

Abstract
Purpose
Pancreatic neuroendocrine tumors (pNETs) are uncommon malignancies noted for their propensity to metastasize and comparatively favorable prognosis. Although both the treatment options and clinical outcomes have improved in the last decades, most patients will die of metastatic disease. New systemic therapies are needed.
Experimental Design
Tissues were obtained from 43 patients with well-differentiated pNETs undergoing surgery. Gene expression was compared between primary tumors versus liver and lymph node metastases using RNA-Seq. Genes that were selectively elevated at only one metastatic site were filtered out to reduce tissue-specific effects. Ingenuity Pathway Analysis (IPA) and the Connectivity Map (CMap) identified drugs likely to antagonize metastasis-specific targets. The biological activity of top identified agents was tested in vitro using two pNET cell lines (BON-1 and QGP-1).
Results
902 genes were differentially expressed in pNET metastases compared to primary tumors, 626 of which remained in the common metastatic profile after filtering. Analysis with IPA and CMap revealed altered activity of factors involved in survival and proliferation, and identified drugs targeting those pathways, including inhibitors of mTOR, PI3K, MEK, TOP2A, PKC, NF-kB, CDK and HDAC. Inhibitors of MEK and TOP2A were consistently the most active compounds.
Conclusions
We employed a complementary bioinformatics approach to identify novel therapeutics for pNETs by analyzing gene expression in metastatic tumors. The potential utility of these drugs was confirmed by in vitro cytotoxicity assays, suggesting drugs targeting MEK and TOP2A may be highly efficacious against metastatic pNETs. This is a promising strategy for discovering more effective treatments for pNET patients.
Keywords: Pancreas, Neuroendocrine Tumor, Metastasis, Gene Expression, Chemotherapy
INTRODUCTION
Pancreatic neuroendocrine tumors (pNETs) are uncommon neoplasms which account for less than 3% of all pancreatic tumors.1 However, the annual incidence of all neuroendocrine tumors (NETs) has increased steadily over the last several decades, and pNETs now have an approximate annual incidence of 0.8 per 100,000 persons.2 They are characteristically slow-growing malignancies associated with a comparably favorable prognosis, especially when contrasted against the significantly more common pancreatic adenocarcinoma. The indolent nature of the disease often delays diagnosis such that metastases, predominantly to the liver, are present in approximately 60% of patients at diagnosis.3,4 Despite advances in knowledge and management of NETs, the majority of patients with metastatic pNETs will die of their disease, and current therapies have not been shown to definitively improve overall survival. Greater understanding of the biology of these tumors, particularly the changes associated with metastasis, is needed to identify new therapeutic targets and more effective treatments.
Clinical management of metastatic PNETs is multimodal and rapidly evolving. Historically, surgical excision of the primary tumor, debulking of metastatic lesions and somatostatin analogues (SSAs) were the main therapeutic options. While surgery remains the standard of care for localized tumors and an important component of the treatment of metastatic disease, several systemic agents have emerged for treating pNETs. Streptozocin (STZ) was one of the first drugs found to be active against pNETs, although low response rates and significant toxicity limit its use today.5 SSAs have long been used to reduce the symptoms of excess hormone secretion associated with many NETs, and have more recently been shown to have antiproliferative effects as well.6,7 Currently SSAs are considered the first line therapy for metastatic NETs expressing somatostatin receptors. In the last decade, the options for treatment have expanded further with evidence of improved progression-free survival (PFS) using the tyrosine kinase inhibitor, sunitinib, and the mTOR inhibitor, everolimus.8,9 Other tyrosine kinase inhibitors, including cabozantinib and sulfatinib, have shown promise in phase II trials, and phase III trials are underway.10 The combination of the antimetabolite capecitabine and alkylating agent temozolomide (CAPTEM) can be given orally and has less toxicity as compared to STZ-based treatment.11–13 A phase II study has shown significant PFS benefits of this combination over temozolomide alone.14 Peptide receptor radionuclide therapy (PRRT), which uses a radiolabeled SSA to target NETs, was recently approved for the treatment of gastroenteropancreatic NETs after impressive improvements in PFS of treated patients with grade 1 and 2 small bowel NETs relative to those receiving a high dose SSA.15 Therapies that extend overall patient survival, however, are still needed.
Pancreatic neuroendocrine tumors have been recognized as a distinct clinical entity for nearly a century, yet our understanding of their genetic basis is incomplete. Several hereditary cancer syndromes are associated with pNET development, including multiple endocrine neoplasia type 1 (MEN1), von Hippel-Lindau disease (VHL), neurofibromatosis type 1 (NF-1), and tuberous sclerosis complex (TSC1 and 2), but the majority of pNETs occur sporadically (>90%).16 Among sporadic pNETs, the most commonly altered gene is MEN1, with mutations found in 41–44% of cases. Other commonly mutated genes include DAXX or ATRX, genes in the mTOR pathway, genes involved in DNA damage repair such as MUTHY, and genes involved in chromatin modification.17,18 Although mutations in the mTOR pathway are seen in only 12–15% of PNETs, expression and activity level analyses have revealed alterations of PI3K/Akt/mTOR signaling involving both upstream and downstream regulators in the majority of these tumors.16,19,20
In this study, we investigated gene expression patterns in nodal and liver metastases versus primary pNETs (most of which were patient-matched) to identify drug-targetable regulators of metastasis. We uncovered prominent alterations in mitogen-activated protein kinase (MAPK), cyclin-dependent kinase (CDK), topoisomerase (TOP2A), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ĸB) signaling along with expected changes in PI3K and mTOR pathways. Analyses of the metastatic tumor transcriptional changes using two complementary bioinformatic tools identified drugs that displayed significant in vitro efficacy against pNET cell proliferation and survival. The approach, identification of NET metastatic pathways, and discovery of new anti-NET drugs described herein offer unique insights and promising therapies for treating advanced pNETs.
METHODS
RNA Extraction
Tumor samples were obtained from 43 patients undergoing resection of well differentiated, grade 1 and 2 pNETs between 12/1½006 and 7/19/2016 (Supplemental Table 1). Tissue samples from the primary tumor, liver, and lymph node metastases were collected from all patients where available. Informed written consent was provided by all patients in accordance with a protocol approved by the University of Iowa Institutional Review Board (IRB Number 199911057), and studies were conducted in accordance with the Belmont Report. Following surgery, tissue samples were stored at −20°C in RNALater (Thermo Fisher Scientific, Waltham, MA, USA). Total RNA was extracted using the RNeasy Plus Universal Mini Kit (Qiagen, Hilden, Germany) per the protocol recommended by the manufacturer. Tumor cellularity was estimated by a pathologist with NET expertise (AB) from paraffin embedded tissue stained with haemotoxylin and eosin, and samples with less than 70% neuroendocrine cellularity were excluded. The Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) was used to assess RNA quality and assign RNA integrity numbers (RIN).
