude E, The molecular biology of chronic wounds and delayed healing in diabetes. Diabetic Medicine 2006, 23, 594–608, DOI: 10.1111/j.1464-5491.2006.01773.x. [DOI] [PubMed] [Google Scholar]
- 4.Zahedi P; Rezaeian I; Ranaei-Siadat SO; Jafari SH; Supaphol P, A review on wound dressings with an emphasis on electrospun nanofibrous polymeric bandages. Polymers for Advanced Technologies 2010, 21, 77–95, DOI: 10.1002/pat.1625. [DOI] [Google Scholar]
- 5.Khil MS; Cha DI; Kim HY; Kim IS; Bhattarai N, Electrospun nanofibrous polyurethane membrane as wound dressing. Journal of Biomedical Materials Research Part B: Applied Biomaterials 2003, 67, 675–679, DOI: 10.1002/jbm.b.10058. [DOI] [PubMed] [Google Scholar]
- 6.Sabitha M; Rajiv S, Preparation and characterization of ampicillin-incorporated electrospun polyurethane scaffolds for wound healing and infection control. Polymer Engineering & Science 2015, 55, 541–548, DOI: 10.1002/pen.23917. [DOI] [Google Scholar]
- 7.Yao C; Li X; Neoh K; Shi Z; Kang E, Surface modification and antibacterial activity of electrospun polyurethane fibrous membranes with quaternary ammonium moieties. Journal of Membrane Science 2008, 320, 259–267, DOI: 10.1016/j.memsci.2008.04.012. [DOI] [Google Scholar]
- 8.Yücedag F; Atalay-Oral C; Erkal S; Sirkecioglu A; Karasartova D; Sahin F; Tantekin-Ersolmaz SB; Güner FS, Antibacterial oil-based polyurethane films for wound dressing applications. J. Appl. Polym. Sci. 2010, 115, 1347–1357, DOI: 10.1002/app.30788. [DOI] [Google Scholar]
- 9.Andersen KE; Franken CPM; Gad P; Larsen AM; Larsen JR; Franciscus PA; Van Neer A; Vuerstaek J; Wuite J; Martino HA, A randomized, controlled study to compare the effectiveness of two foam dressings in the management of lower leg ulcers. Ostomy Wound Management 2002, 48, 34–41, DOI: 35400010829860.0020. [Google Scholar]
- 10.Payne WG; Posnett J; Alvarez O; Brown-Etris M; Jameson G; Wolcott R; Dharma H; Hartwell S; Ochs D, A prospective, randomized clinical trial to assess the cost-effectiveness of a modern foam dressing versus a traditional saline gauze dressing in the treatment of stage II pressure ulcers. Ostomy Wound Management 2009, 55, 50–55. [PubMed] [Google Scholar]
- 11.Jørgensen B; Price P; Andersen KE; Gottrup F; Bech-Thomsen N; Scanlon E; Kirsner R; Rheinen H; Roed-Petersen J; Romanelli M, The silver-releasing foam dressing, Contreet Foam, promotes faster healing of critically colonised venous leg ulcers: a randomised, controlled trial. International Wound Journal 2005, 2, 64–73, DOI: 10.1111/j.1742-4801.2005.00084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lo SF; Chang CJ; Hu WY; Hayter M; Chang YT, The effectiveness of silver-releasing dressings in the management of non-healing chronic wounds: a meta-analysis. Journal of Clinical Nursing 2009, 18, 716–728, DOI: 10.1111/j.1365-2702.2008.02534.x. [DOI] [PubMed] [Google Scholar]
- 13.Lo SF; Hayter M; Chang CJ; Hu WY; Lee LL, A systematic review of silver-releasing dressings in the management of infected chronic wounds. Journal of Clinical Nursing 2008, 17, 1973–1985, DOI: 10.1111/j.1365-2702.2007.02264.x. [DOI] [PubMed] [Google Scholar]
- 14.Staso MA; Raschbaum M; Slater H; Goldfarb IW, Experience with Omiderm-A new burn dressing. Journal of Burn Care & Research 1991, 12, 209. [DOI] [PubMed] [Google Scholar]
- 15.Yari A; Yeganeh H; Bakhshi H, Synthesis and evaluation of novel absorptive and antibacterial polyurethane membranes as wound dressing. Journal of Materials Science: Materials in Medicine 2012, 23, 2187–2202, DOI: 10.1007/s10856-012-4683-6. [DOI] [PubMed] [Google Scholar]
- 16.Bhardwaj N; Kundu SC, Electrospinning: a fascinating fiber fabrication technique. Biotechnol. Adv. 2010, 28, 325–347, DOI: 10.1016/j.biotechadv.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 17.He CL; Huang ZM; Han XJ; Liu L; Zhang HS; Chen LS, Coaxial electrospun poly (L-lactic acid) ultrafine fibers for sustained drug delivery. Journal of Macromolecular Science, Part B 2006, 45, 515–524, DOI: 10.1080/00222340600769832. [DOI] [Google Scholar]
