

Abstract
Metabolic heart disease (MHD), which is strongly associated with heart failure with preserved ejection fraction (HFpEF), is characterized by reduced mitochondrial energy production and contractile performance. In this study, we tested the hypothesis that an acute increase in ATP synthesis, via short chain fatty acid (butyrate) perfusion, restores contractile function in MHD.
Isolated hearts of mice with MHD due to consumption of a high fat high sucrose (HFHS) or on control diet (CD) for 4 months were studied using 31P NMR spectroscopy to measure high energy phosphates and ATP synthesis rates during increased work demand. At baseline, HFHS hearts had increased ADP and decreased free energy of ATP hydrolysis (ΔG~ATP), though contractile function was similar between the two groups. At high work demand, the ATP synthesis rate in HFHS hearts was reduced by over 50%. Unlike CD hearts, HFHS hearts did not increase contractile function at high work demand, indicating a lack of contractile reserve. However, acutely supplementing HFHS hearts with 4mM butyrate normalized ATP synthesis, ADP, ΔG~ATP, and contractile reserve.
Thus, acute reversal of depressed mitochondrial ATP production improves contractile dysfunction in MHD. These findings suggest that energy starvation may be a reversible cause of myocardial dysfunction in MHD, and opens new therapeutic opportunities.
Keywords: Metabolism, mitochondria, ATP synthesis, NMR spectroscopy, contractile function, metabolic syndrome, heart failure
Graphical Abstract
Obesity induced by high fat high sucrose (HFHS) feeding negatively impacted cardiac energetics (decreased ATP synthesis and decreased |Δ GATP|) and cardiac function which was unmasked further by increasing demand for cardiac workload (rate pressure product). Acute perfusion of HFHS hearts with butyrate increased the rate of ATP synthesis and improved cardiac energetics and contractile function thus correcting the lack of contractile reserve in energetically impaired HFHS hearts and indicating a causal relationship between cardiac energetics and cardiac function.
Introduction
While energetic deficits are well described in the failing heart, the temporal and causal relationship between a chronic energetic deficit and contractile dysfunction is debated.1–3 Moreover, little information is available on whether energetic defects are reversible, or if they contribute to decreased cardiac performance in chronic myocardial disease.1,4,5 Thus, the hemodynamic consequences of myocardial energetic abnormalities are incompletely understood. In addition, it remains unclear whether targeted strategies for improving mitochondrial ATP synthesis may reverse chronically depressed contractile performance.1,2,6,7
The free energy of ATP hydrolysis (ΔG~ATP) is an important measure of myocardial energetics and describes the chemical potential in a molecule of ATP to perform work.8–12 A decline in ΔG~ATP is a consequence of an increase in ADP,4 and precedes contractile dysfunction induced by pharmacologic13 or ischemic14 inhibition of ATP synthesis. Therefore, ΔG~ATP decreases when ADP increases and can be diminished even when [ATP] is preserved, as is frequently found in chronic heart disease.1,13,15 Once ΔG~ATP is decreased, regardless of the cause, the heart has reduced contractile reserve and is at risk for acute mechanical failure during challenges such as an abrupt increase in work demand, hypoxia, ischemia, or arrhythmia.1,12 Worsening of energetics in diseased hearts is frequently thought to be due to diminished contribution of fatty acids to oxidative metabolism.2,6,16–18 Genetic models that prevent this decrease19–21 or chronic interventions that increase utilization of other substrates, such as glucose20,22 or ketones23, improve pathologic cardiac remodeling and function in models of myocardial hypertrophy19,22,24 and progressive heart failure.18,25
Metabolic heart disease (MHD), a consequence of metabolic syndrome, is characterized by LV hypertrophy (LVH), contractile dysfunction, and mitochondrial dysfunction.26 Mice fed a high fat high sucrose (HFHS) diet mimic human MHD27–29 and display decreased myocardial ATP synthesis, increased ADP and decreased ΔG~ATP. Moreover, they are unable to appropriately increase contractile performance in response to increased work demand, indicating a lack of contractile reserve.26 In our prior experiments, supplementation with long chain fatty acids (LCFA) did not rescue contractile or energetic dysfunction in hearts from HFHS mice; thus we were not able to establish causality between energy production and contractile dysfunction.26 Butyrate, a short chain fatty acid (SCFA), is an efficient mitochondrial fuel that bypasses the highly regulated LCFA import mechanisms, directly enters the mitochondrial matrix and undergoes β-oxidation.30–32 Here, we test the hypothesis that cardiac contractile performance in MHD can be improved by rapidly normalizing ATP synthesis, ADP and ΔG~ATP via butyrate perfusion.
Methods
Experimental animals.
WT male C57BL/6J mice 8 weeks of age were fed ad libitum either a control diet (CD) or HFHS diet for 4 months as previously described.26,33,34 The protocol was approved by the Institutional Animal Care and Use Committee of the Boston University School of Medicine.
Simultaneous measurement of LV contractile function and high energy phosphates by 31P nuclear magnetic resonance (NMR) spectroscopy.
