

A strong, tough, and fast-recovery hydrogel is engineered by cooperative interactions between histidine-rich peptides and Zn2+.
Abstract
Many load-bearing tissues, such as muscles and cartilages, show high elasticity, toughness, and fast recovery. However, combining these mechanical properties in the same synthetic biomaterials is fundamentally challenging. Here, we show that strong, tough, and fast-recovery hydrogels can be engineered using cross-linkers involving cooperative dynamic interactions. We designed a histidine-rich decapeptide containing two tandem zinc binding motifs. Because of allosteric structural change-induced cooperative binding, this decapeptide had a higher thermodynamic stability, stronger binding strength, and faster binding rate than single binding motifs or isolated ligands. The engineered hybrid network hydrogels containing the peptide-zinc complex exhibit a break stress of ~3.0 MPa, toughness of ~4.0 MJ m−3, and fast recovery in seconds. We expect that they can function effectively as scaffolds for load-bearing tissue engineering and as building blocks for soft robotics. Our results provide a general route to tune the mechanical and dynamic properties of hydrogels at the molecular level.
INTRODUCTION
With every step of walking, our muscles, cartilage, and tendon are subjected to substantial mechanical loads; yet, these biological soft tissues can quickly recover in a time scale of seconds and function reliably for multiple mechanical cycles. Exploring soft hydrogels with similar mechanical properties to these natural load-bearing tissues may have broad applications for biomechanical actuators (1), synthetic cartilage (2), artificial muscle (3), ionic skin (4), and soft robotics (5). Many efforts have been devoted to enhancing the mechanical strength and toughness of hydrogels by introducing sacrificial bonds/polymer networks or other special energy dissipation mechanisms (6). Successful examples include double network (DN) hydrogels (7), dual–cross-linked hydrogels (8), nano/microcomposite hydrogels (9), and slide-ring hydrogels (10). In addition to mechanical strength and toughness, the quick recovery is a unique trait for load-bearing soft tissues. For example, our musculoskeletal system is constantly being stressed at a frequency higher than 1 Hz for many cycles during exercise, without showing any measurable fatigue. However, most synthetic strong hydrogels lack a mechanism to recover quickly. Traditional DN or hybrid network (HN) hydrogels using short polymer chains as sacrificial networks usually cannot self-recover (7). Strong hydrogels using physical cross-linking, including hydrophobic interactions (11, 12), ionic paring (13–15), hydrogen bonding (16), coordination interactions (17, 18), chain entanglement (19), host-guest interactions (8), and microcrystallization (20) can only slowly recover their mechanical properties at a time scale of minutes to days, even at high temperatures. Although some weak hydrogels using physical cross-linking can recover quickly, they are limited by low strength due to the relatively high intrinsic interaction dynamics (8, 14). It remains challenging to engineer hydrogels with combined high strength, toughness, and fast recovery.
The bulk mechanical properties of hydrogels have been correlated with the properties of cross-linkers at the molecular level, providing tremendous insight into rational design of the mechanical properties of hydrogels (3, 21). The strength of a hydrogel depends on the lifetime of the cross-linkers (22). Slow binding/unbinding kinetics often lead to strong hydrogels, whereas fast exchange rates yield soft ones. The toughness of a hydrogel depends on how much mechanical energy can be dissipated by bond rupture and is related to the mechanical strength of the bonds (3, 23). To possess high strength and toughness, the dissociation of cross-linkers should be slow and require a high activation barrier under force, as evidenced by many studies (24). Fast recovery requires the cross-linker to be dynamic, with high dissociation and association rates (8, 14). Such contradicting requirements make the design of hydrogels with combined high strength, toughness, and fast recovery fundamentally challenging. Intriguingly, naturally occurring load-bearing materials have overcome this issue by using the cooperativity of weak interactions. For example, some folded proteins exhibit remarkable mechanical stability yet can fold quickly after force is released to restore the mechanical properties of the materials (3), which is achieved through concerted multiple weak interactions in the folded protein domains. It remains to be explored whether such a concept can be extended to the design of synthetic hydrogels with integrated high mechanical strength, toughness, and fast recovery using simple building blocks.
We illustrated this idea by engineering HN hydrogels using a specially designed peptide-metal complex as the physical cross-linker. Metal coordination complexes are well known for their fast binding/unbinding kinetics but are often too dynamic to produce hydrogels of high mechanical strength (22, 25). In many load-bearing structural proteins, this problem is circumvented by specifically evolved metal coordination sites (26), which contain multiple ligands in each binding site and can bind metal ions with considerably improved stability. Moreover, the load-bearing proteins often contain multiple binding sites arranged in tandem (27), entailing cooperativity among different binding sites for strong binding. Thus, we anticipate that, by engineering the cooperativity of ligands to form efficient metal binding sites in a peptide sequence, it is possible to obtain hydrogels with high mechanical stability, toughness, and fast-recovery.
RESULTS
Design and molecular mechanism of the cooperative Zn2+ binding
We designed three short histidine-rich peptides (HR-peptides) as ligands to bind with zinc ions (Zn2+) to construct HN hydrogels (Fig. 1A): Their sequences are Gly-Gly-His (denoted as PH1), Gly-His-His-Pro-His (denoted as PH3), and Gly-His-His-Pro-His-Gly-His-His-Pro-His (denoted as PH6). PH1 is the control group that contains only a single metal ion ligand (the histidine residue). PH3 is the Zn2+-binding sequence derived from an HR glycoprotein that contains three histidine residues forming a specific metal ion binding site (28). PH6 comprises two tandem repeats of PH3, which can form two closely arranged complexes with Zn2+ ions. The peptides were synthesized using solid-phase peptide synthesis and purified by high-performance liquid chromatography. The formation of Zn2+-histidine coordination complexes of the peptides was confirmed by ultraviolet (UV)–vis (fig. S1, A to C) and Raman spectroscopy (fig. S1, D to F). The binding stoichiometry of Zn2+ to PH6 was confirmed by x-ray fluorescence spectrometry (fig. S1G). The Zn2+-binding constants were 1.39 × 102 M−1 for PH1, 2.17 × 103 M−1 for PH3, and 2.79 × 104 and 9.45 × 106 M−1 for the two sites of PH6, respectively, as determined by isothermal titration calorimetry (ITC; Fig. 1, B to D). Obviously, the binding affinity is increased by forming a specific Zn2+-binding site in PH3 and further enhanced by tandemly arranging the binding sites in PH6.
Fig. 1. Cooperativity engineering, binding constants, and molecular mechanism of the metal ion coordination interactions at the molecular level for load bearing.
