

Feedback SMLM achieves ultrahigh localization precision and in situ distance measurements on the biological relevant scales.
Abstract
Single-molecule localization microscopy (SMLM) has the potential to quantify the diversity in spatial arrangements of molecules in intact cells. However, this requires that the single-molecule emitters are localized with ultrahigh precision irrespective of the sample format and the length of the data acquisition. We advance SMLM to enable direct distance measurements between molecules in intact cells on the scale between 1 and 20 nm. Our actively stabilized microscope combines three-dimensional real-time drift corrections and achieves a stabilization of <1 nm and localization precision of ~1 nm. To demonstrate the biological applicability of the new microscope, we show a 4- to 7-nm difference in spatial separations between signaling T cell receptors and phosphatases (CD45) in active and resting T cells. In summary, by overcoming the major bottlenecks in SMLM imaging, it is possible to generate molecular images with nanometer accuracy and conduct distance measurements on the biological relevant length scales.
INTRODUCTION
Super-resolution methods such as (direct) stochastic optical reconstruction microscopy (STORM) (1) and DNA point accumulation for imaging in nanoscale topography (DNA-PAINT) (2) are anticipated to revolutionize biology, as they can localize individual molecules in the cellular context. Single-molecule localization microscopy (SMLM) approaches have the potential to capture the naturally occurring diversity in molecular complexes, structures, and spatial organizations. In SMLM, single fluorophores are temporally separated so that a sparse set of point emitters can be captured in each camera frame. The temporal separation is achieved via photoactivation (3), stochastic switching of fluorophores between a fluorescent and nonfluorescent state (1), or the reversible binding of fluorophore to the target site (2). The sequential imaging of individual fluorophores, however, requires long acquisition times, as typically tens of thousands of frames are needed to map a given protein species in a cell. In the ideal case, the localization precision of each molecule primarily depends on the number of photons collected at each point (4). Each fluorescent emission originating from the same molecule undergoing photoswitching/rebinding can be regarded as a separate position measurement and theoretically merged for greater position accuracy as long as the molecule does not move during the acquisition and central-limit statistics applies (5). However, drift during camera exposure affects the localization precision and the accuracy of localization microscopy data analysis (6). Furthermore, at high densities such as proteins in clusters, insufficient localization precision render it challenging to correctly assign the collective stream of photons or positions to the appropriate molecule (Fig. 1A). In practice, the resolution is reduced to tens of nanometers (7), making it unfeasible to conduct distance measurements on biological relevant scales in situ.
Fig. 1. Feedback SMLM.
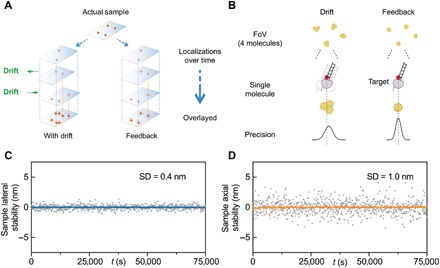
(A) Principle of active stabilization for SMLM. Residual drift compounded from multiple fluorescent detections degrades the final precision of the target molecule. (B) Drift within the on-time of an imaging strand negatively affects the precision of a single emitter. Our active stabilization is performed at faster rates than the on-time of the molecules, thus aligning a single emitter within a single frame. (C and D) Drift is reduced below the photon-limited localization precision. Lateral (C) and axial (D) stability of the sample is reflected in the SDs of 0.4 and 1 nm, respectively. In (C) and (D), gray symbols represent sample deviation, and blue and orange lines represent a 10-point average (1/1000 points plotted).
Distance measurements below 10 nm are typically determined with Föster resonance energy transfer (FRET) experiments, where energy transfer between two fluorophores is interpreted as a distance. However, FRET efficiency is also influenced by factors such as dipole alignment and the local microenvironment (8). Furthermore, separation distances >10 nm cannot be measured with FRET, because the absence of a FRET signal cannot be interpreted as the absence of molecular interactions. Measuring the Euclidian distance between two proteins to identify molecular interactions and separation distances is thus advantageous, particularly when applicable to a broad range of distances and cellular conditions.
To date, direct distance measurements with nanometer precision can be broadly grouped into two types of experiments. In the first type of experiment, the imaging setup is used to control the position of the fluorophores in the samples. For example, Pertsinidis et al. (9) achieved subnanometer distance measurements by optically trapping molecules with a feedback-controlled system. This approach typically involves highly specialized optical setups and is only applicable to a small subset of sample formats. In the second type of experiment, the same molecule is imaged many times to improve the imaging resolution (10, 11). These approaches are designed for recombinant samples (11, 12) and require extensive post-acquisition processing (12, 13). These methods can become inaccurate when distances are similar to the localization precision (<30 nm) (12) and often rely on averaging data over multiple structures and samples (11, 13). Data averaging assumes that each structure and complex is identical. While this is true for synthetic biology structures such as DNA origamis (11, 13) or well-characterized protein complexes such as the nuclear pore complex (14), it is not true for less well-defined samples. Because it is desirable to directly measure distances between two individual proteins in a wide range of sample formats, including intact cells, to capture sample heterogeneity as well as rare events, there is an unmet need for a microscope that can achieve ultrahigh localization precisions in heterogeneous biological environments.
Distance measurements are particularly important for our understanding of cellular signaling networks, as the spatial organization of signaling molecules regulates signaling outcomes. In receptor signaling, it is often known which proteins can interact but measuring which proteins do interact in intact cells is substantially more challenging. This is because only a small fraction of a given protein participates in signaling events and a few signaling events can drive signaling outcomes. For example, fewer than 10 agonist ligands binding to only a few of the ~50,000 copies of the T cell receptor (TCR) is sufficient to induce signals to activate a T cell (15, 16). To identify rare events, however, requires a technology that can determine the (re)arrangement of the molecular machinery discreetly and with high accuracy. Typically, rare events and subtle changes go undetected and tend to be overlooked or “averaged out” during postprocessing. If SMLM could be improved to achieve ultraprecision without postprocessing, it should be possible to directly measure discrete structural changes or separation distances (<30 nm) within cellular environments.
To address these challenges, we take a relatively simple approach: If we can make unprocessed SMLM data sufficiently accurate and robust, we can measure the distances between two protein species directly and thus determine the spatial scale that controls signaling reaction. We first developed an engineering solution for SMLM, termed Feedback SMLM, which can capture molecular emissions in a complex and unknown cellular environment with equal probability and high precision. We then used Feedback SMLM and DNA-PAINT to accurately measure distances between molecular species, specifically, early TCR signaling events. By recording high-quality raw data with ultrahigh localization precision and increased accuracy, we could determine separation distances directly without resorting to post-acquisition processing including drift correction, grouping, filtering, averaging, or summation.