Left ventricular (LV) contractile function was assessed in a Langendorff heart preparation as previously described.20,26,35 Hearts were perfused with Krebs-Henseleit (KH) buffer containing 10 mM glucose, 0.5mM pyruvate, +/− 4mM butyrate and +/− 200 μM 4-bromocrotonic acid (BCA) to inhibit ß-oxidation. A water-filled balloon was inserted into the LV to measure LV pressure and adjust LV volume. LV developed pressure (DevP) was calculated as: DevP = systolic pressure (LVSP) – end diastolic pressure (LVEDP). Perfused hearts were placed in a 10 mm glass tube in a 9.4T vertical bore magnet and maintained at 37°C throughout the protocol. After stabilization, balloon volume was adjusted to achieve an LVEDP of 8 to 9 mmHg, and held constant during the protocol. LV workload was changed by increasing the concentration of CaCl2 in the KH from 2 to 4 mM, increasing the pacing rate from 450 to 600 bpm, or both. Work performed was estimated as the rate pressure product (RPP) = DevP × heart rate.26 [ATP], [phosphocreatine] (PCr), and [inorganic phosphate] (Pi), were measured using 31P NMR Varian spectrometer at 161.4 MHz and corrected for saturation.20,26,35 Each 31P NMR spectrum resulted from the average of 208 free induction decay signals over 8 min. Intracellular pH (pHi), cytosolic free [ADP], and the free energy of ATP hydrolysis (ΔG~ATP) was calculated (Eq 1: |ΔG~ATP | (kJ/mol) = |ΔG0 + RT ln ([ADP][Pi]/[ATP]) where ΔG0 (−32 kJ/mol) represents the ΔG~ATP at standard conditions of molarity and temperature. The free [ADP] was calculated using the creatine kinase equation which was assumed to be at equilibrium and where Keq = 1.66 × 109M−1 as previously described26 (Eq 2: [ADP] = ([ATP][Cr])/([PCr][H+])Keq).
After NMR spectroscopy, while still beating, the hearts were immediately freeze clamped and stored at −80°C for subsequent analysis of total creatine concentration by HPLC36 to allow for ADP and ΔG~ATP calculations.26
ATP synthesis rate.
The rate of ATP synthesis was measured with the 2-site saturation transfer technique by applying a low-power, narrow-band radiofrequency pulse to saturate γ-ATP resonance and measure changes in the Pi resonance. The Pi is solely in the intracellular space as there is no phosphate in K-H buffer. These measurements were performed at increased work demand (hearts paced at 450 bpm, 4mM Ca2+ in the perfusate). Spectra were acquired with (M∞) and without (M0) a selective 4.8-second saturating pulse.8,20,26 The unidirectional pseudo–first-order rate constant of ATP synthesis was calculated as kf = (M0-M∞)/T1 M0 and flux= kf [Pi], where T1 is the intrinsic longitudinal relaxation time for Pi,37 M0 and M∞ are magnetizations of Pi at 0 and 4.8 seconds of γ-ATP saturation, respectively. Each spectrum resulted from averaging a total of 256 scans, interleaved between blocks of 8M0 and 8M∞ scans. The measurements were acquired in 44 min.26 To eliminate the effect of the direct attenuation of the observed resonance by the gamma-ATP targeted saturation pulse, during the M0 acquisition, an RF pulse of the same power was used and targeted at an equal frequency offset downfield from the observed resonance. Therefore, the symmetrical irradiation that was targeted at the same offset on the opposite side of the Pi resonance allowed us to control for “spillover” RF effects.
Statistical analysis.
Results are presented as mean ± SD. Comparisons between groups were performed using unpaired t-tests, Mann-Whitney non-parametric tests or 2-way or repeated measures ANOVA, as appropriate. All statistical analyses were performed using GraphPad Prism software. P-value < 0.05 was considered significant.
Results
Butyrate improves the rate of ATP synthesis in HFHS hearts.
At increased work demand (4mM Ca2+, 450 bpm), the rate of ATP synthesis in HFHS hearts was only 44% that of CD hearts (Figure 1A). Perfusion of HFHS hearts with KH + 4mM butyrate increased the ATP synthesis rate by 2-fold compared to HFHS hearts not perfused with butyrate (p<0.001) and 1.3× that of KH perfused CD hearts without butyrate (p<0.05). To test the specificity of the butyrate effect, ATP synthesis was measured during butyrate infusion under the same workload conditions with and without pretreatment with BCA, an inhibitor of β-oxidation.30 The butyrate-induced improvement in ATP synthesis was completely prevented by pretreatment with BCA (Figure 1A). Representative nuclear magnetic resonance spectra of the 2-site saturation transfer technique show the disappearance of the saturated γ-ATP peak at M∞ (Figure 1B–D). Subsequent exchange between the saturated γ-ATP and Pi results in a reduction in the Pi peak at M∞. In CD and HFHS + butyrate hearts, the reduction in the Pi peak at M∞ is greater (Figure 1B, D) than in HFHS hearts without butyrate (Figure 1C) indicative of higher ATP synthesis rates in CD and HFHS + butyrate hearts. An observed reduction in the β-ATP peak between M0 and M∞ is likely due to nuclear Overhauser effects as previously described.38
Figure 1: 31P NMR magnetization transfer measured ATP synthesis in CD and HFHS hearts during high work demand.

CD and HFHS hearts were perfused with KH buffer containing 10 mM glucose, 0.5 mM pyruvate and 4 mM Ca2+ while paced at 450 bpm. (A) ATP production was reduced in HFHS hearts. Adding 4mM butyrate to HFHS KH perfusion increased ATP production and this increase was abolished with the addition of 4-bromocrotonic acid (BCA), an inhibitor of β-oxidation. Representative 31P NMR magnetization transfer spectra collected from CD (B), HFHS (C) and HFHS+butyrate (D) hearts at 0 seconds (M0) and 4.8 seconds (M∞) after a saturating pulse was applied to γ-P of the ATP. The peaks are assigned (from left to right) as Pi, PCr, γ-ATP, α-ATP and β-ATP. Note the disappearance of the γ-ATP peak (saturated) at M∞. The exchange of the saturated [γ-P] between ATP and Pi resulted in reduced Pi peak area. Both CD hearts and HFHS + butyrate hearts have a larger decrease (reflecting higher rates of ATP synthesis) than HFHS hearts. CD (n = 7), HFHS (n = 8), HFHS + butyrate (n = 4), HFHS + butyrate + BCA (n = 4); ** p < 0.01 vs CD, ### p <0.001 vs HFHS.
