

Abstract
Objective
To evaluate the diagnostic yield of an 89-gene panel in a large cohort of patients with suspected muscle disorders and to compare the diagnostic yield of gene panel and exome sequencing approaches.
Methods
We tested 1,236 patients from outpatient clinics across Canada using a gene panel and performed exome sequencing for 46 other patients with sequential analysis of 89 genes followed by all mendelian genes. Sequencing and analysis were performed in patients with muscle weakness or symptoms suggestive of a muscle disorder and showing at least 1 supporting clinical laboratory.
Results
We identified a molecular diagnosis in 187 (15.1%) of the 1,236 patients tested with the 89-gene panel. Diagnoses were distributed across 40 different genes, but 6 (DMD, RYR1, CAPN3, PYGM, DYSF, and FKRP) explained about half of all cases. Cardiac anomalies, positive family history, age <60 years, and creatine kinase >1,000 IU/L were all associated with increased diagnostic yield. Exome sequencing identified a diagnosis in 10 (21.7%) of the 46 patients tested. Among these, 3 were attributed to genes not included in the 89-gene panel. Despite differences in median coverage, only 1 of the 187 diagnoses that were identified on gene panel in the 1,236 patients could have been potentially missed if exome sequencing had been performed instead.
Conclusions
Our study supports the use of gene panel testing in patients with suspected muscle disorders from outpatient clinics. It also shows that exome sequencing has a low risk of missing diagnoses compared with gene panel, while potentially increasing the diagnostic yield of patients with muscle disorders.
Inherited muscle disorders form a heterogeneous group characterized by different patterns of muscle weakness. Some have a unique phenotype allowing a diagnostic with single-gene testing. Others, like the limb-girdle weakness pattern, do not have a specific gestalt and are associated with numerous genes. Muscle biopsy has long been considered the gold standard test but is not without limitations. Indeed, it is particularly invasive, and specific antibodies are not available for all proteins encoded by causal genes.1 Therefore, genetic testing is increasingly used and can be obtained in a noninvasive manner. Across different countries such as Canada, the United States, China, Korea, Germany, the United Kingdom, Egypt, Poland, Australia, and Japan, gene panel sequencing has a yield varying from 16% to 65%,2–7 depending on subgroups of patients' selection, whereas exome sequencing has a yield in between 13% and 69%8–15 in different settings. Its superiority over gene panel, in diagnosing common etiologies, remains to be quantified.
Reaching a molecular diagnosis is becoming increasingly important for patients management, including participation in clinical trials and treatment eligibility, such as enzyme replacement in Pompe disease.16
Here, we report on the diagnostic yield of an 89-gene panel in a large cohort of Canadian patients with suspected muscle disorders from outpatient clinics, and we compare the diagnostic yield of gene panel and exome sequencing approaches in a single-center cohort.
Methods
Recruitment of patients
We analyzed DNA samples of 1,236 patients (201 children and 1,035 adults; 574 females and 662 males) seen by 187 physicians in outpatient clinics (general neurology, specialized neuromuscular, genetics, physiatry, and general practice) at 61 locations across Canada (figure e-1, links.lww.com/NXG/A233). DNA samples were extracted from blood samples. The clinical gene panel test was performed in the laboratory of Sherbrooke Genomic Medicine (a not-for-profit organization), and the cost of the test was covered by a special program with financial support from Sanofi Genzyme. To be eligible, patients were required to show any type of muscle weakness or symptoms suggestive of muscle involvement (i.e., myalgia, rhabdomyolysis, exercise intolerance, and unexplained respiratory insufficiency), at least 1 abnormal laboratory finding suggestive of muscle involvement (plasma creatine kinase [CK], EMG, muscle biopsy, or MRI), and no reported diagnosis. Demographics and clinical information were obtained from the laboratory requisition, which included specific sections for muscle phenotype, respiratory symptoms, cardiac anomalies, and clinical laboratory results. Age at onset and previous genetic testing were not systematically mentioned. Furthermore, complete muscle biopsy reports were not available to us. Respiratory and cardiac phenotypes were described in 83.4% and 71.0% of patients, respectively. Cardiac anomalies included hypertrophic or dilated cardiomyopathies and arrhythmias for 82.2% of patients and were unspecified for the remaining. CK, EMG, muscle biopsy, and MRI results were available for 89.5%, 81.6%, 32.7%, and 11.7% of patients, respectively. In addition, 46 patients were recruited using the above-mentioned criteria from neuromuscular and genetics outpatient clinics at Centre Hospitalier Universitaire de Sherbrooke. There was no overlap with the 1,236 patients above. Fifty-four percent had single gene or panel testing prior performing exome sequencing. DNA was than analyzed by exome sequencing on a research basis, following genetic counseling. Because patients come from outpatient clinics, follow-up was made by treating physicians.
Standard protocol approvals, registrations, and patient consents
The study was approved by the institutional ethics review board at Université de Sherbrooke (project ##MP-31-2013-533, 12-208). All participants (or their legal guardians) who underwent exome sequencing provided participants' written consent.
