

Summary
Background A proportion of nasal epithelial cells (NEC) in patients with allergic rhinitis (AR) are known to express the major histocompatibility complex Class II molecule (HLA‐DR).
Objective We hypothesized that NEC may play a role in antigen presentation to T cells. To elucidate the possible role of NEC in antigen presentation, we examined the expression of HLA‐DR, CD80 and CD86 in NEC, their regulation by cytokines and the capacity of NEC to induce antigen‐specific proliferation of T cells.
Methods We examined the expression of HLA‐DR, CD80 and CD86 in nasal epithelial scrapings of patients with seasonal allergic rhinitis (SAR) to Japanese cedar pollen pre‐season and in‐season, by immunohistochemistry. Next, we examined the effect of IL‐1β, TNF‐α, (IFN‐γ), IL‐4 α, IL‐13 and diesel exhaust particles (DEP) on the HLA‐DR, CD80 and CD86 expression in cultured nasal epithelial cells (CNEC), by flow cytometry. Further, we analysed the capacity of mite antigen (Der f II)‐pulsed mitomycin‐C‐treated CNEC to induce proliferation of autologous T cells from patients with perennial allergic rhinitis.
Results NEC constitutively expressed HLA‐DR and CD86, but not CD80. The expression of HLA‐DR and CD86 in NEC was significantly increased in‐season, in patients with SAR as compared with that of pre‐season. While IFN‐γ up‐regulated the expression of HLA‐DR, IL‐1β and TNF‐α up‐regulated the expression of CD86 in CNEC. Furthermore, in the presence of mite antigen, CNEC induced the proliferation of autologous peripheral blood T lymphocytes. Anti‐CD86 and anti‐HLA‐DR monoclonal antibody but not anti‐CD80 inhibited the epithelial cell‐induced T cell proliferation. Stimulation with a combination of DEP and mite antigen significantly up‐regulated HLA‐DR and CD86 expression in CNEC.
Conclusions These studies suggest that NEC in patients with AR may play a role in antigen presentation through the enhanced expression of HLA‐DR and CD86. Furthermore, these results suggest the possibility that DEP may enhance the antigen‐presenting function of CNEC.
Keywords: allergic rhinitis, APC, CD86, DEP, HLA‐DR, nasal epithelial cells
Introduction
For several years, nasal epithelial cells (NEC) were considered to play a simple role of a mucosal barrier while being involved in the secretion of mucous or removal of foreign agents by their cilia [1]. However, recent studies have shown that epithelial cells have a much wider range of activities including the release of eicosanoids [2, 3], endopeptidases [4], cytokines and chemokines [5, 6, 7, 8, 9]. Epithelial cells in allergic individuals (asthmatics and rhinitics) are in an activated state, as shown by the increased expression of adhesion molecule‐like intercellular adhesion molecule‐1 (ICAM‐1) and vascular cell adhesion molecule‐1 (VCAM‐1) [10, 11], and the increased production of inflammatory mediators like IL‐6, IL‐8, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), TNF‐α [8, 9] thus contributing to the enhancement of the allergic reaction. However, epithelial cells may also play other important roles via direct cell‐to‐cell interaction. A proportion of NEC has been reported to express the major histocompatibility complex (MHC) Class II molecule (HLA‐DR) [9, 12, 13, 14, 15]. We therefore hypothesized that NEC may probably play a role in antigen presentation.
In addition to HLA‐DR, CD80 and CD86 (B7‐1 and 2) are key co‐stimulatory molecules involved in antigen presentation [16]. Optimal activation of T cells requires both co‐stimulation and T cell receptor engagement. Antigen presentation in the absence of co‐stimulation may lead to T cell anergy. Co‐stimulatory interactions between the B7 family ligands expressed on antigen‐presenting cells (APC) and their receptors on T cells play critical roles in the growth, differentiation and death of T cells [17, 18, 19, 20, 21]. Engagement of the T cell co‐stimulatory receptor CD28 by its ligands CD80 and CD86 augments the activation of T cells and promotes T cell survival. The question as to whether NEC expresses co‐stimulatory molecules is relevant because epithelial cells express MHC Class II molecules. However, little is known about the expression of co‐stimulatory molecules on NEC. To elucidate the possible role of NEC in antigen presentation, we examined the expression of HLA‐DR, CD80 and CD86 in NEC and its regulation by cytokines. We also examined the potential of cultured nasal epithelial cells (CNEC) in inducing antigen‐specific proliferation of autologous T cell.
In recent years, epidemiological studies have shown a significant association between exposure to various pollutants and an increase in allergic diseases. In fact, short‐term exposure to diesel exhaust in healthy human volunteers has demonstrated an increase in the release of inflammatory mediators, increased IgE synthesis and up‐regulation of leucocyte adhesion molecule expression [22]. In vitro studies have shown that diesel exhaust particles (DEP) can enhance the cytokine secretion from epithelial cells [23], but its effects on cell surface adhesion molecules like HLA‐DR and CD86 have not yet been studied. We therefore examined the effect of DEP on the expression of HLA‐DR and CD86 in CNEC.
Materials and methods
Patients
Thirteen patients with seasonal allergic rhinitis (SAR) to Japanese cedar pollen (JCP) (M : F 9 : 4; mean age 29.7 years) who presented with typical symptoms of SAR, and were diagnosed on the basis of clinical history, anterior rhinoscopic examination and RAST for allergen‐specific IgE in the serum were included in the SAR study. None of the SAR patients included in the study were on topical steroids or immunotherapy. In the second study, 10 patients with perennial allergic rhinitis (PAR) to house dust mite (HDM) (M : F 6 : 4; mean age 29.0 years) were selected on the basis of their typical nasal symptoms of sneezing, rhinorrhoea and nasal congestion and allergy tests (nasal provocation test, skin test and RAST). None of the PAR patients were on any medications for at least 2 weeks before collecting the specimens and none were on immunotherapy. All patients were symptomatic at the time of taking specimens. All studies had been approved by the Human Protection of Subjects Committee of the Nippon Medical School, Tokyo, Japan.
Collection and preparation of specimens
Nasal epithelial scrapings were obtained in the out‐patient clinic using a small sterile surgical curette measuring 2.5 × 3.5 mm in cup size, as described previously [24]. The pre‐season scrapings were collected in the first 2 weeks of January (early January) before the onset of the pollen scattering. The in‐season scrapings were collected at the peak of the season from the middle of February to the middle of March of 2001, which was a heavy pollen season. Approximately six to eight scrapings were collected from each patient. The scrapings were fixed in periodate lysine paraformaldehyde (PLP), washed in phosphate‐buffered saline with a graded series of sucrose (10–15%) cytospinned onto silane‐coated slides and stored at −80°C until further use.
Monoclonal antibodies
The primary antibodies used in this study, the mouse anti‐human HLA‐DR monoclonal antibody (mAb) (Becton Dickinson, Mountain View, CA, USA), the mouse anti‐human CD86 and the mouse anti‐human CD80 mAbs (Pharmingen, San Diego, CA, USA), the fluorescein isothiocyanate (FITC)‐conjugated mouse anti‐human HLA‐DR (Becton Dickinson) and the phycoerythrin (PE)‐conjugated mouse anti‐human CD86 and the PE‐conjugated mouse anti‐human CD80 mAbs (Pharmingen), were purchased as indicated. The isotype‐matched immunoglobulins used as negative controls in this study, the mouse IgG1 (Dako, Denmark, UK) and the mouse IgG2a (Dako), the respective FITC/PE‐conjugated mouse isotype‐matched IgG (Becton Dicknison) were purchased as indicated.