RESULTS
Feedback SMLM
We have developed an SMLM system that performs real-time drift correction in three dimensions (3D) (fig. S1). The approach realigns molecular emissions independently from the fluorescence, therefore readjusting the point-spread function (PSF) of the molecules while these are still emitting (Fig. 1B). The system, termed Feedback SMLM, uses three types of corrections. First, there is a feedback loop between the sample and stage position. A key feature is that nonfluorescent fiducials are used outside the field of view (FoV) of the camera for SMLM, providing great flexibility in sample positioning and ensuring that fiducials do not interfere with data acquisition (fig. S1). Thus, stage corrections occur during the acquisition of fluorescence data and are completely independent from the sample nature (i.e., structures of interest) and density or distribution of fluorophores. The nonfluorescent fiducials are 3-μm polystyrene beads that are illuminated with an infrared light-emitting diode (LED) to create diffraction rings. The interference pattern is imaged onto a separate complementary metal-oxide semiconductor (CMOS) camera (112 μm × 70 μm FoV, about fivefold larger than the fluorescence camera) at a maximum speed of 370 frames per second. The diffraction rings of the 3-μm beads provide information of both the x-y and z position (17, 18). The position is calculated sufficiently fast to facilitate stage corrections at 15 Hz (fig. S2). The active stage correction provides a stabilization of 0.4 and 1 nm (SD) in the lateral (x-y) and axial (z) directions, respectively, over hours and days (Fig. 1, C and D). Unlike other SMLM instruments, the accuracy of the Feedback SMLM correction is not limited by the sample properties (19), fiducial characteristics (11, 13, 20), or fluorophore density (9, 21). An additional advantage of our autonomous optical feedback loop for stage positioning is that the sample position is maintained during buffer exchanges, for example, during multicolor DNA-PAINT experiments (fig. S3).
In addition to the stage/sample stabilization, we also use an autonomous optical feedback loop in the emission path. A white LED, integrated in the microscope body, creates an optical fiducial on the electron-multiplying charge-coupled device (EMCCD) camera. The position of the LED is localized with a precision of 0.05 nm. A piezoelectric mirror is used to account for long-term drift of the microscope (i.e., mechanical instabilities) and reduces the image drift to 0.22 nm (fig. S4). Structural instability of the microscope is generally negligible over short-term experiments (seconds to minutes) but can have an influence over the course of long-term experiments (hours to days). The piezoelectric mirror correction ensures that the remaining fluorescence path is stable, removes the need for any post-acquisition drift correction, and guarantees reproducibility across experiments. The third and final correction reduces the variation registered across the EMCCD ~10-fold (fig. S5).
Ultrahigh resolution without post-acquisition processing
It is advantageous to generate raw (unprocessed) SMLM data with ultrahigh localization precision rather than relying on post-acquisition corrections. The latter can introduce errors in the quantification including blurring of the localization. Here, we demonstrate that our technique is particularly useful for DNA-PAINT, where the improvement by Feedback SMLM is twofold. First, DNA-PAINT relies on repeated binding of the imaging strand to the target molecule (2). The longer the acquisition period, the more reliable a single-molecule detection by DNA-PAINT becomes. Therefore, long experimental times are often used to ensure that all target molecules are detected (i.e., ~6 hours per target), which makes DNA-PAINT experiments particularly susceptible to long-term drift. Second, each binding event in DNA-PAINT is, on average, ~200 ms. Movement within this millisecond window also degrades the localization precision (demonstrated in fig. S6). Here, Feedback SMLM improves DNA-PAINT images because each binding event is localized more precisely.
We achieved ultrahigh resolution (~1 nm localization precision) of individual fluorescent accumulation points with DNA origami structures that consist of three docking sites, which are separated by 20 nm (Fig. 2, A and B). Even without averaging, summation, individual correction of binding sites, or any assumption regarding the number and relative position of the binding sites, the reconstruction of a single origami ruler is possible. We demonstrate an about eightfold improvement in resolution of the Feedback SMLM over standard SMLM imaging using the same DNA origami structures (Fig. 2, C and D) (22).
Fig. 2. Feedback SMLM achieves ultrahigh resolution.
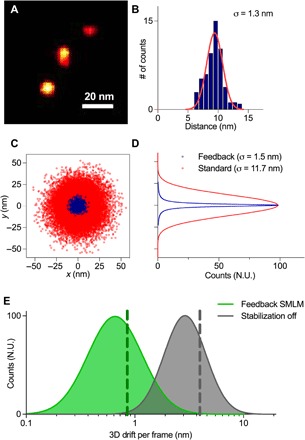
(A) DNA-PAINT image of a single origami ruler. (B) Cross-sectional profile of a single binding site. Red line represents Gaussian fit. (C) xy distributions of localization points for individual binding sites. The distributions for each binding site were aligned by their respective center and superimposed. (D) Cross-sectional fits of (C). In (C) and (D), blue symbols and lines represent data from Feedback SMLM, and red symbols and lines represent data from standard SMLM with post-acquisition drift correction. (E) The mean 3D drift registered per fluorescent frame is 0.84 nm (green dotted line) using the Feedback SMLM (green curve) and 3.54 nm (gray dotted line) without our stabilization (gray curve). N.U., normalized units.
The improvement in resolution in Feedback SMLM stems from the rapid and accurate drift corrections (sample/stage stabilization of ≤1 nm in 3D). Without active stabilization, the sample shows an average 3D displacement of 3.5 nm after 200 ms (Fig. 2E), a time period that is equivalent to the mean binding time of a fluorescent DNA-PAINT imaging strand. Because drift does not occur in a straight line, an average distance of 5.7 nm remains uncorrected within each binding time when the active stabilization is switched off (fig. S6). This is a much larger position uncertainty than the 1-nm uncertainty that is needed to accurately reconstruct densely packed molecules (11). This is because residual drift compounded from multiple fluorescent detections significantly degrades the final precision of the target molecule. With the Feedback SMLM, the correction is performed in real-time and faster than the fluorescence acquisition. The drift during the average binding time of a single strand is reduced to 0.8 nm. Thus, our approach decreases drift below the photon-limited localization precision and causes the fluorescent positions to be prealigned. This eliminates the need for assumptions about the sample geometry or additional drift correction methods and makes Feedback SMLM suitable for both short-term (binding time) and long-term (hours) acquisition time scales.