Gene panel sequencing
We designed a panel of 89 genes targeting diverse patterns of muscle weakness that could be encountered in outpatient clinics and covering the following groups of disorders: limb-girdle muscular dystrophies (LGMDs), congenital muscular dystrophies, congenital myasthenic syndromes, nemaline myopathy, myofibrillar myopathy, centronuclear myopathy, collagen VI–related myopathies, inclusion myopathies, metabolic myopathies, rigid spine syndromes, and scapuloperoneal syndromes. The list of the 89 genes is provided in table e-1 (links.lww.com/NXG/A234) and supplementary file e-1 (links.lww.com/NXG/A240). For each patient, DNA libraries were prepared following a standard protocol (Kapa Biosystems, Roche, MA), followed by target enrichment (Seq Cap EZ-Custom) and sequenced on a MiSeq (Illumina, San Diego, CA) or a NextSeq (Illumina) with a 150-bp paired-end protocol. A total of 383 and 853 patients were sequenced on MiSeq and NextSeq, respectively.
Exome sequencing
Exome sequencing was performed as previously described.8 Briefly, it was performed at the McGill University and Génome Québec Innovation Centre (Montreal, Canada) or Fulgent (Temple City, CA). DNA libraries were prepared for each patient (TruSeq; Illumina), followed by target enrichment (Agilent SureSelect All Exon kit v4 or v5 or Illumina Truseq Exome) and sequenced on a HiSeq 2000 (Illumina) with a 100-bp paired-end protocol or HiSeq 4000 (Illumina) with a 150-bp paired-end protocol.
Bioinformatics and statistical analysis
We analyzed the sequencing data using a Linux-based bioinformatics pipeline based on the one developed by the McGill University and Génome Québec Innovation Centre (bitbucket.org/mugqic/mugqic_pipelines) as previously described.6 Briefly, (1) raw reads were trimmed using Trimmomatic17 (version 0.32); (2) sequence alignment was performed with Burrows-Wheeler Aligner18 (version 0.7.10); (3) genetic variants (single nucleotide polymorphisms and indels) were called with the HaplotypeCaller using the Genome Analysis Toolkit19 (version 3.2.2) with prior local realignment, base recalibration, and removal of polymerase chain reaction (PCR) duplicates using Picard (version 1.123, broad institute.github.io/picard/); (4) gene annotation was performed with SnpEff/SnpSift20 (versions 3.6 and 4.2, including SIFT, Polyphen2, and MutationTaster predictions) with an additional in-house script to annotate variants present in the ClinVar21 database; and (5) a filtering process removed variants outside targeted sequences, with population frequency >1% (dbSNP 138 and ExAC 0.322) and genotype quality less than Q30. Coverage depth was calculated using BED Tools.23 Filtered variant lists obtained from the bioinformatics pipeline were then interpreted with an in-house script and manual revision. For both gene panel and exome sequencing, deletion and duplication analysis were performed using the CoNVaDING software24 and manual review of binary alignment map files before quantitative PCR confirmation using Taqman Copy Number Assay (Thermo Fisher Scientific, Montreal, Canada). Variants were revised manually and were reported according to the American College of Medical Genetics and Genomics guidelines.25 For exome sequencing, a subset of 89 genes (included in the gene panel) was analyzed before the whole-exome data. Variants of uncertain significance (VUS) were reported only if related to the patient's phenotype. Sanger sequencing confirmation was only performed if any of the following criteria were not fulfilled: minimum genotype quality >Q40, quality score >500, strand bias score <60, and heterozygous read ratio >60/40.
Odds ratios (figure 4) were calculated for clinical criteria by using standard statistics (χ2 test). When data were missing on a patient for a specific clinical criterion, we removed the patient from the analysis and calculated among patients with complete data.
Figure 4. Odds ratios of the different clinical criteria.

Age is separated in 2 categories (<60 and ≥60 years). Diagnostic yield is higher in patients aged <60 years. Some values could not be included because the results were not available for all patients (familial history = 938/1,236, creatine kinase [CK] = 1,106/1,236, quantitative CK = 642/1,236, EMG = 1,008/1,236, muscle biopsy = 404/1,236, MRI = 146/1,236, respiratory insufficiency = 1,031/1,236, cardiac anomaly = 878/1,236). Error bars represent 95% confidence interval.
Data availability
Anonymized data will be shared by request from any qualified investigator. When not possible, given the risk to identify rare patients, additional aggregate data in table form will be produced to address specific questions.
Results
Diagnoses identified by the 89-gene panel in outpatient clinics
Samples from a total of 1,236 patients with a suspected muscle disorder were analyzed by gene panel. Adults and children younger than 18 years accounted for 83.7% and 16.3%, respectively. A third of adult patients were aged 60 years and older. Ethnic background was reported to be European non-Finnish in 67.5%, when data were available. Other ethnicities included South and East Asians (14.7%), Mixed Ethnicities (5.1%), Middle East (3.5%), Native Americans and Canadians (3.4%), African Americans (3.2%), Hispanic (1.7%), Ashkenazi Jewish (0.5%), and European Finnish (0.4%). Patients who were recruited from specialized neuromuscular outpatient clinics represented 61.7% of the total cohort, whereas other general neurology, clinical and biochemical genetics, and other clinics accounted for 23.9%, 10.4%, and 4.0%, respectively. More than half of our cohort presented with a limb-girdle weakness, in both children and adults (figure 1).