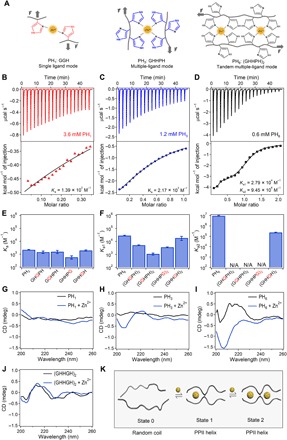
(A) The metal ion coordination complexes formed by single ligands (PH1, left) are dynamic and weak. When forming a metal chelation site made of multiple ligands (PH3, middle), the metal ion binding becomes much stronger and less dynamic than that of single ligands. Furthermore, when arranging two metal chelation sites in tandem (PH6, right), the binding affinity, mechanical strength, and association rate can be improved due to cooperativity between the two sites. (B to D) ITC titration data of PH1 (left), PH3 (middle), and PH1 (right) peptides with ZnCl2 in 1 M tris buffer (pH 7.60, containing 300 mM KCl) at 25°C. (E) Zn2+-binding constants (Ka) of PH3 and mutated PH3 peptides. The mutated amino acids are highlighted in red. The error bars represent the fitting errors. (F) Zn2+-binding constants of PH6 and mutated PH6 peptides. Left and right panels correspond to Ka1 and Ka2 for the two binding sites of PH6. Only PH6 and (GHHGH)2 peptides exhibited two binding constants. The rest of the peptides showed single-site binding characteristics. The error bars represent the fitting errors. (G to J) CD spectra of (G) PH1: GGH; (H) PH3: GHHPH; (I) PH6: (GHHPH)2; and (J) (GHHGH)2 peptides in the absence and presence of Zn2+ ions. The relative contents of PPII structures of PH1 and PH3 are 9.6 and 34.2% based on the height of the major CD peak at 205 nm, assuming that the PH6-Zn2+ complex shows 100% PPII helical structure. (K) Schematic illustration of the cooperative Zn2+-binding mechanism of PH6. The conformational change of the first coordination site leads to structural changes of the second one to a conformation more favoring Zn2+ binding. N/A, not applicable.
To understand how Zn2+ binding is notably enhanced by the uniquely designed peptide sequence, we mutated each amino acid in PH3 and PH6 to glycine to evaluate its contribution to Zn2+ binding. Glycine was chosen because it is the amino acid lacking a functional side chain, making it possible to evaluate the contribution of the imidazole group of histidine to Zn2+ binding and the effect of the unique torsion angle of proline to the structural change of the peptides. The mutated peptide sequences included GHGPH, GGHPH, GHHPG, GHHGH, (GHGPH)2, (GGHPH)2, (GHHPG)2, and (GHHGH)2. As shown in Fig. 1E and fig. S2, the Zn2+-binding constants of GHGPH, GGHPH, and GHHPG were smaller than that of PH3, indicating that every histidine in PH3 contributes to the coordination with Zn2+ ions. Moreover, the Zn2+-binding constants of GHHGH were almost the same as that of PH3, suggesting that the proline probably does not affect the coordination of PH3 to Zn2+ ions. In contrast, the Zn2+ binding to (GHGPH)2, (GGHPH)2, and (GHHPG)2 showed only a single binding constant, suggesting that they cannot form two binding sites properly. The Zn2+-binding constants of these three peptides were much smaller than that of PH6, indicating that the synergistic Zn2+ ion binding of PH6 is important for the enhanced binding affinity (Fig. 1F and fig. S2). Moreover, the proline residue was also critical for the synergistic Zn2+ binding of PH6. After mutating proline to glycine, although the peptide still showed two Zn2+-binding constants, the second one was much smaller than that of PH6 (Fig. 1F). All together, these results suggest that the specially designed peptide sequence ensures synergistic and cooperative Zn2+ binding, and PH6 exhibits much improved Zn2+-binding affinity than the peptides with histidine residues randomly on their sequences.
To further understand the molecular mechanism of the cooperative Zn2+ binding of PH6, we characterized the structural change of PHn peptides and mutations upon Zn2+ binding using circular dichroism (CD; Fig. 1, G to J, and fig. S3, A to C). PH1 did not show any characteristic peaks of secondary structures both in the absence and presence of Zn2+, presumably because the peptide sequence is too short. However, both PH3 and PH6 peptides showed a new CD peak at 208 nm in the presence of Zn2+, which can be assigned to the poly-proline type II helix (PPII) structure (29). The formation of the PPII structure of Zn2+-bound PH3 and PH6 peptides was further confirmed by Fourier transform infrared spectroscopy (fig. S3, D to F). As mentioned before, formation of the PPII structure did not change the Zn2+-binding affinity of PH3 but greatly affected the binding affinity of PH6, suggesting that the secondary structure is important for cooperative Zn2+ binding of the two tandem binding sites in PH6. Without proline, (GHHGH)2 cannot adopt a PPII structure, and the Zn2+ binding–induced structural change was absent (Fig. 1J). Therefore, the binding affinity was greatly reduced. On the basis of these experimental evidence, we suggest that the conformational change of the first coordination site of PH6 upon Zn2+ binding is critical to cooperative binding, as it can lead to structural changes of the second site to a conformation more favoring Zn2+ binding (Fig. 1K). This mechanism resembles the allosteric effect widely found in large proteins and has not been reported in small peptides.
Mechanical properties of HR-peptide–Zn2+ cross-linkers
As the mechanical stability of the cross-linkers is important to toughness of hydrogels, we then measured the mechanical stability of the HR-peptide–Zn2+ complexes at the molecular level using atomic force microscopy (AFM)–based single-molecule force spectroscopy (SMFS). The experimental scheme is illustrated in Fig. 2A. We linked the N terminus of the peptides to the silicon nitride cantilever tip and the glass substrate using a polyethylene glycol (PEG) linker (MW, 5000 Da). Peptide-Zn2+ complexes were formed when the cantilever approached the substrate in the presence of Zn2+ ions. Retracting the cantilever led to stretching of the PEG linker and eventual breakage of the peptide-Zn2+ complex. Figure 2 (B to D) shows the representative traces with clear single-rupture peaks for the three peptides. Worm-like chain fitting to each single peak yields a persistence length of ~0.35 to 0.4 nm, consistent with the mechanical features of PEG (30). The rupture force histograms at a pulling speed of 1000 nm s−1 are shown in Fig. 2 (E to G). The average rupture force of PH6 (135 ± 41 pN) is distinctly higher than those of PH1 and PH3 (90 ± 29 and 87 ± 24 pN, respectively). Control experiments in the presence of the metal ion chelation agent, ethylenediaminetetraacetic acid (EDTA), showed much reduced pickup rates, confirming that the force peaks measured in these experiments were indeed due to the formation of peptide–metal ion coordination bonds (fig. S4, A to D).
Fig. 2. Single-molecule force spectroscopy of the metal ion coordination complexes.
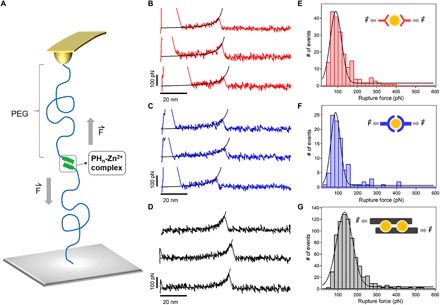
(A) Schematic diagram of the AFM-based single-molecule force spectroscopy experiments. The peptide ligands were linked to the cantilever tip and the substrate via a PEG linker (MW, 5 kDa). (B to D) Typical force-extension curves for the rupture of PH1-Zn2+ (red), PH3-Zn2+(blue), and PH6-Zn2+ (black) complexes at a pulling speed of 1000 nm s−1. Worm-like chain fitting of the force-extension curves (black lines) confirmed that the peak at an extension of ~50 nm corresponds to the rupture of a single metal ion chelation bond. (E to G) The rupture force histograms for PH1-Zn2+ (red), PH3-Zn2+(blue), and PH6-Zn2+ (black), respectively. The Gaussian fitting shows the average rupture forces of 90 ± 29, 87 ± 24, and 135 ± 41 pN, respectively. The proposed Zn2+ ion binding modes for the three peptides are shown in the insets.