- 18.Jannesari M; Varshosaz J; Morshed M; Zamani M, Composite poly (vinyl alcohol)/poly (vinyl acetate) electrospun nanofibrous mats as a novel wound dressing matrix for controlled release of drugs. International Journal of Nanomedicine 2011, 6, 993–1003, DOI: 10.2147/IJN.S17595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Unnithan AR; Barakat NA; Pichiah PT; Gnanasekaran G; Nirmala R; Cha Y-S; Jung C-H; El-Newehy M; Kim HY, Wound-dressing materials with antibacterial activity from electrospun polyurethane–dextran nanofiber mats containing ciprofloxacin HCl. Carbohydrate Polymers 2012, 90, 1786–1793, DOI: 10.1016/j.carbpol.2012.07.071. [DOI] [PubMed] [Google Scholar]
- 20.Choi JS; Choi SH; Yoo HS, Coaxial electrospun nanofibers for treatment of diabetic ulcers with binary release of multiple growth factors. Journal of Materials Chemistry 2011, 21, 5258–5267, DOI: 10.1039/C0JM03706K. [DOI] [Google Scholar]
- 21.Huang ZM; He CL; Yang A; Zhang Y; Han XJ; Yin J; Wu Q, Encapsulating drugs in biodegradable ultrafine fibers through co-axial electrospinning. Journal of Biomedical Materials Research Part A 2006, 77, 169–179, DOI: 10.1002/jbm.a.30564. [DOI] [PubMed] [Google Scholar]
- 22.Thakur R; Florek C; Kohn J; Michniak B, Electrospun nanofibrous polymeric scaffold with targeted drug release profiles for potential application as wound dressing. International Journal of Pharmaceutics 2008, 364, 87–93, DOI: 10.1016/j.ijpharm.2008.07.033. [DOI] [PubMed] [Google Scholar]
- 23.Dongargaonkar AA; Bowlin GL; Yang H, Electrospun blends of gelatin and gelatin–dendrimer conjugates as a wound-dressing and drug-delivery platform. Biomacromolecules 2013, 14, 4038–4045, DOI: 10.1021/bm401143p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lalani R; Liu L, Electrospun zwitterionic poly (sulfobetaine methacrylate) for nonadherent, superabsorbent, and antimicrobial wound dressing applications. Biomacromolecules 2012, 13, 1853–1863, DOI: 10.1021/bm300345e. [DOI] [PubMed] [Google Scholar]
- 25.Landsdown A; Williams A, Bacterial resistance to silver in wound care and medical devices. Journal of Wound Care 2007, 16, 15–19, DOI: 17334141. [DOI] [PubMed] [Google Scholar]
- 26.Percival S; Bowler P; Russell D, Bacterial resistance to silver in wound care. Journal of Hospital Infection 2005, 60, 1–7, DOI: 10.1016/j.jhin.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Boucher HW; Talbot GH; Bradley JS; Edwards JE; Gilbert D; Rice LB; Scheld M; Spellberg B; Bartlett J, Bad bugs, no drugs: No ESKAPE! Clinical Infectious Diseases 2009, 48, 1–12, DOI: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 28.Hetrick EM; Shin JH; Stasko NA; Johnson CB; Wespe DA; Holmuhamedov E; Schoenfisch MH, Bactericidal efficacy of nitric oxide-releasing silica nanoparticles. ACS Nano 2008, 2, 235–246, DOI: 10.1021/nn700191f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schäffer MR; Tantry U; Gross SS; Wasserkrug HL; Barbul A, Nitric oxide regulates wound healing. Journal of Surgical Research 1996, 63, 237–240, DOI: 10.1006/jsre.1996.0254. [DOI] [PubMed] [Google Scholar]
- 30.Shekhter AB; Serezhenkov VA; Rudenko TG; Pekshev AV; Vanin AF, Beneficial effect of gaseous nitric oxide on the healing of skin wounds. Nitric Oxide 2005, 12, 210–219, DOI: 10.1016/j.niox.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Carpenter AW; Schoenfisch MH, Nitric oxide release: Part II. Therapeutic applications. Chem. Soc. Rev. 2012, 41, 3742–3752, DOI: 10.1039/C2CS15273H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riccio DA; Schoenfisch MH, Nitric Oxide Release: Part I. Macromolecular scaffolds. Chem. Soc. Rev. 2012, 41, 3731–3741, DOI: 10.1039/C2CS15272J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carpenter AW; Worley BV; Slomberg DL; Schoenfisch MH, Dual action antimicrobials: Nitric oxide release from quaternary ammonium-functionalized silica nanoparticles. Biomacromolecules 2012, 13, 3334–3342, DOI: 10.1021/bm301108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y; Slomberg DL; Shah A; Schoenfisch MH, Nitric oxide-releasing amphiphilic poly (amidoamine)(PAMAM) dendrimers as antibacterial agents. Biomacromolecules 2013, 14, 3589–3598, DOI: 10.1021/bm400961r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Worley BV; Schilly KM; Schoenfisch MH, Anti-biofilm efficacy of dual-action nitric oxide-releasing alkyl chain modified poly(amidoamine) dendrimers. Molecular Pharmaceutics 2015, 12, 1573–1583, DOI: 10.1021/acs.molpharmaceut.5b00006. [DOI] [PubMed] [Google Scholar]