Figure 1. Clinical presentation of the pediatric (A) (n = 201) and adult (B) (n = 1,035) cohorts.

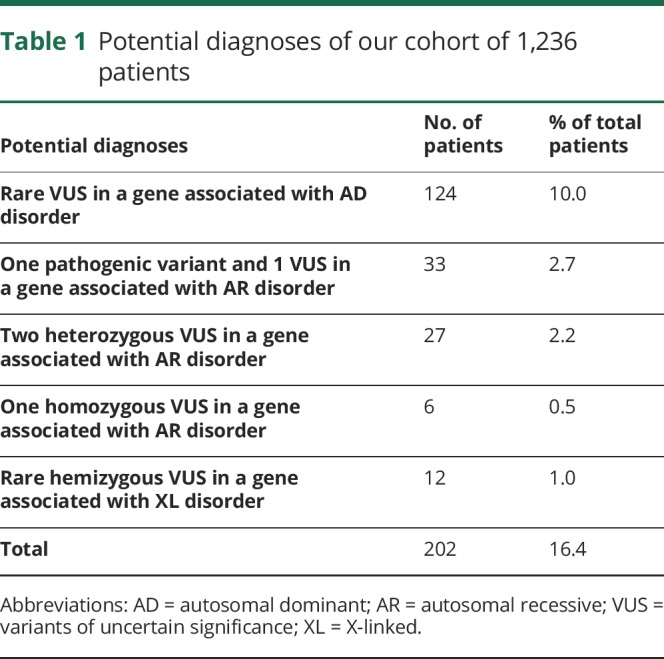
A total of 187 (15.1%) patients had a diagnosis identified (figure 2 and table e-2, links.lww.com/NXG/A235), with a diagnostic rate of 22.4% in children and 13.7% in adult patients. A higher diagnostic rate (27.8%) was observed within males of the pediatric cohort due to DMD, a gene causing an X-linked disorder. Otherwise, there was not any gender bias. A likely carrier status of a recessive disorder was identified in 9.9%. A potential diagnosis was suspected in 16.4% of the cases (table 1). In particular, 66 patients had 2 variants in a gene associated with an autosomal recessive disorder compatible with the patient's phenotype, including 33 patients with 1 pathogenic variant and 1 VUS. A biopsy was performed in 18 of 33 patients, of which 17 were abnormal. Among these 17 abnormal biopsies, 14 showed nonspecific findings. Two patients harboring NEB variants showed nemaline bodies on muscle biopsy, which supported a diagnosis of nemaline myopathy, but parents were not tested to document whether variants were inherited in trans (patients 336 and 372, see table e-3, links.lww.com/NXG/A236). One patient harboring SGCA variants showed absent alpha-sarcoglycan on muscle biopsy, which supported a diagnosis of autosomal recessive LGMD type 2D, but parents were not tested to document whether variants were inherited in trans (patient 356, see table e-3). Overall, information was insufficient to confirm the diagnosis in these 66 patients. Parental testing and additional specific immunochemistry, when applicable, were therefore recommended.
Figure 2. Proportion of the confirmed diagnoses (n = 187) and potential diagnoses (n = 202) among our 1,236 patients.

Table 1.
Potential diagnoses of our cohort of 1,236 patients
The 187 confirmed diagnoses were distributed across 40 different genes, but 6 explained approximately 50% of all cases (figure 3C and table e-2, links.lww.com/NXG/A235). DMD was the most common etiology (figure 3). Among the patients with causal variants in DMD, all the 17 adult patients had Becker muscular dystrophy, and 4/11 pediatric patients had Duchenne muscular dystrophy. The latter were all due to a single nucleotide variation (table e-5, links.lww.com/NXG/A238). Half of patients with Becker muscular dystrophy had an exon deletion. Besides DMD, the genes most commonly found to be responsible for muscular disease in our cohort were RYR1, CAPN3, PYGM, DYSF, and FKRP (figure 3). Among the 187 diagnoses, we identified 248 causal variants: 8.1% were large deletions (exon or whole gene deletion), 23.0% small indels, and 68.9% substitutions. Twenty-six percent of the causal variants are new variants not previously reported (64 variants). Of the 20 large deletions, the majority were hemizygous DMD deletions. We also observed 1 heterozygous KLHL41 whole gene deletion, 1 heterozygous GAA whole gene deletion, 2 heterozygous PYGM exon deletions, 1 heterozygous DYSF exon deletion, and 1 heterozygous ANO5 exon deletion (table e-2, links.lww.com/NXG/A235).
Figure 3. Genes that accounted for the most diagnoses in (A) the pediatric cohort (n = 45), (B) the adult cohort (n = 142), and (C) the whole cohort (n = 187).