Immunohistochemical analysis
The PLP‐fixed scrapings that were cytospinned onto silane‐coated glass slides were immunostained with the anti‐human HLA‐DR mAb, anti‐CD80 mAb or anti‐CD86 mAb using the alkaline phosphatase‐anti‐alkaline phosphatase method (APAAP Kit; DAKO). Briefly, air‐dried slides were fixed for 10 min in acetone. After rehydration in 0.05 m Tris‐buffered saline, pH 7.6 (TBS), and blocking with 10% normal rabbit serum (DAKO), the cells were incubated overnight with the relevant primary antibody anti‐HLA‐DR/anti‐CD80 or anti‐CD86 mAb at optimal concentrations, at 4°C. After rinsing thrice in TBS, the cell specimens were incubated in the rabbit anti‐mouse immunoglobulin (DAKO) for 30 min at room temperature. Finally, the reaction was developed using a two‐step APAAP method and a Fast Red substrate, in accordance with the manufacturer's instructions. Cell specimens were then counterstained with Mayer's haematoxylin (Sigma Chemical, St Louis, MO, USA) for 10 s, rinsed in distilled water and mounted in Dako gel. For negative control, the primary antibodies were substituted with isotype‐matched mouse IgG1.
Cell counting
Cell counting was performed by counting the positively stained NEC per 100 epithelial cells in five randomly selected fields using an objective micrometer and at a magnification × 200). In separate studies, we have demonstrated that more than 90% of the freshly isolated cells in the nasal scraping samples were epithelial cells as detected by cytokeratin staining.
Isolation and culture of NEC
NEC were isolated and cultured from biopsies of nasal inferior turbinates as described previously [5]. Briefly, inferior turbinates were collected at surgery carried out for the treatment of severe nasal obstruction in the PAR patients due to hypertrophied nasal turbinates. The specimens were collected in McCoy's 5A medium (GIBCO, Grand Island, NY, USA) supplemented with 1% penicillin–streptomycin (GIBCO) and 5 × 10−5 mol/L of 2‐mercaptoethanol (McCoy's 2+medium) and then incubated in McCoy's 2+medium containing 0.1% protease (type 14) (Sigma) for 16 h at 4°C. After incubation, 10% fetal calf serum (FCS) was added and the epithelial cells were detached by gentle scraping. The cell suspension was centrifuged at 200 g for 10 min, and the pellet was resuspended in McCoy's 2+medium. The cells were then plated onto collagen‐coated 24‐well culture plates for 24 h at 37°C in a humidified CO2 incubator (5% CO2). At 24 h, the supernatant was discarded and the adherant cells were cultured to confluence for 5 days in McCoy's 2+medium with 10% FCS. In separate studies, we have demonstrated that all cells in this culture system were cytokeratin positive [5].
Flow cytometric analysis
The culture medium was changed at confluency and the NEC were stimulated for a further period of 5 days with recombinant human IFN‐γ (R&D, Minneapolis, MN, USA) (dose from 1 ng to 1 μg/mL), IL‐1β (Sigma) (doses from 1 ng to 100 ng/mL), TNF‐α (Genzyme, Cambridge, MA, USA) (doses from 1 ng to 100 ng/mL), IL‐4 (Genzyme) (doses from 10 ng to 100 ng/mL) or IL‐13 (Chemicon International, Temecula, CA, USA) (doses from 10 ng to 100 ng/mL). After stimulation with respective recombinant cytokines for 5 days, CNEC were harvested with Trypsin‐EDTA (Gibco), washed and stained with the FITC‐conjugated mouse anti‐human HLA‐DR mAb (Becton Dickinson), PE‐conjugated mouse anti‐human CD80 mAb (Pharmingen) or the PE‐conjugated mouse anti‐human CD86 mAb (Pharmingen) and analysed by flow cytometry using a FACScan® (Becton Dickinson). Gating for live epithelial cells was performed as shown in Fig. 3a (upper panel) and dead cells were excluded by propidium iodine (PI) staining. Negative control was performed by substituting the FITC/PE‐conjugated primary antibody with isotype‐matched FITC/PE‐conjugated mouse IgG. A shift to the right in the histogram after stimulation with relevant cytokines as compared with unstimulated CNEC, indicates the increase in mean flourescence intensity (MFI). Results are expressed as the percentage positive CNEC expressing CD86 or HLA‐DR. An increase in the percentage positive CNEC expressing CD86 or HLA‐DR relative to unstimulated (control) indicates an up‐regulation of CD86 and HLA‐DR.
Figure 1.
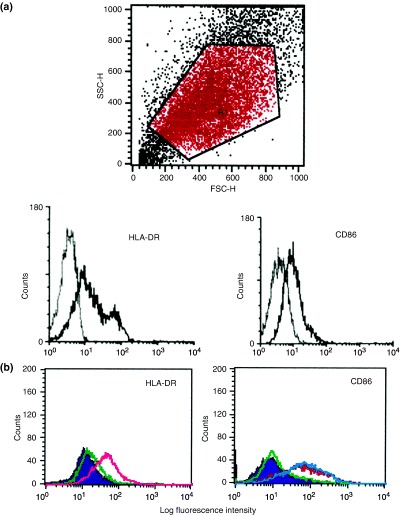
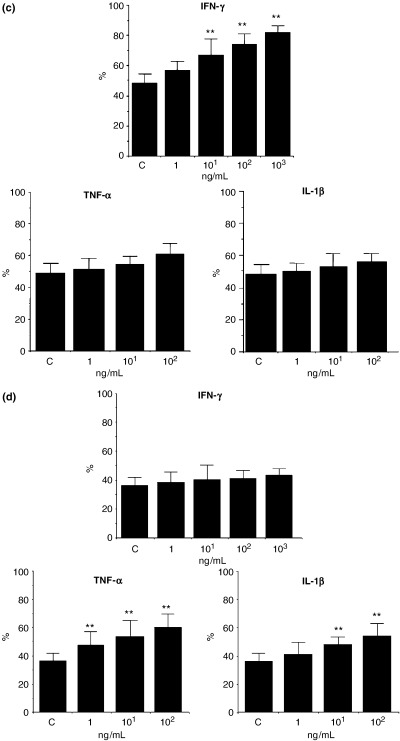
Fig. 3. Effect of IFN‐γ, TNF‐α and IL‐1β on HLA‐DR and CD86 expression in cultured nasal epithelial cells (CNEC). Expression of HLA‐DR and CD86 in CNEC was analysed by flow cytometry as described in the text. Upper panel: forward and side scatter showing gating for epithelial cells. Lower panel: Left: histogram of a representative sample of unstimulated CNEC from a patient with PAR showing the expression of HLA‐DR the (grey line shows the negative control, and the black line shows the HLA‐DR stained one). Right: histogram of a representative sample of unstimulated CNEC from the same patient with PAR showing the expression of CD86 (the grey line shows the negative control, and the black line shows the CD86‐stained one). (b) Left: histogram of a representative sample of CNEC from the same patient with PAR showing the effect of IFN‐γ and TNF‐α on HLA‐DR expression in CNEC. HLA‐DR expression in unstimulated CNEC (filled violet shaded); effect of IFN‐γ on HLA‐DR expression in CNEC (pink line); effect of TNF‐α on HLA‐DR expression in CNEC (green line). A shift to the right in the histogram can be seen after stimulation with IFN‐γ (but not TNF‐α) as compared with unstimulated CNEC. IFN‐γ but not TNF‐α up‐regulated the expression of HLA‐DR. Right: histogram of a representative sample of CNEC from the same patient with PAR showing the effect of IFN‐γ, TNF‐α and IL‐1β on the expression of CD86 in CNEC. CD86 expression in unstimulated CNEC, (filled violet shaded); effect of IFN‐γ on CD86 expression in CNEC, (green line); effect of TNF‐α on CD86 expression in CNEC (blue line) and effect of IL‐1β on CD86 expression in CNEC (red line). A shift to the right in the histogram can be seen after stimulation with IL‐1β and TNF‐α (but not IFN‐γ) as compared with unstimulated CNEC. IL‐1β and TNF‐α but not IFN‐γ up‐regulated the expression of CD86. (c) Effect of IFN‐γ, TNF‐α and IL‐1β on HLA‐DR expression in CNEC. IFN‐γ but not IL‐1β and TNF‐α up‐regulated the expression of HLA‐DR in CNEC in a dose‐dependent manner. Results are expressed as the percentage positive CNEC expressing HLA‐DR. An increase in the percentage positive CNEC expressing HLA‐DR can be seen after treatment with IFN‐γ but not TNF‐α or IL‐1β. (n=6); ** P<0.01. (d) Effect of IFN‐γ, TNF‐α and IL‐1β on CD86 expression in CNEC. TNF‐α and IL‐1β but not IFN‐γ up‐regulated the expression of CD86 in CNEC in a dose‐dependent manner. Results are expressed as the percentage positive CNEC expressing CD86. An increase in the percentage positive CNEC expressing CD86 can be seen after treatment with TNF‐α and IL‐1β but not with IFN‐γ. (n=6); ** P<0.01.
