

Abstract
A significant therapeutic challenge for cancer patients is resistance to chemotherapies such as taxanes. Overexpression of LIN9, a transcriptional regulator of cell cycle progression, occurs in 65% of patients with triple negative breast cancer (TNBC), a disease commonly treated with these drugs. Here we report that LIN9 is further elevated with acquisition of taxane resistance. Inhibiting LIN9 genetically or by suppressing its expression with a global BET inhibitor restored taxane sensitivity by inducing mitotic progression errors and apoptosis. While sustained LIN9 is necessary to maintain taxane resistance, there are no inhibitors that directly repress its function. Hence, we sought to discover a druggable downstream transcriptional target of LIN9. Using a computational approach, we identified NIMA-related Kinase 2 (NEK2), a regulator of centrosome separation that is also elevated in taxane-resistant cells. High expression of NEK2 was predictive of low survival rates in patients who had residual disease following treatment with taxanes plus an anthracycline, suggesting a role for this kinase in modulating taxane sensitivity. Like LIN9, genetic or pharmacologic blockade of NEK2 activity in the presence of paclitaxel synergistically induced mitotic abnormalities in nearly 100% of cells and completely restored sensitivity to paclitaxel, in vitro. In addition, suppressing NEK2 activity with two distinct small molecules potentiated taxane response in multiple in vivo models of TNBC, including a patient-derived xenograft, without inducing toxicity. These data demonstrate that the LIN9/NEK2 pathway is a therapeutically targetable mediator of taxane resistance that can be leveraged to improve response to this core chemotherapy.
Keywords: Taxane, NEK2, LIN9, Mitosis, Triple Negative Breast Cancer
INTRODUCTION
Taxanes such as paclitaxel are a standard of care therapy for many malignancies. These drugs function as microtubule stabilizing agents and while they can be highly effective, resistance is common. Identifying therapeutically-targetable mechanisms underlying therapeutic resistance will be necessary to improve long-term survival. At clinically-relevant doses, paclitaxel induces multi-polar spindles and chromosomal missegregation, leading to chromosomal instability (CIN) and mitotic catastrophe1,2. These errors manifest as micro- and multi-nucleated cells, often with abnormally shaped nuclei1,3. Paclitaxel also directly inhibits centrosome separation during mitosis, preventing chromosome detachment from microtubule minus-ends2,4. This promotes movement of centrosomes away from nucleating chromosomes and disrupts the proper positioning of spindle poles during mitosis4,5. As a result of these defects, taxanes induce extensive mitotic abnormalities resulting in mitosis-associated cell death or mitotic catastrophe.
Mechanisms of taxane resistance are not fully understood. They include β-tubulin overexpression or mutations6, overexpression of P-glycoprotein and other transport or efflux proteins7, abnormalities in mitotic and spindle checkpoints8, modulation of pro- or anti-apoptotic proteins9, and overexpression of Aurora kinase A (AURKA)10. Thus far, none of these targets have produced effective therapeutic approaches in patients for preventing resistance or restoring sensitivity to taxanes. It is notable that combining atezolizumab, an anti-PD-L1 antibody that acts as a checkpoint inhibitor, with nab-paclitaxel has recently been shown to improve outcomes of patients with Triple Negative Breast Cancer (TNBC)11, but the basis for this improvement likely extends beyond the specific mechanisms underlying taxane resistance and may be attributable to generation of neoantigens. In the current study, we used TNBC as a model for identifying a therapeutically targetable mediator of taxane resistance.
TNBC is associated with poor patient survival. While the mean time to recurrence of all breast cancer subtypes is 5 years, the mean for TNBC is 2.6 years12. Taxanes are one of the most commonly used cytotoxic chemotherapies for TNBC patients in neoadjuvant, adjuvant, and metastatic disease settings, alone or in combination with other anti-neoplastics such as gemcitabine or anthracyclines13. While initially effective in this disease, taxanes are hampered by dose-limiting toxicities and rapid development of resistance. Of the few treatments available for TNBC patients who have developed taxane resistance, less than 25% of resistant tumors will respond, with the average response lasting less than 6 months14. Identifying therapeutically targetable mechanisms of taxane resistance should significantly expand the number of patients that can be effectively treated with these drugs.
As indicated above, taxanes primarily function by disrupting the cell cycle, particularly the mechanical events of mitosis. On a transcriptional level, cell cycle progression is controlled by the MuvB complex. This complex is comprised of five subunits (LIN9, LIN54, LIN52, LIN37, and RBBP4) that together bind and regulate expression of cell cycle genes15. We previously reported that overexpression of LIN9, which encodes the core scaffold of MuvB, is associated with worse outcomes in breast cancer and that LIN9 is overexpressed in ~65% of TNBC cases16. Moreover, suppressing LIN9 induces multinucleation and subsequent apoptosis or senescence of TNBC cells. Herein, we report the discovery of a novel, druggable mechanism underlying taxane resistance in TNBC that involves upregulation of LIN9 and its downstream transcriptional target, NEK2, a centrosomal kinase. Genetically suppressing LIN9 or NEK2 causes profound mitotic defects that synergize with taxanes to induce cell death. Most importantly, therapeutically targeting the LIN9/NEK2 pathway restores taxane sensitivity in resistant cells and xenografted tumors. These data provide a new mechanism-based, two-pronged approach to induce excessive mitotic progression errors in TNBC and ensure taxane response that may be useful for improving patient outcomes.
MATERIALS AND METHODS
Additional methodological details may be found in Supplemental Materials.
Cell culture and reagents
All cell lines were acquired from the American Type Culture Collection (ATCC) and cultured at 37°C with 5% CO2. Cells were authenticated using STR profiling (BDC Molecular Biology Core Facility, University of Colorado) or were purchased within six months from ATCC. MDA-MB-231, MDA-MB-468, HCC70, HCC38, and HCC1143 cell lines were maintained in RPMI-1640 with 10% FBS. Insulin (0.023 IU/mL) was added to this media for the BT-549 cell line. SUM159 cells were cultured in Ham’s F12 with 10% FBS, insulin (10mg/mL), and hydrocortisone (1mg/mL). SK-BR-3 cells were maintained in McCoy’s 5A medium with 10% FBS. MCF7 cells were cultured in DMEM with 10% FBS. All cell lines were tested monthly for Mycoplasma pulmonis and Mycoplasma spp. according to manufacturer protocol (Bimake, B39032). Cells never exceeded ten passages after thawing. Paclitaxel (Selleckchem, S1150), docetaxel (LC Laboratories, D-1000), JQ1 (Cayman Chemical, 1268524-70-4), CMP3a (MedKoo, 2225902-88-3), and INH1 were dissolved in DMSO. Transient mRNA silencing was conducted using 100nM non-targeting siRNA (Dharmacon, D-001810-02-20) or siRNA targeting LIN9 (L-018918-01), NEK2 (L-004090-00-0020), and LIN37 (L-013311-02-0005) with Lipofectamine-2000 (Invitrogen, 11668-027) in Opti-MEM media (Invitrogen, 31985088) for six hours after which they were maintained in complete media for 24 hours. For paclitaxel and docetaxel dose response curves, cells were treated with the indicated concentration of drug in addition to 250nM JQ1 for 4 days. Viable cells were counted by Trypan blue exclusion on a Countess II FL (Thermo Fisher, AMQAF1000).
RNA analysis
LIN9 (Hs00542748_m1), NEK2 (Hs05021038_g1), ABCB1 (Hs00184500_m1), ABCC2 (Hs00960489_m1), ABCC3 (Hs00978452_m1), ABCG2 (Hs01053790_m1), TUBB1 (Hs00917771_g1), TUBB3 (Hs00801390_s1), and GAPDH (Hs02758991_g1) TaqMan Gene Expression Assays (Thermo Fisher) were used.
Western blot analysis
Primary antibodies are LIN9 (Thermo Fisher, PA5-43640), NEK2 (Bioss, bs-5732R and BD Biosciences, 610594), BcL-XL (Cell Signaling, 2764), β-actin (Sigma, A5316), PARP (Cell Signaling, 9542), and α-actin (Sigma, A1978 clone AC-15).
Immunofluorescence
Cells were grown on coverslips and were fixed with 3.7% formaldehyde for 10 min and permeabilized with 0.1% Triton X-100. They were stained with Texas Red-X phalloidin (Invitrogen, T7471) in 1% BSA/PBS for 20 min. The slides were blocked for 1 hr in PBS containing 1% BSA, 10% normal goat serum, 0.3M Glycine and 0.1% Tween. γ-tubulin primary antibody (Abcam, ab205475) was added at a 1:500 dilution in blocking solution overnight. Vectashield mounting medium with DAPI (Vector Labs, H-1500) was used to counterstain the nuclei. Cells were imaged using an inverted Leica fluorescence microscope.
Gene-specific chromatin immunoprecipitation
ChIP-PCR was performed as previously reported in MDA-MB-231 cells17.
Flow cytometry
Cell cycle analysis was performed as previously reported18 with the following modifications: cells were fixed in 70% ethanol and analyzed using the Attune NxT Flow Cytometer (Thermo Fisher). Gating was performed during the analysis to remove doublets.
Colony formation assay
MDA-MB-231 cells were transfected with siNS, siLIN9, or siNEK2 (described above) and after 1 day, 1,000 live cells were seeded in 24 well plates. Each transfection was plated in duplicate. Cells were grown for 7 days before being fixed and stained (0.05% crystal violet, 1% formaldehyde, 1% methanol, and 10% 10x PBS) for 20 min at RT. To quantify staining, 10% acetic acid was added for 15 min. Sterile water was added 1:4. 100μL of this solution was read at 590 absorbance in duplicate.
Hoescht staining
Cells were treated for 4 days with either vehicle, 250 nM JQ1, 6 nM paclitaxel, or the combination. Cells were then stained with 10μM Hoescht (Thermo Scientific, 62249) for 10 min. at RT. Cells with and without pyknotic nuclei were quantified to calculate the percent of apoptotic cells.
Live cell imaging
MDA-MB-468 cells were transfected with siNS, siLIN9, or siNEK2 and were imaged using the Incucyte Zoom System (Essen BioScience) starting at 42 hr after transfection. Cells were imaged every 20 min. from 36-96 hr after the start of image collection at 20x magnification. Individual cells were tracked from the beginning to end of mitosis and assessed for the time needed to traverse through mitosis.