Clinical characteristics influencing the diagnostic yield
Decreasing diagnostic yield was observed with increasing age, ranging from 22.4% in the 0–17-year-old group, 16.4% between 18 and 59 year olds, and dropping to 9.9% in patients aged 60 years and older. Beside age <60 years, which was significantly associated with a higher diagnostic yield, other clinical criteria were investigated for their impact on the probability of identifying a molecular diagnosis on the gene panel (figure 4). Notably, the probability was significantly higher in patients having a known cardiac anomaly, a positive family history, or showing elevated CK, and in particular those with CK > 1,000 IU/L, who showed a diagnostic yield of 24.9%. Among patients with diagnoses, CK value or cardiac information was available on 179 patients, of whom only 10.7% had normal CK value and no cardiac anomaly. There was no significant difference in diagnostic yield between muscle weakness patterns. Finally, although it did not reach statistical significance, the medical specialty of the ordering physician showed some differences in the diagnostic rate: 17.2% (22/128) for geneticists, 17.0% (130/763) for neuromuscular specialists, and 9.5% (28/295) for general neurologists. Other medical specialties accounted for only a small proportion of patients tested (50/1,236), but 7 diagnoses were made among those 50 patients.
Diagnostic yield of exome sequencing compared with gene panel
A total of 46 patients from 1 center were recruited for exome sequencing and analysis of 3,857 mendelian genes (list of genes provided in supplementary files e-2 and e-3, links.lww.com/NXG/A241, links.lww.com/NXG/A242). We first analyzed the 89 genes included in the gene panel and identified a diagnosis in 7/46 patients (15.2%). Three additional diagnoses were attributed to genes not included in the muscle disorders 89-gene panel: 1 gene causing metabolic myopathies (HADHA), 1 gene causing a mixed nerve and muscle pathology (MYH14), and 1 gene associated with oculopharyngeal muscular dystrophy (PABPN1). In addition, we identified a homozygous pathogenic variant in ABCA1 (Tangier disease), but it could not fully account for the patient's symptoms (table e-4, links.lww.com/NXG/A237). Another patient was found to harbor a pathogenic variant in ANO5 along with a variant of unknown significance, but information was insufficient to confirm this potential diagnosis. Overall, the diagnostic rate of exome was increased to 21.7% compared with 15.2% in the (virtual) gene panel for this single center cohort (patients' details in table e-4).
Although use of exome sequencing could potentially increase the diagnostic rate by analyzing more genes, lower base coverage could lead to missed diagnoses in the 89 genes included in the muscle disorder gene panel. To estimate the proportion of potentially missed diagnoses, we compared the base coverage obtained for these 89 genes from our exome sequencing cohort with the gene panel cohort. On average, the median coverage was 179x (range: 109x–333x) among the 46 patients analyzed by exome sequencing in comparison to 555x (MiSeq, range: 244x–992x) and 1654x (NextSeq, range: 674x–3730x), in 1,236 patients investigated by gene panel according to the sequencing platform used. Among the 3,857 mendelian genes analyzed on exome, 99.61% of bases had adequate coverage as defined by ≥10x. With respect to the 89 genes selected for the muscle disorder gene panel, 99.92% of bases were covered at ≥10x on exome sequencing, similar to what was observed with gene panel on both sequencing platforms (99.92% with MiSeq and 99.95% with NextSeq). Exon 1 of SELENON (SEPN1) showed poor coverage in both exome and gene panel (irrespective of platform used) in all samples owing to extremely high GC content (86.89%). This exon accounts for ∼0.06% of bases of the 89 muscle disorder genes selected. The small difference in base coverage between exome and gene panel was attributed to some specific exons (mostly GC-rich exons 1) showing recurrent borderline to poor coverage in a minority of patients' samples (ranging from 2.7% to 36.1%) on exome sequencing: B3GALNT2 (exon 1), ISPD (exon 1), LMOD3 (exon 1), PLEC (exon 1), SEPN1 (exon 3), SGCB (exon 1), TNNT1 (exons 4 and 5), and VCP (exon 1). Likely pathogenic or pathogenic variants (substitutions and indels) have been previously reported in the ClinVar database for 4 of these 8 exons. However, none of the 228 causal substitutions and indels found among the 187 diagnoses on gene panel localized to these exons, and thus, no diagnosis would have been missed. In addition, 14 of the 20 large deletions identified among the 187 diagnoses would probably have been correctly identified by exome sequencing, as they are hemizygous or homozygous. However, the heterozygous exon deletion involving the last 2 exons of ANO5 could potentially have been missed, as some patients showed base coverage below 50x, which is associated with decreased performance of the CoNVaDING algorithm.24 Notably, this deletion has been identified successfully in another patient on exome sequencing (patient EX11, table e-4, links.lww.com/NXG/A237). The other 5 deletions were all in regions over 100X. At worst, among the patients investigated by gene panel, exome sequencing could have potentially missed 1/248 causal variants (0.4%) or 1 of 187 diagnoses (0.5%).