To examine the influence of DEP on the expression of HLA‐DR, CD80 and CD86 on the NEC, the CNEC were treated for 5 days with DEP (doses from 5 ng to 1 μg/mL) or DEP plus 10 μg/mL purified mite antigen Dermatophagoides farinae II (Der f II) (Torii Pharmaceutical, Tokyo, Japan). DEP was obtained by collecting the exhaust from a light‐duty diesel passenger car into a cyclone impactor equipped with dilution tunnel constant volume sampler systems as described previously [25, 26]. [30 mg of DEP was dissolved in 1 mL of DMSO (Sigma); subsequently, it was dissolved in the medium to a dilution of 1 mg/mL and diluted further for use]. After treatment with DEP or DEP and purified mite antigen (Der f II), the cells were harvested and analysed for the expression of HLA‐DR, CD80 and CD86, by flow cytometry as described above.
T cell proliferation assay
Peripheral blood samples were obtained from the same patients with PAR to HDM. Peripheral blood mononuclear cells were isolated by Ficoll–Hypaque density gradient centrifugation. Autologous T cells were purified from the peripheral blood mononuclear cells by depletion of monocytes and B cells using plastic adherence and passage through nylon wool columns. The purity of T cells was assessed by staining with the anti‐CD3 mAb. Purified T cells (1 × 105/well) were co‐cultured with equal cell numbers of mitomycin‐C (Wako Pure Chemical, Osaka, Japan)‐treated CNEC in the presence or absence of 0.1 μg/mL of phytohaemagglutinin (PHA) (Wako Pure Chemical) (as a positive control) or 1, 5 and 10 μg/mL of purified mite antigen Der f II in RPMI 1640 (GIBCO) supplemented with 10% FCS, 2 mmol/L glutamine and 1% penicillin/streptomycin. For the blocking studies, purified anti‐HLA‐DR mAb, anti‐CD80 mAb, anti‐CD86 or the relevant mouse IgG (as control) were added at a final concentration of 5 μg/mL at the start of the culture. The cultures were incubated for 72 h and pulsed with 1 μCi/well of (3H)‐thymidine for the last 16 h. The cells were then harvested and counted with a Liquid Scintillation System LSC‐3000 scintillation counter (Aloka, Tokyo, Japan). Proliferation data were expressed as a stimulation index (the counts per minute of antigen‐stimulated cells divided by the counts per minute of unstimulated cells).
Measurement of cytokines by enzyme‐linked immunosorbent assay (ELISA)
The levels of GM‐CSF, IL‐8 and IL‐6 in antigen‐stimulated CNEC were measured by the respective IL‐6‐, IL‐8‐ and GM‐CSF‐specific ELISA kits (R&D Systems).
Statistical analysis
All results are expressed as the mean±SD. Statistical analysis for the data from SAR patients was performed using the Wilcoxon signed‐rank test, and the statistical analysis for the remaining data was performed using the Mann–Whitney U‐test. A P‐value <0.05 was considered to be statistically significant. The software program used for the statistical analysis was SPSS for Windows 11.5J, SPSS Japan Inc., Tokyo, Japan.
Results
HLA‐DR, CD80 and CD86 expression in freshly isolated NEC of SAR patients to Japanese cedar pollen pre‐ and in‐season
The proportion of HLA‐DR‐positive NEC in the pre‐season and in‐season nasal epithelial scrapings of patients with JCP is as shown in Fig. 1a. Although in five out of 13 patients the baseline numbers of NEC expressing HLA‐DR were modestly elevated even pre‐season (probably due to minimal persistent inflammation), the percentage of NEC expressing HLA‐DR was significantly increased in‐season as compared with pre‐season (pre‐season: 27.4±12.7%, in‐season: 54.3±6.8%, P<0.01). The proportion of CD86‐positive NEC pre‐season and in‐season of patients with JCP is as shown in Fig. 1b. In the same five patients, there was a marginal baseline increase in CD86 expression in NEC even pre‐season. However, CD86 expressing NEC were significantly increased in‐season as compared with pre‐season (pre‐season: 23.7±8.4%, in‐season: 51.5±7.8%, P<0.01). Representative photographs of the staining are shown in Figs 2a–f. Moreover, the intensity of staining was marked in‐season as compared with weak staining pre‐season. NEC from the SAR patients did not express CD80 either pre‐season or in‐season. Our results demonstrated that a proportion of NEC constitutively expressed HLA‐DR and CD86 and that the proportion of HLA‐DR and CD86 expressing NEC was increased in‐season in SAR patients. Morphologically, the epithelial cell types that expressed HLA‐DR and CD86 appeared to be ciliated cells and basal cells.
Figure 2.
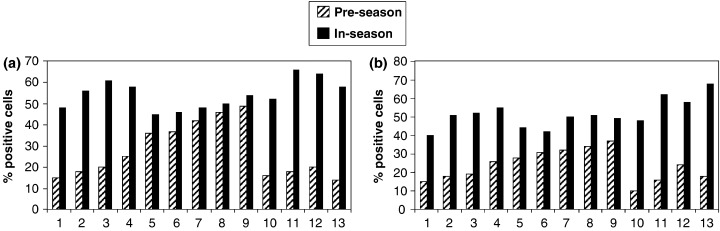
Fig. 1. HLA‐DR and CD86 expression in freshly isolated nasal epithelial cells (NEC) pre‐season and in‐season of patients with seasonal allergic rhinitis (SAR) to Japanese cedar pollen. The proportion of HLA‐DR‐ and CD86‐positive NEC pre‐season and in‐season of the SAR patients was analysed by immunohistochemistry, as described in the text. (a) The proportion of HLA‐DR‐positive NEC pre‐season and in‐season. (b) The proportion of CD86‐positive NEC pre‐season and in‐season. The percentage of NEC expressing HLA‐DR and CD86 was increased in‐season as compared with pre‐season. (n=13).
Figure 3.