Immunohistochemistry
Tissue was fixed in 4% paraformaldehyde and paraffin-embedded. Antigen retrieval with citrate buffer pH 6.0 was performed before incubation with the Ki67 primary antibody (Abcam, ab66155) at 1:400 dilution. Slides were incubated in secondary antibody and DAB stained using the EnVision Detection Systems Peroxidase/DAB kit (Agilent, K406511-2), followed by hematoxylin counterstain (Vector, H-3401). Scoring was blinded for all slides.
Caspase Assay
Cells were plated on glass cover slips in 6 well plates and transfected with siNS or siLIN9 as described above. After 72 hr, 100μM Z-VAD-FMK (550377) or the negative control 100μM Z-FA-FMK (550411) was added. Vectashield mounting medium with DAPI (Vector Labs, H-1500) was used to counterstain nuclei. Cells were quantified using an inverted Leica microscope.
In Vivo Efficacy Assessment
All in vivo experiments were performed with approval from the Institutional Animal Care and Use Committee at Case Western Reserve University, which is certified by the American Association of Accreditation for Laboratory Animal Care. Mice were housed in microisolator units, given standard sterile chow and water ad libitum, and maintained on a 12-h light/dark cycle. MDA-MB-231 or MDA-MB-468 paclitaxel-sensitive cells, MDA-MB-231 or MDA-MB-468 paclitaxel-resistant cells, or PDX TNBC models (TM00091 and TM00098-from The Jackson Laboratory) were xenografted into the two inguinal mammary fat pads of adult female NOD/scid/γ (NSG) mice. At least 10 tumors were evaluated per xenograft/treatment group. Once measurable tumors formed (~15 mm3), mice were randomized into treatment groups of at least 4 mice per group. For the MDA-MB-231 sensitive model, these groups were: vehicle (1:1 propylene glycol:water), INH1 (100 mg/kg IP every other day), paclitaxel (15 mg/kg IP bi-weekly), or the combination, or vehicle (1:1 propylene glycol:water), JQ1 (50 mg/kg IP daily), paclitaxel (15 mg/kg IP bi-weekly), or the combination. For all other models, the treatment groups were: vehicle (1:1 propylene glycol:water), CMP3a (10 mg/kg, IP 5 days/week), paclitaxel (15 mg/kg IP bi-weekly), or the combination. Tumor size was measured twice per week using calipers. Mouse weight was measured once per week to assess toxicity. After various treatment times (MDA-MB-231 sensitive INH1 model-16 days; MDA-MB-231 sensitive JQ1 model-15 days; MDA-MB-231 resistant model-21 days, MDA-MB-468 sensitive model-17 days of treatment; MDA-MB-468 resistant model-28 days; PDX TM00091-27 days; PDX TM00098-29 days) that were limited by the extent of tumor growth occurring in vehicle-treated mice, tumors were removed and processed for analysis.
Statistical methods
Statistical analyses were performed using two-tailed Student’s t-test for all in vitro data and Mann-Whitney U test for all in vivo data. Significance was concluded if the p-value was less than 0.05. Unless noted otherwise, all in vitro data are represented as means with standard deviations of three independent experiments each completed in triplicate.
RESULTS
LIN9 regulates mitotic progression and dictates taxane sensitivity in models of acquired and intrinsic resistance.
We previously reported that LIN9 is overexpressed in the majority of TNBCs, and its suppression induces multinucleation16. Further examination of the expression pattern of LIN9 across breast cancer subtypes revealed that it is most highly expressed in the basal subtype with a median increase of 3.1-fold compared to the normal-like subtype in the TCGA dataset19 (Figure 1A). While suggesting that cancers with high LIN9 may depend on it for maintaining ploidy, the specific roles of this protein in sustaining the growth and aggressiveness of TNBC are unknown. We used live cell imaging to quantify both the duration of mitosis and cell fate with and without LIN9 silencing in TNBC cells. Reducing LIN9 expression led to a significant (p<0.05) increase in the duration of mitosis (Figure 1B and S1A). LIN9 silencing also resulted in an 8.6-fold increase in the number of cells that enter a prolonged interphase and, after successfully completing one round of mitosis, did not divide again (Figure 1C). These data indicate that sustained expression of LIN9 is necessary for normal mitotic progression of TNBC cells.
Figure 1. LIN9 regulates mitotic progression and dictates taxane sensitivity in models of acquired and intrinsic resistance.
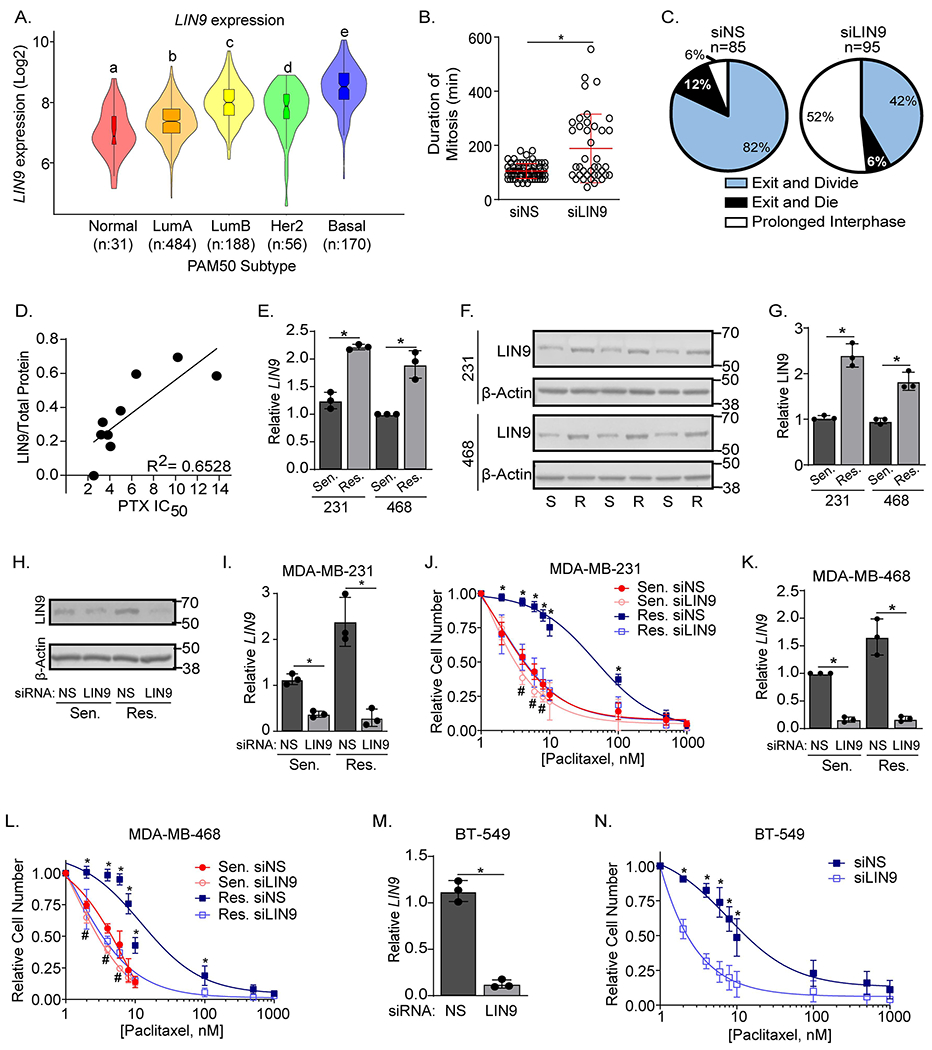
A. LIN9 expression (Log2) in the five PAM50 subtypes from the TCGA dataset (LumA=Luminal A, and LumB=Luminal B). Boxplot lines are quartiles with distinct letters indicating significantly different values from all groups with a different letter (p<0.05). B. MDA-MB-468 cells were transiently transfected with siRNA to LIN9 (siLIN9) or a non-silencing siRNA (siNS) and monitored using live cell imaging for 96 hr. Shown is the length of time for each cell to complete mitosis after LIN9 silencing (*p< 0.05) with the red line indicating the mean. C. Pie charts show the percent of MDA-MB-468 cells that undergo three different mitosis-associated cell fates: exit mitosis and divide (blue), exit mitosis and die (black), or prolonged interphase (white) following transfection with siNS or siLIN9. Panels B-C were completed in one technical replicate for which 85 siNS cells and 95 siLIN9 cells were tracked. D. Relationship between LIN9 protein expression and paclitaxel resistance where each dot represents a single breast cancer cell line in order of increasing paclitaxel (PTX) IC50 value: HCC38, SKBR3, HCC1143, MCF7, HCC70, MDA-MB-468, MDA-MB-231, BT-549, and SUM-159PT. Linear regression was utilized to generate a best fit line and R2 value. These data represent a single biological replicate for each cell line. E. RT-qPCR analysis of LIN9 expression in PTX sensitive/parental or resistant MDA-MB-231 and MDA-MB-468 cell lines, data are the means ± SD of 3 independent experiments performed in triplicate, *p<0.05 resistant compared to sensitive. F. Western blot comparing LIN9 protein levels in PTX sensitive/parental and resistant MDA-MB-231 and MDA-MB-468 cell lines. Each sensitive and resistant pair represents an independent experiment, normalized to total protein. G. Quantitation of panel F with LIN9 protein normalized to total protein. Data are means ± SD, *p<0.05 resistant compared to sensitive H. Western blot analysis confirming LIN9 silencing in MDA-MB-231 cells. I. RT-qPCR confirming LIN9 silencing in MDA-MB-231 sensitive/parental and resistant cells 4 days post-transfection with siNS or siLIN9. *p<0.05. J. After siRNA transfection, cells were treated for 4 days with increasing concentrations of paclitaxel, and live cells counted using trypan blue exclusion. K. RT-qPCR confirming LIN9 silencing in MDA-MB-468 sensitive and resistant cells after transfection with siNS or siLIN9. L. MDA-MB-468 cells were treated and counted as in panel J. M. RT-qPCR confirming LIN9 silencing after transfection in BT-549 cells. N. BT-549 cells were treated and counted as in panel J. I, K, and M data are mean ± SD, *=p<0.05 siLIN9 compared to control. J, L, and N data are mean ± SD, *=p<0.05 comparing resistant siNS to siLIN9, and #=p<0.05 comparing sensitive siNS to siLIN9. All data represent 3 independent experiments performed in triplicate.
