

Colonic commensal-derived antigens support expansion and differentiation of immunosupressive CD4 T cells.
Abstract
The gut microbiome is the largest source of intrinsic non–self-antigens that are continuously sensed by the immune system but typically do not elicit lymphocyte responses. CD4+ T cells are critical to sustain uninterrupted tolerance to microbial antigens and to prevent intestinal inflammation. However, clinical interventions targeting commensal bacteria–specific CD4+ T cells are rare, because only a very limited number of commensal-derived epitopes have been identified. Here, we used a new approach to study epitopes and identify T cell receptors expressed by CD4+Foxp3+ (Treg) cells specific for commensal-derived antigens. Using this approach, we found that antigens from Akkermansia muciniphila reprogram naïve CD4+ T cells to the Treg lineage, expand preexisting microbe specific Tregs, and limit wasting disease in the CD4+ T cell transfer model of colitis. These data suggest that the administration of specific commensal epitopes may help to widen the repertoire of specific Tregs that control intestinal inflammation.
INTRODUCTION
Microbiota-specific CD4+ cells have been described as part of the healthy T cell repertoire in both mice and humans, but, so far, only a handful of microbiota-derived antigens have been identified that are specifically recognized by these cells in vivo or in vitro. Overall, identification of commensal-derived antigens recognized by CD4+ T cells is challenging because mucosal CD4+ cells remain tolerant to these antigens as compared to foreign antigens from infectious microbes. Human regulatory T cells (Tregs) target antigens relevant for mucosal tolerance are currently unknown (1). The identities of microbiome-specific epitopes are important for the future development of intestinal vaccines and therapeutic control of intestinal inflammation caused by microbial dysbiosis. Although murine Tregs specific for commensal strains were described by several independent reports (2, 3), the discovery of microbial epitopes recognized by these cells has been impeded by technical limitations. For example, the identification of microbial epitopes recognized by effector versus immunosuppressive regulatory CD4+Foxp3+ cells has been challenging.
So far, only a few antigenic peptides from commensal proteins have been described, which are recognized by mucosal effector CD4+ T cells in the context of class II major histocompatibility complex (MHC) molecules. Reportedly, fecal antigens from mice monocolonized with segmented filamentous bacteria (SFB) stimulate over 60% of T helper 17 (TH17) cells as compared with negligible reactivity to fecal material from germ-free (GF) mice. These data suggest that the majority of TH17 in SFB-colonized mice recognize SFB-derived antigens (4). Subsequent analysis identified two nine–amino acid peptides that stimulated several different T cell receptors (TCRs) expressed by TH17-derived T cell hybridomas, indicating that these TCRs recognized the same epitope (5). A different microbial antigen from flagellin-expressing Clostridia, CBir (1464–72), has been described as an inducer of naïve CD4+ T cell differentiation to peripheral Tregs (pTregs) (6). Another study described two antigenic peptides derived from the pathobiont Helicobacter hepaticus that induce differentiation of pTregs and follicular (TFH) CD4+ cells (7). Last, a recent report demonstrated that colonization of gnotobiotic mice with Akkermansia muciniphila led to the identification of two epitopes that activate TFH CD4+ cells in Peyer’s patches or multiple (TH1, TH17, Tregs, and TFH) CD4+ subsets in the intestinal lamina propria, contingent upon the absence or presence of other commensals (8). All previously described commensal-derived epitopes were shown to elicit responses of specific CD4+ hybridomas and stained-specific TH17 or pTregs when integrated with class II MHC/peptide in fluorescent tetramers. Although these studies demonstrated that identification of specific antigens derived from commensal flora is feasible, a more universal approach is needed to effectively assess CD4 T cell reactivity to microbiota-derived antigens.
Here, we have used an improved variant of the BW thymoma (BW5147 cell line) that is stably transfected with Nur77GFP to produce CD4+ T cell hybridomas with an elevated sensitivity for subthreshold TCR activation ligands. We then used high-throughput flow cytometry to identify specific hybridomas for several previously unknown epitopes from two commensal consortia established from a normal mouse microbiome. Our report shows that several common commensal bacteria including Clostridium spp. ASF356, Lactobacillus murinus ASF361, Eubacterium plexicaudatum ASF492, and A. muciniphila contain antigens recognized by hybridomas established from Tregs. We provide evidence that some of these antigens induce de novo pTregs or expand thymus-derived Tregs (tTregs) in the colon and are capable of ameliorating intestinal inflammation in a mouse colitis model.
RESULTS
Production of CD4+TCR+ hybridomas with improved sensitivity for low-affinity antigens
We have been researching the activation of regulatory CD4+Foxp3+ cells and noted that their antigenic specificities could be studied using hybridomas produced by the fusion of Tregs and BW5147α−β− thymoma (9). Treg-derived hybridomas cease Foxp3 expression and produce interleukin-2 (IL-2) following TCR activation similar to antigen-activated hybridomas derived from conventional CD4+Foxp3− T cells (10). In the past, this approach was successfully used to decipher the specificities of hundreds of different Tregs for host self and non–self-antigens (11). However, hybridoma responses to microbiota-derived antigens have been consistently difficult to detect using the IL-2 assay (2). Likewise, the expression of NFATGFP increased only in a fraction of Treg-derived hybridomas activated by bacterial isolates or epitopes, suggesting that this reporter does not optimally convey the antigen-induced activation of a homogenous population of CD4+TCR+ cells. A similar bimodal expression of NFATGFP was reported from the 58α−β−NFATGFP hybridoma line transfected with commensal-specific TCRs cloned from Tregs (5, 7, 12–14). To overcome these limitations, we stably transfected an original BW5147α−β− thymoma with bacterial artificial chromosome (BAC) cDNA encoding a Nur77GFP reporter, which expression reflects TCR functional avidity for cognate MHC/peptide ligand (15, 16). Figure 1A shows the expression of Nur77GFP and NFATGFP in two typical hybridomas following activation by aCD3 monoclonal antibody (mAb). All activated hybridoma cells unvaryingly increased expression of the Nur77GFP reporter, whereas corresponding hybridomas with the NFATGFP reporter increased expression only in a portion of stimulated cells. Increase in Nur77GFP expression was observed in response to very low concentrations of activating aCD3 mAb, which did not elicit detectable IL-2 (Fig. 1, C and D). Nur77GFP induction was reliably detectable below 0.1 ng/ml of aCD3 mAb, and reporter expression decayed slowly over time after removal of aCD3 mAb (Fig. 1E). Furthermore, although all Treg-derived hybridomas stimulated by aCD3 mAb (at 10 ng/ml or higher concentration) made comparable amounts of IL-2, only expression of Nur77GFP but not NFATGFP reporter correlated tightly with the amount of secreted IL-2 (Fig. 1B). Overall, these data demonstrate that TCR activation on CD4+ hybridomas can be detected with high sensitivity by changes in Nur77GFP expression.
Fig. 1. Activation of CD4+ T cell hybridomas produced using new BWNur77GFP thymoma.
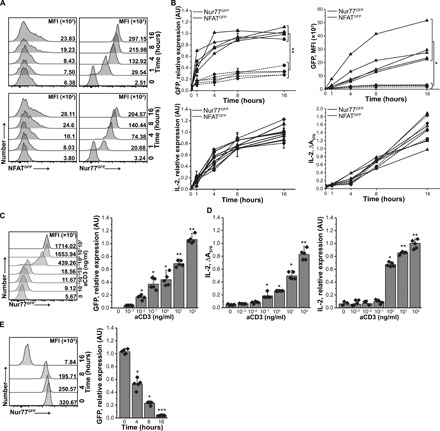
(A) Expression of GFP in NFATGFP (left) and Nur77GFP (right) reporters in response to aCD3 (1 μg/ml) in two representative hybridomas. MFI, mean fluorescence intensity. (B) Up-regulation of NFATGFP or Nur77GFP reporters and IL-2 secretion by aCD3 activated hybridomas. Reporter and IL-2 expression was measured by mRNA (left) and protein level (right). Each symbol marks individual hybridoma (n = 5 of each kind). The experiment was repeated three times, and representative results are shown. The statistic was calculated for t = 16 hours. (C) Expression of Nur77GFP reporter by representative hybridoma activated by titrated aCD3 mAb. Nur77GFP (C) and IL-2 (D) expression by hybridoma from CD4+TCR+Foxp3+ Tregs. GFP and IL-2 expression were measured by FACS/RT-qPCR or by HT-2 assay/RT-qPCR, respectively, from five randomly selected hybridomas. (E) Nur77GFP expression in hybridomas declines gradually following antigen withdrawal. Hybridomas were stimulated with plate-bound aCD3 for 16 hours and then moved to uncoated wells. GFP expression was measured by FACS (left) or RT-qPCR (right) at indicated time points. Means ± SD are shown, and each symbol represents an individual hybridoma. RT-qPCR data were normalized to β-actin. Paired t test; *P < 0.05, **P < 0.01, ***P < 0.001. For statistical analysis, data points in (C) were compared to unstimulated (aCD3 = 0 ng/ml) samples, and for (D), to samples of t = 0 hours.
