

Summary
Background
Since March, 2013, an avian-origin influenza A H7N9 virus has caused severe pneumonia in China. The aim of this study was to investigate the pathogenesis of this new virus in human beings.
Methods
We obtained ex-vivo cultures of the human bronchus, lung, nasopharynx, and tonsil and in-vitro cultures of primary human alveolar epithelial cells and peripheral blood monocyte-derived macrophages. We compared virus tropism and induction of proinflammatory cytokine responses of two human influenza A H7N9 virus isolates, A/Shanghai/1/2013 and A/Shanghai/2/2013; a highly pathogenic avian influenza H5N1 virus; the highly pathogenic avian influenza H7N7 virus that infected human beings in the Netherlands in 2003; the 2009 pandemic influenza H1N1 virus, and a low pathogenic duck H7N9 virus that was genetically different to the human disease causing A H7N9 viruses.
Findings
Both human H7N9 viruses replicated efficiently in human bronchus and lung ex-vivo cultures, whereas duck/H7N9 virus failed to replicate in either. Both human A H7N9 viruses infected both ciliated and non-ciliated human bronchial epithelial cells and replicated to higher titres than did H5N1 (p<0·0001 to 0·0046) and A/Shanghai/1/2013 replicated to higher titres than did H7N7 (p=0·0002–0·01). Both human A H7N9 viruses predominantly infected type II alveolar epithelial cells and alveolar macrophages in the human lung and replicated to higher titres than did H5N1 (p<0·0001 to 0·0078); A/Shanghai/1/2013 replicated to higher titres than did H1N1 (p=0·0052–0·05) and H7N7 (p=0·0031–0·0151). Human H7N9 viruses were less potent inducers of proinflammatory cytokines compared with H5N1 virus.
Interpretation
Collectively, the results suggest that the novel H7N9 viruses are better adapted to infect and replicate in the human conducting and lower airways than are other avian influenza viruses, including H5N1, and pose an important pandemic threat.
Funding
Area of Excellence Scheme of the University Grants Committee (AoE/M-12/96), Hong Kong Special Administrative Region.
Introduction
As of July 7, 2013, 132 laboratory-confirmed human infections with a novel influenza A H7N9 virus have been reported from ten provinces and municipalities in China by the National Health and Family Planning Commission, and one additional case has been reported from Taipei Centres for Disease Control. There have been 43 deaths so far.1, 2 The source of human infection appears to be poultry.1, 3, 4, 5 There is so far no evidence of sustained human-to-human transmission within the community. Genetic analysis of these viruses revealed that all eight gene segments were of avian origin; six internal gene segments were derived from avian influenza A H9N2 viruses found in poultry in Asia, and the haemagglutinin and neuraminidase genes were derived from influenza viruses circulating in ducks and wild birds, respectively.1
Human infections with other H7 influenza viruses (H7N2, H7N3, and H7N7) have previously been reported in the Netherlands, Canada, USA, and UK6 in association with outbreaks in poultry and resulted in conjunctivitis with mild upper respiratory symptoms. However, there was one fatal case in the Netherlands.6, 7
Cytokine dysregulation contributes to the pathogenesis of human disease caused by highly pathogenic avian influenza A H5N1 as well as the 1918 pandemic H1N1 viruses.8, 9, 10, 11, 12 By comparison with seasonal influenza viruses, some highly pathogenic avian influenza H5N1 viruses induced higher concentrations of proinflammatory cytokines from human alveolar epithelial cells and macrophages.8, 9, 11, 12, 13
We previously used ex-vivo cultures of human conjunctiva, nasopharynx, tonsil, bronchus and lung to investigate influenza14, 15 and coronavirus tropism16 and used in-vitro cultures of polarised primary human alveolar epithelial cells and peripheral blood monocyte derived macrophages to compare innate immune responses elicited by different influenza viruses.8, 9, 12, 13 In this study, we used these ex-vivo and in-vitro models of cultured human tissues and cells to compare the virus tropism and host innate immune responses of the novel A H7N9 influenza virus with that of the pandemic H1N1 virus, highly pathogenic avian influenza H5N1 and H7N7 viruses, and a low pathogenic duck H7N9 virus.
Methods
Viruses
We used avian influenza A H7N9 viruses, A/Shanghai/1/2013 (Sh1/H7N9) and A/Shanghai/2/2013 (Sh2/H7N9); a duck H7N9 virus, A/Duck/Jiangxi/3286/2009 (duck/H7N9), with a different genetic derivation from the two H7N9 viruses as a control; a highly pathogenic avian influenza H7N7 virus isolated from a human patient with fatal disease, A/Netherlands/219/2003 (NL/219/H7N7); a highly pathogenic avian influenza H5N1 virus isolated from a fatal human infection, A/Hong Kong/483/1997 (H5N1); and a 2009 pandemic influenza virus, A/California/07/2009 (H1N1pdm). See appendix for further details. All experiments were done in a biosafety level 3 facility and the use of human tissues had been previously approved by the local institutional review board.