To quantitatively measure the unbinding kinetics, we measured the dynamic force spectroscopy of different peptides (fig. S4, E to G). Notably, the average rupture forces of PH6 were much higher than that of PH1 and PH3 at different loading rates. By fitting the data to the Bell-Evans model (31, 32), the off rate at zero force (koff) and the potential width (Δx) along the force direction were obtained. The off rate of PH6 and Zn2+ (0.56 s−1) is much smaller than that of PH3 (2.91 s−1) and even one order of magnitude lower than that of PH1 (9.49 s−1), indicating the much higher unbinding barrier height of PH6-Zn2+ complexes. In contrast, there is no substantial difference in the potential width, Δx, suggesting that the mechanical dissociation pathways of the three peptides are similar.
All these results confirm that the mechanical stability of the metal-ligand complexes can be considerably enhanced by forming tandem binding sites. This is likely due to the simultaneous rupture of the two metal ion-binding sites in PH3-Zn2+ complexes. In contrast, PH3 shows similar rupture forces as PH1, suggesting that formation of a single binding site cannot enhance the mechanical stability, as the Zn2+-His coordination bonds may be ruptured sequentially, instead of cooperatively, in the PH3-Zn2+ complexes.
Energetics of Zn2+ binding to HR-peptides
Assuming the binding-unbinding of the PH1-Zn2+ and PH3-Zn2+ complexes follow a two-state model and the binding-unbinding of the PH6-Zn2+ complexes follow a three-state model (Fig. 3A), the free energy landscape underlying the mechanical dissociation and reassociation of the peptide–metal ion complexes can be obtained on the basis of the thermodynamics data from ITC and kinetics data from single-molecule AFM (see the Supplementary Materials for the calculation details). The free energy values are summarized in table S1 and Fig. 3B. The metal ion binding ligand of PH1 is in isolation, and the peptide-metal ion complexes show both a low-association free energy barrier, ΔGa, and a low-dissociation free energy barrier, ΔGd. We expect that the corresponding hydrogels can recover quickly but of low mechanical strength. For PH3, the unique sequence forms metal coordination sites with increased binding affinity, so the metal-ligand complexes become more stable. As each imidazole-Zn2+ coordination bond in the complexes is ruptured sequentially upon mechanical rupturing, ΔGd was only slightly higher than that for PH1-Zn2+. We expect that the corresponding hydrogels exhibit slightly improved mechanical strength and retain highly dynamic. For PH6, the two metal ion binding sites are arranged in tandem, and the mechanical stability of the coordination bond is even higher because the two sites must simultaneously break upon mechanical fracture, which led to an increased overall ΔGd (33). The association rate for the second binding site is much faster due to the cooperative Zn2+ binding between two binding sites.
Fig. 3. State model and free energy landscape of the peptide-Zn2+ interactions.
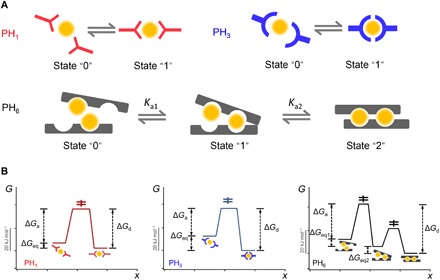
(A) Two states are assigned for PH1 within HN-PH1 or PH3 within HN-PH3, denoted as state “0” and state “1”, respectively, while three states are assigned for PH6 within HN-PH6, denoted as state “0”, state “1”, and state “2”, respectively. Ka1 and Ka2 correspond to the equilibrium constants between states “0” and “1” and between states “1” and “2”, respectively. (B) The free energy landscape of the peptide-Zn2+ interactions. (Left) PH1. (Middle) PH3. (Right) PH6. ΔGa and ΔGd correspond to the free energy barriers for association and dissociation reactions, respectively. ΔGeq corresponds to equilibrium thermodynamic stability of the peptide-Zn2+ complexes. ΔGeq1 and ΔGeq2 at the far right are the free energy change upon binding the first and the second Zn2+ for PH6, respectively.
Preparation and structure of HN hydrogels
We next explored whether the changes of the intrinsic properties of the cross-linkers could indeed lead to distinct macroscopic mechanical properties of the hydrogels. To this end, we prepared a series of HN hydrogels using HR-peptide–Zn2+ complexes as the sacrificial cross-linkers and covalent bonds as the permanent cross-linkers (Fig. 4A). The hydrogels were synthesized in a single step by copolymerization of acrylamide with various ratios of acrylate-terminated 4-armed PEG (4-Armed PEG-Aclt), which served as the permanent cross-linkers, and arcylate-terminated peptides in deionized water. The resulting hydrogels were named HN-PH1, HN-PH3, and HN-PH6 depending on the peptide sequence used (Fig. 4B). To enable comparison, the concentrations of histidine residues in all hydrogels were kept constant. The concentrations of peptides in the HN-PH1, HN-PH3, and HN-PH6 hydrogels were 0.30, 0.10, and 0.05 M, respectively. The mesh sizes, sol/gel fractions, and the actual peptide being incorporated to the hydrogel network were measured prior to the addition of zinc ions. As shown in fig. S5, no obvious difference can be observed for all the HN-PHn gels, suggesting that the network structures were similar in all three hydrogels. Then, these hydrogels were immersed in a tris buffer [1 M, containing 50 mM ZnCl2 and 300 mM KCl (pH 7.6)] to allow the formation of the peptide-Zn2+ cross-linkers in the hydrogels. All the HN gels remained transparent despite integration of the second network cross-linked by metal coordination complexes (Fig. 4B). Moreover, the swelling ratio of the gels was significantly reduced due to the presence of peptide-Zn2+ interactions (fig. S6). The lowest swelling ratio of the HN-PH6 gel indicated that the PH6-Zn2+ complexes were more stable than the PH3-Zn2+ and PH1-Zn2+ complexes. The network structure of the HN-PH6 gels was studied by scanning electron microscopy (fig. S7). Formation of the PH6-Zn2+ complexes led to the presence of additional fibrous structures in the hydrogel network. The HN-PH6 gel was more compressible than the HN-PH1 and HN-PH3 hydrogels (Fig. 4B). The HN-PH6 hydrogels can function effectively under stressful mechanical environments. Continuously compressing (to ~70% strain) or stretching the hydrogels (to ~150% stain) 100 times at a frequency of ~1.6 Hz did not cause much damage to the gels (Fig. 4, C and D, and movies S1 and S2). Moreover, the HN-PH6 gel can be twisted into a spiral shape (Fig. 4E) and compressed with a sharp blade without causing permanent damage to the hydrogel (Fig. 4F and movie S3).
Fig. 4. Structure and properties of HN-PHn HN hydrogels cross-linked by the peptide-Zn2+ coordination complexes.
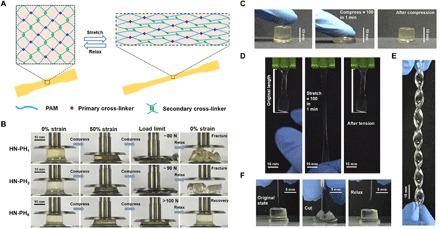
(A) Schematic illustration of the network structure of HN-PHn hydrogels. The network comprises covalent bonds as the primary cross-linkers and ligand-metal interactions as the secondary cross-linkers. (B) Optical images of the HN-PH1 (top), HN-PH3 (middle), and HN-PH6 (bottom) hydrogels under a compression-relaxation cycle. The HN-PH1 and HN-PH3 gels were fractured, whereas the HN-PH6 gel was almost fully recovered. (C) Optical images of the HN-PH6 gel under an extreme compressive condition (compressed to >70% strain for 100 times at 1.6 Hz). (D) Optical images of the HN-PH6 gel under an extreme tensile condition (stretched to >150% strain for 100 times at 1.6 Hz). (E) Optical image of the HN-PH6 gel twisted into a spiral shape. (F) Optical images of the HN-PH6 gel compressed with a sharp blade and relaxed. No detectable cut was observed on the gel. PAM, polyacrylamide. Photo credits: Wenxu Sun, Nanjing University.