Raw image data resolve individual actin filaments in cells
We next used the Feedback SMLM to image F-actin (Fig. 3), an abundant protein that organizes into various structures. The width of actin can be used as a subdiffraction imaging benchmark to demonstrate the in situ resolution of a microscope. Even without post-acquisition processing, averaging, filtering, or customized filament analysis (23), it was possible to identify single filaments and determine their widths as 5 to 9 nm (Fig. 3, B to D). The width of the actin filament agrees with previous measurements using electron microscopy, whereas dSTORM measurements resulted in a greater width of ~20 nm, presumably because of insufficient localization precision (24). Grouping of fluorescent events, which belong to the same molecule as frequently used in (d)STORM, would further improve the localization accuracy. Furthermore, we did not use any filtering to refine localizations along the actin fiber as previously used in single-molecule imaging (25). To monitor residual drift, gold nanoparticles were deposited onto the coverslips and drift-corrected post-acquisition. Performing post-acquisition correction did not significantly reveal any drift during acquisition or improve the imaging resolution of our microscope (Fig. 3E).
Fig. 3. Feedback SMLM resolves individual actin filaments in cells without postprocessing.
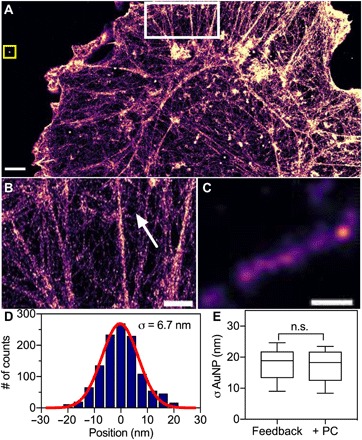
(A) DNA-PAINT image of phalloidin in a COS-7 cell without post-acquisition processing or filtering. Scale bar, 2 μm. (B) Zoomed region of F-actin structures highlighted (white square) in (A). Scale bar, 1 μm. (C) Zoomed region of a single actin filament indicated by the arrow in (B). Scale bar, 50 nm. (D) Cross-sectional profile of five filaments aligned by the center of each filament. Red line, Gaussian fit with σ of 6.7 nm. (E) Size distribution of gold nanoparticles that were embedded in the sample [yellow square in (A)]. Data are mean and standard deviations of n = 10 nanoparticles; n.s., not significant (P > 0.05, t test assuming equal variance). Post-acquisition drift correction was performed by using gold nanoparticles as fiducial markers, followed by redundant cross-correlation algorithm (RCC). Post-acquisition drift correction did not improve the resolution of actin or reduce drift.
Distance measurements in active and resting T cells in situ
To demonstrate the utility of the new microscope for distance measurements, we imaged individual signaling proteins in T cells. T cells make so-called fate decisions based on the quality and quantity of antigens displayed on the surface of antigen-presenting cells (26). T cell fates are determined by antigen binding to the TCR and the resulting intracellular signaling activities. The process begins with the phosphorylation of the TCR-associated CD3 dimers, a process termed TCR triggering (27, 28). An unsolved question in T cell biology is how antigenic peptides bound to major histocompatibility complex (pMHC) molecules initiate TCR signaling. It is generally assumed that TCR triggering requires the exclusion of phosphatases such as CD45 (29). However, so far, it has not been possible to measure the separation distance between signaling TCR-CD3 complexes and CD45.
Here, we activated Jurkat-ILA T cells on a supported lipid bilayer with antigenic pMHC-I molecules (30) and performed DNA-PAINT acquisitions of CD45 and phosphorylated CD3ζ (pCD3ζ) with Feedback SMLM (Fig. 4A and fig. S7). As above, even unprocessed images revealed that CD45 was spatially excluded from pCD3ζ nanoclusters (Fig. 4B). To measure spatial separations, we performed a nearest neighbor distance (NND) analysis between the phosphatase CD45 and signaling CD3 complex (pCD3ζ) in both directions. In activated T cells, we found broad distributions with a median distance of 19.6 nm (pCD3ζ to CD45) and 15.7 nm (CD45 to pCD3ζ) (Fig. 4C). NND on selected regions of high density of nanoclusters (n = 40 regions, 10 per cell) show comparable distributions (fig. S8). In resting cells where pCD3ζ is hardly detectable, CD45 and the CD3 complex (CD3ε) appear intermixed with mean distances of 12.5 nm (CD3ε to CD45) and 11.3 nm (CD45 to CD3ε), respectively (Fig. 4D and fig. S9). Thus, if spatial separation of the phosphatase from the TCR-CD3 complex is the main initiator of TCR triggering, as recently suggested (31), signaling reactions are exquisitely sensitive to 4- to 7-nm differences in spatial separations.
Fig. 4. Direct distance measurements between signaling proteins in active and resting T cells.
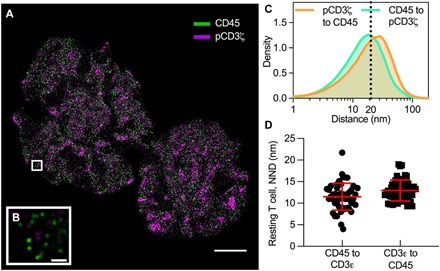
(A) DNA-PAINT image of CD45 (green) and phosphorylated CD3ζ (pCD3ζ, magenta) in an activated Jurkat-ILA. The cell was stimulated with a supported lipid bilayer presenting pMHC molecules and the adhesion protein ICAM-1. Scale bar, 4 μm. (B) Zoomed region of the highlighted area (white box) in (A) shows that pCD3ζ nanoclusters were spatially separated from CD45. Scale bar, 200 nm. (C) Density plot of the nearest neighbor distance (NDD) for pCD3ζ to CD45 (orange line) and for CD45 to pCD3ζ (green line) with medians of 19.6 and 15.7 nm, respectively. The dotted line indicates 20 nm. (D) NND analysis in resting T cells between the CD3 complex (CD3ε) and CD45. CD45 and CD3ε appear intermixed with mean distances of 12.5 nm (CD3ε to CD45) and 11.3 nm (CD45 to CD3ε), respectively. Each symbol represents the mean NND per ROI (n = 50 regions, 10 per cell). Horizontal and vertical bars represent the mean and SD.