Ex-vivo cultures
Fresh bronchus, lung, nasopharynx, and tonsil tissues were obtained from patients aged 51 to 65 years undergoing elective surgery in Hong Kong and were removed as part of clinical care but surplus for routine diagnostic requirements as detailed previously.14, 15, 16 Methods of culture, infection, and analysis are detailed in the appendix.
In-vitro cultures
The effect of desialylation of turkey red blood cells on virus haemagglutination was compared (appendix). Sialidase DAS181 (NexBio, San Diego, CA, USA), which removes both α-2,3-linked and α-2,6-linked sialic acids, and Sialidase S (Prozyme, Hayward, CA, USA), which removes only α-2,3-linked sialic acids, were compared with untreated turkey red blood cells as controls.
To assess innate immune responses, primary human pneumocytes and peripheral blood monocyte-derived macrophages were derived and used for infection with influenza virus as previously described.8, 9 They were infected with influenza A viruses at a multiplicity of infection of two. The primers and methods used for these assays have been reported previously8, 9, 14, 16 and detailed methods for quantification of cytokine mRNA protein are available in the appendix.
Statistical analysis
Experiments with the human ex-vivo cultures and in-vitro cultures of macrophages and pneumocytes were done independently with at least three different donors, each in duplicate. Results shown in figures are the calculated mean and SEM. Mock infected tissues served as negative controls. The differences in log10 transformed viral titres and quantitative cytokine and chemokine mRNAs between viruses and over time were compared with two-way ANOVA followed by a Bonferroni multiple-comparison test. Differences were deemed significant at p<0·05.
Role of the funding source
The sponsors had no role in study design, data collection, analysis, or interpretation, or in the writing of the report. JSMP, YG, and JMN had full access to all data in the study and had final responsibility for the decision to submit for publication.
Results
The human A H7N9 viruses Sh1/H7N9 and Sh2/H7N9 replicated efficiently in ex-vivo cultures of the human bronchus and lung (figure 1 ). Sh1/H7N9 and Sh2/H7N9 viruses replicated similarly in bronchus, but Sh1/H7N9 replicated to significantly higher titres than Sh2/H7N9 in the lung. Sh1/H7N9 and Sh2/H7N9 virus titres in the bronchus and lung were higher than those observed with H5N1 virus. Sh1/H7N9 and Sh2/H7N9 virus titres in bronchus were similar to that observed with the H1N1pdm virus, but Sh1/H7N9 replicated to higher titres in the lung. Sh1/H7N9 replicated to higher titres than NL/219/H7N7 virus, and NL/219/H7N7 replicated to higher titres than H5N1 virus, in bronchus and lung. Limited experimental replicates of ex-vivo nasopharyngeal (n=2) and tonsil (n=1) cultures restrict statistical analysis, but there was a trend suggesting productive replication of Sh1/H7N9 and Sh2/H7N9 in nasopharynx (appendix). Duck/H7N9 failed to replicate in any of these tissues. Sh2/H7N9 replicated in nasopharyngeal tissues at 33°C (appendix).
Figure 1.

Viral replication kinetics of influenza A H7N9 virus in ex-vivo cultures of bronchus (A) and lung (B), infected with 106 TCID50/mL of influenza viruses at 37°C
Barcharts show the mean virus titre pooled from at least three independent experiments. The horizontal dotted line denotes the limit of detection in the TCID50 assay.; error bars show SEM. Tables show statistical significance between virus titres at each timepoint. Red shows that the reference virus titre (listed at the top of the column) is significantly higher than the comparator (viruses listed on the left) and black shows that it is significantly lower. TCID50=tissue culture infective dose. ns=non-significant. *p<0·0001.
Immunohistochemistry showed that Sh1/H7N9 and Sh2/H7N9 viruses extensively infected bronchial epithelium, infecting both ciliated and non-ciliated bronchial epithelial cells (figure 2C, E ), the extent of infection being greater than that seen with H5N1 (figure 2G) and other avian viruses (table 1 ). In the lung, Sh1/H7N9 and Sh2/H7N9 infected mainly type II pneumocytes (figure 3 ) and alveolar macrophages (figure 3 and appendix). There was no evidence of infection of vascular endothelium in the blood vessels of the lung. The duck/H7N9 virus, which differs in its genetic origin from the viruses causing the current human H7N9 outbreak, failed to infect or replicate in any of the human tissues tested, with the exception of occasional cells expressing viral antigen seen in the bronchial epithelium (table 1).
Figure 2.