Transcript Profiling
Transcription profiling using RNA-Seq was performed by the University of Iowa Genomics Division using manufacturer-recommended protocols. Initially, 500 ng of DNase I-treated total RNA was used to enrich for polyA containing transcripts using oligo(dT) primers bound to beads. The enriched RNA pool was then fragmented, converted to cDNA and ligated to sequencing adaptors containing indexes using the Illumina TruSeq stranded mRNA sample preparation kit (Cat. #RS-122–2101, Illumina, Inc., San Diego, CA). The molar concentrations of the indexed libraries were measured using the 2100 Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA) and combined equally into pools for sequencing. The concentrations of the pools were measured using the Illumina Library Quantification Kit (KAPA Biosystems, Wilmington, MA) and sequenced on the Illumina HiSeq 4000 genome sequencer using 150 bp paired-end sequencing by synthesis (SBS) chemistry.
FASTQ files were used to quantify transcript abundance using Salmon version 0.11.3.21 Tximport 1.8.0 was used to perform gene-based quantification of abundance, using Ensembl Genes version 96/Biomart to map gene symbols to Ensembl transcript identifiers,22,23 and voom 24 for library normalization and to generate normalized transcripts per million (TPM) values. StringTie version 1.3.025 was used to assign reads to transcripts using the GFF3 file from Ensembl (http://ftp.ensembl.org/pub/grch37/release-87/gtf/homo_sapiens/Homo_sapiens.GRCh37.87.gtf.gz).
RNA-Seq Processing
RNA-seq count data was normalized using TPM. To identify differentially expressed genes, the nonparametric Wilcoxon rank sum test was used. Log2 fold changes (LFC) between tumors at different sites were estimated using the voom package.24 LFC>0 indicates that a gene was up-regulated in the metastases, whereas LFC<0 indicates downregulation. To filter out results that may result from the confounding influence of comparing different tissue types between primary and metastatic tumors, we carried out two rank sum tests: one comparing primary and metastatic tumors (combining liver and lymph node metastases), and the other comparing liver and lymph node metastases. From the results of these two tests, genes of interest are those which are significantly differentially expressed between primary and metastatic sites (p<0.05), but not differentially expressed between liver and lymph node tumors. Genes were defined as tissue specific and filtered out if expression was 2-fold higher at either metastatic site compared to the other (LFC>1). This tissue specific filtering was designed to remove genes whose overexpression was dependent on the site of metastasis, and thus was applied only to the genes which were upregulated in the metastases. All analyses were performed in R 3.5.0 (www.R-project.org). Unsupervised analysis of the PNET samples was performed using both t-distributed stochastic neighbor embedding (t-SNE) and principal component analysis (PCA).
Drug Prediction
The filtered list of differentially expressed metastatic genes was used to query the Connectivity Map touchstone database.(CMap; Broad Institute, Cambridge, MA)26 This database includes gene expression signatures derived from 9 cancer cell lines treated with 2,429 well-annotated compounds . These expression profiles were compared to our metastatic PNET signature, and compounds were assigned a connectivity score (ranging from −100 to 100) based on the similarity of the expression changes they induced when compared to our metastatic signature. A positive connectivity score indicates similar gene expression changes, while a negative score indicates an opposing pattern. Each compound and cell line are assigned a separate score, and a summary score is provided for each compound across all cell lines. In parallel, the IPA analysis match function was also used to predict potentially therapeutic drugs. The IPA analysis of our metastatic profile was compared to analyses from other cell lines treated with various compounds, and these drugs were assigned a score which ranged from −14.3 to −244.3. A more negative score indicated expression changes resulting in predictions which were in the opposite direction of those based on our metastatic profile. The Molecule Activity Predictor and Upstream Analysis tools in IPA (Qiagen)27 were used to predict the activation state of drug targets from our metastatic PNET expression signature. IPA calculates a z-score in order to predict the activation state of upstream regulators, with a positive score indicating activation, a negative score indicating inhibition, and an absolute value greater than 2 suggesting significance. Drugs were selected for testing in pNET cell lines based on a combination of factors: a highly negative connectivity score from CMap and the IPA analysis match, drug availability, and a mechanism of action which was highly represented on both the CMap and IPA lists.
Gene Validation
A subset of 14 significantly differentially expressed genes were selected for quantitative polymerase chain reaction (qPCR) validation in a separate cohort of PNET primary and metastatic samples not tested by RNA-Seq. RNA was extracted as described above from primary tumors, lymph node metastases and liver metastases from 12 other available patients with well differentiated, grade 1 or 2 pNETs. Total RNA was then reverse transcribed to cDNA using the qScript cDNA Supermix (QuantaBio). qPCR was performed using gene-specific primers from Integrated DNA Technologies (Coralville, IA) and PerfeCTa SYBR Green Supermix dye (QuantaBio) using the 7900HT Fast Real-Time PCR System (Applied Biosystems). Primer sequences were obtained from PrimerBank (https://pga.mgh.harvard.edu/primerbank/). Genes were selected using IPA from a list of genes whose differential expression was used to predict the activation states of our drug targets using the molecule activity predictor. Expression was normalized against the internal control gene RPLP1, which was used to calculate the delta cycle threshold (dCT) for each gene of interest. The average expression of each gene in the metastatic (liver and lymph node) tumors was compared to expression in the primary tumors, and statistically significant differential expression was determined using the Wilcoxon rank-sum test.
Drug Sensitivity Assay
Drugs were purchased from Medchem Express LLC (Monmouth Junction, NJ) and Fisher Scientific (Hampton, NH), dissolved in dimethyl sulfoxide (DMSO), and stock solutions stored at −20°C. BON-1 and QGP-1 pNET cells were seeded at a density of 2,000 cells per well in 96-well flat-bottomed culture dishes. After overnight incubation, each drug was added at the indicated concentrations and incubated for five days; assays were performed in triplicate. Samples were evaluated for relative cell number using alamarBlue (Thermo Fisher Scientific, Waltham, MA). Results were quantified using a fluorescence microplate reader by measuring fluorescence of alamarBlue at an excitation wavelength of 560 nm with fluorescence emission at 590 nm. For simvastatin treated cells, media containing simvastatin was removed and replaced with new media prior to the addition of the alamarBlue reagent since high concentrations of simvastatin interferes with measurement of alamarBlue. Results were analyzed using CompuSyn software (ComboSyn, Inc.) to determine the IC50 for each drug.