CD4+ T cell hybridomas with a Nur77GFP reporter respond to microbiota-derived antigens
Next, we generated clonal hybridomas from intestinal CD4+ T cells and examined their responses to commensal antigens from cecal lysate after overnight coculture with autologous bone marrow–derived dendritic cells (DCs) from specific pathogen–free (SPF) mice. In this study, we used mice in which T cells express the restricted αβTCR repertoire (TCRmini), and Tregs express Foxp3GFP reporter (but not hybridomas derived from these cells) (2). In TCRmini mice, all T cells share the same TCRβ chain (Vβ14Dβ2Jβ2.6), while the introduced TCRα mini-locus naturally rearranges a single Vα2.9 segment to one of the two (Jα2 or Jα26) segments (17). Combinatorial diversity of the TCRmini repertoire is minimal, but insertions and subtractions of random N nucleotides within the CDR3α region generate approximately 105 different αβTCRs on peripheral CD4+ T cells (compared to an estimated 107 TCRs found on these cells in wild-type mice) (18). In TCRmini mice, one can use the sequencing of TCR CDR3α regions to track CD4+ cells that express the same or unique, commensal-specific TCRs in different cell subsets and organs (2).
As shown in Fig. 2(A and B), multiple hybridomas made from colonic CD4+Foxp3+ Tregs and stimulated by cecal lysate made almost no IL-2 (Fig. 2A) but reproducibly elevated Nur77GFP expression (Fig. 2B). Hybridoma activation by cecal antigens depended on class II MHC molecules, because DCs lacking Ab or preincubated with Ab blocking mAb (Y3P) were not stimulatory (Fig. 2C). In contrast, Ab+β2m− DCs lacking class I and nonclassical MHC molecules activated hybridomas as efficiently as native autologous DCs (Fig. 2C). Responses to microbial antigens have not improved upon the blocking of histone methylases or deacetylases (fig. S1, A and B). Similarly, interference with inhibitory signals by blocking programmed cell death protein 1 (PD-1) with mAb (multiple hybridomas expressed PD-1, and tested DCs were PD-L1+) had no detectable impact on stimulation, suggesting that epigenetic regulation or costimulatory inhibition did not restrict hybridoma activation (fig. S1, A and B). Likewise, hybridomas produced from anergic CD4+CD44+Foxp3−FR4+CD73+ cells recognized commensal antigens similarly to CD4+ hybridomas made from CD44+Foxp3−FR4− effector/memory counterparts (fig. S1, C and D). In contrast, we found that the lack of CD4+ T cell exposure to microbial antigens or pTregs measurably improved hybridoma responses to cecal antigens. Typically, hybridomas established from CD4+ cells from GF mice or pTreg-deficient Foxp3CNS1k/o mice (19) expressed higher Nur77GFP following activation by cecal lysate than their equivalents from SPF and CNS1+/+ strains (Fig. 2D). Overall, these experiments showed that TCR-mediated Nur77GFP up-regulation provides an improved sensitivity to identify Tregs specific for microbiota or diet-derived antigens (20) from cecal content.
Fig. 2. An increase in Nur77GFP reporter follows discrete TCR activation by bacterial antigens that do not induce detectable IL-2.
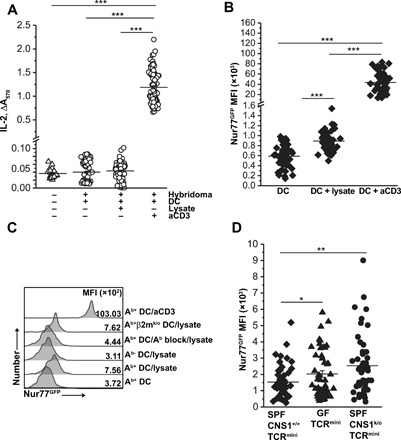
(A and B) A total of 100 hybridomas were individually cocultured with DCs pulsed with bacterial protein lysate or aCD3 mAb, and IL-2 production (A) or Nur77GFP up-regulation (B) was measured 16 hours later. The experiment was repeated twice. (C) Nur77GFP exclusively reports TCR activation of CD4+ hybridoma to antigens presented in the context of class II MHC (Ab). Antigens presented by Ab-negative DCs or DCs with antibody-blocked Ab do not up-regulate Nur77GFP. Lack of MHCI (β2mk/o) but not MHCII does not interfere with GFP up-regulation. aCD3 was used as positive control, and DCs not pulsed with fecal bacteria protein lysate served as a negative control. (D) Expression of Nur77GFP reporter in response to fecal bacteria lysate in hybridomas prepared from TCRmini, GF TCRmini, and CNS1k/oTCRmini CD4+ cells. Fifty hybridomas of each kind were tested and MFIs of GFP were assessed by FACS. One experiment of at least three is shown. Paired t test; *P < 0.05, **P < 0.01, ***P < 0.001.
To discover microbe-derived antigenic epitopes, we next colonized GF TCRminiFoxp3GFP mice with one of two defined microbial mini consortia. The first consortium, the altered Schaedler flora (ASF), encompasses eight microorganisms and has been used for standardization to study the spatial distribution of individual bacterial strains, genome analysis, and microorganism-specific host immune responses (21). The second consortium, Oligo-MM, was assembled using comparative metagenomic analysis to reflect most of the functional features of a fully complex microbiota and consists of 12 common mouse commensals (22). All colonized GF TCRminiFoxp3GFP hosts had increased proportions and total number of intestinal CD4+Foxp3+ T cells as compared with uncolonized controls, suggesting that ASF and Oligo-MM enhanced Tregs induction and expansion (fig. S2A). To identify peptide epitopes derived from specific commensal bacteria, we chose for further testing three stains from ASF (Clostridium spp. ASF365, L. murinus ASF361, and E. plexicaudatum ASF492) and A. muciniphila from Oligo-MM. These individual bacteria were selected on the basis of their dominance in microbiomes isolated from GF C57BL/6 mice colonized with corresponding consortia (21, 22).
Identification of bacterial epitopes that expand intestinal Tregs
To find specific commensal-derived epitopes recognized by intestinal CD4+ T cells, we screened CD4+TCR+ hybridomas from TCRminiFoxp3GFP and TCRminiCNS1k/o mice (latter strain lacks pTregs) against bacterial antigens. For this purpose, we used a whole-genome shotgun library protocol (fig. S2B) (23). Briefly, genomic DNA from selected bacteria was randomly fragmented (size, 100 to 1000 base pairs) and cloned into a pGEX expression vector. Individual bacterial colonies were grown in microtiter tubes, and protein expression was induced with isopropyl-β-d-thiogalactopyranoside (IPTG). Bacteria were then lysed, and protein lysate from 96 wells was used to stimulate CD4+ hybridomas in cocultures with autologous DCs. Clones from the positive pools were screened individually against the hybridoma bait. The inserts of positive clones were sequenced, and their sequences were blasted against the specific bacteria genome, then aligned to annotated open reading frames, and the peptide-binding motifs to Ab were predicted using public data tools [NetMHCII, IEDEB (Immune Epitope Database), and Rankpep]. Only DNA fragments encoding membrane or extracellular but not secretory proteins were considered for further screening. The minimal fragments anticipated to confer antigenicity were further verified by stimulating hybridomas with synthetic peptides (GenScript and ABI Scientific). Overall, using this approach, we identified six recombinant peptides derived from three ASF bacterial components that stimulated a total of 21 hybridomas representing colonic CD4+Foxp3+ cells and another 35 hybridomas established from CD4+Foxp3− cells (Fig. 3, A to C). The strongest responses were elicited by peptides derived from E. plexicaudatum ASF492 and Clostridium spp. ASF356, which together encoded more than 80% of microbial epitopes. These peptides elevated expression of Nur77GFP in hybridomas representing CD4+Foxp3GFP− cells from TCRminiCNS1k/o mice (Fig. 3, C and D). These data indicated that pTregs deficiency correlates with a higher incidence of CD4+Foxp3− clones that were specific for commensal-derived antigens (also see Fig. 2D).
Fig. 3. CD4+ hybridomas recognize specific microbial antigens from selected ASF bacteria.