The mitotic defects induced by LIN9 silencing are similar to those observed in paclitaxel-treated breast cancer cells1–3 and we postulated that LIN9 expression may contribute to paclitaxel response. Supporting this possibility, we found that LIN9 protein expression measured by western blot, is positively correlated (R2=0.6528) with the intrinsic IC50 for paclitaxel across 9 breast cancer cell lines (Figure 1D). We then determined if expression of LIN9 is elevated in two TNBC cell lines representing basal (MDA-MB-468) and mesenchymal (MDA-MB-231) subtypes with acquired paclitaxel resistance that were generated by continually increasing paclitaxel exposure over several months. The MDA-MB-231 and MDA-MB-468 sensitive/parental cell lines had IC50 values of 7nM and 6nM for paclitaxel respectively, while the IC50 of the resistant derivatives was increased to 60nM. Both LIN9 mRNA and protein were increased ~2-fold (Figure 1E–G) in resistant compared to sensitive/parental cells, indicating that resistance is associated with elevated LIN9 expression.
To assess the role of LIN9 in paclitaxel resistance, we used RNAi to determine if changing its expression can alter paclitaxel potency. Both sensitive/parental and resistant cells were transfected with either siLIN9 or a non-silencing control (siNS) and treated with increasing concentrations of paclitaxel. LIN9 silencing shifted the dose response curve of sensitive/parental cells to the left, reducing the IC50 by 50% (from 7 to 4nM) in MDA-MB-231 and 67% (from 6 to 2nM) in MDA-MB-468 cells (Figure 1H–L). More strikingly, silencing LIN9 in the resistant derivatives restored paclitaxel sensitivity to a level that was similar to parental/sensitive cells, with a 3-10 fold shift in the IC50 from 48.2 to 4.06nM in MDA-MB-231 cells and from 10.72 to 3.59nM in MDA-MB-468 cells. LIN9 silencing also increased sensitivity to paclitaxel in an intrinsically resistant TNBC cell line (BT-549, Figure 1M–N). To determine if the impact of LIN9 was generalizable to taxanes as a class, we treated MDA-MB-231 sensitive/parental and resistant cells with docetaxel and found that suppressing LIN9 expression similarly restores sensitivity to this drug (Figure S1B). The effect of LIN9 on taxane sensitivity was not a general response to modulating MuvB because genetically silencing LIN37, another component of the complex, did not alter paclitaxel response (Figure S1C–D). These data indicate that LIN9, but not all members of MuvB, controls paclitaxel sensitivity in TNBC cells. It also suggests that resistant cells may depend on LIN9 expression to minimize catastrophic mitoses induced by taxanes. To determine if elevated LIN9 is sufficient to induce taxane resistance, we used standard and inducible overexpression paradigms in MDA-MB-231 and MDA-MB-468 cell lines; however, all induced rapid cell death. Since LIN9 expression in resistant cells is just 2-fold greater than sensitive/parental cells, and ~3-fold higher in TNBC compared to normal-like tumors, these data suggest that a finely-tuned level of LIN9 is necessary to ensure TNBC cell viability.
Several mechanisms of taxane resistance have been previously identified6–10. We assessed whether these may promote resistance in the models used herein. However, we found that none of the four ABC transporters (ABCB1, ABCC2, ABCC3, or ABCG2) nor TUBB1 or TUBB3 were differentially expressed in resistant versus sensitive/parental cells (Figure S1E–H). Furthermore, none of the previously described mutations in TUBB1 were detected. These data indicate that the role of LIN9 in resistance is independent of these previously characterized factors.
LIN9 maintains chromosomal stability.
Sustained expression of LIN9 is necessary for mitotic progression and taxane resistance. Because taxanes induce tumor cell death by causing chromosomal missegregation20, we postulated that LIN9 may promote proliferation but suppress mitotic progression errors and chromosomal instability even in the absence of taxane exposure. Following transient transfection with siRNA to LIN9 or a non-silencing control, we quantified four characteristics of mitotic dysfunction and chromosomal instability: supernumerary (3+) centrosomes, micronuclei, and multiple and dysmorphic nuclei. LIN9 silencing in sensitive/parental MDA-MB-231 and MDA-MB-468 cells causes a significant increase in all four characteristics of chromosomal instability (Figures 2A–E and S2A–E). In some cases, we also observed fragmented nuclei following LIN9 silencing. We classified these as dysmorphic and not apoptotic because treatment with a caspase inhibitor, Z-VAD-FMK, in the presence of LIN9 silencing did not prevent the acquisition or number of fragmented nuclei (Figure S2F–G). Notably, we also found that taxane-resistant cells have a basal increase in mitotic errors compared to sensitive cells, even when cultured in the absence of paclitaxel and without LIN9 silencing (Figure 2, Res. siNS compared to Sens. siNS), yet these cells are viable. This suggested that resistant cells may acquire an ability to suppress apoptosis as a means to tolerate moderately increased genomic defects. Indeed, BCL-xL, an anti-apoptotic protein, was increased ~3-fold in resistant compared to sensitive cells (Figure S2H–J). BCL-xL was also modestly increased in sensitive cells when LIN9 was silenced, suggesting that BCL-xL upregulation is an intrinsic response to increased genomic instability.
Figure 2. LIN9 maintains chromosomal stability.
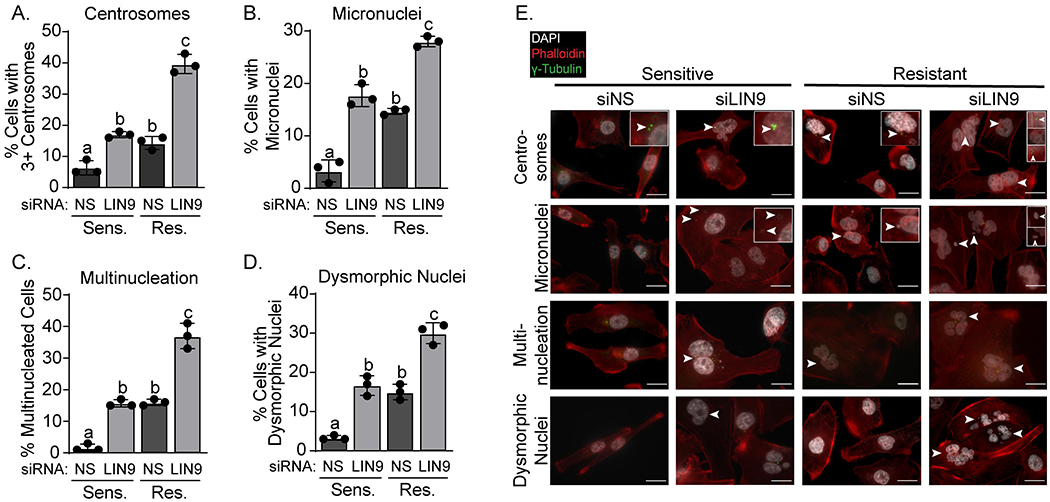
MDA-MB-231 cells were transiently transfected with siLIN9 or siNS. Three days post transfection, cells were stained with DAPI (white, nuclei), Texas Red-X phalloidin (red, actin cytoskeleton), and γ-tubulin (green, centrosomes). The percent of cells with A. 3+ centrosomes, B. micronuclei C. multiple nuclei, and D. dysmorphic nuclei were counted (minimum of 150 cells counted per experiment). Distinct letters above bars indicate significantly different values from all groups with a different letter (p<0.05). E. Representative images (60x) of panels A-D with insets showing centrosome abnormalities and the presence of micro-, multi-, or dysmorphic nuclei denoted by arrowheads. Scale bars are 20μm. All panels were repeated in 3 independent experiments in triplicate and represent means ± SD.
Lastly, we asked whether the increase in LIN9 expression that occurs in resistant cells may be an adaptive response that allows a low level of genomic defects but suppresses accumulation of rampant chromosomal instability. Indeed, we found that reducing LIN9 expression in taxane resistant cells further increased the extent of mitotic progression errors (Figure 2A–E). Together, these data indicate that LIN9 is necessary to sustain cancer cell fitness and suppress genomic defects, both in taxane sensitive and resistant cells.
Pharmacological inhibition of LIN9 expression potentiates paclitaxel-induced cell death.
The ability of LIN9 silencing to restore taxane sensitivity in resistant cells suggests that suppressing its activity may provide a means for ensuring taxane response in patients. However, due to its function as a scaffolding protein that controls transcription15, LIN9 will likely be difficult to therapeutically target. We previously reported that JQ1, a Bromodomain and ExtraTerminal protein inhibitor (BETi), repressed LIN916 expression and confirmed this finding in MDA-MB-231 cells (Figures 3A–B). To determine if BETi-mediated suppression of LIN9 levels can potentiate taxane response, sensitive/parental and resistant TNBC cells were treated with paclitaxel in the presence and absence of JQ1. Using the Chou-Talalay approach, we treated cells with escalating doses of combined JQ1 and paclitaxel. This revealed that JQ1 only minimally synergizes with paclitaxel to inhibit growth of taxane-sensitive cells (CI values of 0.880 and 0.955, respectively). In stark contrast, these two drugs were robustly synergistic in the taxane-resistant lines (CI values of 0.198 and 0.366, respectively) (Figure 3C). To determine if BETi would mimic the effects of LIN9 silencing on the paclitaxel dose-response relationship (Figures 1H–J and S1E), taxane sensitive and resistant TNBC cells were treated with a minimally effective concentration of JQ1 (250nM) and increasing doses of paclitaxel. JQ1 recapitulated the effects of LIN9 silencing by shifting the paclitaxel dose-response curve to the left for both sensitive/parental and resistant cells. However, adding JQ1 was much more impactful when examining paclitaxel resistant cells (Figures 3D and S3A). In addition, treating resistant cells with the combination of paclitaxel (6nM, IC50 of paclitaxel sensitive cells) and JQ1 (250 nM) resulted in a dramatic increase in multi- and micronucleated cells compared to either drug alone (Figures 3E–F and S3B). This result was similar to that observed with LIN9 silencing in the presence of paclitaxel (Figures 2B–C and S2B–C), and further supports the role of LIN9 in repressing excessive genomic instability and ensuring cell fitness and viability. Indeed, treating resistant cells with combined paclitaxel and JQ1 caused a substantial increase in the percent of SubG0 cells compared to either drug alone (Figures 3G and S3C). Hoescht staining further revealed that combining JQ1 with paclitaxel induced greater apopotic cell death than either drug alone (Figures 3H and S3D).