Two human PNET cell lines were used for these studies. BON-1 cells, established from a lymph node metastasis of a pNET,28 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) / F12 containing 10% fetal bovine serum (FBS), 4 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. QGP-1 cells, established from primary somatostatin producing pNET,29 were purchased from the Japanese Collection of Research Bioresources (JCRB0183), were maintained in RPMI 1640 medium containing 10% FBS, 4 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Both cell lines were recently authenticated by Hofving et al. and found to express neuroendocrine markers and harbor mutations associated with NETs.30 Both cell lines were tested for mycoplasma contamination by selective enzymatic assay (MycoAlert Plus, Lonza Inc., Morristown, NJ) and found to be negative (most recently on 1/6/2020). Cryopreserved stocks of each cell line were made at low passage numbers (less than 5–8 passages after receipt) and cells were used in experiments for a maximum of 10–12 passages (roughly 5–6 weeks) after being thawed.
RESULTS
RNA-Seq was performed on 39 primary tumors, 21 lymph node metastases and 17 liver metastases from 43 patients with pNETs (Supplemental Table 1). All patients had grade 1 or 2, well-differentiated tumors. A flowchart summarizing our methods from sequencing to drug testing is presented in Figure 1. A total of 18,103 genes were measurably expressed in the tumor samples. The results of our RNA-Seq are available on the database of Genotypes and Phenotypes (dbGaP) under phs study number phs001772.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gapprev/gap/cgi-bin/preview1.cgi?GAP_phs_code=DrKURjFTCAuySlUS). Of these, 902 genes were significantly differentially expressed in the metastatic tumors compared to the primaries: 718 were overexpressed and 184 underexpressed (Figure 2a). Of the overexpressed genes, 13 showed greater than 2-fold higher expression in the nodal versus liver metastases, while 263 were expressed at greater than 2-fold higher levels in the liver versus nodal metastases. These 276 genes were filtered out to reduce tissue specific effects on the metastatic gene signature, leaving 626 differentially expressed genes which constituted our final metastatic pNET gene signature (Supplemental Table 2, Figure 2b).
Figure 1:

Schematic drawing of our bioinformatic analysis. RNA-Seq was performed on well-differentiated PNET samples from 43 patients. Gene expression in the primary tumors was compared to the metastases to derive a metastatic expression profile which was analyzed using IPA and CMap to produce a list of therapeutic agents. A subset of these drugs were then tested against BON-1 and QGP-1.
Figure 2:
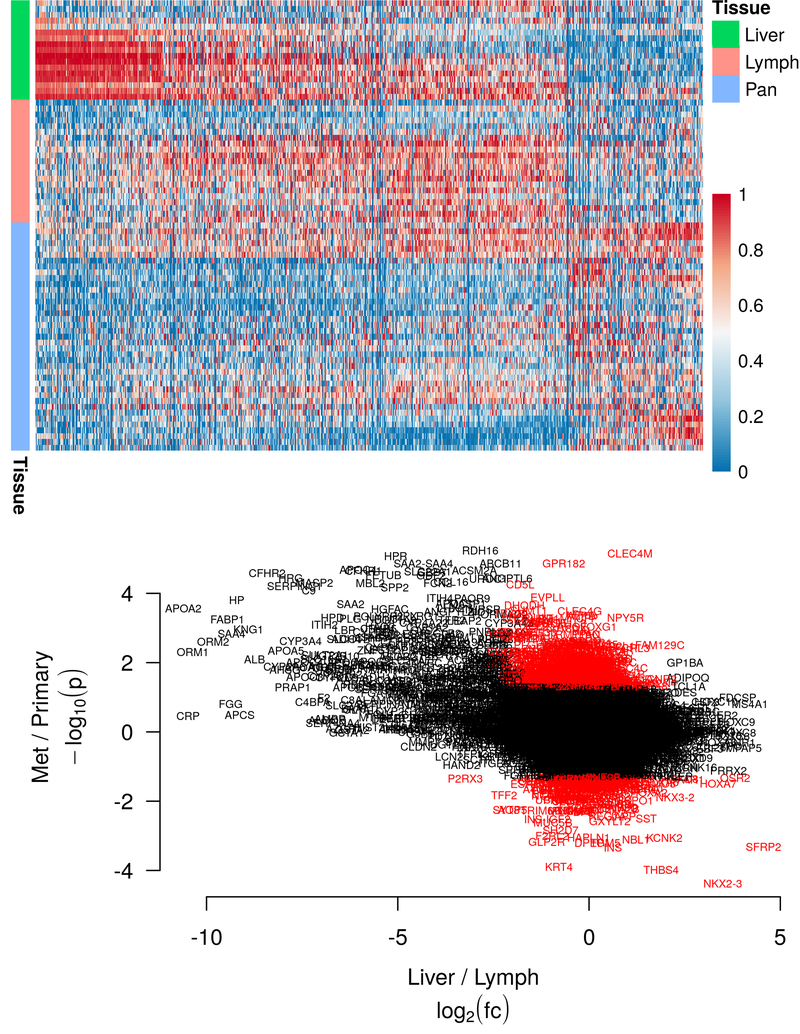
(Top) Heatmap showing the 902 differentially expressed genes in the unfiltered metastatic profile. Red color indicates higher expression, blue indicates lower. Expression values are normalized on a per-gene basis, with a value of 1 indicating the maximum expression in any tissue, and a value of 0 indicating the minimum. Liver = liver metastasis, Lymph = lymph node metastasis, Pan = pancreatic primary tumor
(Bottom) Representation of the filter used to account for tissue specific gene overexpression. The y-axis shows the negative log of the p-value, with more extreme values indicating lower p-value, positive values indicating higher expression in the metastases, and negative values indicating higher expression the primaries. The x-axis shows the log fold change between the liver and lymph node, with positive values indicating higher expression in the lymph nodes, and negative vales indicating higher expression in the liver. Genes included in the filtered metastatic expression profile are shaded in red.
Analysis of the liver genes which were removed by the filter using IPA revealed that they were largely involved in processes associated with normal liver function, including production of coagulation factors and lipid metabolism. Moreover, IPA analysis of the unfiltered metastatic gene set (903 genes) demonstrated the dramatic effect that inclusion of these liver genes had on subsequent analysis; in the unfiltered set, the pathways and cellular processes with the strongest predicted activation were generally associated with normal hepatic function.
Dimensionality reduction using PCA and t-SNE revealed that the primary determinant of clustering was the tissue of origin (Figure 3). This was particularly true for the liver metastases, which tended to cluster separately from the primary tumors and lymph node metastases, which were much closer together. Despite this tendency, there were several outliers for which the clustering was primarily determined by the individual patient. This second trend is most clearly visualized by t-SNE, which also shows that six of the liver metastases cluster tightly with their corresponding primary and lymph node samples, rather than with the other liver metastases.