Discussion
Gene panels and exome sequencing are increasingly being used early in the clinical investigation of patients with suspected muscle disorders, evaluated in outpatient clinics. This is likely to be due in part to decreased sequencing cost, improved availability of molecular testing, its less invasive nature compared with muscle biopsy, and some previous studies reporting high diagnostic yield of such approaches in some study populations.26–28 In our study, we report an overall diagnostic yield of 15.1% (22.4% in children and 13.7% in adults) in a large cohort of patients with suspected muscle disorders from outpatient clinics across Canada. Although this is lower than most previous studies,2–7 the present estimation is more likely to be representative of current clinical practice, as it relies on less restrictive criteria for testing (such as previous abnormal muscle biopsy or limb-girdle weakness pattern), does not focus on suspicion of a specific group of disorders (e.g., LGMDs), and involves a large number of different physicians (187) from the common medical specialties involved in the care of patients with suspected muscle disorders: neuromuscular specialists, general neurologists, and geneticists. As expected, the majority of patients (61.7%) were seen by a neuromuscular specialist. The trends suggesting some difference in diagnostic yield between those specialists could either reflect referral bias or more stringent selection of patients for molecular testing.
Our diagnostic yield could be underestimated, as some additional information on patients with potential diagnoses, which represents up to 16.4% of patients, was not readily available to us. In particular, follow-up molecular testing in other family members and detailed muscle biopsy reports could have enabled the reclassification of some VUS as likely pathogenic and led to additional confirmed diagnoses. Thus, the overall diagnostic yield could be as high as 31.5%, but the 15.1% certainly represents a more conservative estimate. Of note, a recent large sequencing effort in the United States involving more than 4,000 patients with a suspected LGMD observed a 27% diagnostic yield with their 35-gene panel.7 The authors included, among the confirmed diagnoses, patients who were compound heterozygotes for a variant of uncertain significance and a likely or pathogenic variant in recessive genes. This contributed to 5.5% of the 27% yield. Similarly, the yield of the present study would have been 17.8% if we had included such patients. Another factor that could have contributed to this relatively lower diagnostic yield is the significant proportion of patients older than 60 years (a third of adult patients). As observed in our study, age >60 years is associated with a lower diagnostic yield (9.9%). This could be due to an increased contribution of nongenetic disorders in that group.
Similarly to previous studies, we observed a high genetic heterogeneity among diagnoses.29–32 Of interest, the genes most commonly represented among the confirmed diagnoses were similar between children and adults, with DMD, RYR1, CAPN3, PYGM, DYSF, and FKRP accounting for 50% and 58% of diagnoses in children and adults, respectively. The previous large LGMD US study identified CAPN3, DYSF, and FKRP as the 3 major contributing genes, associated with 17%, 16%, and 9% of diagnoses, respectively, but DMD was only found in 4%, whereas RYR1 and PYGM were not tested.7
In our study, variants in DMD were the most frequent cause identified on gene panel in both age groups. This relatively high proportion might reflect some differences between our cohort and other published cohorts as regards prior molecular testing to rule out DMD pathogenic variants when there was a clinical suspicion of Duchenne or Becker dystrophy. We did not require that DMD deletions/duplications be ruled out by specific testing prior performing the gene panel. Despite the wide availability of multiplex ligation-dependent probe amplification-based deletion and duplication analysis across Canada, a total of 12 of 28 causal variants identified in DMD were deletions of one or multiple exons. We suspect that a change in clinical practice might have occurred among the users of our gene panel as a result of its easy access and its ability to identify virtually all DMD variants in males. This would have led to a decrease in the use of MLPA-based deletion and duplication analysis and an increase in the proportion of DMD cases identified by the 89-gene panel. With decreasing cost of gene panel and easier availability, this tendency could occur in other countries as well.
However, all exon deletions were observed in patients with Becker muscular dystrophy and not in patients with Duchenne muscular dystrophy, which may be related to the more variable phenotype of Becker muscular dystrophy. When detailed phenotypic data were available, we could observe suggestive evidence in some patients that atypical clinical presentation likely contributed to the decision not to request DMD MLPA, leading to an increased prevalence of deletions observed in our cohort. For instance, there was a 68-year-old male patient with mild limb-girdle and distal weakness and negative family history (patient 52 on table e-2, links.lww.com/NXG/A235) and two 11-year-old twins with predominant distal weakness (patients 77–78 on table e-2; elevated CK 560–1,100 IU/L, no muscle biopsy performed). Another male patient, in whom a DMD deletion was subsequently identified, presented initially with polyhydramnios and neonatal hypotonia. At age 13 years, he was reported to show diffuse weakness, decreased muscle bulk, and negative family history (patient 2 on table e-2; elevated CK 950 IU/L, no biopsy available). In particular, in this last case, a variant in a second gene (or an additional nongenetic disorder) likely contributed to the atypical phenotype.
Finally, the fact that DMD sequencing is not easily accessible in some Canadian provinces and requires special approval processes that may limit access in some cases may have contributed to an increased proportion of DMD cases in our cohort. Supporting this, all patients diagnosed with Duchenne muscular dystrophy were caused by single nucleotide variation. Those patients are expected to be recognized on a clinical basis, and thus would likely not be sent for gene panel if DMD sequencing was more accessible in some Canadian provinces. This factor presumably contributed to increase the proportion of causal DMD variants observed, especially in the pediatric group compared with other countries having easier access to DMD sequencing. Nevertheless, even if we exclude patients with Duchenne muscular dystrophy, variants in DMD would remain the most common cause of muscle disorders identified in the pediatric group.