Mechanical properties of HN hydrogels
The tensile mechanical properties of the gels were quantitatively measured by standard mechanical tensile tests. The typical tensile stress-strain curves are shown in Fig. 5A, and a summary of the mechanical properties is listed in table S2. The mechanical properties of the hydrogels at the bulk level were correlated with those at the molecular level. The break strain, Young’s modulus, and toughness for the HN-PH6 gel were remarkably higher than those of the HN-PH3 and HN-PH1 gels (Fig. 5A). The Young’s modulus was 59, 71 and 221 kPa for the HN-PH1, HN-PH3, and HN-PH6 gels, respectively. Because all three HN gels contained the same concentrations of the 4-Armed PEG-Aclt and the same covalent cross-linking density, the Young’s moduli for all three HN gels before adding Zn2+ ions were similar (27.6 to 33.6 kPa) (fig. S8, A to C), close to that estimated based on the rubber elasticity theory (34). Therefore, the covalently cross-linked networks were similar for the three hydrogels; the difference in the modulus stems from the additional peptide-Zn2+ cross-linking. The number of peptide-Zn2+ complexes in the hydrogels depends only on the association constants because the concentrations of Zn2+ ions and the peptides were the same in all HN samples. The Young’s moduli of the three gels followed the order of the association constants of the peptide-Zn2+ cross-linkers. The Young’s moduli of the gels can also be reversibly modulated by the removal and addition of Zn2+ (fig. S8, D to K), suggesting that the peptide-Zn2+ binding is reversible. The toughness was 50, 137 and 1328 kJ m−3 for the HN-PH1, HN-PH3, and HN-PH6 gels, respectively. Increasing the concentration of PH6 led to higher toughness of the HN gels, suggesting that the energy dissipation is correlated with the rupture of the PH6-Zn2+ complexes (fig. S9A).
Fig. 5. Mechanical and rapid recovery properties of HN-PHn gels.
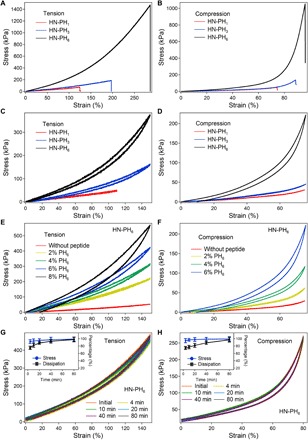
The left panels (A, C, E, and G) and the right panels (B, D, F, and H) correspond to the tensile and compressive mechanical tests, respectively. (A and B) Uniaxial stress-strain curves under tension and compression. (C and D) Uniaxial stretching/compression-relaxation cycles. (E and F) Uniaxial stretching/compression-relaxation cycles of HN-PH6 gels at varied peptide concentrations. (G and H) Consecutive uniaxial stretching/compression-relaxation cycles of HN-PH6 gels with different recovery times. The insets correspond to the mechanical properties (stress and energy dissipation) of HN-PH6 gels after different recovery times in 1 M tris buffer (pH 7.60, containing 300 mM KCl) in the tensile/compressive mechanical tests. The error bars represent the standard deviation, n = 3.
Similar mechanical trends for the HN-PH1, HN-PH3, and HN-PH6 gels were observed in the compressive mechanical tests (Fig. 5B). Notably, the maximum compression strain can be larger than 95%, and the maximum load can reach 1.1 MPa for the HN-PH6 gel, revealing the remarkable mechanical properties of this hydrogel. To further control the mechanical properties of the HN-PH6 gel, we varied the concentrations of 4-Armed PEG-Aclt, which determines the cross-linking density of the covalent network. The tensile stress-strain curves are shown in fig. S9B. The Young’s modulus increased with increasing 4-Armed PEG-Aclt concentration. It is worth mentioning that the fracture stress and toughness of HN-PH6 gels did not always increase at increased cross-linking density because the hydrogels became more brittle and less fracture tolerant at high cross-linking density (11).
Moreover, the mechanical properties of the HN-PH6 hydrogels can be further improved by increasing the acrylamide concentrations. As shown in fig. S9 (C to F), the break strain, fracture strength, and toughness of HN-PH6 gels reached 412%, 3.02 MPa, and 4.03 MJ m−3, respectively, when 450 mg ml−1 of acrylamide was used in the original reaction mixture to prepare the HN-PH6 gels. The enhanced toughness at higher acrylamide concentrations was due to the increase in break strain instead of the increase in Young’s modulus.
The stress-strain curves for the stretching-relaxation and compression-relaxation cycles of HN gels are shown in Fig. 5 (C and D), respectively. In HN hydrogels, the dissipated energy in the loading-unloading cycles is linked to the rupture of the physical cross-linkers. The amplitude of the energy dissipation is correlated with the mechanical strengths of the cross-linkers and the reassociation rates. Little hysteresis was observed between the stretching/compression and relaxation curves for the HN-PH1 and HN-PH3 gels, indicating that either the PH1-Zn2+ and PH3-Zn2+ complexes were mechanically too weak or too dynamic that could reform at a time scale comparable to that of the relaxation rates. In contrast, due to the improved mechanical and thermodynamic stabilities of the PH6-Zn2+ complexes, the HN-PH6 gel showed clear hysteresis in the stress-strain curves. The amount of energy dissipated correlated with the concentration of the peptide-metal ion complexes. With increasing PH6 concentration from 20 to 80 mg ml−1, the dissipated energy increased approximately eight times in the uniaxial stretching-relaxation cycles at 150% strain (Fig. 5E). The same tendency was observed in the uniaxial compression-relaxation cycles at 75% strain (Fig. 5F). The hysteresis for HN-PH6 gels enhanced at increased strain, indicating that more metal chelation bonds were broken at higher forces (fig. S10, A and B). Note that the hysteresis of the HN-PH6 hydrogels is smaller than many other hydrogels with similar toughness. This can be attributed to the fast reassociation of the PH6-Zn2+ complexes even under residual forces. Because of the relatively slow strain rate (5 to 6 mm min−1), some of the complexes were already formed along the relaxation process, leading to the relaxation trace close to the pulling trace. If the hydrogels were pulling and relaxing faster, bigger hysteresis could be observed as less cross-linkers were reformed during relaxation (fig. S10, C and D).
The recovery property of the HN-PH6 gels was studied by applying loading-unloading cycles with different waiting times [Fig. 5, G (for stretching-relaxation) and H (for compression-relaxation)] (35). Although we stretched the gels to only 150% strain, the stress was larger than 400 kPa, which is comparable to the typical mechanical load on many biological tissues (36). The maximum stress and dissipated energy percentage of each cycle are shown in the insets (Fig. 5, G and H). The gel almost totally recovered its macroscopic mechanical properties within several minutes, probably due to the formation of specific metal coordination sites that can bind metal ions more quickly. The recovery of the HN-PH6 gel was rigorously tested by applying continuous stretching-relaxation cycles to the same hydrogel for 20 cycles (fig. S11A) and compression-relaxation cycles to the same hydrogel for 50 cycles (fig. S11C) without waiting between each cycle. There were no large differences between consecutive stress-strain curves, indicating the remarkable macroscopic recovery rate of the HN-PH6 gels. The relative maximum stress of each cycle only slightly decreased and remained greater than 85% in both experiments (fig. S11, B and D). Therefore, the HN-PH6 gel can function effectively in stressful mechanical environments due to its fast recovery rate. However, if the HN-PH6 gels were cut into pieces, the hydrogels cannot self-heal, because covalent cross-linkers cannot reform after fracture.