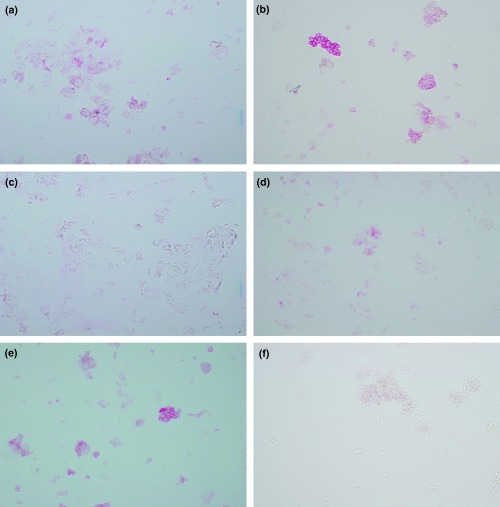
Fig. 2. HLA‐DR and CD86 expression in freshly isolated nasal epithelial cells (NEC) of patients with seasonal allergic rhinitis to Japanese cedar pollen, pre‐ and in‐season. (a) Expression of HLA‐DR in NEC, pre‐season. (b) Expression of HLA‐DR in NEC, in‐season. (c) Negative control with isotype‐matched IgG2a. (d) Expression of CD86 in NEC, pre‐season. (e) Expression of CD86 in NEC, in‐season. (f) Negative control with Isotype‐matched IgG1. The intensity of staining of both HLA‐DR and CD86 in NEC was greater in‐season as compared with pre‐season. Negative control showed no positive staining.
Effects of IFN‐γ, TNF‐α and IL‐1β on HLA‐DR and CD86 expression in cultured nasal epithelial cells
We examined the expression of HLA‐DR, CD80 and CD86 in CNEC and its regulation by cytokines, using flow cytometry. Epithelial cell purity in CNEC was confirmed in previous experiments by cytokeratin staining. Gating for live epithelial cells was performed (Fig. 3a) and dead cells were excluded by propidium iodine (PI) staining. CNEC expressed HLA‐DR and CD86 (Fig. 3a) but not CD80 and negative controls with PE‐ and FITC‐conjugated isotype control showed no staining (Fig. 3a). When we examined the effect of IFN‐γ, TNF‐α and IL‐1β on the HLA‐DR expression in CNEC, as shown in Figs 3b and c, IFN‐γ up‐regulated the expression of HLA‐DR in CNEC in a dose‐dependent manner (P<0.01). However, TNF‐α and IL‐1β did not up‐regulate the expression of HLA‐DR in CNEC (Figs 3b and c). We also studied the effect of IFN‐γ, TNF‐α and IL‐1β on the CD86 expression in CNEC. As shown in Figs 3b and d, TNF‐α and IL‐1β up‐regulated the expression of CD86 in CNEC in a dose‐dependent manner (P<0.01). On the other hand, IFN‐γ did not have any marked effect on the expression of CD86 in CNEC (Figs 3b and d). IL‐4 and IL‐13 did not have any effect on the expression of HLA‐DR or CD86 in CNEC (data not shown).
The increase in the percentage positive NEC expressing CD86 and HLA‐DR in‐season of SAR patients, as compared with off season and as analysed by immunohistochemistry is in concert with the increased expression of CD86 and HLA‐DR in NEC of PAR patients at baseline (unstimulated CNEC) and the increase in percentage positive NEC expressing HLA‐DR or CD86 in response to relevant cytokines, as analysed by FACS.
Proliferative responses of autologous T cells to Der f II pulsed CNEC and inhibitory effects of anti‐CD80, CD86 and HLA‐DR mAbs
We next investigated the ability of CNEC to induce the proliferation of autologous T cells. Antigen‐pulsed (Der f II 1, 5, 10 μg/mL) mitomycin‐treated CNEC induced proliferation on T cells (purity >95%) in a dose‐dependent manner (Fig. 4a). We then looked at the ability of PHA (0.1 μg/mL) pulsed mitomycin‐treated CNEC to induce the proliferation of autologous T cells (Fig. 4b). Further, in order to determine the functional co‐stimulatory molecules involved in the interaction of T cells and CNEC, we examined the effect of mAbs against CD80, CD86 and HLA‐DR. The proliferative response of these T cells co‐cultured with antigen‐pulsed CNEC was significantly (P<0.01) inhibited by anti‐CD86 or anti‐HLA‐DR mAbs, but not by the anti‐CD80 mAb (Fig. 4c). These results suggest that NEC may play a role in antigen presentation through the expression of HLA‐DR and CD86.
Figure 4.
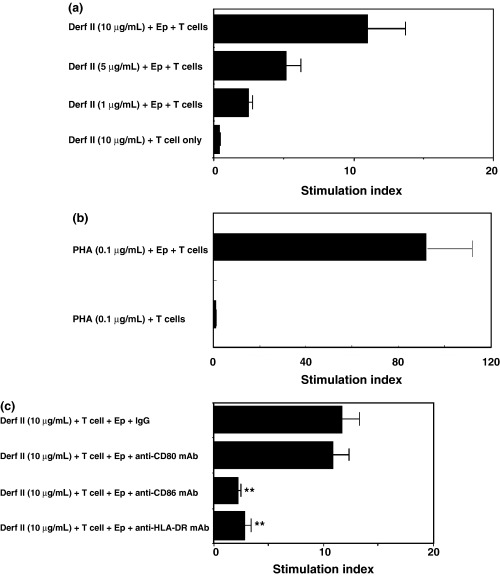
Fig. 4. Proliferation of autologous T cells to mite antigen (Der f II)/PHA‐pulsed mitomycin‐C‐treated cultured nasal epithelial cells (CNEC). Purified T cells (1 × 105/well) from perennial allergic rhinitis patients with house dust mite allergy were co‐cultured for 72 h with mitomycin–C‐treated CNEC in the presence or absence of Der f II (10 μg/mL) or phytohaemaglutinin (PHA) (0.1 μg/mL) and the proliferation of T cells was analysed by (3H)‐thymidine uptake. (a) Der f II (1, 5, 10 μg/mL) pulsed mitomycin‐treated CNEC induced the proliferation of T cells in a dose‐dependent manner, (n=3). (b) PHA (0.1 μg/mL) pulsed mitomycin‐treated CNEC induced the proliferation of autologous T cells, (n=3). (c) Inhibition studies were performed using the anti‐HLA‐DR monoclonal antibody (mAb) anti‐CD80 mAb, and the anti‐CD86 mAb or the relevant mouse IgG (as control) (5 μg/mL). Anti‐CD86 and anti‐HLA‐DR mAbs, but not anti‐CD80 inhibited the epithelial cell‐induced T cell proliferation. (n=6); ** P<0.01.
Effect of DEP on HLA‐DR and CD86 expression in cultured NEC
We also examined the effect of DEP on the HLA‐DR, CD80 and CD86 expression in CNEC, by flow cytometry. Treatment with DEP (100 ng/mL)alone significantly (P<0.01) up‐regulated the expression of HLA‐DR but not CD86 in CNEC in a dose‐dependent manner (Figs 5a and b). However, stimulation of CNEC with a combination of DEP (100 ng/mL) and mite antigen (Derf II) significantly up‐regulated the expression of both HLA‐DR (Figs 6a and b) and CD86 in CNEC. CNEC did not express CD80 in any experiment. Treatment with mite antigen alone did not have a statistically significantly effect on the expression of HLA‐DR or CD86 (data not shown) in CNEC. These findings strongly suggest the possibility that DEP may enhance the antigen presentation function of NEC.
Figure 5.