Butyrate normalizes abnormally elevated ADP in HFHS hearts.
The [ATP] was similar in hearts from HFHS- and CD-fed mice throughout the protocol (Figure 2A). In CD hearts, [PCr] decreased with increasing work demand. In HFHS hearts, [PCr] was lower at all work demand levels (vs. CD hearts) (Figure 2B). The [PCr]/[ATP] was also significantly lower at all work demand levels (Figure 2C), however, total [Cr] (Supplementary Figure 1) and intracellular pH (Figure 2D) were the same for all groups. Calculated cytosolic free [ADP] was higher in HFHS hearts at baseline and at all levels of work demand (Figure 2E). In HFHS hearts, the addition of 4 mM butyrate to the perfusate normalized [PCr], [PCr]/[ATP] and [ADP] such that they were similar to CD hearts at all work levels. Neither the decrement in HFHS heart energetics nor the improvement resulting from acute butyrate perfusion was due to changes in contractile efficiency, as the cost of contraction (amount of ATP synthesized per work performed) was the same in all groups (Figure 2F).
Figure 2: 31P NMR measured energetics in isolated hearts from mice fed CD and HFHS diets.

A stress protocol was performed with incremental increases in pacing rate and calcium content in 4 stages over 64 min. The measured or calculated values for ATP (A), PCr (B), PCr/ATP (C), pH (D) and ADP (E) during this stress protocol are shown. Butyrate corrected all energetic abnormalities in the KH perfused hearts from HFHS fed mice. At the highest workload, the amount of ATP synthesized to perform work (Cost of Contraction) was the same for the three groups (F). This demonstrates that diet and metabolic substrate perfusion did not change the amount of ATP required to perform contractile work. (n=6–7); ** p<0.01 HFHS vs. CD and HFHS + butyrate.
Butyrate increases |ΔGATP | in HFHS hearts.
The absolute value of free energy of ATP hydrolysis |ΔGATP | was lower in HFHS hearts at baseline and at all levels of increased work demand. Perfusion with butyrate corrected |ΔGATP|in HFHS hearts (Figure 3A).
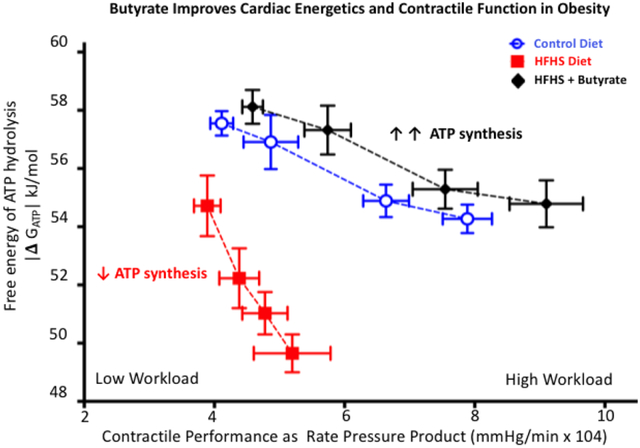
Figure 3: Butyrate improves the free energy of hydrolysis of ATP in HFHS hearts.

The absolute value of free energy of ATP hydrolysis, |ΔGATP |, was lower in HFHS hearts at all levels of work demand. Perfusion with butyrate prevented the detrimental decrease in |Δ GATP| (A). The lack in contractile reserve is revealed when |Δ GATP |is plotted against the work performed (RPP), where, at the highest workloads, the |Δ GATP | fell below the free energy required for sarcoplasmic reticulum ATPase (SERCA) function. This deficit in contractile reserve was fully restored to normal values upon perfusion with butyrate even at the highest work demand (B), n=6–7; **p<0.01.
Butyrate normalizes contractile reserve in HFHS hearts.
In hearts from CD mice, developed pressure and the rate pressure product (RPP) increased progressively with increased work demand, whereas in HFHS hearts these increases were markedly blunted (Figure 4 A, B). LVEDP was manually set to be identical in the HFHS and CD hearts under baseline conditions, but increased more in HFHS hearts at each increased level of work demand (Figure 4C). At the highest work demand (4mM Ca2+, 600 bpm), LVEDP was increased in HFHS hearts compared to CD hearts (21.5 ± 2 mmHg vs. 13.2 ± 2.1 mmHg; p < 0.01). Butyrate perfusion prevented all the abnormalities in contractile function unmasked by high work demand in the HFHS hearts: developed pressure, RPP, and LVEDP were similar to CD hearts at all work levels (Figure 4 A–C).
Figure 4: Butyrate-improved energetics leads to normalization of hemodynamic function in HFHS hearts.