DISCUSSION
The need for direct distance measurement between signaling proteins in intact cells motivated us to develop Feedback SMLM, a technology that can capture individual fluorescent events with ultrahigh precision and uniform detection probability. Because Feedback SMLM does not require filtering, merging, averaging, or other post-acquisition corrections, the molecular emission “landscape” created by successive rebinding or photoactivation/switching events reflects their true spatial and structural arrangement. Previous reports, aimed at resolving structures such as DNA origami or the nuclear pore complex (14), required assumptions about the underlying geometry to enable post-acquisition processing (11). This prevents analyzing structures of unknown shape or diverse structures with an unknown number of subunits as found in cells. With Feedback SMLM, such post-acquisition processing steps are not required, and yet, structures such as individual F-actin filaments and molecular separation distances <20 nm could be resolved. Feedback SMLM can be further combined with advanced analysis for distance measurements without the need to filter out poorly localized molecules (12, 32).
Here, we measured the separation distances between signaling/nonsignaling TCRs and the phosphatase CD45; to our knowledge, the highest-resolution measurement of TCR triggering. We note that these distance measurements cannot be conducted with alternative approaches such as FRET, as the separations distances are outside the dynamic range for FRET (8). Grouping localizations within a designated radius (i.e., 50 nm) can introduce errors in estimating a molecule’s position (33) so that variations in (cross-channel) NND may be lost. High localization precision (1 to 3 nm) and drift below the photon-limited precision (<1 nm) are required to accurately determine the distance between different species; otherwise, the nanoclusters may appear colocalized (34).
Most current strategies to compensate for drift in SMLM use post-acquisition corrections, where the position of individual molecules (and their localization precision) is first calculated before the drift correction (35). The average displacement of multiple fiducial markers in the sample is used to correct the spatial coordinates of the localizations detected. The average displacement is influenced by the precision afforded in each frame and the tracking accuracy and is sensitive to imaging aberrations (36). Tang et al. (37) showed that post-acquisition drift corrections can also be performed using the brightfield images of the sample, rather than fiducials. Although this method is easy to implement, the accuracy is limited by the contrast in the brightfield image (19). Often, post-acquisition corrections make long data acquisitions challenging, for example, for sequential multilabel DNA-PAINT imaging. Advanced post-acquisition methods use synthetic nanostructures with designed geometric patterns as fiducial markers (11, 13). Most samples cannot accommodate sufficient nanostructures (~1500) or binding sites (~30,000) (13), in addition to the biological sample under investigation. These intricate drift corrections are not feasible with many biological sample geometries. Drift correction methods using DNA origami structures are unstable on lipid bilayers, a sample format that is used to mimic cell-cell interfaces, and gold nanorods can create variable emission profiles once attached to the lipid bilayer (fig. S10), highlighting the restrictions some post-acquisition methods place on the sample format. The data quality of Feedback SMLM is unaffected by the inclusion of a supported lipid bilayer in the sample, which highlights the flexibility for sample preparation and SMLM data acquisition.
Dynamic drift correction approaches (9, 19, 20, 38) adjust the position of the sample during the acquisition. In vitro, active correction relies on the optical trapping of molecules, which limits throughput and/or sample formats (9). For cellular imaging, active drift correction has been performed by monitoring the position of the fiducials markers (20, 38) or the brightfield image of the sample (19). Ma et al. (38) used the signal of gold nanoparticles arbitrarily embedded into the cover slide with correction rate of 4 s. Faster sample correction rates (e.g., 1 to 2 s) come at the expense of reduced accuracy (e.g., 10 to 20 nm) (19, 20). Until now, implementations of active stabilization have been performed at slower correction rates of 1 to 10 s (9, 19, 20, 38). Slow and/or inaccurate corrections result in considerably worse in situ resolution, even after post-analysis treatment such as filtering or grouping. Here, we implement rapid graphics processing unit (GPU) calculations to achieve 15-Hz sample correction rate and obtain ≤1-nm sample stabilization in 3D. Our method reduces drift below the photon-limited localization precision within the average DNA-PAINT imaging time. In our setup, the location of the cell does not need to coincide with the 3-μm fiducials used for stage stabilization, and the readout of the position of the fiducials is not based on fluorescence. Hence, the stage correction rate is increased [e.g., 60-fold faster than in Ma et al. (38)] without compromising precision.
A further advantage of our setup is that the number and position of fiducial markers can be uncoupled from the preparation of the biological samples. This opens the door for more sophisticated sample designs such as confluent cell monolayers or 3D cell cultures. Feedback SMLM could further increase the accuracy of single-particle tracking (39), super-resolution imaging techniques in living cells (40), and 3D acquisitions (41). Because of its practical design, Feedback SMLM can be established on existing setups. Improvements could be made with high–numerical aperture (NA) objectives (NAs up to 1.7) and brighter dyes as well as new tag strategies such as nanobodies (42) or aptamers (43). It can be easily adapted to perform high-throughput imaging (i.e., microfluidics) and multiplexed localization analysis.
In summary, our approach advances the current state of the art due to the quick and accurate corrections. This allows an increase in localization precision, considerable sample flexibility, and without the need for complex optical implementations (i.e., optical traps) that limit throughput and sample designs. Feedback SMLM is suitable for direct distance measurements of distances between structures of >2 nm and of rare structures within intact cells. This is an unmet need. We believe that this will be of particular importance in signaling or in samples where cell numbers are limiting. Thus, Feedback SMLM could become the tool of choice to quantify molecular associations and interactions on the nanometer scale.
MATERIALS AND METHODS
Buffers
Imaging buffer A: Phosphate-buffered saline (PBS) with 10 mM magnesium. Cytoskeleton buffer B: 10 mM MES, 150 mM NaCl, 5 mM EDTA, 5 mM glucose, and 5 mM MgCl2. Buffer C: Buffer B with 0.3% glutaraldehyde and 0.25% Triton X-100. Buffer D: Buffer B with 2% glutaraldehyde. Buffer E: Freshly prepared 0.1% NaBH4 in PBS. Buffer F: 10 mM tris-HCl, 100 mM NaCl, and 0.05% Tween 20 (pH 7.5). Buffer G: 5 mM tris-HCl, 10 mM MgCl2, 1 mM EDTA, and 0.05% Tween 20 (pH 8). Buffer H: PBS and 500 mM NaCl (pH 7.2).
Preparation of glass coverslips
All coverslips were cleaned by sonication in ethanol for 15 min, followed by sonication in Milli-Q water for 15 min and plasma cleaning for 3 min, unless stated otherwise.
Buffer exchange chamber
To perform buffer exchanges, we modified the lid of an eight-well chamber to accommodate an inlet and outlet port. Silicon tubing connected to syringes was inserted into the well via right angle brackets. Buffer present in the chamber was first removed via the outlet, and the sample was washed before adding fresh buffer.
