

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is highly resistant to radiation therapy (RT), chemotherapy, or a combination of these modalities, and surgery remains the only curative intervention for localized disease. Although cancer-associated fibroblasts (CAFs) are abundant in PDAC tumors, the effects of RT on CAFs and the response of PDAC cells to RT are unknown. Using patient samples and orthotopic PDAC biological models we showed that RT increased inducible nitric oxide synthase (iNOS) in the tumor tissues. Mechanistic in vitro studies showed that, although undetectable in RT-activated tumor cells, iNOS expression and nitric oxide (NO) secretion were significantly increased in CAFs secretome following RT. Culture of PDAC cells with conditioned media from RT-activated CAFs increased iNOS/NO signaling in tumor cells through nuclear factor kappa B (NFĸB), which in turn elevated the release of inflammatory cytokines by the tumor cells. Increased NO after RT in PDAC contributed to an acidic microenvironment that was detectable using the radiolabeled pH (low) insertion peptide (pHLIP®). In murine orthotopic PDAC models pancreatic tumor growth was delayed when iNOS inhibition was combined with RT. These data show the important role that iNOS/NO signaling plays in the effectiveness of RT to treat PDAC tumors.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) accounts for 90% of all the pancreatic cancer cases and it remains one of the most lethal malignancies due to limited treatment options and resistance to therapy [1]. Notably, limited improvements in overall survival have been achieved for PDAC in comparison to other tumor types, in part because more than half of PDAC cases are diagnosed at an advanced and metastatic stage of the disease [1]. Early detection of PDAC remains a challenge due to nonspecific presenting symptoms, difficulty of imaging early-stage tumors, and lack of tumor biomarkers with both good specificity and sensitivity. The poor prognosis for PDAC can be attributed to the rapid metastasis and resistance to conventional therapeutic approaches (chemotherapy, radiotherapy, and molecular targeted therapy). Clearly, a need exists to identify new therapeutic strategies, including effective combinations of existing therapies to expand treatment options for PDAC. Emerging therapeutic approaches for PDAC include immunotherapies, molecular targeted therapies, and strategies targeting the tumor microenvironment [2].
While pancreaticoduodenectomy (Whipple procedure) or distal pancreatectomy is performed in patients with resectable PDAC, fewer than 20% of all patients fall into these categories, and only cures about 10% of this subset [3]. Patients with unresectable PDAC are treated with chemotherapy or chemotherapy/radiation combination therapies [4]. Technological improvements in radiation therapy (RT) approaches, such as intensity-modulated RT (IMRT), and image-guided RT, have dramatically improved the conformality of the radiation dose deposition in the tumor area while increasingly sparing healthy tissues [5].
RT induces alterations in the secretory profile of TME that results in changes in tumor invasion, tumor growth, inflammatory mediators, and regulators of angiogenesis [6–8]. Non-cancer cells including immune cells, fibroblasts, pericytes, and endothelial cells contribute to the stromal compartment of the TME, and, in PDAC, outnumbers the cancer cells [2, 9]. The TME of PDAC is highly inflammatory [10]; RT alters the inflammatory TME by contributing to a fibroinflammatory desmoplasia [11] and to an increase in the expression of TNFα [12]. The pro-inflammatory cytokines interferon-α (IFN-α), interferon-β (IFN-β), and interferon-γ (IFN-γ) are further induced in RT-treated cells [13, 14]. Cancer-associated fibroblasts (CAFs) expressing both vimentin and α-smooth muscle actin (α-SMA) are the predominant cellular components of the PDAC stroma [2]. Previous studies have demonstrated that irradiated pancreatic CAFs exhibit enhanced tumor-stroma interactions, which impact tumor growth and invasion [7, 15]. Pancreatic CAFs survive severe stress, including damage induced by RT single-fraction doses up to 20 Gy [16].