Figure 3:

Unsupervised analysis of gene expression in the tumor samples using PCA (right) and t-SNE (left). For most samples, the primary determinant of clustering was tumor location, with a few notable exceptions, for example the triplet of samples in the top left of the t-SNE plot, all of which were from patient 210.
Selection of Potentially Therapeutic Compounds
We queried the CMap touchstone database with expression signatures from the filtered metastatic PNET gene expression profile. The top 100 compounds producing expression changes which were the reverse of those seen in our metastatic profile are shown in supplementary Table 3. Among the drugs with the lowest connectivity scores, inhibitors of mTOR and PI3K were the most highly represented. In parallel, the analysis match functionality of IPA was used to identify compounds that featured related pathways with an opposing activation state for prioritization. The most highly rated compounds predicted by IPA analysis match are shown in supplemental Table 4, with numerous inhibitors of mTOR, PI3K, BRAF/MEK and CDK ranked highly. Fifteen compounds were selected from these two lists using the criteria previously described (Table 1). These compounds included inhibitors of CDK, HDAC, NFkB signaling, DNA topoisomerase, RNA-Polymerase, DNA protein kinase, MEK, HMGCR and the PI3K/AKT/mTOR pathway.
Table 1: Drugs selected for in vivo testing in BON-1 and QGP-1.
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| Drug | Class | Summary CMAP Score | Analysis Match Score | BON-1 | QGP-1 |
| PD-0325901 | MEK inhibitor | −88.72 | −124.92 | 0.001 | 0.006 |
| Triptolide | NFkB inhibitor, MDM2, HSF1 | −97.64 | NA | 0.003 | 0.001 |
| Selumetinib | MEK inhibitor | −96.79 | −88.16 | 0.006 | 0.01 |
| Daunorubicin | Topoisomerase inhibitor | −96.05 | NA | 0.016 | 0.009 |
| Doxorubicin | Topoisomerase inhibitor | −96.48 | NA | 0.028 | 0.006 |
| Sirolimus | mTOR inhibitor | −92.86 | −34.51 | 0.01 | 0.03 |
| PIK-75 | PI3K Inhibitor, DNA PK inhibitor | −96.83 | NA | 0.02 | 0.04 |
| PD-184352 | MEK inhibitor | −98.38 | −150.24 | 0.03 | 0.04 |
| Alvocidib | CDK inhibitor | −95.86 | −108.09 | 0.05 | 0.08 |
| Mocetinostat | HDAC Inhibitor | −95.00 | NA | 0.07 | 0.14 |
| Entinostat | HDAC Inhibitor | −85.54 | NA | 0.19 | 0.17 |
| Apitolisib | mTOR inhibitor, PI3K inhibitor | NA | −132.59 | 0.19 | 0.17 |
| PI-103 | mTOR inhibitor, PI3K inhibitor, DNA PK inhibitor | −96.97 | −96.57 | 0.19 | 0.31 |
| bisindolylmaleimide-IX | PKC inhibitor, Topoisomerase inhibitor | −97.44 | NA | 0.13 | 0.4 |
| Simvastatin | HMGCR Inhibitor | −98.93 | NA | 8 | 9 |
IC50 = half maximal inhibitory concentration. CDK = cyclin dependent kinase, HDAC = histone deacetylase, NFkB = nuclear factor kappa-beta, PKC = protein kinase C, PI3K = Phosphoinositide 3-kinase, DNA PK = DNA protein kinase, mTOR = mammalian target of rapamycin, MEK = Mitogen-activated protein kinase kinase, HMGCR = Hydroxymethylglutaryl-coenzyme A Reductase. For drugs listed multiple times in IPA or CMap, only the lowest score is displayed.
The upstream analysis tool in IPA was used to analyze the metastatic gene expression signature, and a number of regulators were predicted to be activated using a z-score cutoff of 2: OSCAR, MEK, IL27, NFE2L2, CD40LG, CAMP, TREM1, PRKCD, collagen type II, and NANOG (Supplemental Table 5). Three upstream regulators were also predicted to be inhibited with z-scores less than −2: CST5, miR-146, and PDCD1. With the exceptions of MEK and protein kinase C delta (PRKCD), none of the other drug targets suggested by CMap were independently predicted to be activated by the IPA upstream analysis tool. By comparison, the molecule activity predictor tool in IPA was used to make predictions as to the activation state of several targets of the drugs predicted by CMap, using a more expansive set of connections that includes data derived from non-human studies. Using this analytical approach, CDK, NFkB, mTOR, MEK, HDAC1, PI3K and PKC were all predicted to be activated in the metastases. These predictions are shown in graphical form in Figure 4.
Figure 4:

(Right) Graphical representation of IPA predictions for the activation states of several drug targets. Genes from the metastatic expression profile which interact with any of these 7 drug targets are arrayed around the outside of the circle. Red shading indicates increased expression in the metastases, while green shading indicates decreased expression. Relationships to drug targets are shown by solid (direct) or dotted (indirect) lines. Activating relationships are shaded orange, while inhibiting relationships are in blue. Yellow arrows indicate a relationship which argues against the overall prediction. The orange shading of the upstream regulators indicates predicted activation. (Left) Validation of differential gene expression predicted by RNA-Seq in PNET primaries and metastases using qPCR. *** indicates p<0.001, ** indicates p<0.01, * indicates p<0.05, # indicates p<0.1
Gene Expression Validation
Fifteen genes found to be differentially expressed in our RNA-Seq data were selected for validation using RT-qPCR. Genes were selected based on relationships with our drug targets, as shown in Figure 4a, and included SST, CXCL8, NAMPT, TNF, NFKBIZ, BIRC3, TNFSF14, APOE, NKX3–1, HBA1, GPR182, SPDEF, STAB2 and DMBT1. Each of these genes either leads to increased activation of one or more of our drug targets or is transcriptionally regulated by one or more drug targets. Unexpectedly, the differential expression seen in our RNA-Seq was confirmed by qPCR validation in just 3 out of the 14 selected genes (Figure 4b). For 2 additional genes, there was significant differential expression between the primary tumor and liver metastasis, but not in the primary tumor and pooled metastasis comparison, while 4 genes displayed a trend toward higher expression in the liver metastases (0.05 < p < 0.1). Overall, 9 of 14 genes trended toward or were significantly altered in PNET metastases versus primary tumors.