DNA origami rulers on glass coverslips
A single well of an eight-well chamber (80841; ibidi) was attached to a clean coverslip and washed with 500 μl of PBS. The well was incubated with 200 μl of bovine serum albumin (BSA)–biotin solution (1 mg/ml in PBS) for 5 min. Excess BSA-biotin was removed by washing with 500 μl of PBS. The surface was incubated with 200 μl of neutravidin (1 mg/ml in PBS) for 5 min and washed with buffer A. Biotin-coated polystyrene beads (TP-30-5; Spherotech) (40 μg/ml) were incubated for 1 hour, and the excess beads were removed. The well was incubated with the DNA origami ruler (HiRes 20R; GATTA-PAINT) diluted 40 times in buffer A to get ~100 rulers per FoV. Excess DNA origamis were removed by washing with buffer A. The imaging strand was a 9–base pair (bp) complementary target strand with Atto 655, with a concentration of 5 nM in buffer A.
Cell culture
COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (11885-084; Life Technologies) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Jurkat-ILA1 T cells were cultured in RPMI 1640 (21870-076; Life Technologies) supplemented with 10% FBS, 2 mM l-glutamine, 1 mM penicillin, and 1 mM streptomycin (all from Life Technologies). Characterization of the Jurkat-ILA1 T cells was performed as previously described (30).
Actin imaging
A coverslip with gold nanorods (600-30 AuF; HESTZIG) was cleaned, and a well chamber (80841; ibidi) was attached to the surface. The surface was coated with PLL-PEG-biotin (10 μg/ml in PBS, 20 min) to prevent nonspecific binding, followed by streptavidin (0.09 μM). COS-7 cells were added (10,000 to 20,000 cells per well) and incubated for 24 hours at 37°C. Biotin-coated 3-μm polystyrene beads (TP-30-5; Spherotech) were mixed with serum-free DMEM (Life Technologies) and deposited onto the surface for 1 hour at 37°C and washed to remove excess beads. Cells were fixed and permeabilized with 300 μl of buffer C for 1 to 2 min, followed by 600 μl of buffer D for 10 min. Cells were treated with 600 μl of buffer E for 7 min and washed with PBS. The surface was passivated using biotin in 5% BSA (1 μM) for 1 hour and washed with PBS. Cells were incubated overnight at 4°C with 300 μl of Biotin-XX phalloidin (B7474; Thermo Fisher Scientific) (0.5 μM) in 5% BSA and then washed with buffer F. Buffer F with streptavidin (3.45 μM) was added to the cells at room temperature for 30 min and washed with PBS. Buffer D was added for 10 min at room temperature. We then washed with PBS, buffer F, and buffer G. Biotinylated DNA docking strands (2 μM) in buffer G were incubated for 30 to 60 min at room temperature and washed with buffer G. Imaging strands (500 pM) in buffer H were added, and the sample was imaged.
Bilayer preparation
Glass coverslips were cleaned with 1 M KOH for 10 min, rinsed with Milli-Q water, placed in 100% ethanol for 20 min, and plasma-cleaned for 5 min. Eight-well silicone chambers (80841; ibidi) were then attached to the cleaned coverslip. A liposome solution of 1 mg/ml with a lipid ratio of 96.5% DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), 2% DGS-NTA(Ni) [2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt)], 1% biotinyl-Cap-PE [1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (sodium salt)], and 0.5% PEG5000-PE [1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-5000] (ammonium salt)] [mole percent; all available from Avanti Polar Lipids (DOPC, 850375C), (DGS-NTA(Ni), 790404C), (Biotinyl-Cap-PE, 870273C), (PEG5000-PE, 880220C)] was created by vesicle extrusion, as described in detail elsewhere (44). The lipid solution was added to the wells at a 1:5 ratio with Milli-Q water along with 10 mM CaCl2 for 15 min and washed repeatedly with PBS, followed by 0.5 mM EDTA in Milli-Q water to remove the excess CaCl2. The well was then washed with PBS and incubated with 1 mM NiCl2 to recharge the NTA groups. Disruption of the lipid bilayer was avoided by maintaining 100 to 150 μl of PBS in the wells.
Bilayer decoration for DNA origami rulers
To decorate the bilayer with fiducials and DNA origami rulers, 200 μl of neutravidin (1 mg/ml in PBS) was incubated in the well for 5 min and washed with buffer A. Biotinylated polystyrene beads (TP-30-5; Spherotech) were then added at 40 μg/ml for 30 min and then washed with buffer A to remove excess beads. The well was then incubated with biotinylated nanorods (C12-25-650-TB-DIH-50; Nanopartz) at 1:20,000 ratio, and DNA origami rulers (HiRes 20R; GATTA-PAINT) were diluted 40 times in buffer A and washed with buffer A to remove any unbound fiducials and rulers. The well was fixed with 4% paraformaldehyde (PFA) for 15 min and washed with buffer A. The imaging strand, which consists of Atto 655 on a 9-bp complementary target strand, was added to the chamber at a concentration of 5 nM in buffer A.
Bilayer decoration for T cell activation
To decorate the bilayer with fiducials and proteins, streptavidin (100 μg/ml) (SNN1001; Life Technologies) was incubated for 10 min and washed with PBS. Biotinylated polystyrene beads (TP-30-5; Spherotech) were then added at 40 μg/ml ratio in PBS for 20 min and washed with PBS to remove excess beads. Biotinylated pMHC 3G (500 ng/ml), His-tagged ICAM-1 (200 ng/ml), and biotinylated nanorods (C12-25-650-TB-DIH-50; Nanopartz) at a 1:20,000 ratio were combined with 5% BSA in PBS. Biotinylated and His-tagged proteins and biotinylated nanorods were then added to the well for 30 min. Last, the surfaces were washed with PBS to remove unbound fiducials or proteins.
T cell activation on bilayer and immunostaining
The wells were washed with RPMI 1640 culture medium and warmed to 37°C for 30 min. Jurkat-ILA cells were added to the bilayer at a density of 250,000 cells in 50 μl for 4 min at 37°C and fixed using 4% PFA in PBS for 20 min at 37°C. For immunostaining, cells were permeabilized with Triton X-100 (T8787; Sigma-Aldrich) at 0.1% for 5 min at room temperature and washed with PBS. The cells were blocked with 5% BSA in PBS for 1 hour. Cells were labeled with primary antibodies against pCD3ζ (pY142) (558402; BD Pharmingen) and CD45 (ab10559; Abcam) for 30 min and then washed with PBS. Primary antibodies were previously conjugated with DNA strands. Cells were then labeled with a secondary antibody against pCD3ζ (115-547-003; Jackson ImmunoResearch) for 30 min at room temperature and then washed with PBS. The sample was fixed using 4% PFA in PBS for 15 min and then washed with buffer A. DNA target strands conjugated in buffer A were added to the sample at a 5 nM concentration. CD45 and pCD3ζ docking strands are all complementary to the individual docking strands on the primary labeled antibodies. Imaging was performed sequentially after performing buffer exchanges.