Prediction from multiscale constitutive theory
To further understand how the outstanding mechanical properties of the HN-PH6 hydrogels were connected with the unique thermodynamic, kinetic, and mechanical properties of the peptide-Zn2+ complexes at the molecular level, we developed a multiscale constitutive theory for fabricated hydrogels (Fig. 6, A and B). This theoretic model is similar to that in our previous work, except that the cross-linkers comprise both the covalent and the peptide-Zn2+ coordination cross-linkers (3). The details of the theory are described in the Supplementary Materials. In the theory, three states were assigned for PH6 in HN-PH6, while two states were assigned for PH1 in HN-PH1 or PH3 in HN-PH3, as illustrated in Fig. 3A. The theory was subsequently used to simulate the stress-strain curves of HN-PH1, HN-PH3, and HN-PH6. In our simulations, exact association rates between zinc and ligands measured in ITC and off rates measured in single molecule AFM, as well as many other experimentally obtained physical quantities, were adopted. The parameters used in the theoretical prediction are listed in table S3. Our simulation results are displayed in Fig. 6 (C to F).
Fig. 6. Theoretical calculation of the mechanical response of the hydrogels.
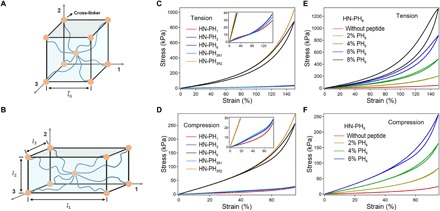
(A) The synthetic material at the dry state is represented with a cube of dimension, , where eight chains cross-linked at the cubic center extend from the cubic center to each corner of the cube. (B) At the current state, the dimensions of representative volume element (REV) become l1, l2, and l3, due to the solvent absorption, metal ion binding, or mechanical loading. (C) Theoretical prediction of stress-strain curves of HN-PH1, HN-PH3, HN-PH6, HN-PH3R1, and HN-PH3R2 when subjected to uniaxial stretching-relaxation. (D) Theoretical prediction of stress-strain curves of HN-PH1, HN-PH3, HN-PH6, HN-PH3R1, and HN-PH3R2 when subjected to uniaxial compression-relaxation. (E) Theoretical prediction of stress-strain curves of HN-PH6 with different concentration of PH6 when subjected to a uniaxial stretching-relaxation cycle. (F) Theoretical prediction of stress-strain curves of HN-PH6 with different concentrations of PH6 when subjected to a uniaxial compression-relaxation cycle. Photo credits: Wenxu Sun, Nanjing University.
As seen from Fig. 6 (C and D), HN-PH6 is much stiffer than HN-PH1 or HN-PH3, and its stress level is much higher than those of the other two types of gel network, when subjected to either uniaxial stretching-relaxation or uniaxial compression-relaxation. This is because of the much higher cross-linking density within HN-PH6 induced by much stronger metal ion binding complex of PH6 than either that of PH1 or that of PH3, even though cross-linking density by covalent bonds would be exactly the same for three different gels in our analysis. Our analysis also indicates that the swelling ratio of HN-PH6 is less than that of HN-PH3, which is also less than that of HN-PH1. As seen from Fig. 6 (E and F), increasing the concentration of PH6 within HN-PH6, the gel network becomes stiffer, its energy dissipation becomes higher, and it also recovers fast when subjected to either uniaxial stretching-relaxation or uniaxial compression-relaxation. These results are semiquantitatively consistent with the experiments.
Our theoretical analysis also suggested that the cooperative zinc binding for PH6 can be important for strong, tough, and also fast-recovery hydrogels. Virtually replacing kinetic rates of PH3 at state “1” with those of PH6 at state “1” in the analysis, with results displayed in Fig. 6 (C and D) being termed as PH3R1, only led to soft and fast-recovery gels but with little energy dissipation. Virtually replacing kinetic rates of PH3 at state “1” with those of PH6 at state “2” in the analysis, with results displayed in Fig. 6 (C and D) being termed as PH3R2, only led to stiff and fast-recovery gels but with little energy dissipation. With two zinc binding motifs of PH6, the rebinding of a broken PH6 within a loading-unloading cycle would firstly experience a relatively low binding rate from state “0” to state “1”, leading to notable energy dissipation, as indicated in our analysis. Meanwhile, a very high binding rate from state “1” to state “2” of PH6 leads to a relatively high cross-linking density within gels at the free-swelling state, so that gels are initially very stiff and also recover fast at relatively low load, as indicated in our analysis. This analysis further confirmed that the unique “three-state” free energy landscape of PH6 is critical to the outstanding mechanical properties of the corresponding hydrogels.
DISCUSSION
Inspired by the rich contents of histidine residues found in natural load-bearing materials, synthetic histidine-containing polymers have been widely explored to increase the strength and toughness of materials. However, it remains a challenge to combine these outstanding mechanical properties with fast recovery. In most of these polymers, histidine residues or imidazole groups were randomly distributed in the polymer chains (37) or linked to the very end of multiarmed polymers (38). The histidine residues or imidazole groups did not form special binding sites and bound to metal ions independently. The unique metal ion binding properties encoded in natural protein sequences were rarely harnessed in those polymers. The resulting hydrogels often did not exhibit high mechanical strength and toughness. Here, using the bio-inspired Zn2+-binding peptide as the cross-linkers, we achieved combined high strength, ultratoughness, and fast recovery by tuning the Zn2+-binding kinetics and thermodynamics at the molecular level. Our results highlight the importance of the formation of cooperative metal coordination sites to achieve desirable mechanical properties.
The correlation of the cross-linker dynamics and the macroscopic mechanical properties of HN hydrogels have previously been studied theoretically and experimentally (39, 40), which were largely based on a “two-state” binding/unbinding scheme. Therefore, the mechanical strengths and association rates are correlated: Weak means fast, and strong means slow (22, 24). For weak and rapid cross-linkers, the fracture strain and toughness increased remarkably as the strain rate decreased, while the Young’s modulus remained almost unchanged. For strong and slow cross-linkers, the Young’s modulus increased, while the fracture strain and toughness decreased alongside the stain rate. On the basis of the strain rate–dependent tensile mechanical properties of the HN-PHn gels (fig. S12), we found that both PH1-Zn2+ and PH3-Zn2+ can be considered as weak and rapid cross-linkers as the corresponding gels showed constant Young’s modulus but largely increased fracture strain and toughness with the decrease in strain rates. In contrast, by forming the tandem Zn2+-binding sites in PH6, PH6-Zn2+ becomes a strong cross-linker. Therefore, the Young’s modulus for HN-PH6 gels increased, and the fracture strain decreased markedly with the increase in strain rates. PH6-Zn2+ complex is probably the first example of a strong and fast cross-linker. In the model proposed by Tito and co-workers (39), ΔGa and ΔGd are correlated by the same transition state. Increasing the strength of the cross-linker requires the increase in ΔGd, which inevitably leads to the increase in ΔGa and the slowdown of the association rates. Therefore, the hydrogels are stiffer and more brittle. However, for PH6-Zn2+ complexes, ΔGa and ΔGd can be decoupled in a complicated three-state free energy landscape to achieve combined high mechanical strength and fast binding (Fig. 3B). The hydrogels are stiffer and also tougher. Our theoretical models further demonstrated the unique advantage of the PH6-Zn2+ binding energetics to the outstanding mechanical properties of the resulting HN gels (Fig. 6).