Drug Sensitivity Assay
Compounds predicted by IPA and CMap to have suppressive activity against PNETs were tested using a quantitative, cell viability assay in two pNET cell lines, BON-1 and QGP-1. Fifteen drugs were selected for testing: triptolide, alvocidib (flavopiridol), mocetinostat, entinostat, bisindolylmaleimide-IX (RO 31–8220), PIK-75, PI-103, sirolimus (rapamycin), simvastatin, doxorubicin, daunorubicin, PD-184352 (CI-1040), PD-325901, selumetinib (AZD-6244) and apitolisib (GDC-0980). For each drug tested, the PNET cell lines displayed similar sensitivities in proliferation/viability assays although in most cases BON-1 cells were slightly more sensitive, as indicated by the lower IC50s (Table 1 and Figure 5). All drugs significantly inhibited the proliferation and survival of PNET cells at increased concentrations although differential responsiveness to the various compounds was observed. Drugs with the highest activities had IC50 values ranging between 1 to 80 nM and included three different MEK inhibitors (PD-325901, selumetinib and PD-184352), two TOP2A inhibitors (daunorubicin and doxorubicin) as well as triptolide, PIK-75, and alvocidib. By comparison, drugs with the lowest anti-proliferative activity in PNETs had IC50 values ranging between 130 nM to 8–9 μM, which included HDAC inhibitors (mocetinostat and entinostat), PI3K and mTOR inhibitors (apitolisib and PI-103), and the least potent drug tested, simvastatin, an HMG-CoA inhibitor. Nine other drugs with low CMap scores were tested, of which five showed very low activity against these cell lines, and four showed moderate activity (Supplemental Table 6).
Figure 5:

Differential in vitro responsiveness of PNET cell lines to anti-cancer drugs suggested by the metastatic gene signature in patient PNETs. BON-1 (closed circles) and QGP1 (open circles) cells were exposed to increasing concentrations of the indicated drugs for five days. The proportion of viable cells is shown on the y-axis, with the dosage on the x-axis. The IC50 values (μM) are denoted for each drug.
DISCUSSION
The incidence of metastatic pNETs is increasing, and although there are a wide variety of treatment options for patients with these tumors, response rates and survival benefits have been modest.6,8,9 An improved understanding of the transcriptomic changes underlying the metastatic process may help to identify novel therapies to improve treatment outcomes. We present a complementary bioinformatics approach to identify novel therapies for pNETs. A previous study demonstrated the utility of a similar approach using pooled gastroenteropancreatic NET data, and identified the HDAC inhibitor entinostat as a potential treatment for metastatic NETs.31 In our study, fifteen compounds identified through our analysis pipeline demonstrated significant antiproliferative activity against pNET cell lines in vitro.
A critical step in identifying the metastatic expression profile involved filtering out normal liver and lymph node genes. We frequently observed that metastases to the liver and lymph nodes overexpress genes associated with the site of metastasis. For example, several of the most highly expressed genes in the liver metastases are related to normal hepatic metabolism or coagulation. Several explanations for this observation exist: 1) hormonal influence from the tumor microenvironment induces expression of these genes; 2) activation of certain hepatic pathways is important for metastatic cells to colonize and survive in the liver; or 3) these genes may result from contamination of normal hepatocytes in our tumor specimens. The tissue specific expression was significantly more pronounced in the liver metastases than in the lymph nodes. This may reflect a stronger transcriptional “reprogramming” effect in the liver or likelihood that, while tumors from all sites have some degree of lymphocytic infiltrate, hepatocytes would only be predicted to be found in liver metastases. Histological review of the tumors in this study revealed that neuroendocrine cells were the most prevalent cell type, with tumor cells making up 70–95% of the samples, and samples with less than 70% cellularity excluded from the analysis. Nonetheless, lymphocytic infiltrate was seen in almost all samples, and projections of hepatocytes were occasionally observed to extend within the gross tumor margin. Regardless of the etiology of this tissue-specific expression, we were interested in defining gene expression changes common to metastases regardless of the site, which required filtering out tissue-specific genes. The preponderance of genes removed by the filter were normal hepatic genes, and their removal significantly influenced subsequent analysis in IPA and CMap.
The filtered, common metastatic expression profile included 626 differentially expressed genes when the significance threshold was set at p<0.05 using non-parametric analysis. A number of software tools are available to analyze RNA-Seq data including DESeq2, voom and others,6,23 each of which employs different statistical methods and makes different assumptions about the data to determine differential gene expression. For our analysis, differential gene expression was determined using the Wilcoxon rank sum test, which makes fewer assumptions about the distribution of data and is relatively insensitive to outliers as compared to DESeq or voom.
Due to the large number of statistical tests performed in any RNA-Seq experiment, correction for multiple testing is typically performed to reduce the incidence of false positive results, resulting in an adjusted p-value, also known as the corrected p-value, false discovery rate (FDR) or q-value. When such a correction was performed for our data, the calculated FDR for nearly all genes in our metastatic PNET expression profile exceeded 0.9. These values may reflect both the broad similarity in gene expression between primary tumors and metastases as well as within group heterogeneity in gene expression. Analysis using PCA and t-SNE showed that for most samples, the primary driver of differential expression was the tissue of origin. However, for a minority, clustering was primarily determined by characteristics specific to individual patients. As illustrated in Figure 3b, there were 6 liver metastases which clustered with their corresponding primary and lymph node samples, rather than with the larger group of liver metastases. This observation, coupled with the relatively indistinct borders between the clusters, highlights the heterogeneous nature of the disease. Nonetheless, the overall expression pattern proved remarkably capable of predicting drugs with activity against pNET cell lines. Moreover, there was significant overlap in the drug classes and specific agents predicted to be efficacious using two separate prediction tools, the IPA analysis match and CMap.
To evaluate the reproducibility of the metastatic gene expression profile, we evaluated several genes by RT-qPCR in a different group of pNET samples. These genes were selected due to their relationships to several important cancer pathways and drug targets, including NF-kB, PI3K, mTOR, CDK, MEK, PKC, and HDAC1, as indicated by IPA. Confirmation of these specific genes, which were used to predict the activation states of drug targets, would help to bolster the predictions made based on their differential expression. Though most genes selected could be validated using qPCR, it should be noted that the fold changes in the RNA-Seq data were near 2-fold, which corresponds to approximately a single PCR cycle. Ideally, we would have tested more tumors, but had our validation set was limited by the number of tumors with primaries, nodal, and/or liver metastasis samples available that were not tested by RNA-Seq. Thus, due to the high calculated FDR and the failure of many genes to validate using qPCR, one should be cautious to not give undue weight to any one gene in the common metastatic profile. Instead, the overall patterns seen in the data provide more relevant, biologically useful information.