In hearts from CD fed mice, there was an increase in developed pressure (A) and rate pressure product (B) at progressively higher work demand. This was blunted in hearts from mice fed a HFHS diet, indicative of a lack of contractile reserve. LV end diastolic pressure (LVEDP) increased with each progressive increase in workload in HFHS hearts compared to CD hearts indicative of impaired relaxation (C). Acute perfusion with butyrate prevented the detrimental contractile and relaxation changes seen in HFHS hearts. Representative raw tracings at high workload (4mM Ca2+ and paced at 450 bpm) show no significant change in cardiac contractility in CD hearts with butyrate (D) but a significant improvement in HFHS hearts within 2 minutes of initiation of butyrate (E). Inhibition of β-oxidation with BCA prevented the acute butyrate-induced improvement in contractility (F). n= 6–7; **p < 0.01.
To test the rapidity of the butyrate effect, acute infusion of butyrate in CD did not further increase developed pressure at high work demand (4mM Ca2+, 450 bpm), (Figure 4D). In HFHS hearts, acute infusion of butyrate rapidly (within 2 minutes) increased developed pressure to the level of CD hearts (Figure 4E). Additionally, as in Figure 1, to test the specificity of the butyrate effect, HFHS hearts were perfused with and without pretreatment with BCA, an inhibitor of β-oxidation.30 The butyrate-induced improvement in developed pressure was prevented by pretreatment with BCA (Figure 4F).
The combined effect of butyrate on energetics and contractile function is particularly striking when |ΔGATP | is plotted against work performed (RPP) (Figure 3B). Butyrate perfusion moved the |ΔGATP |/ RPP relationship in HFHS hearts upwards and rightwards by simultaneously improving both energetics and contractile function and correcting the lack of contractile reserve in energetically impaired HFHS hearts.
Discussion
The main finding of this study is that an immediate doubling of ATP synthesis is sufficient to cause an improvement in contractile function in mice with MHD. Perfusion with butyrate, an efficient mitochondrial energy substrate, rapidly corrected mitochondrial ATP synthesis, high energy phosphate concentration including ADP and importantly, |ΔGATP |. Moreover, doubling in ATP supply resulted in a complete restoration of contractile reserve and normalization of diastolic function at high work demand. The ability of butyrate to acutely and rapidly improve myocardial function suggests a direct causal relationship between the impairment in energetics and cardiac dysfunction.
Failing heart is energy starved.
It has been well established that the failing heart is energy starved39–41. However, whether the energetic changes that are observed in chronic myocardial disease are the primary cause of pump dysfunction or a contributing factor secondary to chronic metabolic remodeling remains unanswered.1,2,4,15 As we have previously shown, an increase in ADP drives the decrease in |ΔG~ATP| in HFHS hearts at baseline and the deficit worsens with increased work demands.26,35 Whether the energy deficit reflected by the decrease in |ΔG~ATP| is sufficient to contribute to contractile dysfunction is unclear. This question is salient given that [ATP] and [Pi] remained unchanged in HFHS hearts compared to CD, thus making increased [ADP] the driver of the decrease in |ΔG~ATP| (see methods Eq 1).
Decreased |ΔGATP | in MHD is too low to support normal diastolic and systolic function.
In CD hearts, increased work demand is matched by a progressive increase in contractile function. In contrast, HFHS hearts have a markedly blunted developed pressure and RPP response across a range of work demand. Thus, an increase in work demand revealed a marked impairment in contractile reserve in HFHS hearts as evidenced by failure to appropriately increase systolic function. The inability to increase contractility in HFHS hearts was even more notable given that HFHS hearts had a higher LV end-diastolic pressure (LVEDP) with increasing work demand. These results are relevant to the clinical setting in which the lack of contractile reserve and increased diastolic filling pressures are observed in patients with heart failure with preserved ejection fraction (HFpEF)28, a clinical syndrome that is frequently caused by MHD, and contributes to exertional or workload-related symptoms in these patients.28,42,43 In our study, with increased work in HFHS hearts, |ΔG~ATP|decreased below the value of 52 kJ/mol; a critical value for normal Sarcoplasmic Reticulum Calcium ATPase (SERCA) function (Figure 3B).13,26,44 SERCA is a major regulator of calcium homeostasis and contractility in the heart and thus SERCA dysfunction may contribute to both diastolic dysfunction and loss of contractile reserve with increased work demand.12,45
Butyrate rapidly enters mitochondria and supports ß-oxidation.
To test the relationship between decreased |ΔG~ATP| and contractile dysfunction, we sought to acutely improve ATP synthesis and |ΔG~ATP|by perfusing hearts with the SCFA, butyrate, an efficient substrate for energy production. Although the substrates in the perfusion conditions (glucose and pyruvate) do not entirely reflect the in vivo substrate selection and hormonal milieu, we had previously observed that perfusion with a mixture of LCFAs did not rescue contractile function in HFHS hearts thus suggesting a “bottleneck” for LCFA utilization by the mitochondria.26 Nevertheless, since the perfusion conditions do not precisely match the in vivo conditions, it may be difficult to predict the effect of increased circulating butyrate in this model. The observed difference between LCFA and SCFA likely reflects the ability of butyrate to bypass the highly regulated LCFA transport mechanisms of the mitochondria which are altered in chronic heart failure.19,30 Therefore, unlike LCFA, butyrate enters the mitochondria directly to undergo ß-oxidation and produce reducing equivalents (NADH and FADH2). Thus, butyrate can rapidly replenish flux through the electron transport chain resulting in improved ATP synthesis. The beneficial effect of butyrate perfusion was evident within approximately 2 minutes, and was associated with increased ATP production which corrected [PCr], [ADP] and ΔG~ATP. The rapid effect of butyrate was due to increased ß-oxidation since concomitant BCA perfusion prevented the butyrate-induced increase in ATP synthesis rate.