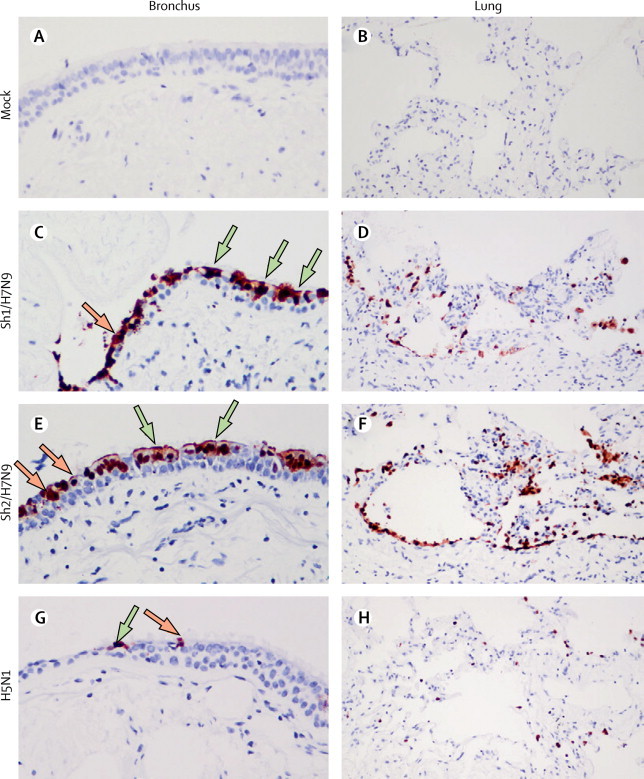
Tissue tropism of influenza A H7N9 virus in ex-vivo cultures of bronchus and lung
Formalin-fixed paraffin-embedded sections of bronchus (A, C, E, G) and lung (B, D, F, H) after 24 h infection with mock (A, B), Sh1/H7N9 (C, D), Sh2/H7N9 (E, F), and H5N1 (G, H) viruses. Sections were stained with a monoclonal antibody against the influenza nucleoprotein with positive cells identified as a red-brown colour. Ciliated and non-ciliated cells are identified with green and orange arrows, respectively.
Table 1.
Tissue tropism of influenza virus infection of human bronchus and lung as assessed by immunohistology
| Bronchus | Lung | |
|---|---|---|
| Sh1/H7N9 | ++ | ++ |
| Sh2/H7N9 | ++ | ++ |
| NL/219/H7N7 | + | + |
| H5N1 | +/− | + |
| H1N1/pdm | + | ++ |
| Duck/H7N9 | Sparse | − |
Sections were stained with a monoclonal antibody to influenza nucleoprotein and scored in a semiquantitative manner: ++=more than 20% of epithelial cells have positive antigen. +=5–20% of epithelial cells have positive antigen. +/−=less than 5% of epithelial cells have positive antigen. Sparse=only 1–3 positive cells identified per section. −=no positive cells identified.
Figure 3.
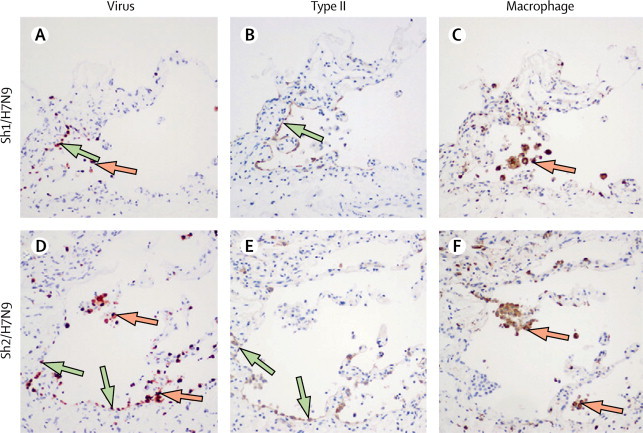
Cellular localisation of influenza antigen in human lung tissues infected with influenza A H7N9 virus
Serial sections of ex-vivo cultures of human lungs infected with Sh1/H7N9 (A–C) and Sh2/H7N9 viruses (D–F). Sections were stained with monoclonal antibodies to influenza nucleoprotein (A, D), surfactant protein D (B, E), and CD68 (C, F). Green arrows show type II pneumocytes and orange arrows show alveolar macrophages.
Treatment of turkey red blood cells with a sialidase that specifically cleaves the α-2,3-linked sialic acid with galactose did not have any effect on haemagglutination of either Sh1/H7N9 or Sh2/H7N9, whereas it completely abolished haemagglutination of H5N1 virus (table 2 ). As a control, DAS181, a sialidase that cleaves both the α-2,3 and α-2,6 links of sialic acid, abolished the haemagglutination of human and avian influenza viruses including that of Sh1/H7N9 and Sh2/H7N9.
Table 2.
Effect of desialylation on virus haemagglutination of turkey red blood cells
| Control | DAS181 treated | Sialidase S treated | |
|---|---|---|---|
| Sh1/H7N9 | 128 | 0 | 128 |
| Sh2/H7N9 | 64 | 0 | 64 |
| H5N1 | 64 | 0 | 0 |
| H1N1pdm | 32 | 0 | 32 |
The reciprocal of the haemagglutination titre is denoted. Three independent experiments were done with identical results.