Compounds that inhibit a variety of distinct signaling pathways were identified by IPA and CMap as potential therapeutic options for patients with metastatic pNETs. Some of the drug targets identified were the usual suspects implicated in pNET tumorigenesis, including PI3K and mTOR.9,17–19 Inhibitors of both kinases were the most highly represented drug classes with connectivity scores less than −90 in CMap and highly negative scores in the IPA analysis match, and the combined PI3K and DNA PK inhibitor PIK-75 was among the more efficacious drugs tested. This drug has shown anticancer activity in preclinical studies.32,33 It is poorly soluble and has not progressed to clinical trials, but improvements in drug delivery may allow for human trials in the future.34 Other inhibitors of mTOR and/or PI3K that were tested in this study included apitolisib, PI-103 and rapamycin (sirolimus). Rapamycin had overlapping IC50s with PIK-75 in the PNET cells but never achieved maximal efficacy, while apitolisib and PI-103 were about 8–10 fold less potent than PIK-75. Even so, apitolisib and rapamycin have both been used clinically. In a phase I trial, apitolisib demonstrated modest anti-tumor activity in patients with advanced solid tumors.35 However, in a phase II trial it was less effective than everolimus for the treatment of renal cell carcinoma, with a significantly higher rate of treatment-limiting adverse effects.36 Interestingly, while there were 12 mTOR inhibitors with connectivity scores below −90, the widely used everolimus was not among the drugs suggested by either platform, with a connectivity score of −15.18 on the CMap.
Other targets, such as HDACs and CDKs, have recently generated significant interest as potential targets for NET therapy:31,37 the HDAC inhibitor, entinostat, and the CDK inhibitor, ribociclib, are both currently being investigated in phase II trials in NETs (trial numbers NCT03211988 and NCT02420691, respectively). One CDK inhibitor, alvocidib, was tested in our study and was among the drugs which showed the highest antiproliferative activity. Although systemic toxicity is a concern, it has been shown to have activity against a wide variety of hematologic and solid malignancies in phase I and II trials.38 We tested two HDAC inhibitors, entinostat and mocetinostat, and found they had moderate antiproliferative activity in the pNET cell lines. Entinostat is currently being investigated in clinical trials for NETs, however mocetinostat has not yet been studied in NETs. To date, phase II trials using mocetinostat for other malignancies, either as monotherapy or as part of a combined regimen, have demonstrated modest antitumor activity accompanied by high rates of adverse events.39–42
Inhibitors of the mitogen-activated protein kinase (MAPK) pathway, which includes mitogen-activated protein kinase kinase (MEK) and the proto-oncogene BRAF, were also well-represented among the compounds suggested by CMap and IPA. Three MEK inhibitors, PD-0325901, PD-184352 and selumetinib were selected for testing. All effectively inhibited proliferation of both pNET cell lines, with PD-0325901 having the lowest IC-50 of any drug tested. MEK inhibitors have not been previously used for the treatment of NETs, however aberrant activation of the MAPK pathway has been implicated in NET tumorigenesis.16 MEK inhibitors have shown excellent activity against BRAF- or NRAS- mutated melanoma as well as Ras-driven plexiform neurofibromas 43 and studies investigating their use in combination with targeted or immune-modulating therapy in melanoma and other solid tumors are ongoing.44 PD-184352, also known as CI-1040, was the first MEK inhibitor to enter clinical trials, however its activity in humans was limited by poor solubility and rapid clearance.45 The antitumor activity of PD-0325901 and selumetinib has been supported in phase I and II trials including patients with melanoma and other solid tumors.46–48
Four HMG-CoA reductase inhibitors, also known as statins, were suggested by CMap, although none were highly ranked by IPA, and simvastatin was selected for testing. The anti-proliferative and anti-metastatic effects of simvastatin have previously been demonstrated in QGP-1 and BON-1, 49 and our testing confirmed its activity against both cell lines. However, high concentrations in the μM range were needed to achieve an anti-proliferative effect.
Among the better acting anti-NET drugs identified in our study, triptolide is a naturally occurring compound that exerts its antitumor effects through a variety of mechanisms, including inhibition of NF-kB signaling and induction of apoptosis.50,51 Although preclinical studies with the drug have been promising, triptolide is poorly soluble in water, limiting its clinical application. The development of a water-soluble analog, Minnelide,52 has allowed for clinical testing, and a phase II trial in pancreatic adenocarcinoma (NCT03117920) is currently in progress. Several inhibitors of DNA topoisomerase were suggested by CMap, and daunorubicin and doxorubicin were selected for testing. Although cytotoxic chemotherapy is less commonly used for well-differentiated NETs, doxorubicin in combination with streptozocin is among the standard treatment options for well-differentiated PNETs.53 Daunorubicin is another prototypical topoisomerase inhibitors that is commonly used as a first line treatment for acute myeloid leukemia.54
The final drug selected for testing was bisindolylmaleimide-IX. This drug is part of the bisindolylmaleimide class of PKC inhibitors and is described as belonging to this class within CMap. Unlike other drug classes which are highly represented in our analysis (e.g. mTOR inhibitors), only two PKC inhibitors received scores below −90, one of which is bisindolylmaleimide-IX, and the other (CGP-60474) is also a CDK inhibitor. Additionally, there were three PKC inhibitors with scores greater than 90, indicating that treatment with these compounds was associated with expression changes mirroring (rather than opposing) those seen in metastatic PNETs. This fact combined with the observation that many PKC isozymes may act as tumor suppressors rather than tumor promoters,55 suggests that bisindolylmaleimide-IX’s anti-tumor activity may occur through a different mediator. Although it has not been noted in CMap, bisindolylmaleimide-IX has also been shown to inhibit DNA-topoisomerase.56 Given the number of topoisomerase inhibitors suggested by CMap, it is possible that bisindolylmaleimide-IX’s predicted activity against NETs reflects its inhibition of topoisomerase, rather than PKC.
Fourteen of fifteen drugs selected for testing in BON-1 and QGP-1 showed significant activity against both cell lines (IC50 <1 μM). For those compounds that have been studied in humans (entinostat, mocetinostat, alvocidib, apitolisib, PD-0325901, selumetinib, daunorubicin, doxorubicin), significant inhibition of proliferation was seen at concentrations that are readily attained in phase I or II trials. By comparison, the mTOR inhibitor, sirolimus (rapamycin), which has been used in numerous human trials plateaued at 60–80% efficacy even at the highest doses tested; simvastatin only showed sensitivity when doses of 10 μM were reached. Nine additional compounds were tested that had low CMap scores (Supplemental Table 6), five of which showed little to no activity and four showed at least moderate activity, including Vinblastine (Vinca alkaloid), Thapsigargin (Calcium ATPase), MLN-2238 (proteasome inhibitor), and Rigosertib (PI3K and PLK1 inhibitor).