- 36.Worley BV; Slomberg DL; Schoenfisch MH, Nitric oxide-releasing quaternary ammonium-modified poly(amidoamine) dendrimers as dual action antibacterial agents. Bioconjugate Chem. 2014, 25, 918–927, DOI: 10.1021/bc5000719. [DOI] [PubMed] [Google Scholar]
- 37.Cottarel G; Wierzbowski J, Combination drugs, an emerging option for antibacterial therapy. Trends Biotechnol. 2007, 25, 547–555, DOI: 10.1016/j.tibtech.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Eliopoulos G; Eliopoulos C, Antibiotic combinations: should they be tested? Clinical Microbiology Reviews 1988, 1, 139–156, DOI: 10.1128/CMR.1.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomalia D; Baker H; Dewald J; Hall M; Kallos G; Martin S; Roeck J; Ryder J; Smith P, A new class of polymers: starburst-dendritic macromolecules. Polym. J. 1985, 17, 117–132, DOI: 10.1295/polymj.17.117. [DOI] [Google Scholar]
- 40.Tomalia DA, Birth of a new macromolecular architecture: dendrimers as quantized building blocks for nanoscale synthetic polymer chemistry. Progress in Polymer Science 2005, 30, 294–324, DOI: 10.1016/j.progpolymsci.2005.01.007. [DOI] [Google Scholar]
- 41.Coneski PN; Nash JA; Schoenfisch MH, Nitric oxide-releasing electrospun polymer microfibers. ACS Appl Mater Interfaces 2011, 3, 426–432, DOI: 10.1021/am101010e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koh A; Carpenter AW; Slomberg DL; Schoenfisch MH, Nitric oxide-releasing silica nanoparticle-doped polyurethane electrospun fibers. ACS Appl Mater Interfaces 2013, 5, 7956–7964, DOI: 10.1021/am402044s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coneski PN; Schoenfisch MH, Nitric oxide release: Part III. Measurement and reporting. Chem. Soc. Rev. 2012, 41, 3753–3758, DOI: 10.1039/C2CS15271A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pham QP; Sharma U; Mikos AG, Electrospun poly (ε-caprolactone) microfiber and multilayer nanofiber/microfiber scaffolds: characterization of scaffolds and measurement of cellular infiltration. Biomacromolecules 2006, 7, 2796–2805, DOI: 10.1021/bm060680j. [DOI] [PubMed] [Google Scholar]
- 45.Savoji H; Rana D; Matsuura T; Tabe S; Feng C, Development of plasma and/or chemically induced graft co-polymerized electrospun poly (vinylidene fluoride) membranes for solute separation. Separation and Purification Technology 2013, 108, 196–204, DOI: 10.1016/j.seppur.2013.02.013. [DOI] [Google Scholar]
- 46.Sun B; Slomberg DL; Chudasama SL; Lu Y; Schoenfisch MH, Nitric oxide-releasing dendrimers as antibacterial agents. Biomacromolecules 2012, 13, 3343–3354, DOI: 10.1021/bm301109c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallant-Behm CL; Yin HQ; Liu S; Heggers JP; Langford RE; Olson ME; Hart DA; Burrell RE, Comparison of in vitro disc diffusion and time kill-kinetic assays for the evaluation of antimicrobial wound dressing efficacy. Wound Repair Regen 2005, 13, 412–421, DOI: 10.1111/j.1067-1927.2005.130409.x. [DOI] [PubMed] [Google Scholar]
- 48.Soto RJ; Privett BJ; Schoenfisch MH, In vivo analytical performance of nitric oxide-releasing glucose biosensors. Analytical Chemistry 2014, 86, 7141–7149, DOI: 10.1021/ac5017425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bryers JD, Medical biofilms. Biotechnology and Bioengineering 2008, 100, 1–18, DOI: 10.1002/bit.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Costerton JW; Stewart PS; Greenberg E, Bacterial biofilms: a common cause of persistent infections. Science 1999, 284, 1318–1322, DOI: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 51.Vogt C; Xing Q; He W; Li B; Frost MC; Zhao F, Fabrication and characterization of a nitric oxide-releasing nanofibrous gelatin matrix. Biomacromolecules 2013, 14, 2521–2530, DOI: 10.1021/bm301984w. [DOI] [PubMed] [Google Scholar]
- 52.Hall RE; Bender G; Marquis RE, Inhibitory and cidal antimicrobial actions of electrically generated silver ions. Journal of Oral and Maxillofacial Surgery 1987, 45, 779–784, DOI: 10.1016/0278-2391(87)90202-3. [DOI] [PubMed] [Google Scholar]
- 53.Wold KA; Damodaran VB; Suazo LA; Bowen RA; Reynolds MM, Fabrication of biodegradable polymeric nanofibers with covalently attached NO donors. ACS Appl Mater Interfaces 2012, 4, 3022–3030, DOI: 10.1021/am300383w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.