We also discovered the molecular mechanism underlying the cooperative binding of the designed peptide sequence. Using glycine mutagenesis, we found that all histidine residues in PH3 contribute to Zn2+ binding, despite that Zn2+ forms tetrahedral coordination geometry (41) with four imidazole rings of two PH3 peptides. This is consistent with recent work by Kozłowski and co-workers (42), showing that Zn2+, Ni2+, and Cu2+ can form dynamic polymorphic binding states with polyhistidine peptides. Moreover, we found that the proline residue is not critical for Zn2+ binding in PH3 but is important for the cooperative Zn2+ binding of the two binding sites in PH6. Such allosteric conformational change–induced cooperative Zn2+ binding of PH6 endows the PH6-Zn2+ complexes both fast association rate and high thermodynamic stability. Moreover, the dissociation barrier is also increased because the two metal ion binding sites are ruptured simultaneously under force. The resulting hydrogels show much better mechanical properties than those constructed with isolated metal coordination bonds or single metal ion binding sites. We propose that such a mechanism may also be adopted by other repetitive metal ion binding sequences found in naturally occurring load-bearing proteins. Such a design is advantageous over directly increasing cross-linking density without cooperative metal ion binding, as we have shown that the mechanical strength and toughness of HN-PH6 hydrogels are much higher than that of HN-PH3 hydrogels containing the same number of Zn2+-binding sites (Fig. 5).
Hydrogels with high mechanical strength and toughness have been widely used for hard tissue engineering (43). Gong and co-workers elegantly showed that DN hydrogels are promising candidates for the regeneration of cartilage (2, 44). However, most DN hydrogels reported so far cannot recover their mechanical properties quickly in multiple load-unload cycles and show obvious mechanical fatigue. The existence of energy-dissipative secondary network in hydrogels is typically considered as a limiting factor to achieve fast recovery of hydrogels, despite that they can greatly enhance the mechanical strength and toughness of hydrogels. A few studies indicated that, using reversible and fast dynamic cross-linkers (45) or loosely formed second network (46), the recovery rate can be greatly improved, although the underlying physical mechanism has not been well understood. Lin and co-workers (47) showed that simply prestraining a conventional DN hydrogel can lead to less hysteresis and fast recovery but also reap all of the benefits of delayed fracture. In our approach, by tailoring the metal ion binding kinetics, thermodynamics, and mechanics at the molecular level, fast recovery was achieved, yet the hysteresis was not diminished. Instead, the fracture stress and toughness were both increased in our hydrogels. Compared with the mechanical properties of other hydrogels reported in literature, the fracture strength of the HN-PH6 gel is higher than the majority of the hydrogels (table S4). The toughness is larger than many hydrogels with similar water contents and is only slightly lower than some of the hydrogels with lower water contents. The recovery rate is also faster than most hydrogels. Note that the HN-PH6 hydrogel is probably the only one that shows the combined high strength, ultratoughness, and fast recovery, making it a potential candidate for cartilage regeneration. However, other mechanical features, such as the adhesion strength to other tissues (48), should be seriously considered before their practical applications in tissue engineering.
MATERIALS AND METHODS
ITC titration
All titrations were performed using a Microcal ITC200 apparatus at 298 K. The solutions for the titrations were prepared in 1 M tris buffer (pH 7.60, containing 300 mM KCl). Metal halide solution (6 mM ZnCl2) was added into the solution of HR-peptides [3.6 mM PH1, 1.2 mM PH3, 0.6 mM PH6, 1.8 mM GHGPH, 1.8 mM GGHPH, 1.8 mM GHHPG, 1.2 mM GHHGH, 0.9 mM (GHGPH)2, 0.9 mM (GGHPH)2, 0.9 mM (GHHPG)2, and 0.6 mM (GHHGH)2] during the titrations. Blank titrations in 1 M tris buffer (pH 7.60, containing 300 mM KCl) were performed, and the result was subtracted from the corresponding titrations to account for the effect of the dilution. The fitting was performed using Origin software provided by Microcal.
CD spectroscopy measurements
CD spectra were recorded to investigate the secondary structure of the peptides in the absence and presence of Zn2+ ions. To avoid the effects of Cl− in the CD spectra measurements, Zn(NO3)2 instead of ZnCl2 was used. Zn(NO3)2 stock solutions at the concentration of 4.0 mM were prepared with deionized water, respectively. Then, the Aclt-HR-peptides were dissolved into Zn(NO3)2 solutions to concentrations of 3.0, 1.0, and 0.5 mM for PH1, PH3, and PH6 peptides, respectively. The pH of the final solutions was about 7.8. CD spectra of all samples were recorded using a J-815 (JASCO Inc., Japan) spectrophotometer. The cuvette width was 1 cm, and the bandwidth was 0.2 nm.
Preparation of the HR-peptide–coated cantilevers and glass substrates
Standard silicon nitride (Si3N4) cantilevers were obtained from Bruker (type, MLCT). The cantilevers and glass cover slides were cleaned with Milli-Q water and placed in a chromic acid at 95°C for 20 min. The cantilevers and glass substrates were then immersed in 1% (v/v) (3-mercaptopropyl) trimethoxysilane (MPTMS) methylbenzene for 2 hours to introduce thiol groups to the surface. They were rinsed with Milli-Q water, dried under nitrogen, and immersed in a dimethyl sulfoxide (DMSO) solution containing Mal-PEG–N-hydroxysuccinimide (NHS) (1 mg ml−1) (MW, 5 kDa) for 2 hours. The resulting NHS-coated cantilevers and glass substrates were immersed in a DMSO solution containing 0.1 mM HR-peptides for approximately 12 hours to react with the N-terminal amino groups of the HR-peptides. The resulting HR-peptide–coated cantilevers and substrates were rinsed with Milli-Q water and stored at 4°C for the single-molecule experiments.
Single-molecule AFM experiments
The SMFS experiments were carried out on a commercial AFM (JPK ForceRobot 300) at room temperature (~25°C). The HR-peptide–functionalized D cantilevers (spring constant of ~0.05 or ~0.1 N m−1, respectively) were used in all experiments (see the Supplementary Materials for details). The spring constant was calibrated using the equipartition theorem for each experiment. The experiments were conducted in 1 M tris buffer (pH 7.60, containing 300 mM KCl and 50 μM ZnCl2). During each SMFS experiment, the cantilever was brought into contact with the surface at a contact force of ~500 pN for 500 ms to trigger formation of the peptide-Zn2+ complexes. Then, the cantilever was pulled back to obtain the force-extension curves of the polymers. All force curves were collected by commercial software from JPK and analyzed offline using a custom-written protocol in Igor 6.0 (WaveMetrics Inc.).
Preparation of the HN-PHn gels
In a typical preparation of hydrogels, acrylamide and 4-Armed PEG-Aclt (MW, 20 K) were dissolved in Milli-Q water at 225 (3165.5 mM) and 50 mg ml−1 (2.5 mM). Then, the Aclt–HR-peptides were dissolved into the mixture to concentrations of 84.3 (313.2 mM), 60.9 (104.4 mM), and 60.0 mg ml−1 (52.2 mM) for HN-PH1, HN-PH3, and HN-PH6, respectively. The molar concentration of PEG cross-linkers was far more less than that of PH6 peptide in the HN-PH6 hydrogels, and the Zn2+-PH6 cross-linkers contribute more to the modulus. The reaction mixture was degassed with argon protection and sonicated three times (each time for 15 min) to remove dissolved oxygen. Ammonium persulfate was added as the photo-initiator, and the polymerization was initiated under UV (285 nm) illumination at room temperature for 20 hours. Transparent pre-gels were obtained and extensively washed with deionized water to remove the unreacted monomers. The gels were dialyzed in 1 M tris buffer (pH 7.60, containing 300 mM KCl and 50 mM ZnCl2) for 24 hours to trigger formation of the coordinate complexes.