We investigated whether any clinical criteria could be used to select patients who would more likely benefit from gene panel testing while having minimal risk of missing a diagnosis. As expected, positive family history and younger age were correlated with an increased likelihood of finding a genetic etiology, but the yield remained significant in patients older than 60 years (9.9%). We had to use age at testing as a proxy for age at onset because accurate historical information was not systematically available. Other criteria that were associated with increased diagnostic yield were high serum CK (especially if above 1,000 IU/L), the presence of a cardiac anomaly, and a positive familial history. These likely reflect better diagnostic yield in muscular dystrophies.31,33,34 Nevertheless, excluding patients with normal CK results and no cardiac anomaly from testing would have resulted in missing 10.7% of the diagnoses, including 3/6 patients with MYOT causal variants who were aged >55 years. Some other patients with such conditions may have been excluded from testing by our protocol, which required 1 abnormal laboratory for testing to be performed. Evaluation of more patients would be needed to analyze the impacts of more complex combinations of clinical criteria.
Finally, with decreasing sequencing cost, many clinical laboratories are considering moving to larger sequencers that would enable analysis of exome-based virtual panels instead of gene panels. Both methods have their advantages and limitations (table e-6, links.lww.com/NXG/A239). In our study, we investigated how this applies in the context of suspected muscle disorders and the genes that were included in our analysis. First, there was only minimal difference (0.03%) in terms of percentage of bases with adequate coverage (>10x), even if median coverage was more than 3–10 times higher for gene panel (depending on the sequencing platform used) compared with exome-based virtual panel. In a minority of patients' samples analyzed by exome, some exons showed borderline or low coverage that could impede detection of single nucleotide variations or indels, although it would not be expected to affect significantly the diagnostic yield based on causal variants found in the gene panel cohort. Moreover, detection of large heterozygous deletions and duplications could be limited by lower coverage of the exome-based panel, especially when dropping below 50x.24 Nonetheless, even with optimal coverage, detection of small exon deletions (<3 exons) remains challenging.35,36 This applies to DMD heterozygous deletions in female and duplications in both male and female. In our study, we did not systematically study the performance of both MLPA and next-generation sequencing method to detect those variants. As deletions and duplications remain a rare type of causal variant, apart from the X-linked DMD gene exon deletions that can still be detected with decreasing coverage in males, the impact on the diagnostic yield would have been limited. Still, it would have possibly resulted in 1 missed diagnosis (1/187, 0.5%). This emphasizes the need to carefully validate each virtual gene panel to ensure sufficient coverage to detect all variant types with optimal performance. Alternatively, performing additional deletion analysis in parallel by exon array or MLPA for specific genes could be recommended when a single pathogenic variant is found or for DMD in all cases. Nevertheless, despite these limitations, our study shows that an exome-based panel with analysis extended to other mendelian genes increased the diagnostic yield from 15.2% to 21.7% in the exome cohort. On patients selected with the same criteria, the 3 additional diagnoses emphasize the fact that all gene panel designs are subject to omission of genes that may represent more atypical cause of weakness (HADHA), the difficulty sometimes encountered in differentiating myopathic from neuropathic pathologies on a clinical basis (MYH14), and accuracy for triplet repeats might be limited for next-generation sequencing compared with PCR (PABPN1).37 In particular, our panel did not provide comprehensive coverage of metabolic myopathies, given that the primary indication of our panel was weakness and not rhabdomyolysis. It is not possible to transpose and generalize this increase of diagnostic yield to the group of patients recruited from various Canadian outpatient clinics (cohort of 1,236 patients), although overall diagnostic yield of the 89 genes is similar between the 2 groups of patients studied (15.1% and 15.2%). Various bias could influence the proportion of cases harbouring variants in genes outside the 89-gene panel. This includes a different population of recruiting physicians with different level of expertise in muscle disorders and a different proportion of abnormal muscle biopsies or prior negative genetic tests, which are both not accurately known in the cohort of 1,236 patients. We also observed a modest increase of variants of uncertain clinical significance related to patient phenotype when extending the analysis beyond the virtual 89-gene panel. However, we were quite conservative in reporting and had access to the complete patient file in the exome cohort. The number of VUS might be much more increased in the regular laboratory setting with limited clinical information. Overall, our results support the use of an exome-based panel for muscular disorders, although several considerations must be taken into account when introducing exome-based panel in the laboratory38–40 (table e-6, links.lww.com/NXG/A239).
Establishing a molecular diagnosis in patients with muscle disorders is becoming increasingly important, as the potential to alter the disease course with gene specific treatments is expected to increase in the upcoming years. Specific treatments are currently limited for hereditary muscle disorders, and enzyme replacement therapy represents 1 example. However, gene therapy approaches are undergoing rapid development for the treatment of LGMDs and dystrophinopathies, in particular. Indeed, several studies are in clinical trials and have shown the potential of this method.41–46 This shows the importance of having a diagnosis for these patients.