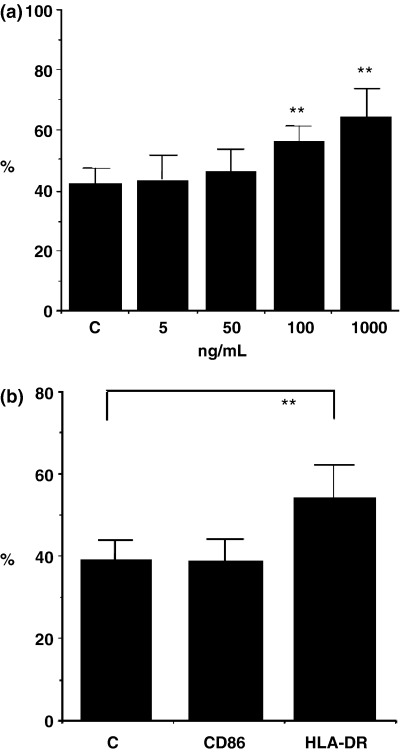
Fig. 5. Effect of diesel exhaust particles (DEP) on HLA‐DR and CD86 expression in cultured nasal epithelial cells (CNEC) from perennial allergic rhinitis patients. The effect of DEP on HLA‐DR and CD86 expression on CNEC was analysed by flow cytometry after treatment of CNEC with DEP (5 ng −1 μg/mL) for 5 days, as described in the text. Results are expressed as the percentage positive CNEC expressing HLA‐DR. (a) Effect of DEP on HLA‐DR expression in CNEC. An increase in the percentage positive CNEC expressing HLA‐DR can be seen after treatment with DEP, in a dose‐dependent manner. Treatment with DEP alone significantly up‐regulated the expression of HLA‐DR in CNEC in a dose‐dependant manner.n=6; ** P<0.01. (b) Effect of DEP on HLA‐DR and CD86 expression in CNEC. An increase in the percentage‐positive CNEC for HLA‐DR but not CD86 can be seen after treatment with DEP. Treatment with DEP alone 100 ng/mL up‐regulated HLA‐DR expression in CNEC but not the expression of CD86. (n=7); ** P<0.01.
Figure 6.
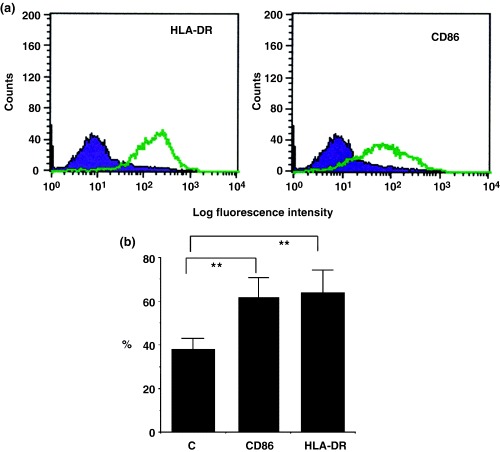
Fig. 6. Effect of diesel exhaust particles (DEP) and mite antigen on HLA‐DR and CD86 expression in cultured nasal epithelial cells (CNEC) from perennial allergic rhinitis (PAR) patients. The effect of DEP and mite antigen on HLA‐DR and CD86 expression on nasal epithelial cells was analysed by flow cytometry after treatment of CNEC with DEP plus purified mite antigen (Der f II) for 5 days, as described in the text. (a) Histogram of a representative sample of a patient with PAR showing up‐regulation of HLA‐DR (left histogram) and CD86 (right histogram) in CNEC after treatment with DEP plus (Derf II) mite antigen (violet full shaded: unstimulated CNEC; green line: after stimulation with DEP plus mite antigen). A shift to the right can be seen after treatment with DEP plus (Derf II) mite antigen as compare with unstimulated CNEC. (b) Results are expressed as the percentage‐positive cells expressing HLA‐DR or CD86. An increase in the percentage positive CNEC expressing HLA‐DR and CD86 can be seen after treatment with DEP plus (Derf II) mite antigen. (n=7); ** P<0.01.
Levels of GM‐CSF, IL‐6 and IL‐8 in culture supernatants of antigen‐stimulated CNEC
The levels of GM‐SCF in antigen‐stimulated CNEC (2.74±0.71 ng/mL), were higher than that of unstimulated control CNEC (1.83±0.38 ng/mL). The level of IL‐8 in antigen‐stimulated CNEC (97.4±10.1 ng/mL), was higher than that of unstimulated control CNEC IL‐8 (66.1±8.7 ng/mL). Similarly the level of IL‐6 in antigen‐stimulated CNEC (7.44±2.17 ng/mL) was higher than that of unstimulated control CNEC (5.96±1.78 ng/mL). Addition of DEP to mite antigen to stimulate CNEC did not further up‐regulate the antigen‐induced cytokine production (data not shown).
Discussion
Airway epithelial cells have been considered to play a simple role as a mucosal barrier while being involved in the secretion of mucous or the removal of foreign agents by their cilia [1]. However, over the recent years, several studies suggest that airway epithelial cells can also act as immune effector cells in response to endogenous or exogenous stimuli and play a key role in the immunologic interaction between the airway and the external environment, being the primary interface for antigenic materials in immune and inflammatory responses.
Epithelial cells are known to play an important role in allergic inflammation by the increased expression of adhesion molecules like ICAM‐1 and VCAM‐1 [10, 11] and the increased production of inflammatory mediators like IL‐6, IL‐8, GM‐CSF and TNF‐α [8, 9]. However, epithelial cells may also play other important roles via direct cell‐to‐cell interaction. In this study, we noticed the expression of HLA‐DR on NEC in patients with AR. HLA‐DR is known to play a significant role in antigen presentation to T cells [16], and CD80 and CD86 are essential to this function as co‐stimulatory molecules. Signals transduced through the T cell receptor after antigen recognition are not sufficient for full T cell activation. Recent studies have demonstrated that at least two signals provided by APC are required to induce effective T cell activation [27]. The first signal is antigen specific and occurs after T cell receptor recognition of the MHC‐antigen complex. The second signal, often termed the co‐stimulatory signal is produced through a set of receptor and co‐receptor interactions between APC and T cells. More specifically, the interaction of CD80 or CD86 on APC with CD28 on T cells is required for full T cell activation and effector function [28, 29]. The importance of these interactions is reflected in the observations that T cell receptor recognition of the MHC–antigen complex in the absence of co‐stimulatory signals can lead to the induction of anergy [18, 27]. Recent studies have suggested that structural cells, such as keratinocytes [30], gastric epithelial cells [31] and colonic epithelial cells, also expressed CD80 and CD86 [32] and could provide co‐stimulatory signals through these molecules. Ye et al. [31] demonstrated that the human gastric epithelial cell line and freshly isolated epithelial cells from gastric biopsies expressed CD80 and CD86, and T cell proliferation by gastric epithelial cell was significantly inhibited by anti‐CD86 mAb. Nakazawa et al. [32] demonstrated that freshly isolated colonic epithelial cells from patients with ulcerative colitis efficiently co‐stimulated the proliferation of CD4+ T cells, and this response was significantly inhibited by anti‐CD86 mAb. In the present study, we demonstrated an increased expression of HLA‐DR and CD86 in freshly isolated nasal scrapings of patients with SAR to JCP in‐season as compared with that off‐season. We also demonstrated an increased expression of HLA‐DR and CD86 in NEC of patients with PAR to HDM. The increase in percentage positive NEC expressing CD86 and HLA‐DR that is increased in‐season in SAR patients, as analysed by immunohistochemistry, is comparable with the increase in percentage‐positive NEC expressing HLA‐DR or CD86 of PAR patients in response to relevant cytokines or DEP plus antigen as analysed by FACS. Furthermore, we also demonstrated that NEC can function as APC through the expression of HLA‐DR and CD86. CD80 expression was not detected in NEC by immunohistochemical staining, and anti‐CD80 mAb could not inhibit the epithelial cell‐induced T cell proliferation. These findings suggest that CD86 is more important as compared with CD80 in regulating antigen presentation by NEC in patients with AR.