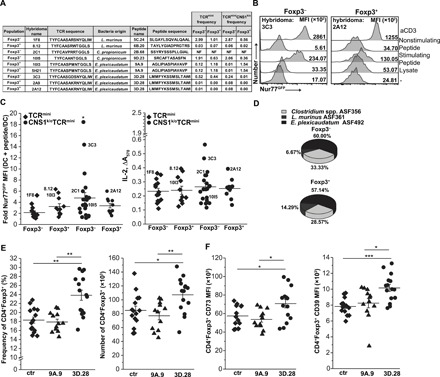
(A) Features of TCRs expressed on specific hybridomas and specific microbial epitopes that activated these hybridomas. The table shows sequences of antigenic peptides and corresponding TCR sequences found on selected reactive hybridomas prepared from indicated CD4+ populations. Frequencies of TCRs in the colonic CD4+ cell repertoire in TCRmini and TCRminiCNS1k/o mice are shown. (B) Representative responses of hybridomas from colonic CD4+Foxp3− and CD4+Foxp3+ cells to commensal’s antigens [activated by bacterial lysate or specific peptide (3D.28)]. Nonstimulatory peptide and aCD3 mAb served as negative and positive controls, respectively. (C) Summary of hybridomas’ responses to identified reactive peptides (TCRmini: Foxp3−, n = 14 and Foxp3+, n = 11; TCRminiCNS1k/o: Foxp3−, n = 21, Foxp3+, n = 10). Some hybridomas showed in (A) are marked on the graph. Each symbol represents an individual hybridoma. (D) Relative occurrence of hybridomas responding to antigens from three ASF strains (percentages show hybridomas responding to specific lysate). (E) Immunization of TCRmini mice with E. plexicaudatum ASF492–derived 3D.28 peptide (n = 15 mice) [but not 9A.9 peptide (n = 12 mice) or control (ctr) mice with cholera toxin only (n = 15 mice)] induces colonic Tregs [(total CD4 numbers: ctr, 290.9 ± 42.5; 9A.9, 296.6 ± 23.7; 3D.28, 312.1 ± 28.5 (×103)]. (F) 3D.28 elicits colonic Tregs with high levels of CD73 and CD39, enhancing these cells suppressor function. (E and F) Each symbol represents an individual mouse. The experiment was repeated three times. Paired t test; *P < 0.05, **P < 0.01, ***P < 0.001.
The 3D.28 peptide from E. plexicaudatum ASF492 (Fig. 3, A and B) stimulated many CD4+Foxp3GFP− hybridomas. E. plexicaudatum ASF492 was one of the two bacterial strains from the ASF consortium that makes butyrate and propionate, possibly supporting the expansion of Tregs (24–27). Thus, it was plausible that this strain is also a source of antigens driving naïve CD4+Foxp3GFP− T cell conversion to pTregs. Therefore, we selected this peptide for immunization of SPF TCRmini mice. The immunization with the 3D.28 peptide, but not another peptide (9A.9) from the same bacteria, increased the proportion and number of colonic CD4+Foxp3+ T cells as compared with control mice (Fig. 3E and fig. S2C). The Tregs expanded or induced by the 3D.28 peptide had elevated levels of CD39 and CD73 ectonucleases (Fig. 3F and fig. S2D), which convert adenosine 5′-triphosphate (ATP)/adenosine 5′-diphosphate (ADP) to adenosine 5′-monophosphate (AMP) and can shift CD4+Foxp3− T cells from a pro- to anti-inflammatory state by altering their metabolism (28–30). To study the impact of the 3D.28 peptide only on tTregs, we immunized TCRminiCNS1k/o mice with this peptide (19). In contrast to pTregs-competent TCRmini strain, TCRminiCNS1k/o mice that lack pTregs showed little change after injection of the 3D.28 or control peptide, suggesting that this epitope is preferentially recognized by CD4+Foxp3− clones and drives their conversion to pTregs. Separate immunization of C57BL/6 (B6) mice with the 3D.28 peptide had little effect on Treg subsets (fig. S2E), perhaps due to low precursor frequency of TCRs specific for this peptide.
A. muciniphila induces pTregs and supports the proliferation of residual colonic Tregs
Previous reports have found that the presence of other bacteria modifies the ability of A. muciniphila to influence colonic CD4+ T cells (31, 32). Therefore, we also examined A. muciniphila–specific responses by CD4+ T cell hybridomas generated from GF TCRminiFoxp3GFP mice that monocolonized with this commensal. Six weeks after colonization, we sacrificed recipient mice, generated hybridomas from mucosal Foxp3GFP− and Tregs, and screened their responses against a bacterial isolate. This assay identified 45 A. muciniphila–specific hybridomas (27 derived from non-Tregs and 18 from Tregs), which were used to screen shotgun expression libraries of genomic DNA from this commensal (fig. S2B). Using open-source MHC-binding prediction software, we identified multiple epitopes with Ab binding motifs that could be recognized by CD4+ T cells. These epitopes were synthesized and used to rescreen hybridomas previously identified as A. muciniphila specific, and to immunize TCRminiFoxp3GFP mice. Overall, four A. muciniphila–derived epitopes that activated at least one of eight hybridomas were found (Fig. 4A). The A. muciniphila–specific hybridomas expressed six different TCRs, which, in our database of TCRs sequenced from TCRmini mice, were found on CD4+Foxp3−, CD4+Foxp3+ cells, and tTregs from TCRminiCNS1k/o mice (Fig. 4A). This result suggested that in the TCRmini model, these epitopes may support both pTregs formation and tTregs expansion. Consequently, we choose the synthetic peptide 2C.1 that was capable of activating three different hybridomas ex vivo (Fig. 4, A to C) for immunization of SPF TCRminiFoxp3GFP mice. Two weeks later, we found that mice primed with the 2C.1 peptide (but not with control peptide 6H.3 from the same bacteria) had increased the proportion and a total number of proliferating (Ki67+) colonic CD4+Foxp3+Nrp-1−Helios− pTregs (fig.S3, A to F) that expressed high levels of CD39 and CD73 (Fig. 4, D to F; and fig. S3, A to E). In B6 mice expressing a wild-type TCR repertoire, a similar analysis was not statistically significant (P = 0.09536) (fig. S3F). A recent report demonstrated that A. muciniphila supports the differentiation of colonic TFH cells in B6 mice (8). We found that following immunization with the 2C.1 peptide in TCRminiFoxp3GFP mice, almost half of colonic Tregs up-regulated markers of follicular T cells (PD-1 and CXCR5) and expanded follicular helper T cells (TFH) as compared with animals immunized with control peptide (fig. S3G). Thus, in both models, exposure to A. muciniphila epitope(s) results in terminal differentiation of CD4+ T cells to the TFH lineage.
Fig. 4. Epitopes from A. muciniphila activate hybridomas derived from colonic CD4+ T cells.
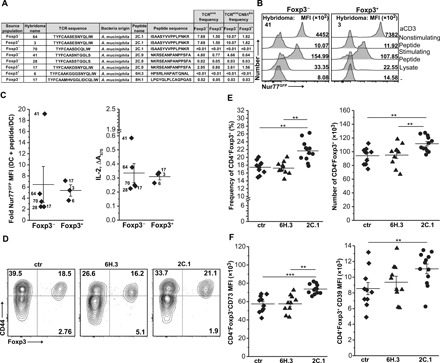
(A) Antigenic peptides and CDR3 sequences from specific TCRα chain expressed on hybridomas. Frequencies of these TCRs on colonic CD4+ cells in TCRmini and TCRminiCNS1k/o mice are also shown. (B) Representative up-regulation of Nur77GFP reporter by colonic CD4+ hybridoma in response to epitope (2C.1 or 2C.9) from A. muciniphila. Nonstimulating peptide and aCD3 served as negative and positive controls, respectively. (C) Summary of hybridoma responses to A. muciniphila epitopes. Each symbol represents an individual hybridoma. Responding hybrids from (A) are marked (Foxp3−, n = 5; Foxp3+, n = 3). (D) Representative dot plots show an expansion of colonic Tregs in TCRmini mice after immunization with A. muciniphila–derived 2C.1 peptide. (E) Frequencies and total number of colonic Tregs in TCRmini mice primed with cholera toxin only (ctr) (n = 10) or with toxin with 6H.3 (n = 10) or 2C.1 (n = 12) peptides [total CD4 numbers: ctr, 350.9 ± 59.2; 6H.3, 296.6 ± 57.5; 2C.1, 340.6 ± 53.3 (×103)]. (F) Tregs elicited by 2C.1 immunization express higher levels of CD73 and CD39. Gated colonic CD4+Foxp3+ cells were tested. (E and F) Each symbol represents an individual mouse. The experiment was repeated twice. Paired t test; **P < 0.01, ***P < 0.001.