Bilayer decoration for inactive T cell imaging
To decorate the bilayer with fiducials and proteins, streptavidin (100 μg/ml) (SNN1001; Life Technologies) was incubated for 10 min and washed with PBS. Biotinylated polystyrene beads (TP-30-5; Spherotech) were then added at 40 μg/ml ratio in PBS for 20 min and washed with PBS to remove excess beads. His-tagged ICAM-1 (200 ng/ml) and biotinylated CD90 at a 1:500 ratio (13-0909-80; Thermo Fisher Scientific) were combined with 5% BSA in PBS. His-tagged proteins were then added to the well for 30 min. The surfaces were then washed with PBS to remove unbound fiducials or proteins.
Inactive T cell deposition and immunostaining
The procedure is the same as described for the active T cell with the additional labeling using an antibody against CD3ε (ab213608; Abcam).
Optical setup
Laser lines of 405 nm (Stradus 405-100; Vortran), 488 nm (Stradus 488-150; Vortran), and 637 nm (Stradus 637-180; Vortran) were passed through cleanup filters (405 nm, LD01-405/10-12.5; 488 nm, LL01-488-12.5; 640 nm, LD01-640/8-12.5; Semrock), combined into a single path using dichroic mirrors (ZT442rdc and ZT594rdc; Chroma), and fiber-coupled (P3-405BPM-FC-2; Thorlabs). A pair of achromatic doublet lenses (f = 30 and 300 mm) was used to expand the lasers. The lasers were focused onto the back aperture of a 100× 1.49 NA total internal reflection fluorescence (TIRF) objective (Nikon, CFI Apochromat) using an achromatic lens (f = 200 mm). TIRF illumination was achieved by displacing the laser beams toward the periphery of the objective. The displacement was performed by moving the focusing lens with a mirror assembled on a translation stage (M-423-MIC; Newport). Lasers were delivered to the objective using a dichroic beam splitter (ZT488/640rpc; Chroma), which reflected all lasers (and infrared LED) but allowed transmission of the fluorescence. The sample was mounted on a nanopositioning stage with 0.1-nm step size in the x/y axis and 0.4 nm in the z axis (LP50-200, Mad City Labs), integrated on an inverted microscope body (RM21; Mad City Labs). The microscope body allowed easy integration of components into the frame. The optical setup was built on an actively stabilized optical table (M-ST-46-8; Newport) and tuned for the payload distribution. Table vibrations were monitored in real time (ST-300; Newport).
Sample fluorescence was filtered using an emission filter (Em01-R405/488/635-25; Semrock). A lens (f = 400 mm) focused the fluorescence onto an EMCCD camera (Andor, iXon 897 Ultra) via a piezoelectric mirror (Polaris-K1S3P; Thorlabs). The EMCCD camera was water-cooled (EX2-1055; Koolance) to avoid fan vibrations and mounted in a custom-made steel holder. The EMCCD camera was set to a temperature of −95°C to minimize camera pixel noise. The final magnification of the EMCCD camera resulted in a 40 μm × 40 μm FoV (individual pixel size, 80 × 80 nm). A white LED (BLS-LCS-4000-03-22; Mightex) illuminated a pinhole and was combined with the fluorescence path using a 0.5% reflective mirror (WG11010-A; Thorlabs). The spot on the EMCCD camera created by the LED was fitted with a two-dimensional (2D) Gaussian, and deviations from the original position were applied to the piezoelectric mirror to correct for drift in the fluorescence path. To avoid disturbance to the fluorescence, the LED spot was positioned on the periphery of the EMCCD camera. Image deviation was calculated as (dx2 + dy2)0.5 for each frame, where dx and dy represent the deviations registered from the set position.
To perform sample stabilization, an infrared LED (LCS-0850-02-22; Mightex) was filtered using a bandpass filter (FF01-842/56-25; Semrock) and directed to the sample via a condenser (ACL25416U-B; Thorlabs). The infrared light was removed from the fluorescence path using the laser dichroic, separated from the lasers using a dichroic beam splitter (FF750-SDi02-25x36; Semrock), filtered using a bandpass filter (FF01-842/56-25; Semrock), and focused (f = 200 mm) onto a CMOS camera (Allied Vision Manta G-235). The final magnification of the CMOS camera resulted in a 112 μm × 70 μm FoV (individual pixel size, 58.6 nm), about fivefold larger than the EMCCD camera. The noncoherent light source did not generate interference speckles and produced a high-contrast diffraction pattern on the CMOS camera. The focus of the CMOS camera is offset by 3 μm with respect to the fluorescence camera. The data acquisition and instrument control were performed using an Xbox One controller (Microsoft) integrated into 64-bit LabVIEW (National Instruments).
Environmental control
An environmental control box was installed around the optical setup to control temperature fluctuations. A series of Peltiers (TEC3-6; Thorlabs) with temperature transducers (AD590; Thorlabs) were regulated individually (TED200C; Thorlabs). Temperature and humidity were registered independently from the temperature control units (TSP01; Thorlabs). The temperature and humidity have a standard deviation of 0.02°C and 0.88%, respectively. Before imaging, we allowed the samples to acclimatize for 15 to 30 min.
Active stabilization
A clean glass coverslip was coated with PLL-PEG-Biotin (10 μg/ml in PBS) to prevent nonspecific binding, followed by streptavidin (0.09 μM). Biotin-coated polystyrene beads (TP-30-5; Spherotech) (40 μg/ml) were incubated for 1 hour, and the excess beads were removed. Surface passivation was performed with biotin (1 μM) and then washed with PBS.