Antigen presentation to T cells is an important intital step in the pathophysiology of allergic diseases like AR and atopic asthma. In a previous study using immunohistochemical analysis, dendritic cells (professional APC) were reported in the nasal epithelial layer, but numerically these cells are very few in number [33, 34]. Antigen‐presenting function by NEC could be important because of their predominance in number in the nasal epithelial layer. Godthelp et al. [34] reported that the number of HLA‐DR‐positive NECs did not increase after 2 weeks of antigen provocation out of season in SAR (grass pollen) patients. However, in the present study, HLA‐DR was up‐regulated in‐season in NEC as the nasal epithelial scrapings were obtained from patients under natural exposure to allergen and during the peak season in a heavy pollen JCP season. These observations suggest that sufficient natural exposure to allergen is more important to induce a significant up‐regulation of HLA‐DR in NEC to of SAR patients.
There is increasing evidence that AR or asthma, chronic airway inflammatory diseases characterized by reversible airway obstruction, hyper‐reactivity and lymphocyte/eosinophil recruitment are driven and maintained by chronically activated T cells with a T‐helper 2 (Th2) phenotype. These T cells promote the activation and recruitment of B cells and regulate the Ig class switch to the development of antigen‐specific IgE responses [35, 36, 37, 38]. We previously reported that a majority of the CD4+ T cells in the nasal mucosa of allergic rhinitics co‐expressed the CD45RO surface molecule, and a predominant proportion of CD4+ T cells in the nasal epithelium were CD45RO+ (memory T cells) [39]. These T cells can be activated more easily as compare with the naïve T cells. Epithelial cells are in direct contact with the inhalant allergens and based on the presence of the memory T cells in the epithelial layer and the capacity of epithelial cells to induce antigen‐specific proliferation of T cells, epithelial cells may play more important roles in on‐going allergic inflammation. While we have shown that mast cell–epithelial cell interaction contributes to enhancing allergic inflammation [40], airway epithelial cell‐induced T cell activation may significantly contribute to the development of chronic airway inflammatory diseases such as AR or asthma. Furthermore, in separate studies, we have shown that NEC express the α and γ chains of the high‐affinity IgE receptor (FcɛRI), further emphasizing their potential role in the recognition of IgE‐complexed allergens. Moreover, we have also shown that IL‐4 can up‐regulate the expression of the FcɛRI α chain in NEC, suggesting an up‐regulation of antigen‐presenting function in an enhanced Th2 microenvironment as in the case of allergic inflammation [41].
In this study, we demonstrated the increased expression of HLA‐DR and CD86 observed in‐season in patients with JCP, and enhanced expression of these molecules on CNEC stimulated by inflammatory cytokines like IFN‐γ, IL‐1β and TNF‐α. Bachert et al. [42] previously reported the increased levels of IL‐1β and TNF‐α in nasal secretions from patients with AR. This finding suggests the possibility that the increased levels of pro‐inflammatory cytokines like IL‐1β and TNF‐α in‐season in patients with JCP may contribute towards the up‐regulation of the CD86 expression in NEC. In contrast, a lower level of IFN‐γ mRNA was observed in the nasal mucosa of patients with PAR [43] and in the nasal mucosa of patients with SAR after the allergen provocation test [44]. We demonstrated modest levels of IFN‐γ in the nasal mucosa of PAR patients even though the levels were lower than in those with chronic infective rhinitis [45]. Linden et al. [46] demonstrated high levels of IFN‐γ in nasal lavage fluid from subjects with acute infectious rhinitis caused by coronavirus inoculation, in contrast, and lower levels of IFN‐γ in subjects with AR after allergen challenge. These findings suggest the possibility that respiratory viral infections can up‐regulate the APC function of NEC through the enhanced expression of HLA‐DR induced by IFN‐γ and this may in part explain the aggravation of allergic symptoms upon viral infection.
Epidemiological studies have shown a significant association between exposure to various pollutants and increase in allergic diseases. DEP has been demonstrated to have an adjuvant activity for IgE synthesis in vivo [26, 47]. Diaz‐Sanchez et al. showed that transnasal challenges of the DEP‐derived extract in humans enhanced local IgE and cytokine production [25, 48, 49], and combined DEP and ragweed allergen challenge enhanced local ragweed‐specific IgE [50]. We demonstrated that DEP up‐regulated the expression of HLA‐DR in CNEC and this up‐regulation was enhanced by a combined stimulation of DEP and mite antigen (Der f II). Although DEP alone did not up‐regulate the expression of CD86, the combined stimulation of DEP and Der f II significantly up‐regulated the expression of CD86 in CNEC. In vitro studies have shown that mite allergens induce the production of proinflammatory cytokines from airway epithelial cells through the activation of protease‐activated receptor (PAR)‐2 [51]. On the other hand, DEP can enhance the cytokine secretion from airway epithelial cells [23] and can up‐regulate the expression of adhesion molecules like ICAM‐1 on bronchial epithelial cells [52], and this process is dependent on p38 mitogen‐activated protein kinase (MAPK) and nuclear factor‐κB (NF‐κB) activation pathways [52, 53]. In the present study, mite antigen up‐regulated the production of IL‐6, IL‐8 and GM‐CSF from NEC but this was not further up‐regulated with addition of DEP. Although the precise mechanism by which it up‐regulates the expression of HLA‐DR or CD86 is not yet known, similar mechanisms may probably regulate the expression of these molecules. One of the possible mechanisms may be via the arylhydrocarbon receptor (AhR) in cytosol [54]. More recently, using TLR‐4 point mutant mice, Inoue et al. [55] demonstrated that DEP exposure significantly induced the lung expression of IL‐1β, keratinocyte chemoattractant (KC) and macrophage inflammatory protein (MIP)‐1α when compared with vehicle challenge. In the presence of DEP, the levels of IL‐1β, KC and fibrinogen were up‐regulated. These researchers suggest that TLR‐4 is one of the recognition receptors against DEP in the airways. In fact, NEC express TLR‐4 and its expression is up‐regulated in patients with PAR (unpublished observations). Furthermore, recently, DEP has been shown to induce antigen‐independent dendritic cell maturation (phenotypic or functional) via epithelial cell–dendritic cell interactions mediated by epithelial cell derived GM‐CSF [56]. On the other hand, DEP can enhance peripheral blood T cell activation directly in severe asthmatics [57]. However, the precise immunological mechanisms by which DEP enhance allergic sensitization and inflammation are not yet well defined.
Taken together, we demonstrated for the first time that NEC from patients with AR can amplify the allergic reaction and contribute to on‐going allergic inflammation via its capacity to act as APC through the increased expression of HLA‐DR and CD86. Furthermore, these molecules are up‐regulated by pro‐inflammatory cytokines like IFN‐γ, TNF‐α, IL‐1β as well as allergen and DEP. These findings also demonstrate a novel role for DEP in enhancing allergic inflammation through the up‐regulation of co‐stimulatory molecules that regulate antigen presentation in NEC and thus modulation of the airway epithelial cell function.