A. muciniphila is prevalent in the microbiome of SPF TCRmini mice, but it is barely detectable in the microbiome of SPF TCRminiFoxp3GFPCNS1k/o mice that lack pTregs (Fig. 5A). In addition, gavage of GF TCRminiFoxp3GFP mice with this strain increased the proportion and total number of colonic Tregs, suggesting that A. muciniphila has a positive effect on the abundance of these cells (Fig. 5B and fig. S4A). To decipher the mechanism(s) behind these effects, we adoptively transferred naïve, congenic CD4+Foxp3GFP− cells from CNS1+/+ donors into TCRminiFoxp3GFPCNS1k/o recipients. Next, half of the recipients received A. muciniphila, and after 8 weeks, we examined colonic CD4+Foxp3GFP+ Tregs in all recipients. In these settings, pTregs could only differentiate from adoptively transferred but not from residual CD4+ T cells. We found that only the recipients that received adoptive transfer and A. muciniphila had a higher frequency and number of pTregs and residual Tregs than corresponding recipients that received only CD4+Foxp3GFP− cells, suggesting that provision of this commensal supports Tregs irrespective of their thymic or peripheral origin (Fig. 5C and fig. S4B). This conclusion correlated with the observation that TCRminiFoxp3GFPCNS1k/o mice gavaged with A. muciniphila had a significantly elevated fraction of proliferating residual tTregs in their colons compared with gavaged control mice, suggesting that colonic tTregs underwent expansion (Fig. 5, D and E, and fig. S4C).
Fig. 5. A. muciniphila supports Tregs in vivo expansion.
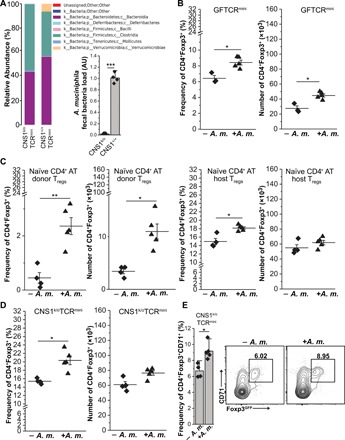
(A) 16S ribosomal RNA analysis of microbiomes from TCRminiCNS1k/o and TCRmini mice. Data were confirmed by RT-qPCR (shown below legend). Reads were first normalized to UniF340/UniR514 and then to CNS1+/+ A. muciniphila signal. (B) Inoculation of GF TCRminiFoxp3GFP mice with A. muciniphila expands colonic Tregs. (C) A. muciniphila induces pTregs and expands tTregs. Naïve CD4+CD44lowFoxp3− cells from congenic TCRmini mice were adoptively transferred into CNS1k/oTCRmini hosts, and half of the recipients received A. muciniphila. Graphs show frequencies (left) and total number (right) of Foxp3+ cells gated according to their origin. (D) TCRminiCNS1k/o mice were colonized with A. muciniphila, and 8 weeks later, their colonic tTregs were analyzed. Graphs show percentage (left) and numbers (right) of CD4+Foxp3+ cells. (E) tTregs up-regulate the CD71 proliferation marker in response to A. muciniphila. Summary and typical dot plots are shown. For (B) to (D), each symbol represents individual animals, and data were obtained in at least two independent experiments. Paired t test; *P < 0.05, **P < 0.01.
The provision of A. muciniphila extends the survival of recipient mice in the adoptive transfer model of colitis
The TCRαk/o mice bred in our facility have negligible amounts of A. muciniphila in their fecal microbiomes (fig. S5, A and B). To study whether A. muciniphila–induced pTregs can limit intestinal inflammation, we transferred naïve CD4+CD44−Foxp3GFP−(CD45RBhigh) T cells into TCRαk/o mice that had been pretreated with a cocktail of antibiotics. Although the use of antibiotics before the transfer of naïve CD4+ T cells delays the onset and progression of colitis in mice, antibiotic treatment of humans has been linked to dysbiosis because partial ablation of the commensal flora enhances susceptibility to colonization with enteric pathogens (33). All tested TCRαk/o recipients received a mix of antibiotics (Abx) in the drinking water for 2 weeks, which was followed by gavage with either A. muciniphila (A.m.), control Escherichia coli BL21DE3 strain (BL21), or phosphate-buffered saline (PBS) only (Fig. 6A and fig. S5B). Two weeks later, all recipients received adoptive transfer of 1 × 106 of CD4+CD44lowFoxp3GFP− cells from CNS1+/+ or CNS1k/o TCRmini mice, and we continuously monitored their weight and proportion of CD4+ T cells in the blood (Fig. 6, B to D). Uncolonized recipients and controls that received E. coli BL21 developed wasting disease and had to be euthanized within 10 weeks (before day 75) after adoptive transfer, whereas mice that received A. muciniphila survived for an additional 7 weeks until the experiment was terminated at day 120 (17 weeks total) (Fig. 6, B and C). During the experimental proceedings, we observed a steady increase in the proportion of CD4+Foxp3+ T cells only in the recipient mice colonized with A. muciniphila, which, upon sacrifice, also had the highest proportion and total number of colonic Tregs that expressed CD73 and CD39 (Fig. 6, D to F, and fig. S5C). Detailed histological analysis showed that recipients of Abx and Abx/BL21 had severe inflammation in their colons, while recipients of Abx/A.m. had minimal lymphocyte infiltration in the colon (Fig. 6G). Furthermore, colons from A. muciniphila–treated recipients had significantly lower levels of the proinflammatory cytokines interferon-γ (IFN-γ), IL-17A, and IL-6 and unchanged levels of the anti-inflammatory cytokine IL-10 (fig. S6, A and B). Overall, these results indicate that A. muciniphila–derived epitopes accelerate the conversion of CD4+ cells to pTregs that reduce intestinal inflammation (fig. S6C). On the contrary, control recipients that received CD4+ cells from CNS1k/o rapidly succumbed to intestinal inflammation, likely because adoptively transferred cells could not convert to pTregs that suppressed activation of colitogenic CD4+ effectors (Fig. 6, B and C, and fig. S5D). Nevertheless, colonization with A. muciniphila extended the life span of recipient mice by almost 4 weeks (Fig. 6, B and C), suggesting that the anti-inflammatory effect of this bacteria can prevent/delay advanced stages of intestinal inflammation. Overall, these data showed that colonization with A. muciniphila could protect the host from colitogenic effector cells, possibly by converting some of them to pTregs.
Fig. 6. A. muciniphila–induced pTregs prevent colitis in the adoptive transfer model.
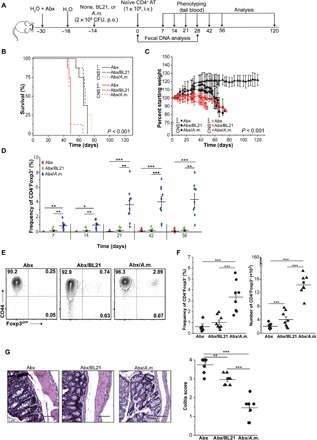
(A) Outline of the experiment. Mice were treated with a cocktail of antibiotics (Abx) and inoculated with indicated bacteria (n = 8 for each condition). At indicated time points, tail blood and feces samples were collected. i.v., intravenously. (B) Survival curves of adoptive transfer recipients precolonized with indicated bacteria (Kaplan-Meier curves and log-rank test). (C) Loss of initial body weight following adoptive transfer. Each point represents the mean percentage of initial body weight for the cohort ± SEM. (D) Frequency of pTregs in the blood at indicated time points for CNS1+/+ cell recipients. (E) Representative staining of analysis of colonic CD4+CNS1+/+ cells in recipient mice. (F) Proportions and total number of colonic CD4+ cells in individual mice after colonization and adoptive transfer. (G) Histological analysis of colonic sections. Representative H&E staining (×40 magnification) and colitis scores (scores are presented as an average of three fragments of each tissue) are shown. Scale bars, 100 μm. The experiment was repeated twice with three to five mice per group. Each symbol depicts individual animal. For survival analysis, the log rank test was performed; otherwise, paired t tests were applied; *P < 0.05, **P < 0.01, ***P < 0.001. BL21-E. coli BL21DE3.
Last, we investigated whether a food supplement that expands A. muciniphila can reproduce an anti-inflammatory effect observed in mice gavaged with live commensal bacteria. Thus, we examined whether prefeeding with grape seed extract can limit inflammation and subsequently prolong the survival of recipient mice in the adoptive transfer model of colitis. Reportedly, grape seed extract contains proanthocyanidins that induce the intestinal bloom of A. muciniphila by limiting the production of IL-17 and enhancing immunosuppression by CD4+Foxp3+ cells (34, 35). For this purpose, we injected intravenously 1 × 106 of naïve CD4+Foxp3− cells from CNS1k/o or CNS1+/+ TCRmini donors into TCRαk/o lymphopenic recipients, of which some received gavage of solubilized grape seed extract three times a week for 3 weeks before and 1 week after the transfer (fig. S7A). As shown in figs. S7B and S6B, mice that received grape seed extract had three times more A. muciniphila detected in their feces than the untreated group. Notably, the level of this bacteria in the grape seed extract–treated group remained below levels observed in naturally colonized TCRmini mice or TCRαk/o recipients gavaged with live bacteria (fig. S5B). Expansion of A. muciniphila correlated with survival, because only recipients fed with grape seed extract that received adoptive transfer survived over 60 days (78% of animals were alive at day 180), whereas other recipients died earlier. The median survival of mice that received grape seed extract and CD4+ cells from CNS1k/oTCRmini donors was only marginally longer than untreated controls, suggesting that gross of the anti-inflammatory effect depended on the formation of pTregs (fig. S7A). Survival of mice also correlated with increased pTreg numbers (fig. S7C) and a reduced level of proinflammatory cytokines (fig. S7D). Multiple hybridomas generated from colonic pTregs in grape seed–treated mice responded to a lysate from A. muciniphila (12.5%; 7 of 56 hybridomas tested) but not to ASF492 lysate, and TCRs expressed on these responder hybridomas accounted for less than 0.5% of all TCRs expressed by colonic CD4+ cells (fig. S7E). Thus, grape seed extract can mimic, although less effectively, the anti-inflammatory effect of oral delivery of live A. muciniphila by supporting Treg expansion.