In human peripheral blood monocyte-derived macrophages, influenza viral matrix (M) gene expression of Sh1/H7N9, Sh2/H7N9, H5N1, and H1N1pdm viruses were generally similar at 3, 8, and 24 h post infection, but Sh2/H7N9 had significantly higher M-gene copy numbers compared with H1N1pdm virus at 8 h after infection and H5N1 had significantly higher M-gene expression than H1N1pdm at 3 h after infection. As expected, H5N1 induced significantly more CCL5, CXCL10, tumour necrosis factor α (TNFα; at 3, 8, and 24 h post infection), CCL2, MX1 (at 8 h post infection), and interferon β (at 3 h post infection) mRNA compared with H1N1pdm virus (figure 4 ). Sh1/H7N9 and/or Sh2/H7N9 viruses induced significantly higher concentrations of interferon β (at 3 h post infection), MX1 and CCL5 (at 8 h post infection), and CXCL10 (at 8 h and 24 h post infection) mRNA than did H1N1pdm virus, but less interferon β and TNFα compared with H5N1 at 3 h post infection (figure 4). In parallel, H5N1 virus elicited more CCL5 and TNFα protein secretion compared with H1N1pdm (figure 5 ). Overall, the induction of proinflammatory cytokines by Sh1/H7N9 or Sh2/H7N9 appeared to be intermediate between those induced by H5N1 and H1N1pdm viruses. Sh1/H7N9 and Sh2/H7N9 viruses both induced significantly higher concentrations of CCL5 and CXCL10 mRNA than did H1N1pdm virus, but less interferon β and TNFα compared with H5N1 (figure 4).
Figure 4.
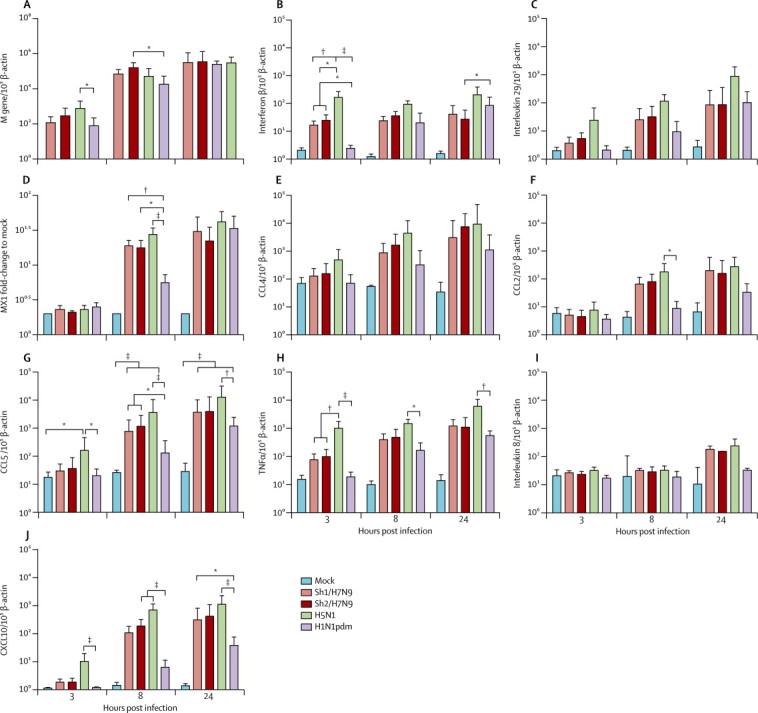
Cytokine and chemokine mRNA expression profile in peripheral blood monocyte-derived macrophages infected with mock, Sh1/H7N9, Sh2/H7N9, H5N1, and H1N1pdm viruses
Expression of the influenza matrix (M) gene (A), interferon β (B), interleukin 29 (C), MX1 (D), CCL4 (E), CCL2 (F), CCL5 (G), tumour necrosis factor α (TNFα; H), interleukin 8 (I), and CXCL10 (J) at 3, 8, and 24 h after infection. Graphs show mean mRNA copies expressed per 105 β-actin copies from three independent experiments; error bars show SEM. MX1=myxovirus resistance 1. CCL=chemokine (C-C motif) ligand. CXCL10=chemokine (C-X-C motif) ligand 10. *p<0·05. † p<0·01. ‡p<0·005.
Figure 5.
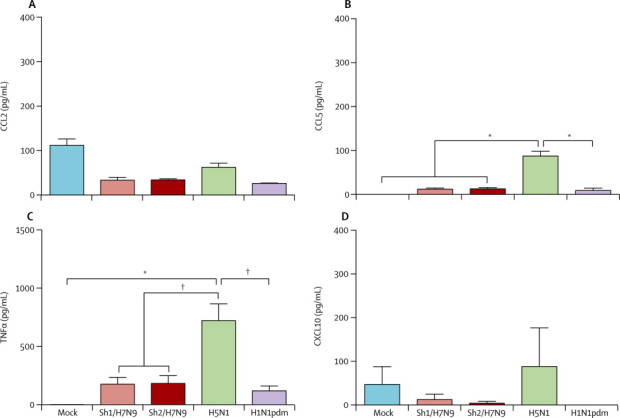
Cytokine and chemokine protein expression in cell culture supernatants of peripheral blood monocyte-derived macrophages infected with mock, Sh1/H7N9, Sh2/H7N9, H5N1, and H1N1pdm viruses
Expression of CCL2 (A), CCL5 (B) tumour necrosis factor α (TNFα; C), and CXCL10 (D) at 24 h post infection by ELISA (R&D Systems, Minneapolis, MN, USA). Graphs show mean protein from three independent experiments; error bars show SEM. CCL=chemokine (C-C motif) ligand. CXCL10=chemokine (C-X-C motif) ligand 10. *p<0·005. † p<0·01.