Figure 3. Pharmacological inhibition of LIN9 expression potentiates paclitaxel-induced cell death.
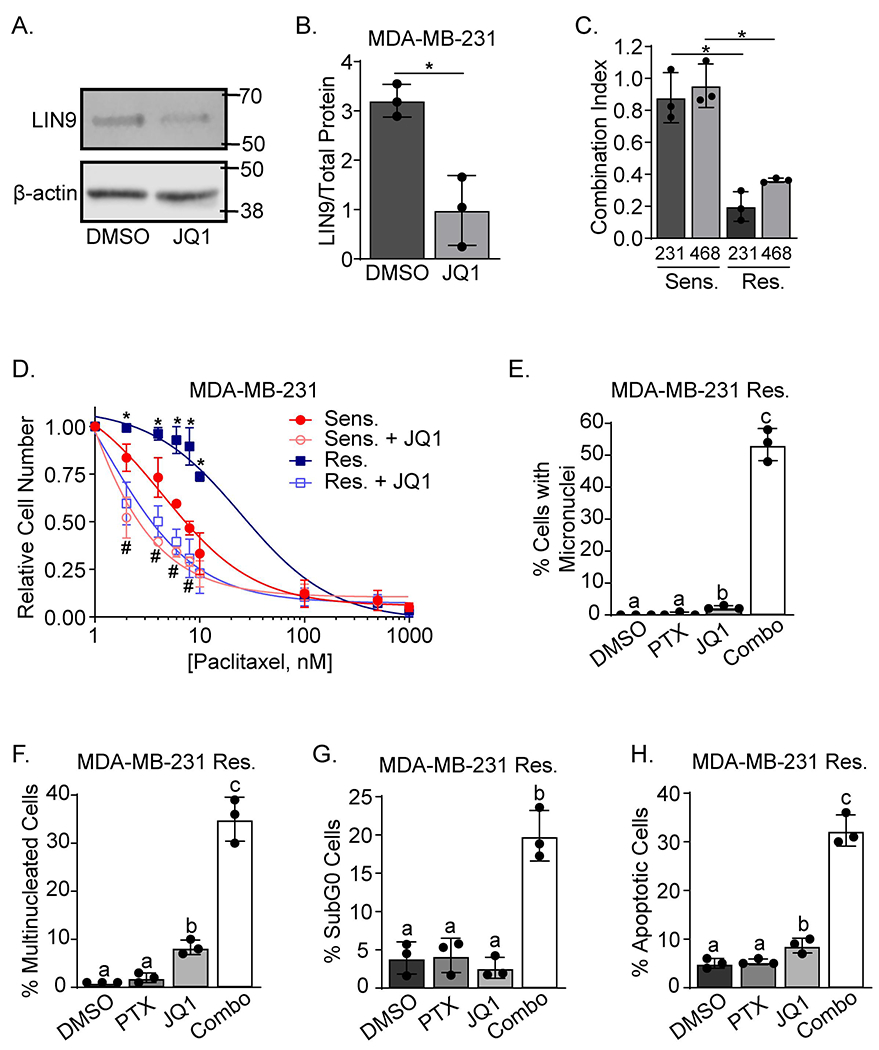
A. Western blot analysis of LIN9 or β-actin expression in MDA-MB-231 cells treated with 250nM JQ1 or vehicle for 24 hr. B. Quantitation of LIN9 protein relative to vehicle treated cells after normalization to total protein, *=p<0.05. C. The Chou-Talalay method was used to assess drug synergy between paclitaxel (6nM) and JQ1 (250nM) after 4 days of treatment in both MDA-MB-231 and MDA-MB-468 PTX sensitive/parental and resistant cell lines. *=p<0.05 comparing the PTX sensitive/parental to PTX resistant cells in each cell line. D. MDA-MB-231 sensitive/parental and resistant cells were treated with 250nM JQ1 and increasing concentrations of paclitaxel for 4 days. Live cells were counted using trypan blue exclusion. *=p<0.05 comparing paclitaxel alone to combination treatment in PTX resistant cells, and #=p<0.05 comparing paclitaxel alone to combination treatment in MDA-MB-231 sensitive/parental cells. E and F. MDA-MB-231 resistant cells were treated with 6nM Paclitaxel and/or 250nM JQ1 for 4 days and stained with DAPI and Texas Red-X phalloidin. The percent of cells with E. micronuclei and F. multinucleated cells were counted. Distinct letters above bars indicate significant differences between groups with different letters (p<0.05). A minimum of 150 cells were counted per experiment. G. MDA-MB-231 resistant cells were treated for 4 days with 6nM paclitaxel and/or 250nM JQ1, stained with propidium iodide, and analyzed using flow cytometry, *p<0.05 combination compared to individual drugs. H. MDA-MB-231 resistant cells were treated for 4 days, stained with Hoescht, and the number of pyknotic relative to normal nuclei were counted. Distinct letters above bars indicate significant differences (p<0.05). All data are means ± SD and were repeated in 3 independent experiments in triplicate.
Having observed the profound ability of BETi to restore paclitaxel sensitivity in resistant TNBC cells, we attempted to assess the efficacy of combining JQ1 with paclitaxel in vivo. However, the drug combination caused substantial mouse weight loss with multiple dosing strategies, indicating toxicity (Figure S3E). This is likely due to adding a global transcriptional inhibitor (BETi/JQ1) with a significant toxicity profile of its own, to a cytotoxic drug (paclitaxel). Thus, we turned to identifying a downstream transcriptional target and mediator of LIN9, with the goal of discovering a more precise and less toxic approach for modulating its impact on paclitaxel response.
NEK2 is a transcriptional target of LIN9 that is associated with taxane resistance.
To identify direct transcriptional targets of LIN9 that may modulate breast cancer progression and be druggable, we used an in silico approach with the following criteria: 1) candidate target mRNAs must correlate with LIN9 mRNA in human breast cancer [Positive Pearson correlation greater than 0.5 with LIN9 in TCGA and METABRIC19,21,22], 2) candidate genes must contain a LIN9 binding site [using the only publicly-available ChIP-seq dataset for LIN923], and 3) high expression of candidate genes must associate with reduced breast cancer survival. This approach yielded nine candidates (NEK2, DTL, CENPF, CKAP2L, RBBP5, NUSAP1, EXO1, DKC1, and ASPM). Because LIN9 silencing causes abnormalities in centrosome composition (Figures 2A and S2A) similar to those observed with taxane treatment2,4, we further refined this list to four candidates that are established regulators of centrosome function (NEK2, DTL, CENPF, and CKAP2L). Of these, only NEK2 (NIMA-related Kinase 2) encodes an enzyme for which inhibitors currently exist, making it readily druggable. NEK2 is a serine-threonine centrosomal kinase that controls mitotic progression by ensuring proper centrosome separation and bipolar spindle formation24,25. Dysregulated NEK2 expression leads to a variety of mitotic failures including abnormal spindle formation, supernumerary centrosomes, micro- and multi-nucleation, aneuploidy, and mitotic catastrophe26,27. Several of these phenotypes are similar to those occurring with LIN9 silencing, suggesting that NEK2 may be a major mediator of the effects of LIN9 on mitosis.
Like LIN9, NEK2 is more highly expressed in the basal subtype of breast cancer compared to normal-like tumors [TCGA data28,29] (Figure 4A). Meta-analyses of a collection of breast cancer gene expression datasets revealed that higher expression of NEK2 correlates with a lower probability of disease-specific and relapse-free survival across the diverse cohort of breast cancer patients (Figure 4B and S4A). Expression of LIN9 and NEK2 are also highly correlated in breast cancers (Figures 4C and S4B), suggesting that LIN9 may regulate NEK2 gene expression. Indeed, transiently silencing LIN9 decreased NEK2 mRNA and protein in parental MDA-MB-231 and MDA-MB-468 cells (Figure 4D–F). Analysis of a LIN9 ChIP-seq dataset indicated that LIN9 binds to the NEK2 gene in HeLa cells (Figure S4C) and chromatin immunoprecipitation/PCR confirmed LIN9 binding to the NEK2 gene in MDA-MB-231 cells similar to its interaction with a positive control, AURKA (Figure 4G). Lastly, we treated cells with JQ1 to reduce LIN9 levels, and this also decreased NEK2 protein expression (Figures 4H–I). To determine if NEK2 is downregulated by JQ1 in a LIN9 dependent manner, we silenced LIN9 and treated with either DMSO or JQ1. We found that JQ1 treatment does not decrease NEK2 to a greater extent than LIN9 silencing alone, indicating it does not independently regulate NEK2 (Figure S4D). These data indicate that NEK2 is a transcriptional target of LIN9 that is associated with worse breast cancer patient outcomes.
Figure 4. NEK2 is a transcriptional target of LIN9 that is associated with taxane resistance.