DISCUSSION
Intestinal CD4+Foxp3+ Tregs help to enforce tolerance toward commensal bacteria while permitting immunoreactivity to harmful pathogens. The development of better therapeutics to control these cellular responses to commensals requires further advances in our knowledge of how epitopes derived from different bacterial strains affect the CD4+ T cell repertoire by reprogramming naïve CD4+ T cells to activated effectors versus tolerogenic pTregs and by expanding residual Tregs (36). Here, we show that discrete specificities of intestinal Tregs to commensal-derived antigens can be effectively recapitulated ex vivo by studying their reactivity using immortalized hybridomas that up-regulate expression of the Nur77GFP reporter following TCR activation. Hybridomas do not inherit parental Tregs ex vivo unresponsiveness but retain these cell TCRs, whereas introduced Nur77GFP reporter provides superior sensitivity and uniformity upon TCR activation as compared with IL-2 or NFATGFP assays. The first feature is particularly important for studying commensal antigens that often elicit responses resembling activation induced by weak agonists. In contrast, activation of TH17-derived hybridomas by SFB induced high levels of IL-2 in hybridomas (37). In addition, hybridomas established from CD4+CCR5+PD1+ follicular cells from gnotobiotic mice colonized with ASF flora supplemented with A. muciniphila recognized commensal antigens as full agonists, and their TCRs bound specific Ab/peptide tetramers (8). In our study, hybridoma activation by commensal epitopes was reproducibly diminished as compared with the same cell responses to aCD3 mAb triggering, even if tested microbial epitopes encoded well-defined Ab binding motifs. Instead, we noted overall stronger responses of hybridoma derived from CD4+ T cells that before colonization had not been exposed to microbial antigens or lacked pTreg “supervision” due to inherent deficit in CNS1 control region in foxp3 locus. Our results suggest that CD4+ cell interactions with microbiota increase the activation threshold induced by microbial antigens, in part due to specific suppression by Tregs. Therefore, mice lacking microbiota or pTregs harbor a higher frequency of CD4+ T cells with higher-affinity TCRs specific for commensal-derived antigens.
We also examined how often colonic CD4+Foxp3− and CD4+Foxp3+ T cells that express identical TCRs are specific for the same, commensal epitopes. In TCRmini mice, thymocytes naturally separate to both CD4+ lineages, and reduced diversity of the TCR repertoire simplifies tracking functionally distinct clones that express identical TCRs. We identified three different epitopes from two different commensal bacteria that activated nine CD4+Foxp3− and CD4+Foxp3+ clones that shared among them six different TCRs. However, only one of the shared TCRs was expressed by two clones representing different CD4+ lineages. This result implied that the same commensal-derived epitopes could specifically activate CD4+ cells with different effector functions and mostly nonoverlapping TCR repertoires (8).
CNS1k/o mice showed a significant deficiency in A. muciniphila compared with CNS+/+ mice. This observation is consistent with a previous report in which transplantation of fresh fecal material from SPF breeders to GF CNS1+/+ or CNS1k/o mice resulted in low abundance of mucus-penetrating Mucispirillum schaedleri and A. muciniphila strains only in the latter strain, suggesting that deficiency in pTregs plays a role in this bacteria stable colonization (38). However, when we examined fine specificities of colonic CD4+Foxp3− and Tregs ex vivo, we found that hybridomas derived from both subsets expressed A. muciniphila–specific TCRs, irrespectively of their origin (CNS1+/+ or CNS1k/o strain). This result agrees with other observations that pTreg deficiency in CNS1k/o mice does not prevent A. muciniphila from being present in the microbiome of these mice, suggesting that preexisting tTregs suffice to sustain its low incidence (38). However, the intestinal prominence of this bacteria depended on the recruitment of specific pTregs, suggesting that the majority of clones expressing specific TCRs are derived from Foxp3− cells. Hybridomas established from CD4+ cells from CNS1k/o mice responded to A. muciniphila isolates and specific epitopes comparably or stronger than hybridomas produced from the corresponding subset from control mice. This observation suggests that the blockade in pTreg induction enhances the sensitivity of naïve CD4+ T cells for this commensal bacterium.
A recent study that used live or pasteurized A. muciniphila to treat patients with obesity, untreated type 2 diabetes mellitus, or inflammatory bowel disease (IBD) concluded that daily administration of dead bacteria provides health benefits including improved insulin sensitivity, reduced levels of insulin, and enhanced gut barrier as compared with controls receiving a placebo (39). Although the involvement of intestinal Tregs in the therapeutic effect of A. muciniphila in humans still has to be determined, the fact that live or dead bacteria had similar therapeutic effects suggests that recognition of specific epitopes may regulate mucosal adaptive immune cells. Because A. muciniphila can degrade the mucus layer, which negatively affects gut wall integrity, the use of pasteurized bacteria is clinically more compelling than its use as live probiotic (40), as also illustrated by ongoing clinical trial NCT01314690 that uses pasteurized A. muciniphila. On the other hand, penetration of mucus enhances access of microbial antigens to the intestinal mucosa and peptide exposure to specific CD4+ T cells. Thus, several approaches to treat colitis in mice have used polyphenol-rich food supplements like resveratrol or syringaresinol to propagate residual A. muciniphila to reduce proinflammatory CD4+ IFN-γ+ and CD4+ IL-17+ subsets and increase CD4+Foxp3+ Tregs (41, 42). In our adoptive transfer model, delivery of similar food supplements also extended survival of host mice that received naïve CD4+ CNS+/+ but not CNS1k/o T cells. The similar therapeutic effect guiding the conversion and expansion of polyclonal Tregs in human patients will likely require delivery of a mix of microbial peptides that TCRs on naïve CD4+ T cells can recognize as weak agonists. Other ongoing clinical trials testing the capacity of A. muciniphila to reduce inflammation in visceral fat, type 1 diabetes, and metabolic syndrome suggest that peptide epitopes derived from this bacteria can be useful in other autoimmune and digestive disorders (43).
In summary, our data are consistent with previous reports that the manipulation of A. muciniphila intestinal content improves colonic health (44, 45). However, we provide evidence that this bacterium is the main source of antigens recognized by Tregs and conventional CD4+ cells that leads to an increase in Treg number and redirection of harmful, colitogenic CD4+ clones, into a protective pTreg lineage. Our findings imply that the identification of commensal epitopes can help improve the treatment of patients with IBD. In aggregate, our data reveal previously unknown information on how Tregs and CD4+Foxp3− clones respond to symbiont-derived antigens and should help in understanding how imbalances in effector and Tregs subsets contribute to intestinal inflammation.
MATERIALS AND METHODS
Mice
TCRmini mice expressed Foxp3GFP reporter (Jax strain no. 023800). In some experiments, congenic Vα2 mice (Vα2+Vβ14wt) were used. CNS1k/o mice were a gift of S. Schlenner (KU Leuven, Belgium) and were crossed with TCRminiFoxp3GFP animals (46). SPF ultraclean TCRαk/o mice were purchased from the Jackson laboratory (strain no. 2116) and were bred in our facility. β2mk/o mice (Jax strain no. 002087) were used as bone marrow donors in some experiments. For all experiments, 8- to 10-week-old littermate or age-matched mice of both sexes rederived to ultraclean status at Georgia State University (GSU) were used. In some experiments, TCRmini mice rederived to GF status (University of Alabama at Birmingham, Alabama) or TCRmini or TCRαk/o mice treated with an antibiotics cocktail [metronidazole (1 mg/ml), vancomycin (0.3 mg/ml), streptomycin (2 mg/ml), and ciprofloxacin (0.15 mg/ml)] all in sterile drinking water [containing 1% w/v sucrose (Sigma-Aldrich) changed twice a week for 2 weeks before experiment] were used. All mice were kept on a sterile chow with a 12-hour light/dark cycle. All presented experiments followed Institutional Animal Care and Use Committee (IACUC) guidelines and were approved by the GSU IACUC.
Antibodies and reagents
All used reagents are listed in table S1. mAbs were purchased from commercial vendors (eBioscience, BD Biosciences, or BioLegend).