The mRNA expression of genes related to interferon and interferon receptor signalling in human peripheral blood monocyte-derived macrophages after Sh1/H7N9, Sh2/H7N9, H5N1, and H1N1pdm virus infection was investigated by cDNA PCR array and expressed as fold-change over mock infected cells (appendix). Only genes that were three or more fold changes overexpressed or underexpressed relative to H1N1pdm were noted. The interferon stimulated genes MX1, ISG15, IFI35, IFI44, IFIH1, and OAS1 were effectively upregulated by Sh1/H7N9 and Sh2/H7N9 infections (data not shown). Expression of IL20RB, IFNAR1, IL20RA, IL22RA2, IL2RB, IL9R, and IRF4 were overexpressed by either Sh1/H7N9 or Sh2/H7N9 infection compared with H1N1pdm infection. IL10RB and IL4R were more downregulated by Sh1/H7N9 or Sh2/H7N9 compared with H1N1pdm.
In primary human pneumocytes, replication of Sh1/H7N9 and Sh2/H7N9 viruses (multiplicity of infection of two) were similar, as assessed by viral M-gene quantitation at 1, 6, and 24 h after infection, whereas Sh2/H7N9 had a significantly higher M-gene level compared with H5N1 (p=0·05) and H1N1pdm (p=0·0346) at 24 h after infection (figure 6A ). As expected, H5N1 virus induced significantly more levels of interferon β, interleukin 29, CCL4, CCL5, and CXCL10 mRNA than did H1N1pdm virus infection. Sh1/H7N9 and/or Sh2/H7N9 elicited significantly more interferon β, interleukin 29, CCL5, and CXCL10 mRNA than did H1N1pdm virus infection. However, Sh1/H7N9 and Sh2/H7N9 viruses both elicited less interferon β, interleukin 29, and CCL5 mRNA than did H5N1 virus (figure 6). In virus-infected cell supernatants, H5N1 virus induced significantly more CCL5 protein than did H1N1pdm, Sh1/H7N9, and Sh2/H7N9 viruses and more CXCL10 protein than did H1N1pdm (figure 7 ).
Figure 6.
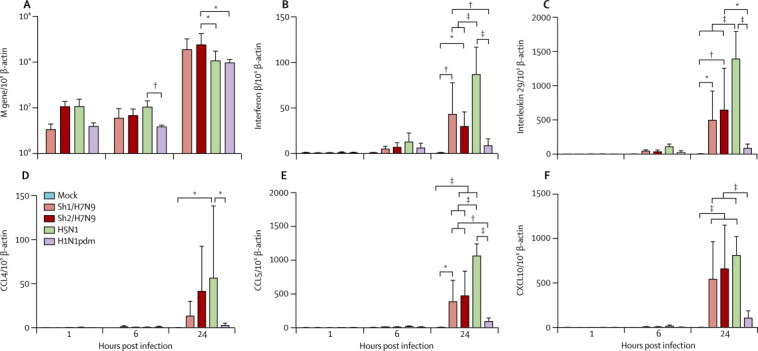
Cytokine and chemokine mRNA expression profile in human pneumocytes infected with mock, Sh1/H7N9, Sh2/H7N9, H5N1, and H1N1pdm viruses
Expression of influenza matrix (M) gene (A), interferon β (B), interleukin 29 (C), CCL4 (D), CCL5 (E), and CXCL10 (F) at 1, 6, and 24 h after infection. Graphs show mean mRNA copies expressed per 105 β-actin copies from three independent experiments; error bars show SEM. CCL=chemokine (C-C motif) ligand. CXCL10=chemokine (C-X-C motif) ligand 10. *p<0·05. † p<0·01. ‡p<0·005.
Figure 7.

Cytokine and chemokine protein expression in cell culture supernatants of pneumocytes infected with mock, Sh1/H7N9, Sh2/H7N9, H5N1, and H1N1pdm viruses
Expression of CCL5 (A) and CXCL10 (B) at 24 h post infection by ELISA (R&D Systems, Minneapolis, MN, USA). Graphs show mean protein from three independent experiments; error bars show SEM. CCL=chemokine (C-C motif) ligand. CXCL10=chemokine (C-X-C motif) ligand 10. *p<0·05. † p<0·01.