Hofving et al. tested 1224 compounds (at 1 μM concentration) from a commercially available inhibitory library against BON1 and QGP1.30 They found a general sensitivity to MEK inhibitors, drugs targeting HSP90 and Aurora kinases, but not to HDAC inhibitors. Comparing these results to our RNA-Seq strategy is of interest. We achieved greater than 50% inhibition for both cell lines at <1 μM concentration with 14 of the 15 (93%) drugs selected. Of these, Hofving et al. confirmed sensitivity of both BON1 and QGP1 cell lines (IC50 at 1 μM) to 3 of the 14 (PD032591, PI-103, and PIK-75), to one but not the other cell line in 4 (Selumetinib, Doxorubicin, PD-184352, PI-103), whereas 5 showed limited or no activity in both cell lines (Daunorubicin, Sirolimus, Mocetinostat, Entinostat, bisindolylmaleimide-IX); 2 of the drugs we found were not in their screen (Triptolide, Apitolisib). For the 12 drugs tested in common, their screen found sensitivity for 25% of those we selected, partial sensitivity (one cell line only) for 33%, and missed 42% of these potentially useful drugs. These results indicate that although these screens can be complementary, screening of these 1224 compounds would have resulted in many of the drugs we found to effectively kill both cell lines being missed, and would have also markedly increased the number of potential false positives that would need further evaluation. Although these results are promising, further studies will be required to confirm the clinical utility of these drugs. As described above, the expression changes between primary and metastatic PNETs are frequently small in magnitude, and this combined with tumor heterogeneity leads to significant difficulty in finding statistically significant differential gene expression. Sequencing of additional samples, and the use of techniques which account for differences in tumor cellularity, such as tissue deconvolution or single cell RNA-Seq, may help to overcome this issue.
While the drugs tested showed activity against BON-1 and QGP-1 pNET cells, both lines have extremely high proliferative indices compared to more typical well-differentiated pNETs.57 A strength of this study is that we identified new NET drugs using gene expression data from patients with well-differentiated tumors rather than cell line-based profiling, but confirmation of drug activity against less proliferative pNET models will ultimately be required. We and others in the NET field are beginning to generate well-differentiated, patient-derived NET spheroid cultures 58, providing an ideal system for future drug testing. Once optimal acting drugs are confirmed in well-differentiated NET cell populations, further pre-clinical testing in a murine model of metastatic NETs would enable measurement of drug tolerability, anti-proliferative effects, and anti-metastatic activity in vivo.
In conclusion, we present a pNET metastatic gene signature that was determined from a comparison of patient-matched metastases and primary tumors. Bioinformatic analyses of the gene expression changes using IPA and CMap identified drugs with predicted activity against metastatic pNETs, which was verified by in vitro cytotoxicity assays in two pNET cell lines. Our methodology uses patient-derived expression data to inform and focus subsequent drug testing, providing mechanistic rationale for compound selection and effectively prioritizing drugs for preclinical screening prior to progression to clinical trials.
Supplementary Material
Statement of Translational Relevance:
Pancreatic neuroendocrine tumors (pNETs) are slow growing malignancies which have metastasized at the time of diagnosis in roughly 60% of patients. Currently, there are a variety systemic treatment options for metastatic PNETs, but improvements in survival are typically modest, and more effective agents are needed. We analyzed differential gene expression associated with metastasis to identify drugs with predicted activity against pNETs. A large number of potential compounds were identified using this technique, many of which have not previously been used for the treatment of NETs. The antiproliferative properties of fifteen of these drugs were demonstrated in vitro using two pNET cell lines. Both the method of drug selection and the novel pharmacologic classes suggested by the analysis have the potential to significantly improve the treatment of patients with metastatic pNETs.
Acknowledgement:
This work was supported by the T32 grant CA148062 (AS) and Neuroendocrine Tumor SPORE grant P50 CA174521 (BD, TB, DEQ, AB, JD, TO, JH). RNA-seq data presented herein were obtained at the Genomics Division of the Iowa Institute of Human Genetics which is supported, in part, by the University of Iowa Carver College of Medicine and the Holden Comprehensive Cancer Center (National Cancer Institute of the National Institutes of Health under Award Number P30CA086862). We thank Dr. David Gordon for his guidance using CompuSyn software for drug response analyses.
T32 grant CA148062 (AS), SPORE grant P50 CA174521 (BD, TB, DEQ, AB, JD, TO, JH), and NCI Core Grant P30 CA086862 (Holden Comprehensive Cancer Center)
Footnotes
Conflicts of interest: The authors declare no potential conflicts of interest.
References:
- 1.Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19(10):1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dasari A, Shen C, Halperin D, et al. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017;3(10):1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–3072. [DOI] [PubMed] [Google Scholar]
- 4.Hallet J, Law CH, Cukier M, Saskin R, Liu N, Singh S. Exploring the rising incidence of neuroendocrine tumors: a population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer. 2015;121(4):589–597. [DOI] [PubMed] [Google Scholar]
- 5.Moertel CG, Hanley JA, Johnson LA. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1980;303(21):1189–1194. [DOI] [PubMed] [Google Scholar]
- 6.Caplin ME, Pavel M, Cwikla JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224–233. [DOI] [PubMed] [Google Scholar]
- 7.Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663. [DOI] [PubMed] [Google Scholar]
- 8.Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513. [DOI] [PubMed] [Google Scholar]
- 9.Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grillo F, Florio T, Ferrau F, et al. Emerging multitarget tyrosine kinase inhibitors in the treatment of neuroendocrine neoplasms. Endocr Relat Cancer. 2018;25(9):R453–R466. [DOI] [PubMed] [Google Scholar]
- 11.Fine RL, Gulati AP, Tsushima D, et al. Prospective phase II study of capecitabine and temozolomide (CAPTEM) for progressive, moderately, and well-differentiated metastatic neuroendocrine tumors. Abstract 179. presented at: Gastrointestinal Cancers Symposium. Journal of Clinical Oncology. 2014;32(3_suppl):179–179. [Google Scholar]