Bacterial strains
Clostridium propionicum ASF356, L. murinus ASF361, and E. plexicaudatum ASF492 were the gifts of J. Fox (Massachusetts Institute of Technology). Oligo-MM microconsortium was a gift of B. Stecher (Ludwig-Maximilians-Universität München, Germany). A. muciniphila [American Type Culture Collection (ATCC BAA-835)] was a gift of B. Chassaing (GSU). E. coli DH5α and BL21DE strains (Invitrogen) were used for plasmid propagation and protein induction. DH5α and BL21DE3 were grown in LB broth. A. muciniphila was grown in an anaerobe BHI broth in an anaerobic chamber (Coy Lab Products). All media were from Fisher Scientific. ASF was introduced to GF mice by cohousing. Oligo-MM was introduced from cryostocks. All GF mice were at 6 weeks of age at the time of colonization and were analyzed 6 to 8 weeks after bacteria introduction. A. muciniphila was grown overnight and washed twice in PBS, and 100 μl (2 × 108 colony-forming units) per mouse was gavaged to 6- to 8-week-old mice.
Bacterial library preparation
Genomic DNA libraries were prepared as reported (5, 7) with some modifications. The overview of the procedure is summarized in fig. S2B. Briefly, genomic DNA from each bacterium was isolated with GenElute bacteria genomic isolation kit (Sigma-Aldrich), followed by ethanol precipitation. Genomic DNA (0.1 mg of each) was digested with Sau3AI, gel purified (Thermo), and cloned into Bam HI cut, calf intestine alkaline phosphatase-treated pGEX-KG vector (ATCC, 77103). More than 5000 clones from each bacterium were obtained. Libraries of 96 single clones were grown overnight at 37°C in microtiter tubes (Thermo) in LB + ampicillin (100 μg/ml; Sigma-Aldrich), diluted 1:100 with fresh LB/ampicillin, and induced with IPTG (0.5 mM; Sigma-Aldrich) for 4 hours at 25°C. Cells were pelleted, washed twice with PBS, pasteurized (70°C, 40 min), and resuspended in sonication buffer [50 mM tris (pH 8.0), 0.05% (v/v) Tween 20 (all from Thermo), 0.1 mg/ml (w/v) lysozyme (Sigma-Aldrich), and 0.1% (v/v) protease inhibitors (ProBlock Gold bacterial, GoldBio)]. Cells were sonicated on ice with a Branson sonicator (duty 30, output 2; 10-s burst, 10-s rest, 3 min total). Insoluble material was removed by centrifugation (4°C, 15 min, 10,000g). Protein content was measured with 660-nm Protein Assay Reagent (Fisher Scientific).
TCR sequencing
TCRVα2 CDR3 regions were amplified as reported (17). Hybridoma TCRs were sequenced by ACGT (Germantown, MD).
Fecal bacteria and bacterial DNA isolation and real-time PCR
Fresh fecal pellets were resuspended in PBS and subjected to a series of low-speed centrifugations to remove debris. Bacteria were then pelleted at 4000g (15 min, 4°C) and washed with PBS. The procedure was repeated three times after which bacteria were pasteurized, resuspended in sonication buffer, and sonicated, and protein extract was cleared as above. For hybridoma testing, nontoxic concentration of proteins (10 to 100 μg/ml) was used (lysates were typically at 100 mg/ml and used at least at 1:1000 dilution). For some experiments, we further purified bacterial fraction as reported (4), but both methods consistently gave similar results. Therefore, all data presented in this manuscript were generated using sonicated bacteria to better account for the protein amount loaded onto DCs.
Fecal DNA was isolated from 100.0 mg of freshly obtained mouse feces with QIAamp PowerFecal DNA Kit (Qiagen) according to the manual. One microliter of each DNA was run in triplicate on QuantStudio 3 real-time polymerase chain reaction (RT-PCR) instrument using SsoAdvanced Universal IT SYBR Green kit (Bio-Rad). The melting curve was run for each reaction. The comparative CT (ΔΔCT) method was used for analysis with data normalized to UniF340/UniR514 universal bacteria primers and then to appropriate control as indicated. For green fluorescent protein (GFP) and IL-2 expression studies, hybridomas were treated as indicated and total DNA-free RNA was isolated with RNeasy kit (Qiagen). One microgram of each RNA was reverse transcribed using SuperScript III kit (Invitrogen) using random hexamers. Reverse transcription quantitative PCR (RT-qPCR) was run as above, and data were first normalized to β-actin and then to the appropriate control. IFN-γ, IL-17, IL-6, and IL-10 transcripts were assessed with TaqMan assays (Invitrogen). All primer sequences and assay numbers are shown in table S1. Primers were from Invitrogen.
Fecal bacteria 16S rDNA sequencing
Sequencing was done at the Genomics Facility at Cornel University, and data analysis was done as reported (47). Briefly, sequences were demultiplexed, quality filtered using QIIME2 software package 39, and forward and reverse Illumina reads were joined using the fastq-join method. QIIME default parameters for quality filtering were used before assigning sequences to operational taxonomic units (OTUs) using UCLUST algorithm with a 97% threshold of pairwise identity and classified taxonomically using the Greengenes reference database. A single representative sequence for each OTU was aligned, and a phylogenetic tree was built using FastTree and used for computing the unweighted UniFrac distances between samples. Data were analyzed with QIIME [reads were classified to taxonomic groups (class), and relative abundance was calculated]. Unprocessed sequencing data have been deposited in the European Nucleotide Archive under accession number PRJEB36240.
Tissue preparations, FACS analysis, and cell sorting
Colon samples were prepared as reported (2). Cells were counted (Z1 Coulter, Beckman), and 1 × 106 cells were plated; Fc receptors were blocked with 2.4G2 antibody for 5 min at room temperature and stained with titrated, dye-, or biotin-conjugated antibodies (20 min on ice in the dark). Intracellular cytokines were detected per the manufacturer’s instructions (eBioscience) after stimulation for 4 hours with phorbol 12-myristate 13-acetate (PMA) (50 ng/ml) and ionomycin (500 ng/ml) in the presence of brefeldin A and monensin (each 1:1000). For intracellular cytokine or Ki-67 analysis, cells were fixed and permeabilized after surface staining and then incubated for 20 min at room temperature with antibodies for IFN-γ, IL-17A, IL-10, or Ki-67. Dilution of proliferation dye eFluor670 (eBioscence) was assessed according to the manufacturer’s instruction. Briefly, cells were loaded with eFluor670 (500 nM) for 15 min at 37°C, and the reaction was terminated with a culture media containing fetal bovine serum. Cells were washed three times with PBS, seeded, activated with an indicated antigen, and analyzed on day 4. Cells were washed and analyzed by a CytoFLEX (B5-R3-V5) flow cytometer (Beckman Coulter). Cells were sorted with an SH800 sorter (four lasers, optical filter pattern 2; Sony) using 100-μm chips. Purity routinely exceeded 98%, and viability was >98% as assessed by 4′,6-diamidino-2-phenylindole (DAPI) or trypan blue staining. For some experiments, CD4+ cells were enriched with MS columns (Miltenyi) to purity >90% before sorting with SH800 sorter. Fluorescence-activated cell sorting (FACS) data were analyzed with FlowJo v.10 (FlowJo, LLC). IL-6 expression was assessed in culture supernatant by a commercial kit (eBioscience) per included instructions. Cells were seeded at 1 × 106 on aCD3 (1 μg/ml)– and aCD28 (0.1 μg/ml)–coated wells and restimulated overnight.
Cell culture
All cells were cultured in CTM (Complete Tumor Media) media. Bone marrow–derived DCs or splenic CD11c+ cells were used for CD4+ cell activation. DCs were prepared from bone marrow by stimulation with granulocyte-macrophage colony-stimulating factor (GM-CSF) (5 ng/ml; PeproTech) for 6 days in CTM. Cells were routinely >80% CD11c+CD80+MHCII+ as assessed by flow cytometry. Further purification (>95% by FACS) did not change CD4+ cell response, and therefore, unsorted cells were used. Likewise, CD11c+ DCs sorted from spleen-stimulated CD4+ hybridomas similarly to GM-CSF–stimulated DCs. Therefore, in all experiments, DCs prepared with GM-CSF were used.