Acknowledgements
This study was supported by Grants‐in‐Aid from the Ministry of Education and Science of Japan.
References
- 1. Jeffery PK. Morphologic features of airway surface epithelial cells and glands. Am Rev Respir Dis 1983; 128:S14–20. [DOI] [PubMed] [Google Scholar]
- 2. Henke D, Danilowicz RM, Curtis JF, Boucher RC, Eling TE. Metabolism of arachidonic acid by human nasal and bronchial epithelial cells. Arch Biochem Byophys 1988; 267:426–36. [DOI] [PubMed] [Google Scholar]
- 3. Daffern PJ, Jagels MA, Saad JJ, Fischer W, Hugli TE. Upper airway epithelial cells support eosinophil survival in vitro through production of GM‐CSF and prostaglandin E2: regulation by glucocorticoids and TNF-α. Allergy Asthma Proc 1999; 20:243–53. [DOI] [PubMed] [Google Scholar]
- 4. Nakano J, Takizawa H, Ohtoshi T et al Endotoxin and pro‐inflammatory cytokines stimulate endothelin‐1 expression and release by airway epithelial cells. Clin Exp Allergy 1994; 24:330–6. [DOI] [PubMed] [Google Scholar]
- 5. Ohnishi M, Ruhno J, Beinenstock J, Dolovich J, Denburg JA. Hematopetic growth factor production by cultured cells of human nasal polyp epithelial scraping: kinetics, cell source and relationship to clinical status. J Allergy Clin Immunol 1989; 83:1091–100. [DOI] [PubMed] [Google Scholar]
- 6. Ohtoshi T, Tsuda T, Vancheri C et al Human upper airway epithelial cell‐derived granulocyte‐macrophage colony‐stimulating factor induces histamine‐containing cell differentiation of human progenitor cells. Int Arch Allergy Appl Immunol 1991; 95:376–84. [DOI] [PubMed] [Google Scholar]
- 7. Cromwell O, Hamid Q, Corrigan CJ et al Expression and generation of interleukin‐8, IL‐6 and granulocyte‐macrophage colony‐stimulating factor by bronchial epithelial cells and enhancement of IL‐1β and tumor necrosis factor‐α. Immunology 1992; 77:330–7. [PMC free article] [PubMed] [Google Scholar]
- 8. Kenney JS, Baker C, Welch MR, Altman LC. Synthesis of interleukin‐1 alpha, interleukin‐6, and interleukin‐8 by cultured human nasal epithelial cells. J Allergy Clin Immunol 1994; 93:1060–7. [DOI] [PubMed] [Google Scholar]
- 9. Nonaka M, Nonaka R, Jordana M, Dolovich J. GM‐CSF, IL‐8, IL‐1R, TNF‐alpha R, and HLA‐DR in nasal epithelial cells in allergic rhinitis. Am J Respir Crit Care Med 1996; 153:1675–81. [DOI] [PubMed] [Google Scholar]
- 10. Tosi MF, Stark JM, Smith CW, Hamdani A, Gruenert DC, Infed MD. Induction of ICAM‐1 expression on human airway epithelial cells by inflammatory cytokines: effects on neutrophil–epithelial cell adhesion. Am J Resir Cell Mol Biol 1992; 7:214–21. [DOI] [PubMed] [Google Scholar]
- 11. Atsuta JS, Sterbinsky A, Plitt J, Schwiebert LM, Bochner BS, Schleimer RP. Phenotype and cytokine regulation of the BEAS‐2B human bronchial epithelial cell: demonstration of inducible expression of the adhesion molecules VCAM-1 and ICAM-1. Am J Resir Cell Mol Biol 1997; 17:571–82. [DOI] [PubMed] [Google Scholar]
- 12. Brandtzaeg P. Immunocompetent cells of the upper airway: functions in normal and diseased mucosa. Eur Arch Otorhinolaryngol 1995; 252:S8–21. [DOI] [PubMed] [Google Scholar]
- 13. Brandtzaeg P, Halstensen TS, Huitfeldt HS et al Epithelial expression of HLA, secretory component (poly‐Ig receptor), and adhesion molecules in the human alimentary tract. Ann NY Acad Sci 1992; 664:157–79. [DOI] [PubMed] [Google Scholar]
- 14. Rossi GA, Sacco O, Balbi B et al Human ciliated bronchial epithelial cells: expression of HLA-DR antigens and of the HLA-DR alpha gene, Modulation of the HLA-DR antigens by gamma-interferon and antigen-presenting function in the mixed lymphocyte reaction. Am J Resir Cell Mol Biol 1990; 3:431–9. [DOI] [PubMed] [Google Scholar]
- 15. Mezzetti M, Soloperto M, Fasoli A, Mattoli S. Human bronchial epithelial cells modulate CD3 and mitogen‐induced DNA synthesis in T cells but function poorly as antigen‐presenting cells compared to pulmonary macrophages. J Allergy Clin Immunol 1991; 87:930–8. [DOI] [PubMed] [Google Scholar]
- 16. Schwartz BD. HLA molecules: sentinel of the immune response. Am J Resir Cell Mol Biol 1991; 5:211–2. [DOI] [PubMed] [Google Scholar]
- 17. Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin‐2 enhancer activity by the T cell accessory molecule CD28. Science 1991; 251:313–6. [DOI] [PubMed] [Google Scholar]
- 18. Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28‐mediated signaling co‐stimulates murine T cells and prevents induction of anergy in T‐cell clones. Nature 1992; 356:607–9. [DOI] [PubMed] [Google Scholar]
- 19. June CH, Ledbetter JA, Gillespie MM, Linsten T, Thompson CB. T cell proliferation involving the CD28 pathway is associated with cyclosporine‐resistant interleukin 2 gene expression. Mol Cell Biol 1987; 7:4472–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chambers CA, Allison JP. Co‐stimulation in T cell responses. Curr Opin Immunol 1997; 9:396–404. [DOI] [PubMed] [Google Scholar]
- 21. Gause WC, Mitro V, Via C, Linsley P, Urban JF, Greenwald RJ. Do effecter and memory T cells also need B7 ligand co‐stimulatory signals? J Immunol 1997; 159:1055–8. [PubMed] [Google Scholar]
- 22. Salvi S, Blomberg A, Rudell B et al Acute inflammatory responses in the airways and peripheral blood after short‐term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med 1999; 159:702–9. [DOI] [PubMed] [Google Scholar]
- 23. Ohtoshi T, Takizawa H, Okazaki H et al Diesel exhaust particles (DEP) stimulate human airway epithelial cells to produce cytokines relevant to airway inflammation in vitro . J Allergy Clin Immunol 1998; 101:778–85. [DOI] [PubMed] [Google Scholar]
- 24. Pawankar R, Okuda M, Hasegawa S et al Interleukin‐13 expression in the nasal mucosa of perennial allergic rhinitis. Am J Respir Crit Care Med 1995; 152:2059–67. [DOI] [PubMed] [Google Scholar]
- 25. Takenaka H, Zhang K, Diaz‐Sanchez D, Tsien A, Saxon A. Enhanced human IgE production results from exposure to the aromatic hydrocarbons from diesel exhaust: direct effects on B-cell IgE production. J Allergy Clin Immunol 1995; 95:103–15. [DOI] [PubMed] [Google Scholar]
- 26. Muranaka M, Suzuki S, Koizumi K et al Adjuvant activity of diesel‐exhaust particulates for the production of IgE antibody in mice. J Allergy Clin Immunol 1986; 77:616–23. [DOI] [PubMed] [Google Scholar]