Tensile and compressive test
The tensile/compressive stress-strain measurements were performed using a tensile-compressive tester (Instron 5944 with a 2-kN sensor) in air at room temperature. In the tension-crack test, the strain rate of stretching was maintained at 5 to 6 mm min−1, while those were maintained at 20 to 30 mm min−1 in the stretching-relaxation cycle test. In the compression-crack test, the rates of compression were maintained at 0.8 to 1.0 mm min−1, while those were maintained at 3.2 to 4 mm min−1 in the compression-relaxation cycle test. The toughness, a parameter that characterizes the work required to fracture the sample per unit, was calculated from the area below the tension stress-strain curve until fracture. In addition, the Young’s modulus comprised the approximate linear fitting values under 100% strain deformation.
Supplementary Material
Acknowledgments
Funding: This research is supported mainly by the National Natural Science Foundation of China (nos. 11934008, 11804148, 11674153, 11804147, 81622033, 21774057, 11872334, and 11572285), the Natural Science Foundation of Jiangsu Province (nos. BK20180320 and BK20180335), and the Fundamental Research Funds for the Central Universities (nos. 020414380080, 020414380118, 020414380130, and 020414380133). Author contributions: Y.C. and W.W. conceived the idea and designed the study. W.S. and B.X. performed the experiments and analyzed the results. B.C. designed the theoretical prediction. Q.F. performed the theoretical prediction. Y.C., W.S., and B.X. wrote and refined the paper. R.T. performed some of the SMFS experiments and analyzed the SMFS data preliminarily. C.W. prepared some of the HN-PHn hydrogel samples and performed part of the mechanical test experiments. B.X., X.W., and Y.L. prepared the HR peptide–coated cantilevers and glass substrates. M.Q. performed the swelling ratio test experiments and analyzed these data. Y.C., M.Q., B.C., and W.W. supervised the project. All the authors discussed the results. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaaz9531/DC1
REFERENCES AND NOTES
- 1.Yuk H., Lin S., Ma C., Takaffoli M., Fang N. X., Zhao X., Hydraulic hydrogel actuators and robots optically and sonically camouflaged in water. Nat. Commun. 8, 14230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao Y., Nakajima T., Yang J. J., Kurokawa T., Liu J., Lu J., Mizumoto S., Sugahara K., Kitamura N., Yasuda K., Daniels A. U. D., Gong J. P., Proteoglycans and glycosaminoglycans improve toughness of biocompatible double network hydrogels. Adv. Mater. 26, 436–442 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Wu J., Li P., Dong C., Jiang H., Bin X., Gao X., Qin M., Wang W., Bin C., Cao Y., Rationally designed synthetic protein hydrogels with predictable mechanical properties. Nat. Commun. 9, 620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun J.-Y., Keplinger C., Whitesides G. M., Suo Z., Ionic skin. Adv. Mater. 26, 7608–7614 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Cvetkovic C., Raman R., Chan V., Williams B. J., Tolish M., Bajaj P., Sakar M. S., Asada H. H., Saif M. T. A., Bashir R., Three-dimensionally printed biological machines powered by skeletal muscle. P. Natl. Acad. Sci. U.S.A. 111, 10125–10130 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Creton C., 50th anniversary perspective: Networks and gels: Soft but dynamic and tough. Macromolecules 50, 8297–8316 (2017). [Google Scholar]
- 7.Gong J. P., Katsuyama Y., Kurokawa T., Osada Y., Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 15, 1155–1158 (2003). [Google Scholar]
- 8.Liu J., Tan C. S. Y., Yu Z., Li N., Abell C., Scherman O. A., Tough supramolecular polymer networks with extreme stretchability and fast room-temperature self-healing. Adv. Mater. 29, 1605325 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Wang J., Lin L., Cheng Q., Jiang L., A strong bio-inspired layered PNIPAM–Clay nanocomposite hydrogel. Angew. Chem. Int. Ed. Engl. 51, 4676–4680 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Okumura Y., Ito K., The polyrotaxane gel: A topological gel by figure-of-eight cross-links. Adv. Mater. 13, 485–487 (2001). [Google Scholar]
- 11.Zhang H. J., Sun T. L., Zhang A. K., Ikura Y., Nakajima T., Nonoyama T., Kurokawa T., Ito O., Ishitobi H., Gong J. P., Tough physical double-network hydrogels based on amphiphilic triblock copolymers. Adv. Mater. 28, 4884–4890 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez M. A., Simon J. R., Ghoorchian A., Scholl Z., Lin S., Rubinstein M., Marszalek P., Chilkoti A., López G. P., Zhao X., Strong, tough, stretchable, and self-adhesive hydrogels from intrinsically unstructured proteins. Adv. Mater. 29, 1604743 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Sun J.-Y., Zhao X., Illeperuma W. R. K., Chaudhuri O., Oh K. H., Mooney D. J., Vlassak J. J., Suo Z., Highly stretchable and tough hydrogels. Nature 489, 133–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y., Wang X., Yang F., Wang L., Wu D., Highly elastic and ultratough hybrid ionic–Covalent hydrogels with tunable structures and mechanics. Adv. Mater. 30, e1707071 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Long T., Li Y., Fang X., Sun J., Salt-mediated polyampholyte hydrogels with high mechanical strength, excellent self-healing property, and satisfactory electrical conductivity. Adv. Funct. Mater. 28, 1804416 (2018). [Google Scholar]
- 16.Chen Q., Zhu L., Zhao C., Wang Q., Zheng J., A robust, one-pot synthesis of highly mechanical and recoverable double network hydrogels using thermoreversible sol-gel polysaccharide. Adv. Mater. 25, 4171–4176 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Zheng S. Y., Ding H., Qian J., Yin J., Wu Z. L., Song Y., Zheng Q., Metal-coordination complexes mediated physical hydrogels with high toughness, stick–slip tearing behavior, and good processability. Macromolecules 49, 9637–9646 (2016). [Google Scholar]
- 18.Lin P., Ma S., Wang X., Zhou F., Molecularly engineered dual-crosslinked hydrogel with ultrahigh mechanical strength, toughness, and good self-recovery. Adv. Mater. 27, 2054–2059 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Yang Y., Wang X., Yang F., Shen H., Wu D., A universal soaking strategy to convert composite hydrogels into extremely tough and rapidly recoverable double-network hydrogels. Adv. Mater. 28, 7178–7184 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Li J., Suo Z., Vlassak J. J., Stiff, strong, and tough hydrogels with good chemical stability. J. Mater. Chem. B 2, 6708–6713 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Long R., Mayumi K., Creton C., Narita T., Hui C.-Y., Time dependent behavior of a dual cross-link self-healing gel: Theory and experiments. Macromolecules 47, 7243–7250 (2014). [Google Scholar]