Discussion
Sh1/H7N9 and Sh2/H7N9 viruses replicated efficiently in ex-vivo cultures of human bronchus and lung with peak viral titres similar to and exceeding that seen with H1N1pdm (panel ). These findings emphasise the relative ease with which H7N9 viruses can infect human beings and is compatible with epidemiological observations. Peak viral titres in the bronchus and lung were higher than those observed with other avian influenza viruses that have infected humans such as highly pathogenic avian influenza H5N1 and H7N7. By contrast, the duck/H7N9 virus, a typically avian virus genetically different from Sh1 and Sh2/H7N9 viruses in all eight gene segments, failed to replicate efficiently in any human respiratory tissue.
Panel. Research in context.
Systematic review
We searched PubMed on May 31, 2013 with the terms “H7N9” and “virus tropism” or “innate immunity” or “autopsy” and found none pertaining to the novel influenza A H7N9 virus. No date or language restrictions were applied. There were papers pertaining to other H7 influenza viruses and one pertaining to the pathogenesis of this virus. Those relevant to the data presented in this Article have been cited in the bibliography. An understanding of the tropism of a novel influenza virus for the human respiratory tract is important to assess the zoonotic and pandemic risk posed by these viruses.
Interpretation
The novel avian origin human influenza A H7N9 viruses replicated more efficiently than did other avian influenza viruses and similarly to the pandemic 2009 H1N1 virus in ex-vivo cultures of the human respiratory tract. Collectively, the results suggest that the novel H7N9 viruses pose a significant zoonotic and pandemic threat.
Immunohistochemistry of ex-vivo cultures of the human bronchus showed Sh1/H7N9 and Sh2/H7N9 virus infection of both ciliated and non-ciliated epithelial cells. H5N1 only led to limited infection in the human bronchus with infection restricted to ciliated bronchial epithelial cells. In the alveoli, H7N9-infected cells were mainly type II pneumocytes and macrophages. Type II alveolar epithelial cells are important in producing surfactant proteins A and D and are crucial in reconstituting damaged type I alveolar epithelium. Loss of type II pneumocytes would lead to impairment of the repair processes after alveolar damage. The tropism of Sh1/H7N9 and Sh2/H7N9 infection in the alveoli is similar to that seen with H5N1 infection,17 but the extent of infection of the bronchus and lung is significantly greater than that seen with H5N1. Although individual genetic and environmental variability might result in diverse phenotypes, that three separate donors yielded very similar trends is interesting (data not shown). Future work needs to address whether genetic (eg, IFITM3) or other host factors play a part in susceptibility to infection of ex-vivo human respiratory cultures.
Sh1/H7N9 and Sh2/H7N9 viruses differ in aminoacid residues Gln226Leu, Ser138Ala, and Gly186Val (H7/H3 numbering) in haemagglutinin, which are expected to enhance binding of Sh2/H7N9 to α-2,6 glycans found in the human upper airways.4 However, Sh1/H7N9 and Sh2/H7N9 infect human bronchus with similar efficiency. A sialidase specific for cleaving the α-2,3 glycosidic abolished the haemagglutination of turkey red blood cells by H5N1 virus, but did not affect haemagglutination by Sh1/H7N9 or Sh2/H7N9 viruses suggesting that both Sh1/H7N9 and Sh2/H7N9 predominantly bind α-2,6 glycans, irrespective of differences in aminoacid residues Gln226Leu, Ser138Ala, and Gly186Val of haemagglutinin. Both Sh1/H7N9 and Sh2/H7N9 have lost a glycosylation site (Thy160Ala) in the 150-loop of haemagglutinin that is known to result in increased affinity for binding α-2,6-linked glycans in H5N1 viruses.4 By contrast, others have reported that a recombinant Sh2/H7N9-like virus haemagglutinin bound poorly to human trachea and a Gly228Ser aminoacid change was needed to confer improved binding to the human upper airways.18 Recombinant viral haemagglutinin might not accurately reflect haemagglutinin binding of native virions where multivalent trimeric haemagglutinin is present together with neuraminidase and fails to take into account any role that the virus neuraminidase (via the catalytic site or the second neuraminidase binding site) might have in virus binding. On the other hand, others have used labelled H7N9 to define virus binding to human respiratory tract tissues, indicating that both Sh1/H7N9 and Sh2/H7N9-like viruses bind very efficiently to the human bronchus (T Kuiken, personal communication).
Recent mass spectrometric data have shown that the bronchus contains sialylated α-2,3 glycans,19 explaining its infection by avian viruses (H5N1 and H7N7) whose glycan array profiles indicate predominant α-2,3 receptor binding. Receptor binding and structural analysis of Sh2/H7N9-like virus haemagglutinin shows efficient binding of both α-2,3 and α-2,6 receptors.20 The functional changes in receptor binding and PB2 Glu627Lys are some of the key functional changes identified by Herfst and colleagues21 in 2012 to be important in adapting an avian H5N1 virus towards transmission in mammals. Since A/Hong Kong/483/97 (H5N1) virus also has lysine at position 627 in PB2, the efficient replication of H7N9 viruses in human ex-vivo cultures of the bronchus is unlikely to be explained solely on this basis.