- 27. Schwaltz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992; 71:1065–8. [DOI] [PubMed] [Google Scholar]
- 28. Freeman GJ, Gribben JG, Boussiotis VA et al Cloning of B7‐2: a CTLA-4 counter–receptor that costimulates human T cell proliferation. Science 1993; 262:909–11. [DOI] [PubMed] [Google Scholar]
- 29. Azuma M, Ito D, Yagita H et al B70 antigen is a second ligand for CTLA‐4 and CD28. Nature 1993; 366:76–9. [DOI] [PubMed] [Google Scholar]
- 30. Fleming TE, Mirando WS, Trefzer U, Tubesing KA, Elmets CA. In situ expression of a B7‐like adhesion molecule on keratinocytes from human epidermis. J Invest Dermatol 1993; 101:754–8. [DOI] [PubMed] [Google Scholar]
- 31. Ye G, Barrera C, Fan X et al Expression of B7‐1 and B7‐2 costimulatory molecules by human gastric epithelial cells. J Clin Invest 1997; 99:1628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakazawa A, Watanabe M, Kanai T et al Functional expression of costimulatory molecule CD86 on epithelial cells in the inflamed colonic mucosa. Gastroenterology 1999; 117:536–45. [DOI] [PubMed] [Google Scholar]
- 33. Fokkens WJ, Broekhuis‐Fluitsma DM, Rijntjes E, Vroom TM, Hoefsmit EC. Langerhans cells in nasal mucosa of patients with grass pollen allergy. Immunobiology 1991; 182:135–42. [DOI] [PubMed] [Google Scholar]
- 34. Godthelp T, Fokkens WJ, Kleinjan A et al Antigen presenting cells in the nasal mucosa of patients with allergic rhinitis during allergen provocation. Clin Exp Allergy 1996; 26:677–88. [PubMed] [Google Scholar]
- 35. Robinson DS, Hamid Q, Ying S et al Predominant TH‐2 like bronchoalveolar T‐lymphocyte population in atopic asthma. N Engl J Med 1992; 326:298–304. [DOI] [PubMed] [Google Scholar]
- 36. Wills‐karp M. Immunologic basis of antigen‐induced airway hyperresponsiveness. Annu Rev Immunol 1999; 17:255–81. [DOI] [PubMed] [Google Scholar]
- 37. Romagnani S. The role of lymphocytes in allergic disease. J Allergy Clin Immunol 2000; 105:399–408. [DOI] [PubMed] [Google Scholar]
- 38. Lordan IL, Jaffer ZH. Role of CD28/B7 co‐stimulation in airway T helper 2 (TH2) immune responses in asthma. Clin Exp Allergy 1998; 28:1317–20. [DOI] [PubMed] [Google Scholar]
- 39. Pawankar R, Okuda M, Okubo K, Ra C. Lymphocyte subjects of the nasal mucosa in perennial allergic rhinitis. Am J Respir Crit Care Med 1995; 152:2049–58. [DOI] [PubMed] [Google Scholar]
- 40. Pawankar R. Epithelial cells as immunoregulators in allergic airway diseases. Curr Opin Allergy Clin Immunol 2002; 2:1–5. [DOI] [PubMed] [Google Scholar]
- 41. Yamagishi S, Pawankar R, Takizawa R, Nonaka M, Yagi T. Nasal epithelial cells express the FceRI: IL-4-induced upregulation of the FceRI and IL-6 production [abstract]. J Allergy Clin Immunol 2003; 111:s347. [Google Scholar]
- 42. Bachert C, Wagenmann M, Hauser U. Proinflammatory cytokines: measurement in nasal secretion and induction of adhesion receptor expression. Int Arch Allergy Immunol 1995; 107:106–8. [DOI] [PubMed] [Google Scholar]
- 43. Lee CH, Rhee CS, Oh SH, Min YG, Lee MS. Increase in expression of IL‐4 and IL‐5 mRNA in the nasal mucosa of patients with perennial allergic rhinitis during natural allergen exposure. Ann Otol Rhinol Laryngol 1997; 106:215–9. [DOI] [PubMed] [Google Scholar]
- 44. Durham SR, Ying S, Varney VA et al Cytokine messenger RNA expression for IL‐3, IL‐4, IL‐5, and granulocyte/macrophage‐colony‐stimulating factor in the nasal mucosa after local allergen provocation: relationship to tissue eosinophilia. J Immunol 1992; 148:2390–4. [PubMed] [Google Scholar]
- 45. Pawankar R, Ra C. Heterogeneity of mast cells and T cells in the nasal mucosa. J Allergy Clin Immunol 1996; 98:248–26. [DOI] [PubMed] [Google Scholar]
- 46. Linden M, Greiff L, Andersson M et al Nasal cytokines in common cold and allergic rhinitis. Clin Exp Allergy 1995; 25:166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Takafuji S, Suzuki S, Koizumi K et al Diesel‐exhaust particulates inoculated by the intranasal route have an adjuvant activity for IgE production in mice. J Allergy Clin Immunol 1987; 79:639–45. [DOI] [PubMed] [Google Scholar]
- 48. Diaz‐Sanchez D, Dotson AR, Takenaka H, Saxon A. Diesel exhaust particles induce local IgE production in vivo and alter the pattern of IgE messenger RNA isoforms. J Clin Invest 1994; 94:1417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Diaz‐Sanchez D, Tsien A, Casillas A, Dotson AR, Saxon A. Enhanced nasal cytokine production in human beings after in vivo challenge with diesel exhaust particles. J Allergy Clin Immunol 1996; 98:114–23. [DOI] [PubMed] [Google Scholar]
- 50. Diaz‐Sanchez D, Tsien A, Fleming J, Saxon A. Combined diesel exhaust particle and ragweed allergen challenge markedly enhances human in vivo nasal ragweed‐specific IgE and skews cytokine production to a T helper cell 2‐type pattern. J Immunol 1997; 158:2406–13. [PubMed] [Google Scholar]
- 51. Asokananthan N, Graham PT, Stewart DJ et al House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Der p 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. J Immnnol 2002; 169:4572–8. [DOI] [PubMed] [Google Scholar]
- 52. Takizawa H, Abe S, Ohtoshi T et al Diesel exhaust particles up‐regulate expression of intercellular adhesion molecule‐1 (ICAM‐1) in human bronchial epithelial cells. Clin Exp Immunol 2000; 120:356–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takizawa H, Ohtoshi T, Kawasaki S et al Diesel exhaust particles induce NF‐kB activation in human bronchial epithelial cells in vitro : importance in cytokine transcription. J Immunol 1999; 162:4705–11. [PubMed] [Google Scholar]
- 54. Nel AE, Diaz‐Sanchez D, Ng D, Hiura T, Saxon A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J Allergy Clin Immunol 1998; 102:539–54. [DOI] [PubMed] [Google Scholar]
- 55. Inoue K, Takano H, Yanagisawa R et al The role of toll‐like receptor 4 in airway inflammation induced by diesel exhaust particles. Arch Toxicol 2006; 80:275–9. [DOI] [PubMed] [Google Scholar]
- 56. Bleck B, Tse DB, Jaspers I, Curotto de Lafaille MA, Reibman J. Diesel exhaust particles‐exposed human bronchial epithelial cells induce dendritic cell maturation. J Immunol 2006; 176:7431–7. [DOI] [PubMed] [Google Scholar]
- 57. Mamessier E, Nieves A, Vervloet D, Magnan A. Diesel exhaust particles enhance T‐cell activation in severe asthmatics. Allergy 2006; 61:581–8. [DOI] [PubMed] [Google Scholar]