Butyrate increases |ΔGATP | sufficiently to support SERCA and normalize diastolic function and contractile reserve.
The improvement in energetics was temporally related to improvements in both systolic and diastolic function with increased developed pressure, rate pressure product, and LVEDP at elevated work demands. The acute effect of butyrate perfusion was not due to an improvement in contractile efficiency as the cost of contraction (amount of ATP synthesized per unit of work performed) was not different between the groups (Figure 2F), suggesting that butyrate did not decrease the demand for ATP. Importantly, butyrate perfusion improved |ΔG~ATP|to > 52 kJ/mol, thereby, potentially, fully satisfying SERCA ΔG~ATP requirements as well as the ΔG~ATP requirements of other ATP dependent contractile and cellular processes such as Na/K ATPase (|ΔG~ATP|~ 48 kJ/mol) and myosin ATPase (|ΔG~ATP|~ 47 kJ/mol).32,46
In addition, the decrease in PCr in HFHS group (Figure 2B, C) did not seem to be due to a decrease in the concentration of creatine which was not different from CD group (Supplementary Figure 1). Using chemical exchange saturation transfer (CEST) of creatine, Pumphrey et al have previously shown decreased cardiac creatine in a mouse model of high fat diet.47 The differences in our results may be due to differences in technique (HPLC vs CEST), diet/model and length of feeding. Further studies into how cardiac creatine changes over time in models of obesity and its relation to energetics are warranted.
Evidence for contractile benefit of butyrate.
The contractile benefit of butyrate perfusion seems to be due to its effect on increased ATP production and lowering [ADP] based on: 1) a rapid increase in contractile function within 2 minutes from the introduction of butyrate to the perfusate 2) a simultaneous increase in both |ΔGATP | and contractile function rather than a reciprocal change that might be expected if other mechanisms were involved (e.g. an inotropic effect or a change in calcium sensitivity) 3) the complete prevention of the butyrate effect on both ATP synthesis and contractile function with BCA, an inhibitor of ß-oxidation.
Causal link for energetics and cardiac function in MHD.
Prior studies have modulated the metabolic remodeling that occurs in heart failure with varying levels of success.6,19,24,48 However, these interventions have occurred over weeks or months introducing the possibility of cardiac remodeling and thus making a temporal and causal relationship between the changes in energetics and cardiac function uncertain.49,50 Our current study, in contrast, is performed over a very brief time frame eliminating the possibility that other adaptive mechanisms, such as changes in protein expression,16,17 could explain the results, and thus providing a causal link between energetics and cardiac function.
Magnitude of our energetic intervention.
In recent years, attention has been given to modulating cardiac metabolism in heart failure by varying the source of energy substrates.6,18,23,51,52 The logic applied in our study was similar: to utilize a carbon source (butyrate) that easily enters the mitochondrial matrix and produces the highest amount of reducing equivalents for ATP synthesis when oxidized. The doubling in the rate of ATP production with butyrate in our study is noteworthy. In prior studies, perfusion of hearts from db/db mice or mice post thoracic aortic constriction with the ketone ß-hydroxybutyrate caused no improvement in cardiac work23,53. However, in these studies the estimated ATP production increased by approximately 20%. The smaller increase in ATP synthesis with ß-hydroxybutyrate in these studies may reflect differences in perfusate concentration and/or that ß-hydroxybutyrate produces a single reducing equivalent (NADH) when converted to acetoacetate18, whereas both NADH and FADH2 are produced from butyrate through β-oxidation prior to entering the TCA cycle30. A subsequent study showed that chronic ketone infusion improved hemodynamics in tachy-mediated cardiomyopathy in canines, though energetics was not assessed.25 Additionally, manipulating pyruvate dehydrogenase complex so as to increase pyruvate flux into the mitochondria may be another beneficial strategy and is being investigated with pyruvate dehydrogenase kinase inhibitors.54 These observations suggest that the magnitude of the increase in ATP production may determine the impact on cardiac function to a greater extent than the choice of provided substrate as long as substrate transport into mitochondria is not limited. The comparison between ketones and SCFA as energy substrates for the heart is also important given their different sites of production in the body (liver for ketones vs. gut microbiome for butyrate)55. Thus, this work widens therapeutic target possibilities for MHD.
Conclusion.
Energetic limitations may be an important cause of contractile dysfunction which leads to exercise intolerance and symptoms in patients with HFpEF due to MHD. As recently noted, the application of mitochondria-targeted therapies for heart failure has lagged.56 Our data provide direct evidence that contractile dysfunction is due to decreased ATP production in the HFHS model of MHD and can be ameliorated by methods which restore ATP production. We have previously shown that reactive oxygen species (ROS) damage mitochondrial electron transport chain proteins contributing to decreased ATP synthesis in the same HFHS model and that quenching ROS with mitochondrial catalase prevents oxidative mitochondrial damage34,35 and both energetic and contractile dysfunction.35 In the short time-frame of the current study, it is unlikely that our acute intervention fixed the adversely remodeled mitochondrial machinery, but rather provided a superior carbon source sufficient to boost ATP synthesis to normal levels. Accordingly, these findings suggest that strategies to improve energy production may be an effective approach for the treatment of MHD. Whether these findings apply more broadly to other forms of heart disease remains to be investigated.
Supplementary Material
There was no difference in the total creatine concentration measured by HPLC between any of the groups indicating that the changes in PCr that were observed were not due to changes in creatine concentration. n = 6–7 per group.