Autopsy data for the lung would provide useful data to understand the pathogenesis of human A H7N9 disease. However, autopsy data usually only provide information about lung pathology at late stages of the illness and thus fail to provide insights into virus tropism in the early stages of infection. Thus data from experimental infection of ex-vivo cultures of the respiratory tract provide unique insights into virus tropism and pathogenesis. Reported patients with H7N9 disease have been older than those with human infections with avian H5N1 or H9N2 viruses, and severe disease appears to be more common with increasing age.3 The biological basis for this unusual epidemiological pattern needs to be understood because it differs from that seen in individuals infected with H5N1. Whether milder cases in young people are unrecognised, thereby skewing the case–fatality ratio as well as the age distribution, is unclear. Paediatric lung and bronchial tissues differ from adult tissues in the balance of sialylated α-2,3 and α-2,6 glycans,19 and this factor might contribute to the increased disease severity with increasing age. However, using similar techniques we found that Sh1/H7N9 and Sh2/H7N9 viruses efficiently infected ex-vivo lung from a 1-year-old child (data not shown). Chronic lung disease increases numbers of type II pneumocytes (the target cells for influenza viruses) and might contribute to an increased disease severity compared with younger, healthier individuals.22
Dysregulation of proinflammatory innate immune responses by highly pathogenic avian influenza H5N1 virus contributed to the unusual severity of human H5N1 disease and investigation of whether Sh1 or Sh2/H7N9 virus infections lead to exaggerated proinflammatory innate immune responses was relevant. Since ex-vivo cultures are heterogeneous in cell type and susceptibility to virus and the infecting dose cannot be precisely controlled, we chose to compare innate immune responses to H7N9 infection with that of highly pathogenic avian influenza H5N1 and H1N1pdm viruses in well defined primary human macrophages and alveolar epithelial cells. By comparison with H5N1 virus (a high cytokine-inducing virus) and H1N1pdm virus (a low cytokine-inducing virus), Sh1/H7N9 and Sh2/H7N9 appear to have an intermediate phenotype in their intrinsic capacity to induce proinflammatory cytokines such as CCL5 and TNFα. The expression of interferon stimulated genes (eg, MX1, ISG15) did not appear to be impaired. Lungs and serum of mice experimentally infected with Sh2/H7N9 also had lower concentrations of proinflammatory chemokines and cytokines compared with those of H5N1-infected mice.23 In a patient with mild disease and one with severe disease in whom cytokine concentrations were studied, there was no difference in proinflammatory cytokines in serum. However, the patient who died had high serum concentrations of the anti-inflammatory cytokine serum interleukin 10.5 Raised plasma or lung cytokine concentration might be secondary to more severe pathology in severely ill patients,24 or secondary to increased replication competence of a virus rather than being the intrinsic cause of such pathology. Therefore, the experiments we have described in which similar virus infecting doses of different viruses are compared for their capacity for cytokine induction provides important data to define the intrinsic viral capacity for proinflammatory cytokine induction, which might be of relevance for pathogenesis and therapy. From our findings, Sh2/H7N9-like viruses appear to be intrinsically more potent inducers of the proinflammatory cytokine responses than are H1N1pdm viruses, but less so than H5N1 viruses. Thus, proinflammatory cytokine responses might contribute in part to the severity of human H7N9 disease, but the unusual tissue tropism of this virus for human bronchus and lung and attendant direct viral cytopathology probably has a more important role in pathogenesis, as was seen with the novel Middle East respiratory syndrome coronavirus.16
Our findings suggest that Sh2/H7N9-like avian viruses are exceptionally fit to infect mammalian species including human beings. Ferrets experimentally infected with Sh2/H7N9 showed efficient virus replication in the respiratory tract with inefficient ferret-to-ferret transmission by airborne droplets.25 Although no sustained human-to-human transmission has been noted to date, the efficient replication of these H7N9 viruses in the human respiratory tract and the detection (albeit inefficient) of ferret-to-ferret transmission by the airborne route by these viruses without previous adaptation increases the pandemic concern associated with these viruses.
Acknowledgments
Acknowledgments
Alan D L Sihoe and staff of the Division of Cardiothoracic Surgery, Department of Surgery, Li Ka Shing Faculty of Medicine, The University of Hong Kong and Queen Mary Hospital provided the human bronchial and lung tissues. Dennis I T Kuok, Iris H Y Ng, Christine H T Bui, and M C Cheung at the Centre of Influenza Research, School of Public Health, and Kevin Fung at the Department of Pathology, The University of Hong Kong, provided technical support. We acknowledge research funding from National Institute of Allergy and Infectious Diseases (NIAID) contract HHSN266200700005C, Area of Excellence Scheme of the University Grants Committee (Grant AoE/M-12/96) and Health and Medical Research Fund (Ref: 12110992) by Research Fund Secretariat, Food and Health Bureau, Hong Kong Special Administrative Region.