The software can automatically identify polystyrene beads within the FoV. The center of the particle was identified (within ~1 pixel) by a background-corrected center of mass algorithm and produced a square region of interest (ROI) of 150 × 150 pixels around each bead. This initial calculation was not critical, as it served as a visual aid that allowed the software to identify beads and register ROIs. CMOS technology allows an increased frame rate by selectively recording a ROI. To correct for drift (and monitor residual drift), the ROIs of two beads were recorded. One bead was referred to as the lock bead, and a second bead was referred to as the reference bead. The software calculated the position of both beads but only acted upon drift registered by lock bead. The reference bead served as an internal control, or an out-of-loop reference, to monitor drift. The average drift over a period of 10 frames was subtracted to the stage position. Applying an equal but opposite drift amplitude suppressed drift movement. The stage was moved at a frequency of 15 Hz to adequately correct for sample drift. Real-time 3D drift correction was performed using a camera-based particle tracking software implemented in LabVIEW. We used real-time data processing in a GPU (GTX1070; Nvidia), which has been shown to achieve 3D particle tracking of fixed beads with a precision of 0.1 nm at kHz rates and down to 0.01 nm at 10-Hz rates in all dimensions (17). The lateral positions of the bead are determined by correlating linear bead intensity profiles with their mirror profiles. The axial position was determined by comparing the radial intensity distribution to a prerecorded look-up table (LUT). The LUT that served as an axial calibration was acquired by performing a z-stack over a 4-μm depth by stepping the nanopositioning stage in 100-nm increments. The software allowed image streaming together with the lateral/axial position determination to be carried out in parallel (17).
Standard SMLM microscope
Standard SMLM imaging was performed on a commercial Zeiss Elyra SMLM microscope. TIRF illumination from a 640-nm laser diode was focused on DNA origami structures via a 100× 1.49 NA Zeiss objective. Axial drift correction was performed in real time using the Zeiss focus-lock module (Zeiss, Definite Focus). Fluorescence was filtered and captured on an EMCCD camera (Andor, iXon 897 Ultra) with a pixel size of 97 nm. The temperature was stabilized using the Zeiss environmental control module (Zeiss, TempModule S). Post-acquisition drift correction was performed by using gold nanoparticles as fiducial markers, followed by redundant cross-correlation algorithm (RCC) (21). For RCC, temporal bins were set at 1000 frames. Performing successive rounds of RCC with sequentially smaller temporal bins did not show an improvement in the data.
Image processing and single-molecule localization
Individual fluorescent images were recorded and saved in a 16-bit TIFF format. The images were automatically split into 4-GB folders, and an ImageJ macro was used to convert the images into a stack format (.OME.TIFF). To localize molecules, the PSF of an individual molecule within each frame was fitted to a 2D Gaussian. Images were analyzed with the Picasso software (13) or ImageJ (National Institutes of Health) with the ThunderSTORM plugin (45). Cross-sectional profiles were fitted with a Gaussian or Lorentzian, according to their distribution (4).
NND analysis
To evaluate the distribution of pCD3ζ, CD3ε, and CD45, the cross NND was calculated using R package “spatstat.” The distance was log-transformed (base 10) before the density was calculated and plotted. Cell boundaries were delimited to eliminate erroneous calculations from the background.
EMCCD characterization and chromatic correction
To reduce the impact of systematic errors in the microscope’s detection path, we adopted a strategy that realigns molecular positions (25, 46). We produced a regular pattern with stable emission by generating a “nanohole array” (fig. S5). The nanohole array consists of a series of circular holes with a diameter of 100 nm nanofabricated on an aluminum-coated coverslip in a 12 × 12 array. The holes were arranged in grid pattern with a vertical and lateral separation of 1 μm. The holes were then filled with red (Alexa Fluor 647) and green (Alexa Fluor 488) dyes at ~100 μM concentration. We registered the predetermined emission pattern in both the green and red channels. The location of the points was determined with nanometer accuracy. The apparent variation between the green and red coordinates was due to the registration errors of the EMCCD and optical and chromatic aberrations. The local deviations of each green-red point were registered as a vector displacement. The pattern was moved in an xy raster pattern in an overlapping approach to characterize the pixels of the EMCCD camera.
Supplementary Material
Acknowledgments
We thank P. Michaux at the ANFF for the nanohole array and T. Boecking, M. Catarino, and J. Goyette for assistance with the manuscript preparation. Funding: This work was supported by the Australia Research Council (CE140100011 to K.G.) and National Health and Medical Research Council of Australia (APP1059278 to K.G.). Author contributions: S.C., J.B., J.J.G., and K.G. wrote the paper. S.C., J.B., P.R.N., and K.G. conceived the project. S.C. and J.B. built the Feedback SMLM. S.C., J.B., and K.F. analyzed the experimental data. S.C., J.B., M.S.G., and J.M.H. performed the experimental work. J.M.H. and H.G. developed the DNA-PAINT antibodies. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaay8271/DC1
REFERENCES AND NOTES
- 1.Rust M. J., Bates M., Zhuang X., Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–796 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jungmann R., Avendaño M. S., Woehrstein J. B., Dai M., Shih W. M., Yin P., Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and exchange-PAINT. Nat. Methods 11, 313–318 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Mortensen K. I., Churchman L. S., Spudich J. A., Flyvbjerg H., Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 7, 377–381 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moerner W. E., New directions in single-molecule imaging and analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 12596–12602 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deschout H., Zanacchi F. C., Mlodzianoski M., Diaspro A., Bewersdorf J., Hess S. T., Braeckmans K., Precisely and accurately localizing single emitters in fluorescence microscopy. Nat. Methods 11, 253–266 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Hell S. W., Sahl S. J., Bates M., Zhuang X., Heintzmann R., Booth M. J., Bewersdorf J., Shtengel G., Hess H., Tinnefeld P., The 2015 super-resolution microscopy roadmap. J. Phys. D Appl. Phys. 48, 443001 (2015). [Google Scholar]