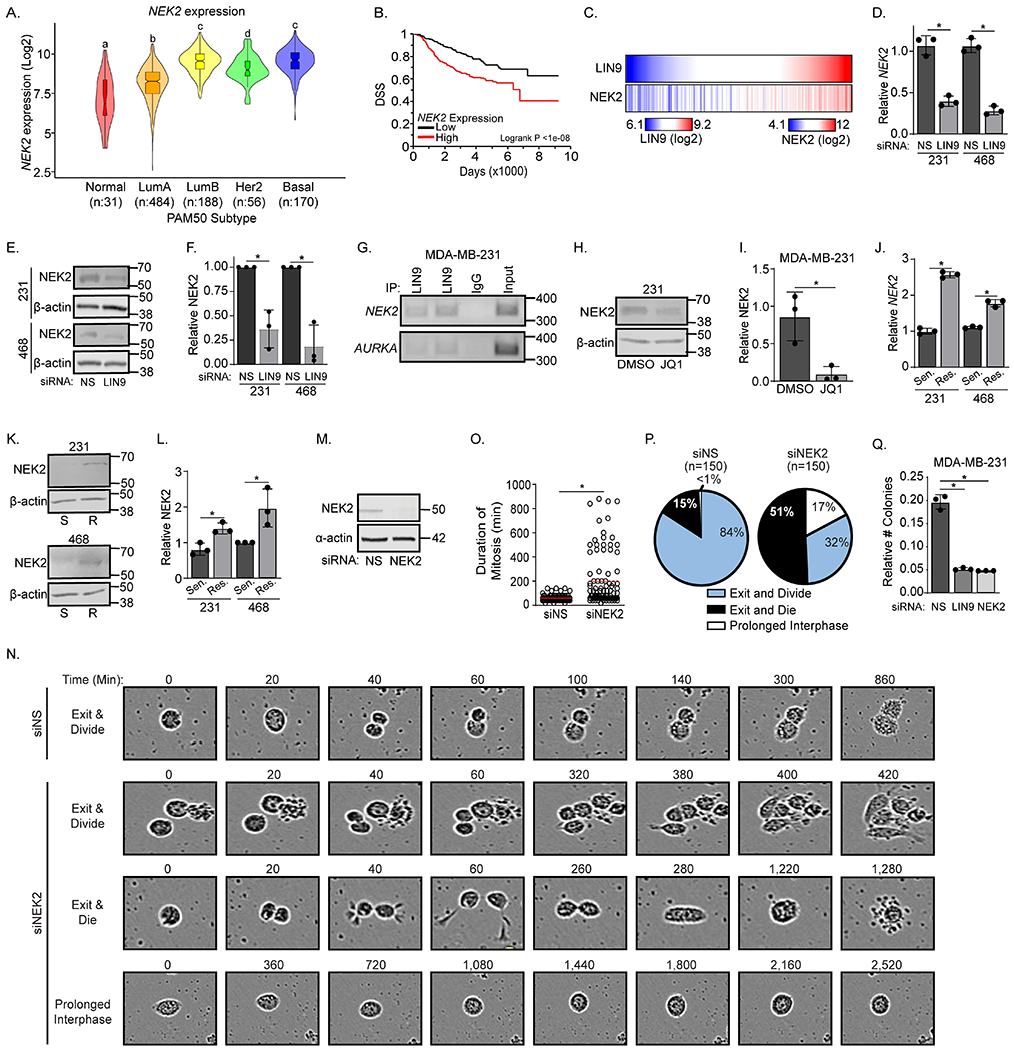
A. NEK2 expression (Log2) in the five PAM50 subtypes after interrogation of the TCGA dataset (LumA=Luminal A, and LumB=Luminal B). Boxplot lines indicate quartiles and distinct letters above bars indicate significantly different values from all groups (p<0.05). B. Disease specific survival curve for breast cancer patients with high or low NEK2 expression using the METABRIC dataset21. C. Heat maps of gene expression indicating a correlation of NEK2 and LIN9 from the TCGA dataset where each vertical line represents one breast cancer patient50. D. RT-qPCR analysis of NEK2 in MDA-MB-231 and MDA-MB-468 cells 3 days after siLIN9 or siNS transfection. E. Western blot of NEK2 expression in MDA-MB-231 and MDA-MB-468 cells 3 days after siLIN9 or siNS transfection. F. Quantification of the western blot in panel E relative to total protein G. Representative gene-specific ChIP-PCR gel of MDA-MB-231 cells assessing the binding of LIN9 to the NEK2 gene and the positive control gene, AURKA. H. Western blot of NEK2 expression in MDA-MB-231 cells after 24-hr of DMSO or 250nM JQ1, normalized to total protein. I. Quantitation of the western blot in panel H relative to total protein. J. RT-qPCR analysis of NEK2 expression in PTX sensitive/parental and resistant MDA-MB-231 and MDA-MB-468 cell lines. K. Western blot of NEK2 expression in PTX sensitive (S) or PTX resistant (R) MDA-MB-231 and MDA-MB-468 cell lines, normalized to total protein. L. Quantitation of NEK2 western blot in panel K normalized to total protein. M. Western blot indicating NEK2 silencing in MDA-MB-468 cells. N. Representative live cell images used for quantitation of cell fates of MDA-MB-468 cells transfected with siNS or siNEK2. Numbers indicate minutes from the first image displayed. O. Quantitation of the length of time for individual MDA-MB-468 cells to complete mitosis when NEK2 is silenced. The red line indicates the mean. P. Pie charts demonstrating the percent of MDA-MB-468 cells transfected with siNS or siNEK2 that underwent different cell fates: exit and divide (blue), exit and die (black), and prolonged interphase (white). Q. MDA-MB-231 sensitive/parental cells were transfected with siNS, siLIN9, or siNEK2 and examined for their ability to form colonies. Colonies were stained with crystal violet, and the absorbance of each well was measured, *p<0.05 siLIN9 and siNEK2 compared to control. For D-L and Q, data are means ± SD, *=p<0.05 and were repeated in 3 independent experiments in triplicate.
Since it is a downstream target of LIN9, it was not surprising that NEK2 mRNA and protein levels were also elevated with the acquisition of taxane resistance in both MDA-MB-231 and MDA-MB-468 TNBC cell lines (Figures 4J–L). Live cell imaging also revealed that similar to LIN9, reducing NEK2 expression increased the duration of mitosis (Figure 4M–O) as well as the percentage of cells that entered prolonged interphase or that died following mitotic exit (Figure 4P). NEK2 silencing did cause a higher rate of death following mitotic exit compared to LIN9 silencing, while LIN9 silencing resulted in more cells that entered prolonged interphase. This difference is likely due to the ability of LIN9 to regulate the expression of many target genes other than NEK2, some of which may suppress cell death such as BCL-xL (Figure S2J). Together, these data indicate that mitotic fidelity is dependent upon the LIN9-NEK2 transcriptional pathway and that its upregulation may convey a level of genomic instability that promotes the tumorigenic potential of cells and taxane resistance while also ensuring cell fitness and viability.
To determine if transient loss of LIN9 or NEK2 has a sustained impact on cell viability and growth, we transfected sensitive/parental cells with siLIN9, siNEK2, or a non-silencing control and assessed colony formation after 8 days. Reducing either LIN9 or NEK2 caused a profound decrease in colonies (Figure 4Q and S4E–F), underscoring the ability of these two proteins to phenocopy one another. In addition, these data indicate that a transient loss of LIN9 or NEK2 activity can have a sustained impact on tumor cell growth, further supporting their potential utility as therapeutic targets.
NEK2 silencing phenocopies the loss of LIN9, inducing mitotic progression errors and restoring paclitaxel sensitivity.
To determine if loss of NEK2 generates similar mitotic dysfunction and chromosomal instability phenotypes as LIN9 silencing in the absence of taxane exposure, paclitaxel sensitive/parental and resistant MDA-MB-231 cells were transiently transfected with siRNA targeting NEK2 or a non-silencing control. Reducing NEK2 (Figure 5A) caused an increase in supernumerary centrosomes as well as micro-, multi-, and dysmorphic nuclei in sensitive and resistant cells, with the effects being more pronounced in resistant cells (Figures 5B–E). Notably, the extents of these changes were identical to those observed with LIN9 silencing (data included from Figures 2A–D to facilitate direct comparison), revealing that NEK2 silencing phenocopies the mitotic defects observed with the loss of LIN9. Comparable results were observed in MDA-MB-468 cells (Figure S5A–E). Notably, these defects occurred in the absence of taxane exposure, again indicating that the reprogramming that occurs during the acquisition of resistance makes cells particularly sensitive to disrupting the LIN9-NEK2 cascade.
Figure 5. NEK2 silencing phenocopies the loss of LIN9, inducing mitotic progression errors and restoring paclitaxel sensitivity.

MDA-MB-231 sensitive/parental and resistant cells were transfected with siNS, siLIN9, or siNEK2 for 3 days before staining with DAPI, Texas Red-X phalloidin, and γ-tubulin. A. RT-qPCR analysis to confirm NEK2 silencing, *=p<0.05 siNEK2 compared to control. The percent of cells with B. 3+ centrosomes, C. micronuclei, D. multinucleation, and E. dysmorphic nuclei were counted (minimum 150 cells per experiment). Graphs B-E are derived from the same experiment as Figure S2 A–D, but with siNEK2 data added. Distinct letters above bars indicate significant differences between groups(p<0.05). F and G. Kaplan-Meier curves of taxane and anthracycline-treated TNBC patients stratified by high or low NEK2 expression (quartiles) who experienced F. a pathological complete response (pCR) or G. residual disease (RD). H. MDA-MB-231 sensitive/parental and resistant cells were transfected with siNS or siNEK2 and then treated with increasing concentrations of paclitaxel for 4 days. Live cells were counted using trypan blue exclusion, *p<0.05 comparing MDA-MB-231 resistant siNS to siNEK2, and #p<0.05 comparing MDA-MB-231 sensitive/parental siNS to siNEK2. Panels A-E and H were repeated in 3 independent experiments in triplicate and are represented as means ± SD.
To evaluate the potential clinical significance of NEK2 in taxane response, we assessed NEK2 levels in a gene expression dataset of breast cancers prospectively collected prior to neoadjuvant treatment with a taxane/anthracycline regimen30. For TNBC tumors that underwent a pathological complete response, NEK2 levels were unable to distinguish long-term clinical outcomes (Figure 5F). In contrast, for TNBC patients that had residual disease following neoadjuvant treatment, basal elevation of NEK2 expression was highly prognostic for shorter relapse-free survival (Figure 5G). Similar results were observed when examining the broader group of breast cancer patients regardless of subtype (Figure S5F–G). These data suggest that NEK2 may be a predictive biomarker for taxane/anthracycline response. They also support the possibility that NEK2 may be a primary effector of LIN9 that controls taxane efficacy in breast cancer.