Sources of Funding
Supported by NIH grants HL-064750 (WSC), K08 HL123744 (MP), EB 014414 (JAB), NIDDK R01 DK103750 (MMB) and the NHLBI-sponsored Boston University Cardiovascular Proteomics Center (Contract No. N01-HV-28178, WSC). Dr. Bachschmid was also funded by American Heart Association “Grant in Aid” 16GRNT27660006. Dr. Luptak is the recipient of an AHA Fellow-to-Faculty Award 15FTF25890062.
Abbreviations
- BCA
bromocrotonic acid
- CD
control diet
- CEST
chemical exchange saturation transfer
- Cr
creatine
- DevP
left ventricular developed pressure
- ΔG~ATP
free energy of ATP hydrolysis
- HFpEF
heart failure with preserved ejection fraction
- HFHS
high fat high sucrose
- KH
Krebs-Henseleit
- LCFA
long chain fatty acid
- LVH
left ventricular hypertrophy
- LVSP
left ventricular systolic pressure
- LVEDP
left ventricular end diastolic pressure
- PCr
phosphocreatine
- Pi
inorganic phosphate
- MHD
metabolic heart disease
- ROS
reactive oxygen species
- RPP
rate pressure product
- SCFA
short chain fatty acid
- SERCA
sarcoplasmic reticulum calcium ATPase
References
- 1.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circulation Research. 2004;95(2):135–145. doi: 10.1161/01.RES.0000137170.41939.d9 [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Abel ED. Effective Metabolic Approaches for the Energy Starved Failing Heart. Circulation Research. 2018;123(3):329–331. doi: 10.1161/JAHA.113.000301.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neubauer S, Horn M, Cramer M, et al. Myocardial Phosphocreatine-to-ATP Ratio Is a Predictor of Mortality in Patients With Dilated Cardiomyopathy. Circulation. 1997;96(7):2190–2196. doi: 10.1161/01.CIR.96.7.2190 [DOI] [PubMed] [Google Scholar]
- 4.Ingwall JS. Phosphotransfer Reactions in the Failing Heart In: Patterson C, Willis MS, eds. Translational Cardiology: Molecular Basis of Cardiac Metabolism, Cardiac Remodeling, Translational Therapies and Imaging Techniques. Totowa, NJ: Humana Press; 2012:39–62. doi: 10.1007/978-1-61779-891-7_2 [DOI] [Google Scholar]
- 5.Liao R, Nascimben L, Friedrich J, Gwathmey JK, Ingwall JS. Decreased Energy Reserve in an Animal Model of Dilated Cardiomyopathy: Relationship to Contractile Performance. Circulation Research. 1996;78(5):893–902. doi: 10.1161/01.RES.78.5.893 [DOI] [PubMed] [Google Scholar]
- 6.Lopaschuk GD. Metabolic Modulators in Heart Disease : Past, Present, and Future. Canadian Journal of Cardiology. 2017;33(7):838–849. doi: 10.1016/j.cjca.2016.12.013 [DOI] [PubMed] [Google Scholar]
- 7.Gupta A, Chacko VP, Schar M, Akki A, Weiss RG. Impaired ATP kinetics in failing in vivo mouse heart. CircCardiovascImaging. 2011;4(1942–0080 (Electronic)):42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bittl JA, Ingwall JS. Reaction rates of creatine kinase and ATP synthesis in the isolated rat heart. A 31P NMR magnetization transfer study. The Journal of Biological Chemistry. 1985;260(6):3512–3517. [PubMed] [Google Scholar]
- 9.Saupe KW, Spindler M, Hopkins JCA, Shen W, Ingwal JS. Kinetic, thermodynamic, and developmental consequences of deleting creatine kinase isoenzymes from the heart: Reaction kinetics of the creatine kinase isoenzymes in the intact heart. Journal of Biological Chemistry. 2000;275(26):19742–19746. doi: 10.1074/jbc.M001932200 [DOI] [PubMed] [Google Scholar]
- 10.Spindler M, Meyer K, Stromer H, et al. Creatine kinase-deficient hearts exhibit increased susceptibility to ischemia-reperfusion injury and impaired calcium homeostasis. American Journal of Physiology-Heart and Circulatory Physiology. 2004;287(3):H1039–H1045. doi:DOI 10.1152/ajpheart.01016.2003 [DOI] [PubMed] [Google Scholar]
- 11.Saupe KW, Spindler M, Tian R, Ingwall JS. Impaired cardiac energetics in mice lacking muscle-specific isoenzymes of creatine kinase. Circulation research. 1998;82(8):898–907. [DOI] [PubMed] [Google Scholar]
- 12.Tian R, Nascimben L, Ingwall JS, Lorell BH. Failure to maintain a low ADP concentration impairs diastolic function in hypertrophied rat hearts. Circulation. 1997;96(0009–7322 (Print)):1313–1319. [DOI] [PubMed] [Google Scholar]
- 13.Tian R, Halow JM, Meyer M, et al. Thermodynamic limitation for Ca2+ handling contributes to decreased contractile reserve in rat hearts. American Journal of Physiology - Heart and Circulatory Physiology. 1998;275(6 Pt 2):H2064–71. [DOI] [PubMed] [Google Scholar]
- 14.Arai AE, Pantely GA, Thoma WJ, Anselone CG, Bristow JD. Energy metabolism and contractile function after 15 beats of moderate myocardial ischemia. Circulation Research. 1992;70(6):1137–1145. [DOI] [PubMed] [Google Scholar]
- 15.Neubauer S, Krahe T, Schindler R, et al. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease. Circulation. 1992;86(6):1810–1818. doi: 10.1161/01.CIR.86.6.1810 [DOI] [PubMed] [Google Scholar]
- 16.Razeghi P, Young ME, Ying J, et al. Downregulation of Metabolic Gene Expression in Failing Human Heart before and after Mechanical Unloading. Cardiology. 2002;97(4):203–209. doi: 10.1159/000063122 [DOI] [PubMed] [Google Scholar]