Our study supports the use of gene panel testing in patients with suspected muscle disorders from outpatient clinics and highlights several relatively common diagnoses identified in adults and children (DMD, RYR1, CAPN3, PYGM, DYSF, and FKRP). Moreover, it shows that exome sequencing has a low risk of missing diagnoses compared with gene panel, while potentially increasing the diagnostic yield of patients with muscle disorders. However, exome sequencing comes with a bigger burden of VUS. It would though be important, with an exome approach, to have elaborated clinical data and to have the data analyzed in a center experimented in variant interpretation in neuromuscular conditions. Combining MLPA-based deletion and duplication analysis for DMD with 1 of the 2 sequencing methods could result in a higher accuracy for Duchenne and Becker muscular dystrophies, especially in females, given lower sensibility for small heterozygous exon deletions.
Acknowledgment
The study was supported by Sanofi Genzyme Canada. The authors are thankful to Génome Québec and Fulgent for the exome sequencing, Dynacare for their contribution in samples' logistics and genetic counselling, Jean-François Lussier for his computing support, and Calcul Québec and Compute Canada for providing part of the computing infrastructure used to analyze the data. They are also grateful to the patients and their families, neurologists, neuromuscular specialists, genetics counsellor, and clinical geneticists across Canada for their participation in this study. F. Thuriot is supported by doctoral scholarship from the Fonds de recherche du Québec–Santé (FRQS).
Glossary
- CK
creatine kinase
- LGMD
limb-girdle muscular dystrophy
- VUS
variants of uncertain significance
Appendix. Authors
Study funding
This study was sponsored by Sanofi Genzyme Canada.
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Ghaoui R, Clarke N, Hollingworth P, Needham M. Muscle disorders: the latest investigations. Intern Med J 2013;43:970–978. 10.1111/imj.12234. [DOI] [PubMed] [Google Scholar]
- 2.Yu M, Zheng Y, Jin S, et al. . Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS One 2017;12:e0175343 10.1371/journal.pone.0175343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuhn M, Gläser D, Joshi PR, et al. . Utility of a next-generation sequencing-based gene panel investigation in German patients with genetically unclassified limb-girdle muscular dystrophy. J Neurol 2016;263:743–750. 10.1007/s00415-016-8036-0. [DOI] [PubMed] [Google Scholar]
- 4.Seong M-W, Cho A, Park HW, et al. . Clinical applications of next-generation sequencing-based gene panel in patients with muscular dystrophy: Korean experience. Clin Genet 2016;88:484–488. 10.1111/cge.12621. [DOI] [PubMed] [Google Scholar]
- 5.Kitamura Y, Kondo E, Urano M, Aoki R, Saito K. Target resequencing of neuromuscular disease-related genes using next-generation sequencing for patients with undiagnosed early-onset neuromuscular disorders. J Hum Genet 2016;61:931–942. 10.1038/jhg.2016.79. [DOI] [PubMed] [Google Scholar]
- 6.Lévesque S, Auray-Blais C, Gravel E, et al. . Diagnosis of late-onset Pompe disease and other muscle disorders by next-generation sequencing. Orphanet J Rare Dis 2016;11:8 10.1186/s13023-016-0390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nallamilli BRR, Chakravorty S, Kesari A, et al. . Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol 2018;5:1574–1587. 10.1002/acn3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thuriot F, Buote C, Gravel E, et al. . Clinical validity of phenotype-driven analysis software PhenoVar as a diagnostic aid for clinical geneticists in the interpretation of whole-exome sequencing data. Genet Med 2018;20:942–949. 10.1038/gim.2017.239. [DOI] [PubMed] [Google Scholar]
- 9.Haskell GT, Adams MC, Fan Z, et al. . Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurol Genet 2018;4:e212 doi:10.1212/NXG.0000000000000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Retterer K, Juusola J, Cho MT, et al. . Clinical application of whole-exome sequencing across clinical indications. Genet Med 2016;18:696–704. 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 11.Harris E, Topf A, Barresi R, et al. . Exome sequences versus sequential gene testing in the UK highly specialised Service for limb girdle muscular dystrophy. Orphanet J Rare Dis 2017;12:151 10.1186/s13023-017-0699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy HM, Cho K-A, Lek M, et al. . The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet 2017;62:243–252. 10.1038/jhg.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghaoui R, Cooper ST, Lek M, et al. . Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol 2015;72:1424–1432. 10.1001/jamaneurol.2015.2274. [DOI] [PubMed] [Google Scholar]
- 14.Fichna JP, Macias A, Piechota M, et al. . Whole-exome sequencing identifies novel pathogenic mutations and putative phenotype-influencing variants in Polish limb-girdle muscular dystrophy patients. Hum Genomics 2018;12:34 10.1186/s40246-018-0167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy HM, Hamed SA, Lek M, et al. . Homozygous nonsense mutation in SGCA is a common cause of limb-girdle muscular dystrophy in Assiut, Egypt: LGMD in Egypt Linked to SGCA. Muscle Nerve 2016;54:690–695. 10.1002/mus.25094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Efthymiou S, Manole A, Houlden H. Next-generation sequencing in neuromuscular diseases. Curr Opin Neurol 2016;29:527–536. 10.1097/WCO.0000000000000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DePristo MA, Banks E, Poplin R, et al. . A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498. 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cingolani P, Platts A, Wang LL, et al. . A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118 ; iso-2; iso-3. Fly (Austin) 2012;6:80–92. 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landrum MJ, Lee JM, Benson M, et al. . ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–D868. 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lek M, Karczewski KJ, Minikel EV, et al. . Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansson LF, van Dijk F, de Boer EN, et al. . CoNVaDING: single exon variation detection in targeted NGS data. Hum Mutat 2016;37:457–464. 10.1002/humu.22969. [DOI] [PubMed] [Google Scholar]