To determine if NEK2 could also phenocopy the impact of LIN9 on taxane sensitivity, we examined the effects of NEK2 silencing on the paclitaxel dose-response relationship in MDA-MB-231 and MDA-MB-468 cells. As with LIN9 suppression, NEK2 silencing modestly shifts the dose-response curve of sensitive/parental cells to the left (Figures 5H and S5H). In contrast, when examining resistant cells, NEK2 silencing has a substantial impact on the IC50, fully restoring taxane responsiveness of resistant cells to an indistinguishable level from the sensitive/parental cells. Together these data support the hypothesis that NEK2 is a transcriptional target of LIN9 that potentiates its effects on mitotic progression and taxane sensitivity.
LIN9 or NEK2 loss potentiates taxane-induced mitotic defects and chromosomal instability.
LIN9 or NEK2 suppression causes profound mitotic progression errors in TNBC cells that are either sensitive or resistant to taxanes (Figures 2 and 4). To determine if these defects would be even more severe in the presence of paclitaxel, sensitive/parental and resistant cells were transfected with siRNAs to LIN9 or NEK2 and then treated with 6nM paclitaxel. As expected, LIN9 or NEK2 silencing in sensitive/parental cells caused significant increases in supernumerary centrosomes as well as micro-, multi- and dysmorphic nuclei (Figure 6A–D). These effects were profoundly increased in paclitaxel-resistant cells, with the vast majority of cells (>90%) undergoing mitotic defects following LIN9 or NEK2 silencing in combination with paclitaxel (Figure 6E–H). These data indicate that the acquisition of taxane resistance causes cells to become wholly dependent on LIN9 and NEK2 to sustain chromosomal stability in the presence of paclitaxel. These data also suggest that pharmacologically targeting these proteins should have a robust ability to ensure taxane responsiveness.
Figure 6. LIN9 or NEK2 loss potentiates taxane-induced mitotic defects and chromosomal instability.
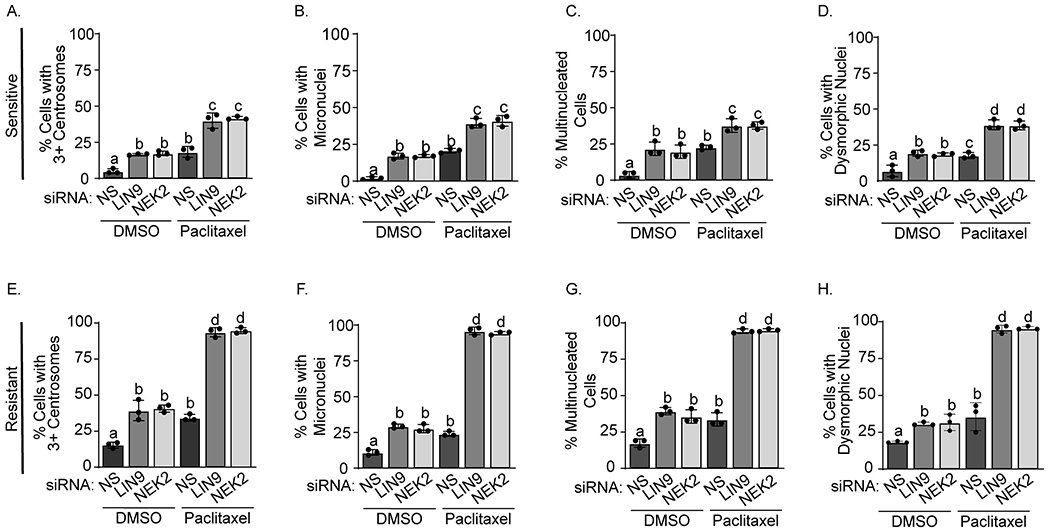
MDA-MB-231 sensitive/parental (panels A-D) and resistant (panels E-H) cells were transfected with siNS, siLIN9, or siNEK2, treated for 4 days with DMSO or 6nM paclitaxel, and then stained with DAPI (nuclei), Texas Red-X phalloidin (actin cytoskeleton), and γ-tubulin (centrosomes). The percent of MDA-MB-231 sensitive/parental and resistant cells with (A and E) 3+ centrosomes, (B and F) micronuclei, (C and G) multinucleation, and (D and H) dysmorphic nuclei were counted. Distinct letters above bars indicate significant differences between groups (p<0.05). A minimum of 150 cells were counted per experiment. All data are represented as means ± SD and were repeated in 3 independent experiments in triplicate.
Pharmacological inhibition of NEK2 potentiates taxane sensitivity and suppression of proliferation
Targeting LIN9 remains challenging. The only inhibitors currently available are global regulators of transcription (BETi) that inhibit the expression of many genes beyond LIN9. Moreover, combining the BETi, JQ1, with paclitaxel displayed significant toxicity in vivo. To more precisely target the mitotic impact of LIN9, we used selective inhibitors of NEK2 to determine if these may provide a viable approach for improving taxane responsiveness. INH1 causes proteasome-mediated decline of NEK2 protein by disrupting its interaction with HEC1/NDC8031. Treatment with 10μM INH1 fully resensitized resistant cells to paclitaxel while having no impact on its own (Figure 7A and S6A). We confirmed the ability of pharmacologically inhibiting NEK2 to restore taxane responsiveness using a second inhibitor with a distinct mechanism of action. CMP3a is an inhibitor of NEK2 catalytic activity that represses glioma growth in mice32 but whose ability to potentiate taxane response has not been assessed. Treating paclitaxel sensitive or resistant MDA-MD-231 cells with a low dose (3nM) of CMP3a alone had no impact on growth (Figure S6B). In contrast, this dose of CMP3a acted similarly as INH1 to modestly sensitize parental cells and profoundly improve the response of resistant cells to paclitaxel (Figure 7B). These effects were nearly identical to that observed with NEK2 silencing (Figure 5K). Notably, NEK2 inhibition was more efficacious at potentiating paclitaxel response than JQ1. This is likely to do differences in the specificity of the targets. While JQ1 regulates the transcription of many genes, including induction of p21, a suppressor of cell cycle entry and inhibitor of taxane efficacy, NEK2 inhibitors are much more selective for processes associated with centrosomes and microtubules.
Figure 7. Pharmacological inhibition of NEK2 potentiates taxane sensitivity, mitotic progression errors, and suppression of proliferation.
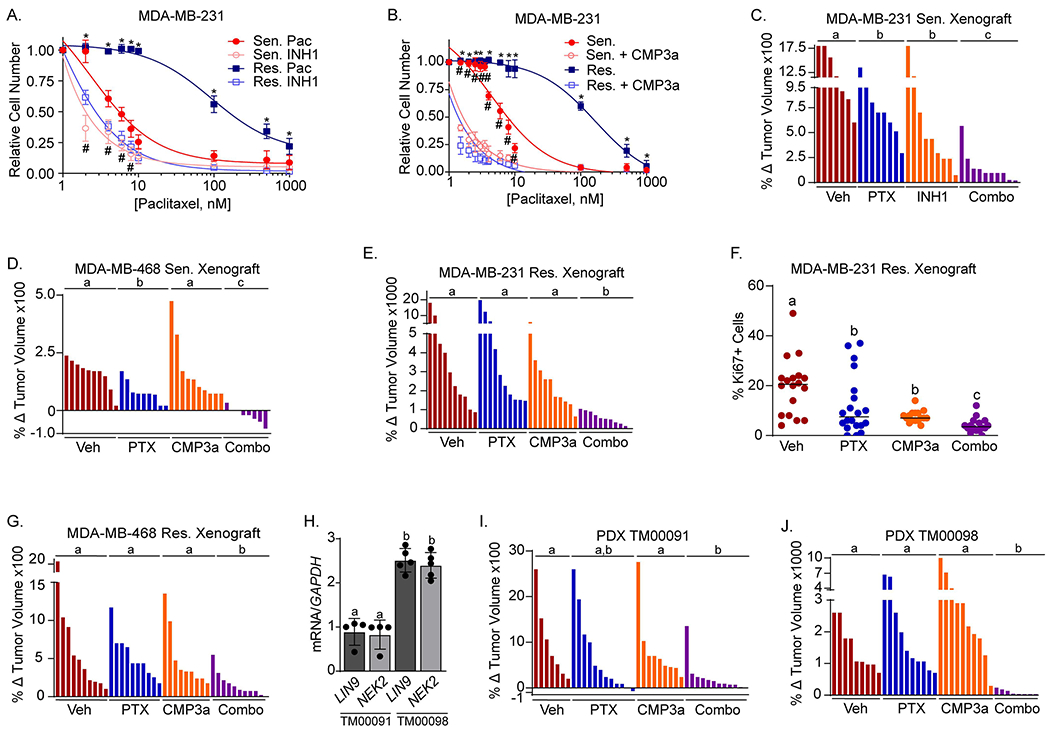
MDA-MB-231 sensitive/parental and resistant cells were treated with increasing concentrations of paclitaxel and/or A. 10uM INH1 or B. 3nM CMP3a. Live cells were counted after 4 days using trypan blue exclusion, *p<0.05 for resistant cells, comparing CMP3a or INH1 treated cells to vehicle treated cells, and #p<0.05 for sensitive/parental cells comparing CMP3a or INH1 treated cells to vehicle treated cells. All data are mean ± SD and were repeated in 3 independent experiments in triplicate. C. MDA-MB-231 sensitive/parental cells were orthotopically xenografted into female NSG mice and tumor-bearing mice were treated with vehicle, paclitaxel (15mg/kg bi-weekly), INH1 (100mg/kg IP every other day), or the combination for 16 days at which point tumors were measured and changes in tumor volume calculated. b=p<0.05, c=p<0.0001. D. Same as C, except with MDA-MB-468 sensitive cells and treatments with vehicle, paclitaxel (15mg/kg bi-weekly), CMP3a (10mg/kg 5 days/week), or the combination for 17 days. b=p<0.0001. E. Same as D except xenografts of MDA-MB-231 resistant cells were treated for 21 days. b=p<0.0001. F. Quantitation of immunohistochemical staining with Ki67 in the MDA-MB-231 resistant tumors from panel E. G. Same as D except MDA-MB-468 resistant cells were treated for 28 days. b=p<0.0001. H. Tumors from each PDX model were harvested and RNA was collected to measure LIN9 and NEK2 expression using RT-PCR. Distinct letters above bars indicate significant differences between groups (p<0.05). I. PDX, TM00091, was orthotopically xenografted into female NSG mice and tumor-bearing mice were treated with vehicle, paclitaxel (15mg/kg qw), CMP3a (10mg/kg 5 days/week), or the combination for 27 days, at which point tumors were measured and changes in tumor volume calculated. J. Same as I, except PDX TM00098 was treated for 29 days.