CD4+ hybridoma production and testing
CD4+ hybridomas were prepared as reported (9). The fusion partner was an improved variant of BW5147α−β− thymoma that expressed Nur77GFP reporter (BWNur77GFP). BWNur77GFP cells were prepared by the transfection of linearized BAC (GENSAT1-BX1262; BACPAC) containing GFP under the Nur77 promoter. BW5147α−β− cells were transfected (FuGENE HD, Promega) with 3 μg of Xho I–linearized BAC together with Bam HI–linearized pcDNA3 (Fisher Scientific) (100:1 BAC:pcDNA3), and cells were selected with G418 (1.5 mg/ml; Cellgro). After 2 weeks, selected cells were pulsed with PMA (10 ng/ml) and ionomycin (250 ng/ml) (both from Sigma-Aldrich) overnight, and single cells expressing the highest level of GFP were sorted with MoFlo sorter (Beckman Coulter). Expanded clones were tested again for GFP up-regulation, sorted into single cells, and expanded. Clone 1.33 was selected as a fusion partner for all experiments. For hybridoma testing, DCs were plated at 2 × 104 cells per well of a round-bottom 96-well plate. Antigen (protein lysate, peptide, or soluble aCD3) was added for 6 hours after, which cells were washed twice with PBS and mixed with CD4+ hybridomas at 1:10. Sixteen hours later, Nur77GFP expression was assessed by FACS in gated CD4+Vα2+ DAPI− cells. Supernatants were taken for IL-2 release assay with HT2 cells. In some experiments, hybridomas were stimulated with plate-bound aCD3. Hybridomas from Tregs were made as reported (9). Hybridomas from anergic CD4+ cells were made essentially like hybrids from Tregs with the exception that aCD28 (1 μg/ml) was used, and IL-2 (1 ng/ml) was added at day 3. Cells were typically fused at days 7 and 8.
For the ASF study, we screened 1485 hybridomas from colonic Foxp3− cells and 858 from the Foxp3+ subset from TCRmini mice and 1158 (Foxp3−) and 515 (Foxp3+) from CNS1k/oTCRmini mice. For the A. muciniphila study, we screened 725 (Foxp3−) and 371 (Foxp3+) hybridomas. For each subset, more than 10 separate fusions of CD4+ cells with BWNur77GFP thymoma were made.
Immunization
Cholera toxin (CT; Sigma-Aldrich) was dissolved in PBS and used at 10 μg per mouse. Antigenic peptides (purity >90%; GenScript or ABI Scientific) were mixed with CT at 30 μg per mouse, and mice were immunized by gavage (CT + peptide in 200 μl PBS/1.5% NaHCO3). For boosting, 15 μg of peptide in PBS was injected intraperitoneally (100 μl per mouse) 1 week later, and mice were sacrificed on day 14. In selected experiments, TCRαk/o mice were first gavaged with grape seed extract (BRI Nutrition) at 12.5 mg/kg three times a week for 1 week, followed by 25 mg/kg for additional 4 weeks, including 1 week after adoptive transfer of naïve CD4+ cells. Control mice received PBS only.
Adoptive transfer colitis model
FACS-purified naïve CD4+Foxp3GFP-CD44−(CD45RBhigh) cells (1 × 106) were adoptively transferred into TCRαk/o mice. Animals were monitored for signs of colitis (weight loss) and euthanized when met humane endpoint or no later than day 180.
Histology
Tissues were excised and placed in 10% buffered formalin (Fisher Scientific) for 24 hours and processed with Excelsior ES tissue Processor (Thermo). Tissues were embedded in paraffin blocks, and 4-μm sections were cut with Shandon Finesse 325 Manual Microtome (Thermo). Slides were stained with hematoxylin and eosin (H&E) (Fisher Scientific) according to a standard protocol. Three fragments of each tissue were photographed [EVOS FL Auto microscope (×40 magnification) equipped with Pearl Scope Software (Fisher Scientific)], and average colitis scores (0, no discernible inflammation; 1, small, focal focus of inflammation; 2, small, multiple foci of inflammation; 3, multiple large foci of inflammation; and 4, significant inflammation with parenchymal destruction) were calculated.
Statistical analysis
Data were analyzed with Origin 2017 (OriginLab) and were presented as the means ± SD or Kaplan-Meier survival plots. Statistical significance of differences was calculated using paired sample Student’s t test or log-rank test where appropriate. Statistical significance was considered as *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank J. Pihkala (Augusta University) for help with cell sorting and H. Ignatowicz (GSU) with maintenance of mouse colony. We thank T. Schoeb (UAB) for rederivation of our mice to GF status and C. Jin (GSU) for rederivation of our mice to ultraclean status. We also thank T. Denning for critically reading the manuscript. Funding: This work was supported by NIH R01DK099264 and 1R01AI121151 research grants and GSU funding to L.I. The work was supported by the German Research Foundation (DFG) Priority Programme SPP1656 (DFG STE1971/4-2), the German Center for Infection Research (DZIF), and the Center for Gastrointestinal Microbiome Research (CEGIMIR) to B.S. J.G.F. is supported by NIH grants P30ES002109, P01CA028842, and R35 CA210088. Author contributions: M.P.K. and L.I. conceived the study, designed experiments, interpreted the data, and wrote the paper. M.P.K. performed the most presented experiments. E.A.S. developed and validated BWNur77GFP thymoma, prepared most CD4 hybridomas, and validated crucial experiments in a blinded manner. A.C. colonized mice with Oligo-MM and examined ASF and Oligo-MM colonic Tregs, prepared ASF hybridomas, and provided GF mice. B.C. analyzed 16S rRNA data and provided A. muciniphila. Y.-J.J. and S.-M.K. helped with histological specimens. B.S. provided Oligo-MM consortium and helped with interpretation of microbiological data. J.G.F. provided ASF isolates. All the authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper can be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaaz3186/DC1
RERERENCES AND NOTES
- 1.Gratz I. K., Campbell D. J., Organ-specific and memory treg cells: Specificity, development, function, and maintenance. Front. Immunol. 5, 333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cebula A., Seweryn M., Rempala G. A., Pabla S. S., McIndoe R. A., Denning T. L., Bry L., Kraj P., Kisielow P., Ignatowicz L., Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 497, 258–262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lathrop S. K., Bloom S. M., Rao S. M., Nutsch K., Lio C. W., Santacruz N., Peterson D. A., Stappenbeck T. S., Hsieh C. S., Peripheral education of the immune system by colonic commensal microbiota. Nature 478, 250–254 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivanov I. I., Atarashi K., Manel N., Brodie E. L., Shima T., Karaoz U., Wei D., Goldfarb K. C., Santee C. A., Lynch S. V., Tanoue T., Imaoka A., Itoh K., Takeda K., Umesaki Y., Honda K., Littman D. R., Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y., Torchinsky M. B., Gobert M., Xiong H., Xu M., Linehan J. L., Alonzo F., Ng C., Chen A., Lin X., Sczesnak A., Liao J.-J., Torres V. J., Jenkins M. K., Lafaille J. J., Littman D. R., Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 510, 152–156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cong Y., Feng T., Fujihashi K., Schoeb T. R., Elson C. O., A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc. Natl. Acad. Sci. U.S.A. 106, 19256–19261 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu M., Pokrovskii M., Ding Y., Yi R., Au C., Harrison O. J., Galan C., Belkaid Y., Bonneau R., Littman D. R., c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ansaldo E., Slayden L. C., Ching K. L., Koch M. A., Wolf N. K., Plichta D. R., Brown E. M., Graham D. B., Xavier R. J., Moon J. J., Barton G. M., Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science 364, 1179–1184 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh N., Pacholczyk R., Iwashima M., Ignatowicz L., Generation of T cell hybridomas from naturally occurring FoxP3+ regulatory T cells. Methods Mol. Biol. 707, 39–44 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Kappler J. W., Skidmore B., White J., Marrack P., Antigen-inducible, H-2-restricted, interleukin-2-producing T cell hybridomas. Lack of independent antigen and H-2 recognition. J. Exp. Med. 153, 1198–1214 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pacholczyk R., Kern J., Singh N., Iwashima M., Kraj P., Ignatowicz L., Nonself-antigens are the cognate specificities of Foxp3+ regulatory T cells. Immunity 27, 493–504 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solomon B. D., Hsieh C.-S., Antigen-specific development of mucosal Foxp3+RORγt+ T cells from regulatory T cell precursors. J. Immunol. 197, 3512–3519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ise W., Kohyama M., Nutsch K. M., Lee H. M., Suri A., Unanue E. R., Murphy T. L., Murphy K. M., CTLA-4 suppresses the pathogenicity of self antigen–specific T cells by cell-intrinsic and cell-extrinsic mechanisms. Nat. Immunol. 11, 129–135 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hooijberg E., Bakker A. Q., Ruizendaal J. J., Spits H., NFAT-controlled expression of GFP permits visualization and isolation of antigen-stimulated primary human T cells. Blood 96, 459–466 (2000). [PubMed] [Google Scholar]