- 25.Richards S, Aziz N, Bale S, et al. . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–423. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volk AE, Kubisch C. The rapid evolution of molecular genetic diagnostics in neuromuscular diseases. Curr Opin Neurol 2017;30:523–528. 10.1097/WCO.0000000000000478. [DOI] [PubMed] [Google Scholar]
- 27.Schofield D, Alam K, Douglas L, et al. . Cost-effectiveness of massively parallel sequencing for diagnosis of paediatric muscle diseases. NPJ Genomic Med 2017;2 10.1038/s41525-017-0006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ankala A, da Silva C, Gualandi F, et al. . A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol 2015;77:206–214. 10.1002/ana.24303. [DOI] [PubMed] [Google Scholar]
- 29.Stehlíková K, Skálová D, Zídková J, et al. . Muscular dystrophies and myopathies: the spectrum of mutated genes in the Czech Republic. Clin Genet 2017;91:463–469. 10.1111/cge.12839. [DOI] [PubMed] [Google Scholar]
- 30.Vasli N, Laporte J. Impacts of massively parallel sequencing for genetic diagnosis of neuromuscular disorders. Acta Neuropathol (Berl) 2013;125:173–185. 10.1007/s00401-012-1072-7. [DOI] [PubMed] [Google Scholar]
- 31.Magri F, Nigro V, Angelini C, et al. . The Italian limb girdle muscular dystrophy registry: relative frequency, clinical features, and differential diagnosis. Muscle Nerve 2017;55:55–68. 10.1002/mus.25192. [DOI] [PubMed] [Google Scholar]
- 32.Park HJ, Jang H, Kim JH, et al. . Discovery of pathogenic variants in a large Korean cohort of inherited muscular disorders. Clin Genet 2017;91:403–410. 10.1111/cge.12826. [DOI] [PubMed] [Google Scholar]
- 33.Te Riele MGE, Schreuder THA, van Alfen N, et al. . The yield of diagnostic work-up of patients presenting with myalgia, exercise intolerance, or fatigue: a prospective observational study. Neuromuscul Disord 2017;27:243–250. 10.1016/j.nmd.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Narayanaswami P. Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2014;83:1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Yu H, Zhang VW, et al. . Capture-based high-coverage NGS: a powerful tool to uncover a wide spectrum of mutation types. Genet Med 2016;18:513–521. 10.1038/gim.2015.121. [DOI] [PubMed] [Google Scholar]
- 36.Yao R, Yu T, Qing Y, Wang J, Shen Y. Evaluation of copy number variant detection from panel-based next-generation sequencing data. Mol Genet Genomic Med 2019;7:e00513 10.1002/mgg3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keogh MJ, Chinnery PF. Next generation sequencing for neurological diseases: new hope or new hype? Clin Neurol Neurosurg 2013;115:948–953. 10.1016/j.clineuro.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertier G, Hétu M, Joly Y. Unsolved challenges of clinical whole-exome sequencing: a systematic literature review of end-users' views. BMC Med Genomics 2016;9:52 10.1186/s12920-016-0213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sie AS, Prins JB, van Zelst-Stams WAG, Veltman JA, Feenstra I, Hoogerbrugge N. Patient experiences with gene panels based on exome sequencing in clinical diagnostics: high acceptance and low distress. Clin Genet 2015;87:319–326. 10.1111/cge.12433. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Shen X, Fang F, et al. . Phenotype-driven virtual panel is an effective method to analyze WES data of neurological disease. Front Pharmacol 2018;9:1529 10.3388/fphar.2018.01529.8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendell JR, Chicoine LG, Al-Zaidy SA, et al. . Gene delivery for limb-girdle muscular dystrophy type 2D by isolated limb infusion. Hum Gene Ther 2019;30:794–801. 10.1088/hum.2019.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelson CE, Robinson-Hamm JN, Gersbach CA. Genome engineering: a new approach to gene therapy for neuromuscular disorders. Nat Rev Neurol 2017;13:647–661. 10.1038/nrneurol.2017.126. [DOI] [PubMed] [Google Scholar]
- 43.Vannoy CH, Xu L, Keramaris E, Lu P, Xiao X, Lu QL. Adeno-associated virus-mediated overexpression of LARGE rescues α-dystroglycan function in dystrophic mice with mutations in the fukutin-related protein. Hum Gene Ther Methods 2014;25:187–196. 10.1088/hgtb.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sondergaard PC, Griffin DA, Pozsgai ER, et al. . AAV.Dysferlin overlap vectors restore function in dysferlinopathy animal models. Ann Clin Transl Neurol 2015;2:256–270. 10.1002/acn3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bengtsson NE, Hall JK, Odom GL, et al. . Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun 2017;8:14454 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nelson CE, Hakim CH, Ousterout DG, et al. . In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016;351:403–407. 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator. When not possible, given the risk to identify rare patients, additional aggregate data in table form will be produced to address specific questions.