Pharmacological inhibition of NEK2 also potentiated taxane response in several mouse models of TNBC, in vivo. Mice with orthotopic xenografts of sensitive/parental MDA-MB-231 cells were treated with vehicle, paclitaxel, INH1, or the combination of both drugs for 16 days. While each drug alone had modest effects on final tumor size, the combination substantially inhibited growth (Figure 7C and S6C) without causing significant weight changes, suggesting minimal toxicity (Figure S6D). Likewise, combining CMP3a with paclitaxel caused tumor regression of orthotopic xenografts of sensitive/parental MDA-MB-468 cells while either drug alone had minimal effects on final tumor size (Figure 7D and S6E). None of the mouse studies combining CMP3a with paclitaxel induced significant weight loss, suggesting this combination is also non-toxic (Figure S6F–I). We then assessed the efficacy of combining paclitaxel and a NEK2 inhibitor in orthotopic xenografts of taxane-resistant MDA-MB-231 cells. CMP3a was used because it appeared that it may be more efficacious than INH1 when combined with paclitaxel in the sensitive/parental xenograft models (Figure 7B vs. 7D). In the resistant xenografts, neither paclitaxel nor CMP3a significantly inhibited tumor growth compared to vehicle. However, combining paclitaxel with CMP3a significantly reduced tumor growth (Figure 7E and S6J). Histologically, no differences were observed in the morphology of viable residual tumor or the percentage of tumor cell necrosis across treatment groups. However, proliferation as measured by Ki67 staining was significantly repressed in tumors treated with combined CMP3a and paclitaxel compared to vehicle or either drug alone (Figure 7F). The combination of paclitaxel and CMP3a was also significantly more efficacious in reducing tumor volume in resistant MDA-MB-468 xenografts than either drug alone (Figure 7G and S6K). To provide a more clinically relevant assessment of the CMP3a/paclitaxel combination, we evaluated its efficacy in two PDX models. The TM00098 model has 2.5 fold higher LIN9 and NEK2 expression than TM00091 (Figure 7H) providing an approach to determine if higher LIN9/NEK2 may distinguish response to combining a NEK2 inhibitor with a taxane. We found that the model with lower LIN9 and NEK2 (TM00091) responded well to the combination (Figure 7I and S6L), but that response was not significantly greater than paclitaxel alone, likely due to variability in tumor response. In contrast, the model with elevated LIN9 and NEK2 (TM00098) was highly responsive to the drug combination versus either drug alone, with near complete suppression of tumor growth in the combination group (Figure 7J and S6M). As another indicator of toxicity, we measured spleen weight and found no differences between groups (Figure S6N). Lastly, we also assessed the ability of the combination to induce apoptosis by measuring PARP cleavage. As expected, combining the NEK2 inhibitor with a taxane greatly increased PARP cleavage compared to either drug alone in the more responsive (high LIN9/NEK2 expressing, TM00098) model but was unable to induce such cleavage in the model (TM00091) with low LIN9/NEK2 (Figure S6O–Q). Taken together, these data support the postulate that elevated LIN9/NEK2 provides a targetable vulnerability that can be exploited in the presence of taxanes and support the future assessment of NEK2 inhibitors for their potential to improve taxane response in patients with TNBC.
DISCUSSION
Taxanes remain one of the most commonly used cytotoxic therapies for breast cancer. However, the high rate of resistance to these drugs necessitates that mechanism-based, multi-faceted combination treatments be developed to ensure therapeutic response. Though combinations with anthracyclines such as doxorubicin, or nitrogen mustards such as cyclophosphamide have been used, resistance is still a critical issue33,34. Here, we report that loss of LIN9 expression thwarts proper mitosis and increases taxane sensitivity in TNBC models. Taxane resistant cells require LIN9 to maintain a functional level of mitotic errors, while also preventing excessive genomic instability that would lead to death upon taxane treatment. We also identified NEK2 as a key transcriptional target of LIN9 that phenocopies the effects of LIN9 on mitotic progression, chromosomal stability, and taxane responsiveness that is more readily druggable than LIN9. Elevated expression of NEK2 is also associated with recurrence in patients that have residual disease following neoadjuvant treatment with combined taxane and anthracycline therapy. These data suggest that NEK2 may be a predictive biomarker of patient outcomes post-neoadjuvant taxane+anthracycline treatment. More importantly, they also provide support for combining NEK2 inhibitors with taxane therapy in the adjuvant setting. Indeed, we found that two different NEK2 inhibitors can potentiate the efficacy of paclitaxel in xenograft mouse models of TNBC.
LIN9 has been reported to be a tumor suppressor in colon carcinoma cell lines and in mouse and human fibroblasts35–37. In contrast, we have identified pro-tumorigenic properties of LIN9 in breast cancer. These include the amplification and overexpression of the LIN9 gene in the majority of TNBCs and the association of high LIN9 levels with worse breast cancer outcomes that we previously described16. In addition, we now report that sustained LIN9 expression is necessary for normal mitotic progression and that reducing LIN9 sensitizes breast cancer cells to taxanes. The ability of LIN9 to have both tumor suppressive and promoting properties is likely due to its role in regulating mitosis, which is exquisitely dependent upon the precise temporal and quantitative expression of specific proteins. Indeed, other proteins that control mitosis and chromosomal stability have also been reported to be tumor suppressive as well as oncogenic. These include Aurora kinase A and Polo-like Kinase38–40, with therapeutic inhibitors to each being clinically evaluated.
There are currently no therapies that directly target LIN9 with or without chemotherapy. However, the AURKA inhibitor, Alisertib, is efficacious in breast cancer patients following cytotoxic therapy41. AURKA is required for mitotic spindle formation, chromosome alignment, centrosome separation, and cytokinesis42. Loss of AURKA activity leads to many mitotic defects that we report also occur with LIN9 suppression. Notably, AURKA is a downstream target of LIN916,43, hence it is likely that inhibition of LIN9 could elicit similar effects in patients as AURKA inhibitors. We previously reported that BETi suppress the expression both AURKA and LIN9 in TNBC, leading to mitotic catastrophe16,44. Thus, a rational approach to inhibiting LIN9/AURKA in TNBC could be the use of BETi. While our studies demonstrated that the BETi, JQ1, does potentiate paclitaxel efficacy in vitro, this led to significant toxicity in vivo. Hence, use of such a broad-scale inhibitor that suppresses the expression of hundreds to thousands of genes_ENREF_56 is unlikely to improve patient outcomes from taxane therapy. More selective targeting of LIN9-regulated genes/proteins may be more appropriate, and an ongoing Phase II clinical trial ( NCT02187991) is currently investigating the potential efficacy of combining Alisertib with paclitaxel in breast cancer patients. We report here that selective inhibition of NEK2, another downstream target of LIN9, did not cause obvious toxicity when combined with paclitaxel but did improve paclitaxel response in vitro and in vivo. This is likely due to the more specific impact of NEK2 inhibition on mitosis. Supporting the potential for selectively targeting NEK2 as a means to improve taxane response in breast cancer patients, NEK2 gene expression correlates with worse patient outcomes overall, is upregulated with the acquisition of taxane resistance, and is associated with disease recurrence following taxane therapy. Notably, combining NEK2 inhibitors appeared to be well-tolerated wherein no changes in mouse body mass or spleen weight were observed. However, it is possible that other systemic effects of this combination could occur that either contribute to suppressing tumor growth or cause undetected toxicity. For example, we have not determined if the combination may have an effect on tumor-associated stroma or the immune system and preclinical studies examining such possibilities would help inform future therapeutic trials.
While selective NEK2 inhibition has not previously been reported to potentiate taxane responsiveness, overexpression of its partnering protein, HEC1/NDC80, was found to be associated with poor prognosis in a small cohort of ovarian cancer patients and to modulate the efficacy of paclitaxel in ovarian cancer cell lines45. Huang, et al. have also reported that a HEC1 inhibitor causes cell death in a variety of breast cancer cell lines46. These data complement the results reported herein demonstrating that selectively targeting the NEK2 kinase can improve taxane response, particularly in resistant TNBC cells, by inducing extensive mitotic defects and pervasive chromosomal instability.
It is well-established that TNBCs exhibit a higher level of aneuploidy and chromosomal instability than other breast cancer subtypes, and such instability is associated with an increased risk of metastasis and worse outcomes47. Our studies revealed that TNBC cells that have acquired paclitaxel resistance have a basal increase in mitotic abnormalities compared to parental cells. Modest genomic changes associated with low levels of instability such as those that we observed in taxane resistant cells may lead to increased tumor cell fitness that promotes growth and metastasis47–49. However, extreme mitotic instability induces cell death stemming from mitotic catastrophe47,49. We found that targeting LIN9 or NEK2 in resistant cells causes greater mitotic defects than in sensitive/parental cells. In the presence of paclitaxel, nearly all resistant cells develop numerous defects associated with extreme chromosomal instability. Thus, combining two insults, ie., the increased instability associated with taxane resistance partnered with suppression of LIN9 or NEK2, causes greater instability and mitotic catastrophe than observed in sensitive/parental cells that have only one insult (LIN9 or NEK2 silencing). We postulate that the increased chromosomal instability associated with the development of resistance provides a specific vulnerability to inhibition of the LIN9/NEK2 pathway. Hence, use of a NEK2 inhibitor should prevent the acquisition of taxane resistance because any cells with a resistance phenotype should be particularly sensitive to such inhibitors. In summary, we found that increased LIN9 expression is correlated with paclitaxel resistance in TNBC cells and that its silencing restores taxane sensitivity though the induction of extensive mitotic abnormalities. We identified NEK2 as a downstream mediator of LIN9 whose suppression also causes chromosomal instability and promotes taxane sensitivity. Importantly, inhibiting LIN9 or NEK2 either pharmacologically (JQ1 or CMP3a/INH1, respectively) or genetically sensitizes cells to paclitaxel and reverses paclitaxel resistance. The ability of NEK2 inhibitors to increase paclitaxel responsiveness was also observed in vivo. These studies provide a rational therapeutic approach for combating and overcoming the major therapeutic challenge of taxane resistance in patients with TNBC through the utilization of NEK2 inhibitors.