- 15.Au-Yeung B. B., Zikherman J., Mueller J. L., Ashouri J. F., Matloubian M., Cheng D. A., Chen Y., Shokat K. M., Weiss A., A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 111, E3679–E3688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashouri J. F., Weiss A., Endogenous Nur77 Is a specific indicator of antigen receptor signaling in human T and B cells. J. Immunol. 198, 657–668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacholczyk R., Ignatowicz H., Kraj P., Ignatowicz L., Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity 25, 249–259 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Vrisekoop N., Monteiro J. P., Mandl J. N., Germain R. N., Revisiting thymic positive selection and the mature T cell repertoire for antigen. Immunity 41, 181–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlenner S. M., Weigmann B., Ruan Q., Chen Y., Von B. H., Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J. Exp. Med. 209, 1529–1535 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim K. S., Hong S.-W., Han D., Yi J., Jung J., Yang B.-G., Lee J. Y., Lee M., Surh C. D., Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Wymore Brand M., Wannemuehler M. J., Phillips G. J., Proctor A., Overstreet A.-M., Jergens A. E., Orcutt R. P., Fox J. G., The Altered Schaedler Flora: Continued applications of a defined murine microbial community. ILAR J. 56, 169–178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brugiroux S., Beutler M., Pfann C., Garzetti D., Ruscheweyh H.-J., Ring D., Diehl M., Herp S., Lötscher Y., Hussain S., Bunk B., Pukall R., Huson D. H., Münch P. C., McHardy A. C., McCoy K. D., Macpherson A. J., Loy A., Clavel T., Berry D., Stecher B., Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat. Microbiol. 2, 16215–16226 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Kim S. V., Xiang W. V., Kwak C., Yang Y., Lin X. W., Ota M., Sarpel U., Rifkin D. B., Xu R., Littman D. R., GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 340, 1456–1459 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atarashi K., Tanoue T., Oshima K., Suda W., Nagano Y., Nishikawa H., Fukuda S., Saito T., Narushima S., Hase K., Kim S., Fritz J. V., Wilmes P., Ueha S., Matsushima K., Ohno H., Olle B., Sakaguchi S., Taniguchi T., Morita H., Hattori M., Honda K., Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Arpaia N., Campbell C., Fan X., Dikiy S., van der Veeken J., deRoos P., Liu H., Cross J. R., Pfeffer K., Coffer P. J., Rudensky A. Y., Metabolites produced by commensal bacteria promote peripheral regulatory T cell generation. Nature 504, 451–455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith P. M., Howitt M. R., Panikov N., Michaud M., Gallini C. A., Bohlooly Y., Glickman J. N., Garrett W. S., The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biggs M. B., Medlock G. L., Moutinho T. J., Lees H. J., Swann J. R., Kolling G. L., Papin J. A., Systems-level metabolism of the altered Schaedler flora, a complete gut microbiota. ISME J. 11, 426–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Telesford K. M., Ochoa-Repáraz J., Haque-Begum S., Christy M., Kasper E. J., Wang L., Wu Y., Robson S. C., Kasper D. L., Kasper L. H., An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat. Commun. 5, 4432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalekar L. A., Mueller D. L., Relationship between CD4 regulatory T Cells and anergy in vivo. J. Immunol. 198, 2527–2533 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulkarni N., Meitei H. T., Sonar S. A., Sharma P. K., Mujeeb V. R., Srivastava S., Boppana R., Lal G., CCR6 signaling inhibits suppressor function of induced-Treg during gut inflammation. J. Autoimmun. 88, 121–130 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Hänninen A., Toivonen R., Pöysti S., Belzer C., Plovier H., Ouwerkerk J. P., Emani R., Cani P. D., De Vos W. M., Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut 67, 1445–1453 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Cekanaviciute E., Yoo B. B., Runia T. F., Debelius J. W., Singh S., Nelson C. A., Kanner R., Bencosme Y., Lee Y. K., Hauser S. L., Crabtree-Hartman E., Sand I. K., Gacias M., Zhu Y., Casaccia P., Cree B. A. C., Knight R., Mazmanian S. K., Baranzini S. E., Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. U.S.A. 114, 10713–10718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sekirov I., Tam N. M., Jogova M., Robertson M. L., Li Y., Lupp C., Finlay B. B., Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect. Immun. 76, 4726–4736 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L., Carmody R. N., Kalariya H. M., Duran R. M., Moskal K., Poulev A., Kuhn P., Tveter K. M., Turnbaugh P. J., Raskin I., Roopchand D. E., Grape proanthocyanidin-induced intestinal bloom of Akkermansia muciniphila is dependent on its baseline abundance and precedes activation of host genes related to metabolic health. J. Nutr. Biochem. 56, 142–151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmad S. F., Zoheir K. M. A., Abdel-Hamied H. E., Ashour A. E., Bakheet S. A., Attia S. M., Abd-Allah A. R. A., Grape seed proanthocyanidin extract has potent anti-arthritic effects on collagen-induced arthritis by modifying the T cell balance. Int. Immunopharmacol. 17, 79–87 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Strauch U. G., Obermeier F., Grunwald N., Gürster S., Dunger N., Schultz M., Griese D. P., Mähler M., Schölmerich J., Rath H. C., Influence of intestinal bacteria on induction of regulatory T cells: Lessons from a transfer model of colitis. Gut 54, 1546–1552 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goto Y., Panea C., Nakato G., Cebula A., Lee C., Diez M. G., Laufer T. M., Ignatowicz L., Ivanov I. I., Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 40, 594–607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell C., Dikiy S., Bhattarai S. K., Chinen T., Matheis F., Calafiore M., Hoyos B., Hanash A., Mucida D., Bucci V., Rudensky A. Y., Extrathymically generated regulatory T cells establish a niche for intestinal border-dwelling bacteria and affect physiologic metabolite balance. Immunity 48, 1245–1257.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Depommier C., Everard A., Druart C., Plovier H., Van Hul M., Vieira-Silva S., Falony G., Raes J., Maiter D., Delzenne N. M., de Barsy M., Loumaye A., Hermans M. P., Thissen J.-P., de Vos W. M., Cani P. D., Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 25, 1096–1103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desai M. S., Seekatz A. M., Koropatkin N. M., Kamada N., Hickey C. A., Wolter M., Pudlo N. A., Kitamoto S., Terrapon N., Muller A., Young V. B., Henrissat B., Wilmes P., Stappenbeck T. S., Núñez G., Martens E. C., A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alrafas H. R., Busbee P. B., Nagarkatti M., Nagarkatti P. S., Resveratrol modulates the gut microbiota to prevent murine colitis development through induction of Tregs and suppression of Th17 cells. J. Leukoc. Biol. 106, 467–480 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho S.-Y., Kim J., Lee J. H., Sim J. H., Cho D.-H., Bae I.-H., Lee H., Seol M. A., Shin H. M., Kim T.-J., Kim D.-Y., Lee S.-H., Shin S. S., Im S.-H., Kim H.-R., Modulation of gut microbiota and delayed immunosenescence as a result of syringaresinol consumption in middle-aged mice. Sci. Rep. 6, 39026–39039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cani P. D., de Vos W. M., Next-generation beneficial microbes: The case of Akkermansia muciniphila. Front. Microbiol. 8, 1765–1772 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez-Siles M., Enrich-Capó N., Aldeguer X., Sabat-Mir M., Duncan S. H., Garcia-Gil L. J., Martinez-Medina M., Alterations in the abundance and co-occurrence of Akkermansia muciniphila and Faecalibacterium prausnitzii in the colonic mucosa of inflammatory bowel disease subjects. Front. Cell. Infect. Microbiol. 8, 281–296 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin N.-R., Lee J.-C., Lee H.-Y., Kim M.-S., Whon T. W., Lee M.-S., Bae J.-W., An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63, 727–735 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Szurek E., Cebula A., Wojciech L., Pietrzak M., Rempala G., Kisielow P., Ignatowicz L., Differences in expression level of helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ regulatory T cells. PLOS ONE 10, e0141161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chassaing B., Koren O., Goodrich J. K., Poole A. C., Srinivasan S., Ley R. E., Gewirtz A. T., Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greer R. L., Dong X., Moraes A. C. F., Zielke R. A., Fernandes G. R., Peremyslova E., Vasquez-Perez S., Schoenborn A. A., Gomes E. P., Pereira A. C., Ferreira S. R. G., Yao M., Fuss I. J., Strober W., Sikora A. E., Taylor G. A., Gulati A. S., Morgun A., Shulzhenko N., Akkermansia muciniphila mediates negative effects of IFNγ on glucose metabolism. Nat Commun. 7, 13329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barman M., Unold D., Shifley K., Amir E., Hung K., Bos N., Salzman N., Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect. Immun. 76, 907–915 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ertesvag A., Austenaa L. M. I., Carlsen H., Blomhoff R., Blomhoff H. K., Retinoic acid inhibits in vivo interleukin-2 gene expression and T-cell activation in mice. Immunology 126, 514–522 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buehr M., Meek S., Blair K., Yang J., Ure J., Silva J., McLay R., Hall J., Ying Q.-L., Smith A., Capture of authentic embryonic stem cells from rat blastocysts. Cell 135, 1287–1298 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Lane S. W., Sykes S. M., Al-Shahrour F., Shterental S., Paktinat M., Lo Celso C., Jesneck J. L., Ebert B. L., Williams D. A., Gilliland D. G., The Apcmin mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood 115, 3489–3497 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaaz3186/DC1