- 17.Osorio JC, Stanley WC, Linke A, et al. Impaired Myocardial Fatty Acid Oxidation and Reduced Protein Expression of Retinoid X Receptor-α in Pacing-Induced Heart Failure. Circulation. 2002;106(5):606–612. doi: 10.1161/01.CIR.0000023531.22727.C1 [DOI] [PubMed] [Google Scholar]
- 18.Aubert G, Martin OJ, Horton JL, et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation. 2016;133(8):698–705. doi: 10.1161/CIRCULATIONAHA.115.017355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolwicz SC, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-Specific Deletion of Acetyl CoA Carboxylase 2 Prevents Metabolic Remodeling During Pressure-Overload Hypertrophy. Circulation Research. 2012;111(6):728–738. doi: 10.1161/CIRCRESAHA.112.268128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005;112(15):2339–2346. doi: 10.1161/CIRCULATIONAHA.105.534594 [DOI] [PubMed] [Google Scholar]
- 21.Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. 2007;116(8):901–909. doi: 10.1161/CIRCULATIONAHA.107.691253 [DOI] [PubMed] [Google Scholar]
- 22.Liao R, Jain M, Cui L, et al. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. 106(16):2125–2131. [DOI] [PubMed] [Google Scholar]
- 23.Ho KL, Zhang L, Wagg C, et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovascular Research. 2019:1–11. doi: 10.1093/cvr/cvz045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okere IC, Young ME, McElfresh TA, et al. Low carbohydrate/high-fat diet attenuates cardiac hypertrophy, remodeling, and altered gene expression in hypertension. Hypertension. 2006;48(6):1116–1123. doi: 10.1161/01.HYP.0000248430.26229.0f [DOI] [PubMed] [Google Scholar]
- 25.Horton JL, Davidson MT, Kurishima C, et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight. 2019;4(4). doi: 10.1172/jci.insight.124079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luptak I, Sverdlov AL, Panagia M, et al. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. Journal of Molecular and Cellular Cardiology. 2018;116(February):106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayalon N, Gopal DM, Mooney DM, et al. Preclinical left ventricular diastolic dysfunction in metabolic syndrome. American Journal of Cardiology. 2014;114(1879–1913 (Electronic)):838–842. doi: 10.1016/j.amjcard.2014.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borlaug BA. The pathophysiology of heart failure with preserved ejection fraction. Nature Reviews Cardiology. 2014;11(9):507–515. doi: 10.1038/nrcardio.2014.83 [DOI] [PubMed] [Google Scholar]
- 29.Rider OJ, Francis JM, Ali MK, et al. Effects of catecholamine stress on diastolic function and myocardial energetics in obesity. Circulation. 2012;125(1524–4539 (Electronic)):1511–1519. [DOI] [PubMed] [Google Scholar]
- 30.Lewandowski ED, Chari MV, Roberts R, Johnston DL. NMR studies of beta-oxidation and short-chain fatty acid metabolism during recovery of reperfused hearts. The American journal of physiology. 1991;261(2 Pt 2):H354–63. [DOI] [PubMed] [Google Scholar]
- 31.Lewandowski ED, Kudej RK, White LT, Donnell JMO, Vatner SF. Mitochondrial Preference for Short Chain Fatty Acid. 2002:367–372. doi: 10.1161/hc0302.102594 [DOI] [PubMed] [Google Scholar]
- 32.Balschi JA, Shen H, Madden MC, Hai JO, Jr ELB, Wolkowicz PE. Model Systems for Modulating the Free Energy of ATP Hydrolysis in Normoxically Perfused Rat Hearts. 1997;3133:3123–3133. [DOI] [PubMed] [Google Scholar]
- 33.Qin F, Siwik DA, Luptak I, et al. The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation. 2012;125(14):1757–1764. doi: 10.1161/CIRCULATIONAHA.111.067801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sverdlov AL, Elezaby A, Behring JB, et al. High fat, high sucrose diet causes cardiac mitochondrial dysfunction due in part to oxidative post-translational modification of mitochondrial complex II. Journal of Molecular and Cellular Cardiology. 2015;78(1095–8584 (Electronic)):165–173. doi: 10.1016/j.yjmcc.2014.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luptak I, Qin F, Sverdlov A, et al. Energetic dysfunction is mediated by mitochondrial ROS and precedes structural remodeling in metabolic heart disease. Antioxidants & Redox Signaling. May 2019:ars.2018.7707. doi: 10.1089/ars.2018.7707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kammermeier H Microassay Extracts of Free and Total of Creatine. Analytical Biochemistry. 1973;345:341–345. [DOI] [PubMed] [Google Scholar]
- 37.Spencer RG, Balschi J a, Leigh JS, Ingwall JS. ATP synthesis and degradation rates in the perfused rat heart. 31P-nuclear magnetic resonance double saturation transfer measurements. Biophysical journal. 1988;54(5):921–929. doi: 10.1016/S0006-3495(88)83028-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nabuurs CI, Hilbers CW, Wieringa B, Heerschap A. Letter to the editor: “Interpretation of 31P NMR saturation transfer experiments: Do not forget the spin relaxation properties.” American Journal of Physiology - Cell Physiology. 2012;302(10):1566–1567. doi: 10.1152/ajpcell.00409.2011 [DOI] [PubMed] [Google Scholar]