Supplementary Material
Significance:
Resistance to chemotherapy is a major hurdle for treating cancer patients. Combining NEK2 inhibitors with taxanes may be viable approach for improving patient outcomes by enhancing mitotic defects induced by taxanes alone.
Acknowledgements
We thank Drs. Kerry Burnstein and Maria Julia Martinez for thoughtful discussions during the execution of this work. This work was supported by R01CA206505 and Velosano Bike to Cure (RAK), F99/K00CA212460 (JMS), R01GM108743 (MKS), P20CA233216 (VV), American Brain Tumor Association Basic Research Fellowship supported by an Anonymous Corporate Partner (MSS), and the Cytometry and Microscopy Shared Resource and the Athymic Animal and Preclinical Therapeutics Shared Resource of the Case Comprehensive Cancer Center (P30CA043703 and S10OD021559).
Financial Support:
R.A. Keri: R01CA206505 and VeloSano Bike to Cure
J.M. Sahni: F99/K00CA212460
M.K. Summers: R01GM108743
M.S. Schrock: American Brain Tumor Association Basic Research Fellowship
V. Varadan: P20CA233216
Footnotes
Conflict of Interest Disclosure: The authors declare no potential conflicts of interest.
REFERENCES
- 1.Zasadil LM et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Science translational medicine 6, 229ra243, doi: 10.1126/scitranslmed.3007965 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu Y, Zhou Y & Shi J Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle 13, 1756–1764, doi: 10.4161/cc.28672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blajeski AL, Kottke TJ & Kaufmann SH A multistep model for paclitaxel-induced apoptosis in human breast cancer cell lines. Experimental cell research 270, 277–288, doi: 10.1006/excr.2001.5349 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Zhu J et al. Centrosome impairments and consequent cytokinesis defects are possible mechanisms of taxane drugs. Anticancer research 25, 1919–1925 (2005). [PubMed] [Google Scholar]
- 5.Hornick JE et al. Live-cell analysis of mitotic spindle formation in taxol-treated cells. Cell motility and the cytoskeleton 65, 595–613, doi: 10.1002/cm.20283 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huzil JT, Chen K, Kurgan L & Tuszynski JA The roles of beta-tubulin mutations and isotype expression in acquired drug resistance. Cancer informatics 3, 159–181 (2007). [PMC free article] [PubMed] [Google Scholar]
- 7.van Ark-Otte J et al. Effects of tubulin-inhibiting agents in human lung and breast cancer cell lines with different multidrug resistance phenotypes. Oncology reports 5, 249–255 (1998). [PubMed] [Google Scholar]
- 8.Sudo T, Nitta M, Saya H & Ueno NT Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer research 64, 2502–2508 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Liu JR et al. Bcl-xL is expressed in ovarian carcinoma and modulates chemotherapy-induced apoptosis. Gynecologic oncology 70, 398–403, doi: 10.1006/gyno.1998.5125 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Anand S, Penrhyn-Lowe S & Venkitaraman AR AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer cell 3, 51–62 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Schmid P et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. The New England journal of medicine 379, 2108–2121, doi: 10.1056/NEJMoa1809615 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Dent R et al. Pattern of metastatic spread in triple-negative breast cancer. Breast cancer research and treatment 115, 423–428, doi: 10.1007/s10549-008-0086-2 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Visconti R & Grieco D Fighting tubulin-targeting anticancer drug toxicity and resistance. Endocrine-related cancer 24, T107–T117, doi: 10.1530/ERC-17-0120 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Porkka K, Blomqvist C, Rissanen P, Elomaa I & Pyrhonen S Salvage therapies in women who fail to respond to first-line treatment with fluorouracil, epirubicin, and cyclophosphamide for advanced breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 12, 1639–1647, doi: 10.1200/JCO.1994.12.8.1639 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Litovchick L et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Molecular cell 26, 539–551, doi: 10.1016/j.molcel.2007.04.015 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Sahni JM et al. Mitotic Vulnerability in Triple-Negative Breast Cancer Associated with LIN9 Is Targetable with BET Inhibitors. Cancer research 77, 5395–5408, doi: 10.1158/0008-5472.CAN-17-1571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yori JL, Johnson E, Zhou G, Jain MK & Keri RA Kruppel-like factor 4 inhibits epithelial-to-mesenchymal transition through regulation of E-cadherin gene expression. J Biol Chem 285, 16854–16863, doi: 10.1074/jbc.M110.114546 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montanez-Wiscovich ME et al. LMO4 is an essential mediator of ErbB2/HER2/Neu-induced breast cancer cell cycle progression. Oncogene 28, 3608–3618, doi: 10.1038/onc.2009.221 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciriello G et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519, doi: 10.1016/j.cell.2015.09.033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weaver BA How Taxol/paclitaxel kills cancer cells. Mol Biol Cell 25, 2677–2681, doi: 10.1091/mbc.e14-04-0916 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curtis C et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352, doi: 10.1038/nature10983 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pereira B et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications 7, 11479, doi: 10.1038/ncomms11479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadasivam S, Duan S & DeCaprio JA The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes & development 26, 474–489, doi: 10.1101/gad.181933.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du J et al. The mitotic checkpoint kinase NEK2A regulates kinetochore microtubule attachment stability. Oncogene 27, 4107–4114, doi: 10.1038/onc.2008.34 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fry AM, Meraldi P & Nigg EA A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. The EMBO journal 17, 470–481, doi: 10.1093/emboj/17.2.470 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cappello P et al. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene 33, 2375–2384, doi: 10.1038/onc.2013.183 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Hayward DG et al. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer research 64, 7370–7376, doi: 10.1158/0008-5472.CAN-04-0960 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 6, pl1, doi: 10.1126/scisignal.2004088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2, 401–404, doi: 10.1158/2159-8290.CD-12-0095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hatzis C et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. Jama 305, 1873–1881, doi: 10.1001/jama.2011.593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu CM et al. Novel small molecules disrupting Hec1/Nek2 interaction ablate tumor progression by triggering Nek2 degradation through a death-trap mechanism. Oncogene 34, 1220–1230, doi: 10.1038/onc.2014.67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J et al. Targeting NEK2 attenuates glioblastoma growth and radioresistance by destabilizing histone methyltransferase EZH2. The Journal of clinical investigation 127, 3075–3089, doi: 10.1172/JCI89092 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Stover DG & Winer EP Tailoring adjuvant chemotherapy regimens for patients with triple negative breast cancer. Breast 24 Suppl 2, S132–135, doi: 10.1016/j.breast.2015.07.032 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Wong NS Primary medical therapy and breast conservation treatment: the medical oncology perspective. Gland surgery 7, 560–575, doi: 10.21037/gs.2018.10.02 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hauser S et al. Loss of LIN9, a member of the DREAM complex, cooperates with SV40 large T antigen to induce genomic instability and anchorage-independent growth. Oncogene 31, 1859–1868, doi: 10.1038/onc.2011.364 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Reichert N et al. Lin9, a subunit of the mammalian DREAM complex, is essential for embryonic development, for survival of adult mice, and for tumor suppression. Molecular and cellular biology 30, 2896–2908, doi: 10.1128/MCB.00028-10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Begley U et al. A human tRNA methyltransferase 9-like protein prevents tumour growth by regulating LIN9 and HIF1-alpha. EMBO molecular medicine 5, 366–383, doi: 10.1002/emmm.201201161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong SB et al. Essential Role of Polo-like Kinase 1 (Plk1) Oncogene in Tumor Growth and Metastasis of Tamoxifen-Resistant Breast Cancer. Molecular cancer therapeutics 17, 825–837, doi: 10.1158/1535-7163.MCT-17-0545 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Tang A et al. Aurora kinases: novel therapy targets in cancers. Oncotarget 8, 23937–23954, doi: 10.18632/oncotarget.14893 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu LY et al. Aurora A is essential for early embryonic development and tumor suppression. J Biol Chem 283, 31785–31790, doi: 10.1074/jbc.M805880200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melichar B et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. The Lancet. Oncology 16, 395–405, doi: 10.1016/S1470-2045(15)70051-3 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Hannak E, Kirkham M, Hyman AA & Oegema K Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. The Journal of cell biology 155, 1109–1116, doi: 10.1083/jcb.200108051 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esterlechner J et al. LIN9, a subunit of the DREAM complex, regulates mitotic gene expression and proliferation of embryonic stem cells. PloS one 8, e62882, doi: 10.1371/journal.pone.0062882 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahni JM et al. Bromodomain and Extraterminal Protein Inhibition Blocks Growth of Triple-negative Breast Cancers through the Suppression of Aurora Kinases. J Biol Chem 291, 23756–23768, doi: 10.1074/jbc.M116.738666 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mo QQ et al. Inhibition of Hec1 expression enhances the sensitivity of human ovarian cancer cells to paclitaxel. Acta pharmacologica Sinica 34, 541–548, doi: 10.1038/aps.2012.197 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang LY et al. Activity of a novel Hec1-targeted anticancer compound against breast cancer cell lines in vitro and in vivo. Molecular cancer therapeutics 13, 1419–1430, doi: 10.1158/1535-7163.MCT-13-0700 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Roylance R et al. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol Biomarkers Prev 20, 2183–2194, doi: 10.1158/1055-9965.EPI-11-0343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silk AD et al. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proceedings of the National Academy of Sciences of the United States of America 110, E4134–4141, doi: 10.1073/pnas.1317042110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swanton C et al. Chromosomal instability determines taxane response. Proceedings of the National Academy of Sciences of the United States of America 106, 8671–8676, doi: 10.1073/pnas.0811835106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldman M et al. The UCSC Cancer Genomics Browser: update 2015. Nucleic acids research 43, D812–817, doi: 10.1093/nar/gku1073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.