- 22.Grindy S. C., Learsch R., Mozhdehi D., Cheng J., Barrett D. G., Guan Z., Messersmith P. B., Holten-Andersen N., Control of hierarchical polymer mechanics with bioinspired metal-coordination dynamics. Nat. Mater. 14, 1210–1216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang J., Mehlich A., Koga N., Huang J., Koga R., Gao X., Hu C., Jin C., Rief M., Kast J., Baker D., Li H., Forced protein unfolding leads to highly elastic and tough protein hydrogels. Nat. Commun. 4, 2974 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Appel E. A., Forster R. A., Koutsioubas A., Toprakcioglu C., Scherman O. A., Activation energies control the macroscopic properties of physically cross-linked materials. Angew. Chem. Int. Ed. 53, 10038–10043 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Fullenkamp D. E., He L., Barrett D. G., Burghardt W. R., Messersmith P. B., Mussel-inspired histidine-based transient network metal coordination hydrogels. Macromolecules 46, 1167–1174 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrington M. J., Masic A., Holten-Andersen N., Waite J. H., Fratzl P., Iron-clad fibers: A metal-based biological strategy for hard flexible coatings. Science 328, 216–220 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee B. P., Messersmith P. B., Israelachvili J. N., Waite J. H., Mussel-inspired adhesives and coatings. Annu. Rev. Mater. Res. 41, 99–132 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgan W. T., The histidine-rich glycoprotein of serum has a domain rich in histidine, proline, and glycine that binds heme and metals. Biochemistry 24, 1496–1501 (1985). [DOI] [PubMed] [Google Scholar]
- 29.Jancsó A., Kolozsi A., Gyurcsik B., Nagy N. V., Gajda T., Probing the Cu2+ and Zn2+ binding affinity of histidine-rich glycoprotein. J. Inorg. Biochem. 103, 1634–1643 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Ott W., Jobst M. A., Bauer M. S., Durner E., Milles L. F., Nash M. A., Gaub H. E., Elastin-like polypeptide linkers for single-molecule force spectroscopy. ACS Nano 11, 6346–6354 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Bell G. I., Models for the specific adhesion of cells to cells. Science 200, 618–627 (1978). [DOI] [PubMed] [Google Scholar]
- 32.Evans E., Ritchie K., Strength of a weak bond connecting flexible polymer chains. Biophys. J. 76, 2439–2447 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y., Di W., Li Y., Huang W., Wang X., Qin M., Wang W., Cao Y., Mg2+-Dependent high mechanical anisotropy of three-way-junction pRNA as revealed by single-molecule force spectroscopy. Angew. Chem. Int. Ed. Engl. 56, 9376–9380 (2017). [DOI] [PubMed] [Google Scholar]
- 34.L. R. G. Treloar, The Physics of Rubber Elasticity (Oxford Univ. Press, New York, 1975).
- 35.Liu J., Tan C. S. Y., Yu Z., Lan Y., Abell C., Scherman O. A., Biomimetic supramolecular polymer networks exhibiting both toughness and self-recovery. Adv. Mater. 29, 1604951 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Stella J. A., D’Amore A., Wagner W. R., Sacks M. S., On the biomechanical function of scaffolds for engineering load-bearing soft tissues. Acta Biomater. 6, 2365–2381 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Enke M., Bode S., Vitz J., Schacher F. H., Harrington M. J., Hager M. D., Schubert U. S., Self-healing response in supramolecular polymers based on reversible zinc–Histidine interactions. Polymer 69, 274–282 (2015). [Google Scholar]
- 38.Yi X., He J., Wang X., Zhang Y., Tan G., Zhou Z., Chen J., Chen D., Wang R., Tian W., Yu P., Zhou L., Ning C., Tunable mechanical, antibacterial, and cytocompatible hydrogels based on a functionalized dual network of metal coordination bonds and covalent crosslinking. ACS Appl. Mater. Interfaces 10, 6190–6198 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Tito N. B., Creton C., Storm C., Ellenbroek W. G., Harnessing entropy to enhance toughness in reversibly crosslinked polymer networks. Soft Matter 15, 2190–2203 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Mayumi K., Guo J., Narita T., Hui C. Y., Creton C., Fracture of dual crosslink gels with permanent and transient crosslinks. Ex. Mechan. Lett. 6, 52–59 (2016). [Google Scholar]
- 41.Zechel S., Hager M. D., Priemel T., Harrington M. J., Healing through histidine: Bioinspired pathways to self-healing polymers via imidazole–metal coordination. Biomimetics 4, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wątły J., Hecel A., Rowińska-Żyrek M., Kozłowski H., Impact of histidine spacing on modified polyhistidine tag – Metal ion interactions. Inorg. Chim. Acta 472, 119–126 (2018). [Google Scholar]
- 43.Huey D. J., Hu J. C., Athanasiou K. A., Unlike bone, cartilage regeneration remains elusive. Science 338, 917–921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitamura N., Yokota M., Kurokawa T., Gong J. P., Yasuda K., In vivo cartilage regeneration induced by a double-network hydrogel: Evaluation of a novel therapeutic strategy for femoral articular cartilage defects in a sheep model. J. Biomed. Mater. Res. A 104, 2159–2165 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Azevedo S., Costa A. M. S., Andersen A., Choi I. S., Birkedal H., Mano J. F., Bioinspired ultratough hydrogel with fast recovery, self-healing, injectability and cytocompatibility. Adv. Mater. 29, 1700759 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Truong V. X., Ablett M. P., Richardson S. M., Hoyland J. A., Dove A. P., Simultaneous orthogonal dual-click approach to tough, in-situ-forming hydrogels for cell encapsulation. J. Am. Chem. Soc. 137, 1618–1622 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Lin S., Zhou Y., Zhao X., Designing extremely resilient and tough hydrogels via delayed dissipation. Ex. Mechan. Lett. 1, 70–75 (2014). [Google Scholar]
- 48.Li J., Celiz A. D., Yang J., Yang Q., Wamala I., Whyte W., Seo B. R., Vasilyev N. V., Vlassak J. J., Suo Z., Mooney D. J., Tough adhesives for diverse wet surfaces. Science 357, 378–381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lau S. J., Sarkar B., Ternary coordination complex between human serum albumin, copper (II), and L-histidine. J. Biol. Chem. 246, 5938–5943 (1971). [PubMed] [Google Scholar]
- 50.Movasaghi Z., Rehman S., Rehman I. U., Raman spectroscopy of biological tissues. Appl. Spectrosc. Rev. 42, 493–541 (2007). [Google Scholar]
- 51.Schmitt C. N. Z., Politi Y., Reinecke A., Harrington M. J., Role of sacrificial protein–metal bond exchange in mussel byssal thread self-healing. Biomacromolecules 16, 2852–2861 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Boyce M. C., Arruda E. M., Constitutive models of rubber elasticity: A review. Rubber Chem. Technol. 73, 504–523 (2000). [Google Scholar]
- 53.Cai S., Suo Z., Mechanics and chemical thermodynamics of phase transition in temperature-sensitive hydrogels. J. Mech. Phys. Solids 59, 2259–2278 (2011). [Google Scholar]
- 54.Fan Q., Chen B., Cao Y., Constitutive model reveals the defect-dependent viscoelasticity of protein hydrogels. J. Mech. Phys. Solids 125, 653–665 (2019). [Google Scholar]
- 55.Marko J. F., Siggia E. D., Stretching DNA. Macromolecules 28, 8759–8770 (1995). [Google Scholar]
- 56.Dong A., Matsuura J., Allison S. D., Chrisman E., Manning M. C., Carpenter J. F., Infrared and circular dichroism spectroscopic characterization of structural differences between β-lactoglobulin A and B. Biochemistry 35, 1450–1457 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Jia H., Huang Z., Fei Z., Dyson P. J., Zheng Z., Wang X., Unconventional tough double-network hydrogels with rapid mechanical recovery, self-healing, and self-gluing properties. ACS Appl. Mater. Inter. 8, 31339–31347 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/16/eaaz9531/DC1