Contributors
MCWC contributed to study design and coordination, analysis and interpretation of results, experiments, and writing of the report. RWYC contributed to study design and coordination, analysis and interpretation of results, and writing of the report. LLYC and CKPM did experiments and contributed to analysis and interpretation of results. KPYH, JHMF, and KPT did experiments and contributed to analysis of results. LLMP developed quantitative PCR assays. YG contributed to study design and critical review of the report. JMN contributed to study design, analysis and interpretation of results, and writing of the report. JSMP contributed to study design and overall coordination, analysis and interpretation of results, and writing of the report.
Conflicts of interest
We declare that we have no conflicts of interest.
Supplementary Material
References
- 1.Gao R, Cao B, Hu Y. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 2.WHO Human infection with avian influenza A(H7N9) virus—update. http://www.who.int/csr/don/2013_07_04/en/index.html (accessed July 7, 2013).
- 3.Li Q, Zhou L, Zhou M. Preliminary report: epidemiology of the avian influenza A (H7N9) outbreak in China. N Engl J Med. 2013 doi: 10.1056/NEJMoa1304617. published online April 24. [DOI] [Google Scholar]
- 4.Kageyama T, Fujisaki S, Takashita E. Genetic analysis of novel avian A(H7N9) influenza viruses isolated from patients in China, February to April 2013. Euro Surveill. 2013;18:20453. [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y, Liang W, Yang S. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet. 2013;381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fouchier RA, Schneeberger PM, Rozendaal FW. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc Natl Acad Sci USA. 2004;101:1356–1361. doi: 10.1073/pnas.0308352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koopmans M, Wilbrink B, Conyn M. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet. 2004;363:587–593. doi: 10.1016/S0140-6736(04)15589-X. [DOI] [PubMed] [Google Scholar]
- 8.Chan MC, Cheung CY, Chui WH. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res. 2005;6:135. doi: 10.1186/1465-9921-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung CY, Poon LL, Lau AS. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360:1831–1837. doi: 10.1016/s0140-6736(02)11772-7. [DOI] [PubMed] [Google Scholar]
- 10.Kash JC, Tumpey TM, Proll SC. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan MC, Chan RW, Yu WC. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir Res. 2009;10:102. doi: 10.1186/1465-9921-10-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu WC, Chan RW, Wang J. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J Virol. 2011;85:6844–6855. doi: 10.1128/JVI.02200-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan MC, Chan RW, Yu WC. Tropism and innate host responses of the 2009 pandemic H1N1 influenza virus in ex vivo and in vitro cultures of human conjunctiva and respiratory tract. Am J Pathol. 2010;176:1828–1840. doi: 10.2353/ajpath.2010.091087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicholls JM, Chan MC, Chan WY. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nature Med. 2007;13:147–149. doi: 10.1038/nm1529. [DOI] [PubMed] [Google Scholar]
- 16.Chan RW, Chan MC, Agnihothram S. Tropism and innate immune responses of the novel human betacoronavirus lineage C virus in human ex vivo respiratory organ cultures. J Virol. 2013 doi: 10.1128/JVI.00009-13. published online April 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinheimer VK, Becher A, Tonnies M. Influenza A viruses target type II pneumocytes in the human lung. J Infect Dis. 2012;206:1685–1694. doi: 10.1093/infdis/jis455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tharakaraman K, Jayaraman A, Raman R. Glycan receptor binding of the influenza A virus H7N9 hemagglutinin. Cell. 2013;153:1486–1493. doi: 10.1016/j.cell.2013.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walther T, Karamanska R, Chan RW. Glycomic analysis of human respiratory tract tissues and correlation with influenza virus infection. PLoS Pathog. 2013;9:e1003223. doi: 10.1371/journal.ppat.1003223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiong X, Martin SR, Haire LF. Receptor binding by an H7N9 influenza virus from humans. Nature. 2013 doi: 10.1038/nature12372. published online June 20. [DOI] [PubMed] [Google Scholar]
- 21.Herfst S, Schrauwen EJ, Linster M. Airborne transmission of influenza A/H5N1 virus between ferrets. Science. 2012;336:1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hocke AC, Berg J, Becher A. Reply to Fujino et al. J Infect Dis. 2013;207:693–695. doi: 10.1093/infdis/jis740. [DOI] [PubMed] [Google Scholar]
- 23.Mok CK, Lee HH, Chan MC. Pathogenicity of the novel A/H7N9 influenza virus in mice. mBio. 2013;4:e00362–e00413. doi: 10.1128/mBio.00362-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.To KK, Hung IF, Li IW. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin Infect Dis. 2010;50:850–859. doi: 10.1086/650581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu H, Wang D, Kelvin DJ. Infectivity, transmission, and pathology of human H7N9 influenza in ferrets and pigs. Science. 2013 doi: 10.1126/science.1239844. published online May 23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.