- 39.Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Molecular and cellular biochemistry. 1998;180(1–2):171–177. [PubMed] [Google Scholar]
- 40.Nascimben L, Ingwall JS, Pauletto P, et al. Creatine kinase system in failing and nonfailing human myocardium. Circulation. 1996;94(8):1894–1901. doi: 10.1161/01.cir.94.8.1894 [DOI] [PubMed] [Google Scholar]
- 41.Beer M, Seyfarth T, Sandstede J, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with 31P-SLOOP magnetic resonance spectroscopy. Journal of the American College of Cardiology. 2002;40(7):1267–1274. doi: 10.1016/S0735-1097(02)02160-5 [DOI] [PubMed] [Google Scholar]
- 42.Lewis GA, Schelbert EB, Williams SG, et al. Biological Phenotypes of Heart Failure With Preserved Ejection Fraction. Journal of the American College of Cardiology. 2017;70(17):2186–2200. doi: 10.1016/j.jacc.2017.09.006 [DOI] [PubMed] [Google Scholar]
- 43.Little WC, Borlaug BA. Exercise intolerance in heart failure with preserved ejection fraction: what does the heart have to do with it? CircHeart Fail. 2015;8(1941–3297 (Electronic)):233–235. [DOI] [PubMed] [Google Scholar]
- 44.Kammermeier H. High energy phosphate of the myocardium: concentration versus free energy change. Basic Research in Cardiology. 1987;82:31–36. doi: 10.1007/978-3-662-11289-2 [DOI] [PubMed] [Google Scholar]
- 45.Tian R, Christe ME, Spindler M, et al. Role of MgADP in the development of diastolic dysfunction in the intact beating rat heart. Journal of Clinical Investigation. 1997;99(0021–9738 (Print)):745–751. doi: 10.1172/JCI119220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sequeira V, Bertero E, Maack C. Energetic drain driving hypertrophic cardiomyopathy. FEBS Letters. 2019;593:1616–1626. doi: 10.1002/1873-3468.13496 [DOI] [PubMed] [Google Scholar]
- 47.Pumphrey A, Yang Z, Ye S, et al. Advanced cardiac chemical exchange saturation transfer (cardioCEST) MRI for in vivo cell tracking and metabolic imaging Ashley. NMR Biomed. 2016;29(1):74–83. doi: 10.1002/nbm.3451.Advanced [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beadle RM, Williams LK, Kuehl M, et al. Improvement in Cardiac Energetics by Perhexiline in Heart Failure Due to Dilated Cardiomyopathy. JACC: Heart Failure. 2015;3(3):202–211. doi: 10.1016/j.jchf.2014.09.009 [DOI] [PubMed] [Google Scholar]
- 49.Murray AJ, Lygate CA, Cole MA, et al. Insulin resistance, abnormal energy metabolism and increased ischemic damage in the chronically infarcted rat heart. Cardiovascular Research. 2006;71(1):149–157. doi: 10.1016/j.cardiores.2006.02.031 [DOI] [PubMed] [Google Scholar]
- 50.Lionetti V, Linke A, Chandler M, et al. Carnitine palmitoyl transferase-I inhibition prevents ventricular remodeling and delays decompensation in pacing-induced heart failure. Cardiovascular Research. 2005;66(3):454–461. doi: 10.1016/j.cardiores.2005.02.004 [DOI] [PubMed] [Google Scholar]
- 51.Board M, Lopez C, van den Bos C, Callaghan R, Clarke K, Carr C. Acetoacetate is a more efficient energy-yielding substrate for human mesenchymal stem cells than glucose and generates fewer reactive oxygen species. International Journal of Biochemistry and Cell Biology. 2017;88(April):75–83. doi: 10.1016/j.biocel.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King LM, Sidell RJ, Wilding JR, Radda GK, Clarke K. Free fatty acids, but not ketone bodies, protect diabetic rat hearts during low-flow ischemia. American Journal of Physiology-Heart and Circulatory Physiology. 2017;280(3):H1173–H1181. doi: 10.1152/ajpheart.2001.280.3.h1173 [DOI] [PubMed] [Google Scholar]
- 53.Verma S, Rawat S, Ho KL, et al. Empagliflozin Increases Cardiac Energy Production in Diabetes. JACC: Basic to Translational Science. 2018;3(5). doi: 10.1016/j.jacbts.2018.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutrition and Metabolism. 2014;11(1):1–9. doi: 10.1186/1743-7075-11-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ríos-Covián D, Ruas-Madiedo P, Margolles A, Gueimonde M, De los Reyes-Gavilán CG, Salazar N. Intestinal short chain fatty acids and their link with diet and human health. Frontiers in Microbiology. 2016;7(FEB):1–9. doi: 10.3389/fmicb.2016.00185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colucci WS, Tian R. NHLBI Working Group Unlocking the Secrets of Mitochondria in the Cardiovascular System : Path to a Cure in Heart Failure - Executive Summary Discussion : In: Colucci WS, Tian R, eds. Bethesda, MD: NHLBI; 2018:1–3. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
There was no difference in the total creatine concentration measured by HPLC between any of the groups indicating that the changes in PCr that were observed were not due to changes in creatine concentration. n = 6–7 per group.