- 8.Jares-Erijman E. A., Jovin T. M., FRET imaging. Nat. Biotechnol. 21, 1387–1395 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Pertsinidis A., Zhang Y., Chu S., Subnanometre single-molecule localization, registration and distance measurements. Nature 466, 647–651 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Mortensen K. I., Sung J., Flyvbjerg H., Spudich J. A., Optimized measurements of separations and angles between intra-molecular fluorescent markers. Nat. Commun. 6, 8621 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai M., Jungmann R., Yin P., Optical imaging of individual biomolecules in densely packed clusters. Nat. Nanotechnol. 11, 798–807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niekamp S., Sung J., Huynh W., Bhabha G., Vale R. D., Stuurman N., Nanometer-accuracy distance measurements between fluorophores at the single-molecule level. Proc. Natl. Acad. Sci. U.S.A. 116, 4275–4284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnitzbauer J., Strauss M. T., Schlichthaerle T., Schueder F., Jungmann R., Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 12, 1198–1228 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Szymborska A., de Marco A., Daigle N., Cordes V. C., Briggs J. A. G., Ellenberg J., Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science 341, 655–658 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Irvine D. J., Purbhoo M. A., Krogsgaard M., Davis M. M., Direct observation of ligand recognition by T cells. Nature 419, 845–849 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Manz B. N., Jackson B. L., Petit R. S., Dustin M. L., Groves J., T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc. Natl. Acad. Sci. U.S.A. 108, 9089–9094 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huhle A., Klaue D., Brutzer H., Daldrop P., Joo S., Otto O., Keyser U. F., Seidel R., Camera-based three-dimensional real-time particle tracking at kHz rates and Ångström accuracy. Nat. Commun. 6, 5885 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gosse C., Croquette V., Magnetic tweezers: Micromanipulation and force measurement at the molecular level. Biophys. J. 82, 3314–3329 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGorty R., Kamiyama D., Huang B., Active microscope stabilization in three dimensions using image correlation. Opt. Nanoscopy 2, 3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grover G., Mohrman W., Piestun R., Real-time adaptive drift correction for super-resolution localization microscopy. Opt. Express 23, 23887–23898 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Wang Y., Schnitzbauer J., Hu Z., Li X., Cheng Y., Huang Z.-L., Huang B., Localization events-based sample drift correction for localization microscopy with redundant cross-correlation algorithm. Opt. Express 22, 15982–15991 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mailfert S., Touvier J., Benyoussef L., Fabre R., Rabaoui A., Blache M.-C., Hamon Y., Brustlein S., Monneret S., Marguet D., Bertaux N., A theoretical high-density nanoscopy study leads to the design of UNLOC, a parameter-free algorithm. Biophys. J. 115, 565–576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters R., Griffié J., Burn G. L., Williamson D. J., Owen D. M., Quantitative fibre analysis of single-molecule localization microscopy data. Sci. Rep. 8, 10418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadegh S., Higgins J. L., Mannion P. C., Tamkun M. M., Krapf D., Plasma membrane is compartmentalized by a self-similar cortical actin meshwork. Phys. Rev. X 7, 011031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pertsinidis A., Mukherjee K., Sharma M., Pang Z. P., Park S. R., Zhang Y., Brunger A. T., Südhof T. C., Chu S., Ultrahigh-resolution imaging reveals formation of neuronal SNARE/Munc18 complexes in situ. Proc. Natl. Acad. Sci. U.S.A. 110, E2812–E2820 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhns M. S., Davis M. M., TCR signaling emerges from the sum of many parts. Front. Immunol. 3, 159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Merwe P. A., Dushek O., Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Brownlie R. J., Zamoyska R., T cell receptor signalling networks: Branched, diversified and bounded. Nat. Rev. Immunol. 13, 257–269 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Cai H., Muller J., Depoil D., Mayya V., Sheetz M. P., Dustin M. L., Wind S. J., Full control of ligand positioning reveals spatial thresholds for T cell receptor triggering. Nat. Nanotechnol. 13, 610–617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pageon S. V., Tabarin T., Yamamoto Y., Ma Y., Nicovich P. R., Bridgeman J. S., Cohnen A., Benzing C., Gao Y., Crowther M. D., Tungatt K., Dolton G., Sewell A. K., Price D. A., Acuto O., Parton R. G., Gooding J. J., Rossy J., Rossjohn J., Gaus K., Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc. Natl. Acad. Sci. U.S.A. 113, E5454–E5463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang V. T., Fernandes R. A., Ganzinger K. A., Lee S. F., Siebold C., McColl J., Jönsson P., Palayret M., Harlos K., Coles C. H., Jones E. Y., Lui Y., Huang E., Gilbert R. J. C., Klenerman D., Aricescu A. R., Davis S. J., Initiation of T cell signaling by CD45 segregation at 'close contacts'. Nat. Immunol. 17, 574–582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Churchman L. S., Flyvbjerg H., Spudich J. A., A non-Gaussian distribution quantifies distances measured with fluorescence localization techniques. Biophys. J. 90, 668–671 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicovich P. R., Owen D. M., Gaus K., Turning single-molecule localization microscopy into a quantitative bioanalytical tool. Nat. Protoc. 12, 453–460 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Deschout H., Shivanandan A., Annibale P., Scarselli M., Radenovic A., Progress in quantitative single-molecule localization microscopy. Histochem. Cell Biol. 142, 5–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henriques R., Lelek M., Fornasiero E. F., Valtorta F., Zimmer C., Mhlanga M. M., QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat. Methods 7, 339–340 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Izeddin I., El Beheiry M., Andilla J., Ciepielewski D., Darzacq X., Dahan M., PSF shaping using adaptive optics for three-dimensional single-molecule super-resolution imaging and tracking. Opt. Express 20, 4957–4967 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Tang Y., Wang X., Zhang X., Li J., Dai L., Sub-nanometer drift correction for super-resolution imaging. Opt. Lett. 39, 5685–5688 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Ma H., Xu J., Jin J., Huang Y., Liu Y., A simple marker-assisted 3D nanometer drift correction method for superresolution microscopy. Biophys. J. 112, 2196–2208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsunoyama T. A., Watanabe Y., Goto J., Naito K., Kasai R. S., Suzuki K. G. N., Fujiwara T. K., Kusumi A., Super-long single-molecule tracking reveals dynamic-anchorage-induced integrin function. Nat. Chem. Biol. 14, 497–506 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Shroff H., Galbraith C. G., Galbraith J. A., Betzig E., Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods 5, 417 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang B., Wang W., Bates M., Zhuang X., Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ries J., Kaplan C., Platonova E., Eghlidi H., Ewers H., A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 9, 582–584 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Strauss S., Nickels P. C., Strauss M. T., Sabinina V. J., Ellenberg J., Carter J. D., Gupta S., Janjic N., Jungmann R., Modified aptamers enable quantitative sub-10-nm cellular DNA-PAINT imaging. Nat. Methods 15, 685–688 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.P. Beemiller, J. Jacobelli, M. Krummel, Imaging and analysis of OT1 T cell activation on lipid bilayers. Protoc. Exch. (2012).
- 45.Ovesný M., Křížek P., Borkovec J., Švindrych Z., Hagen G. M., ThunderSTORM: A comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30, 2389–2390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeWitt M. A., Chang A. Y., Combs P. A., Yildiz A., Cytoplasmic dynein moves through uncoordinated stepping of the AAA+ ring domains. Science 335, 221–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaay8271/DC1