- 12.Kotteas EA, Syrigos KN, Saif MW. Profile of capecitabine/temozolomide combination in the treatment of well-differentiated neuroendocrine tumors. Onco Targets Ther. 2016;9:699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strosberg JR, Fine RL, Choi J, et al. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer. 2011;117(2):268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunz PL, Catalano PJ, Nimeiri H, et al. A randomized study of temozolomide or temozolomide and capecitabine in patients with advanced pancreatic neuroendocrine tumors: A trial of the ECOG-ACRIN Cancer Research Group (E2211)[Abstract]. Journal of Clinical Oncology. 2018;36(15_suppl):4004–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med. 2017;376(2):125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kidd M, Modlin IM, Bodei L, Drozdov I. Decoding the Molecular and Mutational Ambiguities of Gastroenteropancreatic Neuroendocrine Neoplasm Pathobiology. Cell Mol Gastroenterol Hepatol. 2015;1(2):131–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scarpa A, Chang DK, Nones K, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65–71. [DOI] [PubMed] [Google Scholar]
- 19.Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28(2):245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Umesalma S, Kaemmer CA, Kohlmeyer JL, et al. RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth. J Clin Invest. 2019;130:1641–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Narayan R, Corsello SM, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017;171(6):1437–1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kramer A, Green J, Pollard J Jr, Tugendreich S Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evers BM, Townsend CM Jr, Upp JR, et al. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology. 1991;101(2):303–311. [DOI] [PubMed] [Google Scholar]
- 29.Kaku M, Nishiyama T, Yagawa K, Abe M. Establishment of a carcinoembryonic antigen-producing cell line from human pancreatic carcinoma. Gan. 1980;71(5):596–601. [PubMed] [Google Scholar]
- 30.Hofving T, Arvidsson Y, Almobarak B, et al. The neuroendocrine phenotype, genomic profile and therapeutic sensitivity of GEPNET cell lines. Endocr Relat Cancer. 2018;25(3):367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez MJ, Subramaniam PS, Tang LH, et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet. 2018;50(7):979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duong HQ, Yi YW, Kang HJ, et al. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int J Oncol. 2014;44(3):959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holand K, Boller D, Hagel C, et al. Targeting class IA PI3K isoforms selectively impairs cell growth, survival, and migration in glioblastoma. PLoS One. 2014;9(4):e94132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talekar M, Ganta S, Amiji M, et al. Development of PIK-75 nanosuspension formulation with enhanced delivery efficiency and cytotoxicity for targeted anti-cancer therapy. Int J Pharm. 2013;450(1–2):278–289. [DOI] [PubMed] [Google Scholar]
- 35.Dolly SO, Wagner AJ, Bendell JC, et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2016;22(12):2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powles T, Lackner MR, Oudard S, et al. Randomized Open-Label Phase II Trial of Apitolisib (GDC-0980), a Novel Inhibitor of the PI3K/Mammalian Target of Rapamycin Pathway, Versus Everolimus in Patients With Metastatic Renal Cell Carcinoma. J Clin Oncol. 2016;34(14):1660–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aristizabal Prada ET, Nolting S, Spoettl G, Maurer J, Auernhammer CJ. The Novel Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) Alone and in Dual-Targeting Approaches Demonstrates Antitumoral Efficacy in Neuroendocrine Tumors in vitro. Neuroendocrinology. 2018;106(1):58–73. [DOI] [PubMed] [Google Scholar]
- 38.Srikumar T, Padmanabhan J. Potential Use of Flavopiridol in Treatment of Chronic Diseases. Adv Exp Med Biol. 2016;929:209–228. [DOI] [PubMed] [Google Scholar]
- 39.Batlevi CL, Crump M, Andreadis C, et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br J Haematol. 2017;178(3):434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan E, Chiorean EG, O’Dwyer PJ, et al. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother Pharmacol. 2018;81(2):355–364. [DOI] [PubMed] [Google Scholar]
- 41.Choy E, Ballman K, Chen J, et al. SARC018_SPORE02: Phase II Study of Mocetinostat Administered with Gemcitabine for Patients with Metastatic Leiomyosarcoma with Progression or Relapse following Prior Treatment with Gemcitabine-Containing Therapy. Sarcoma. 2018;2018:2068517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grivas P, Mortazavi A, Picus J, et al. Mocetinostat for patients with previously treated, locally advanced/metastatic urothelial carcinoma and inactivating alterations of acetyltransferase genes. Cancer. 2019;125(4):533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dombi E, Baldwin A, Marcus LJ, et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med. 2016;375(26):2550–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grimaldi AM, Simeone E, Festino L, Vanella V, Strudel M, Ascierto PA. MEK Inhibitors in the Treatment of Metastatic Melanoma and Solid Tumors. Am J Clin Dermatol. 2017;18(6):745–754. [DOI] [PubMed] [Google Scholar]
- 45.Barrett SD, Bridges AJ, Dudley DT, et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett. 2008;18(24):6501–6504. [DOI] [PubMed] [Google Scholar]
- 46.LoRusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16(6):1924–1937. [DOI] [PubMed] [Google Scholar]
- 47.Janne PA, Shaw AT, Pereira JR, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14(1):38–47. [DOI] [PubMed] [Google Scholar]
- 48.Robert C, Dummer R, Gutzmer R, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol. 2013;14(8):733–740. [DOI] [PubMed] [Google Scholar]
- 49.Herrera-Martinez AD, Pedraza-Arevalo S, F LL, et al. Type 2 Diabetes in Neuroendocrine Tumors: Are Biguanides and Statins Part of the Solution? J Clin Endocrinol Metab. 2019;104(1):57–73. [DOI] [PubMed] [Google Scholar]
- 50.Hou ZY, Tong XP, Peng YB, Zhang BK, Yan M. Broad targeting of triptolide to resistance and sensitization for cancer therapy. Biomed Pharmacother. 2018;104:771–780. [DOI] [PubMed] [Google Scholar]
- 51.Noel P, Von Hoff DD, Saluja AK, Velagapudi M, Borazanci E, Han H. Triptolide and Its Derivatives as Cancer Therapies. Trends Pharmacol Sci. 2019;40(5):327–341. [DOI] [PubMed] [Google Scholar]
- 52.Chugh R, Sangwan V, Patil SP, et al. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci Transl Med. 2012;4(156):156ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Carbonero R, Rinke A, Valle JW, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasms. Systemic Therapy 2: Chemotherapy. Neuroendocrinology. 2017;105(3):281–294. [DOI] [PubMed] [Google Scholar]
- 54.Economides MP, McCue D, Borthakur G, Pemmaraju N. Topoisomerase II inhibitors in AML: past, present, and future. Expert Opin Pharmacother. 2019:1–8. [DOI] [PubMed] [Google Scholar]
- 55.Newton AC, Brognard J. Reversing the Paradigm: Protein Kinase C as a Tumor Suppressor. Trends Pharmacol Sci. 2017;38(5):438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X, Jia D, Ao J, et al. Identification of Bisindolylmaleimide IX as a potential agent to treat drug-resistant BCR-ABL positive leukemia. Oncotarget. 2016;7(43):69945–69960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benten D, Behrang Y, Unrau L, et al. Establishment of the First Well-differentiated Human Pancreatic Neuroendocrine Tumor Model. Mol Cancer Res. 2018;16(3):496–507. [DOI] [PubMed] [Google Scholar]
- 58.Ear PH, Li G, Wu M, Abusada E, Bellizzi AM, Howe JR. Establishment and Characterization of Small Bowel Neuroendocrine Tumor Spheroids. J Vis Exp. 2019(152). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.