The expression of inducible nitric oxide synthase (iNOS, NOS2) in tumor cells is associated with poor survival in several cancers [17–20]. The iNOS is transcriptionally regulated and induced by inflammatory cytokines [21, 22], contributing to the production of nitric oxide (NO) through conversion of L-arginine into citrulline in the presence of NADPH and oxygen. An increase in NO results in an intrinsic pro-oxidant environment [22], immune escape [23], and resistance to apoptosis [24]. NO shows promise either as a standalone therapeutic agent or as target of cancer therapies. The role of NO in cancer depends on the NO concentration or duration of NO exposure, extracellular conditions, localization of NOS, and cellular sensitivity to NO [21, 25–27]. Pancreatic tumors exhibit higher expression of iNOS compared with non-tumor pancreatic tissue [28]. Pancreatic orthotopic implantation of tumor cells expressing low levels of iNOS result in the formation of pancreatic tumors with metastasis in the liver and formation of ascites; an effect that is not observed in tumor models developed by orthotopic implantation of PDAC cells containing high levels of iNOS [21]. In addition to or independent of iNOS expression in tumor cells, upregulation of iNOS has been detected in stromal fibroblasts and immune cells [22]. Mechanistic studies have demonstrated that in PDAC, CAFs express high amounts of iNOS contributing to the development of tumor chemoresistance via increased NO secretion [29]. Inflammatory cytokines induce NO production by CAFs [29], contributing to PDAC chemoresistance [30] and immunosuppression [31].
Not only does NO contribute to tumor growth and metastasis [21, 26, 32, 33], but mechanistic metabolic studies demonstrated a NO-mediated decrease in mitochondrial respiration, which led cancer cells to undergo higher glycolytic rates to maintain ATP production levels [34]. These results have further supported the role of NO in cancer metabolism by demonstrating that NO regulates the Warburg effect and promotes cancer growth by inhibiting mitochondrial respiration.
We hypothesize that RT induces alterations in the inflammatory TME contributing to an increase in PDAC iNOS expression and NO secretion that results in an increase in tumor growth. In this study, we show that conditioned media from RT-activated CAFs increase NO secretion/iNOS expression by the tumor cells that contributes to PDAC tumor growth, and acidification of the PDAC microenvironment, which can be detected using the radiolabeled pH (low) insertion peptide (pHLIP®). Importantly, we demonstrate that this phenomenon can be mitigated with iNOS inhibition in vitro and in vivo, providing a therapeutic option for overcoming this pro-tumorigenic phenotype.
MATERIALS AND METHODS
Pancreatic cancer cell lines and patient samples
SUIT-2, CAPAN-2, MIA PaCa-2, BxPC-3 human PDACs were purchased from American Type Culture Collection (ATCC) and were grown according to standard procedures. The FC1245 murine PDACs were obtained from the David Tuvenson group and were transformed in Michel Sadelain’s laboratory to generate FC1245luc+ [35]. FC1245 and FC1245luc+ cells were cultured in Dulbecco’s Modified Eagle’s Medium (DME) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin. Cell lines used in this work were purchased in 2016–2019, and they were used within passage number of 15. All the cell lines were mycoplasma free and authenticated at Memorial Sloan-Kettering Cancer Center (MSKCC) integrated genomics operation core using short tandem repeat analysis.
Deidentified patient samples (non-tumor and tumor pancreatic tissues) were obtained before and after RT from David M. Rubenstein Center for Pancreatic Cancer Research following institutional review board (IRB) approval. RT doses, RT time-points, and sample collection time-points are described in Supplementary Table 1.
Murine orthotopic PDAC xenograft model
All experiments involving animals were performed according to the guidelines approved by the Research Animal Resource Center and Institutional Animal Care and Use Committee at Memorial Sloan Keterring Cancer Center, NY, USA. PDAC orthotopic models were developed as previously described [36] and additional details are in Supporting Information.
Contrast injection and computed tomography for pancreatic tumor delineation
Pancreatic tumors were visualized using an image-guided small animal micro-irradiator (XRad225Cx, Precision X-Ray, North Brantford, CT) following a protocol previously described by Thorek and colleagues [37]. Mice were injected with a 3 mL solution of 75 mg Iodine mL−1 (iohexol; Omnipaque, GE Heathcare) at 5 min prior to imaging. Data visualization was performed as previously described [37] and tumor volumes were determined using Amira region-grow tool.
Image guided X-ray radiotherapy of pancreatic tumors
Immediately after visualization of the pancreatic tumor by X-ray CT using a solution of 15 mg Iodine mL−1, radiation Arc therapy was performed in mice bearing orthotopic pancreatic tumors [37]. See Supporting Information for details.
Isolation and expansion of murine and human pancreatic fibroblasts
Fibroblasts were isolated from orthotopic FC1245 PDAC models or human PDAC using methodology previously described by Müerköster and colleagues [29]. Further details are described in Supplementary Information.
Conditioned medium from irradiated and non-irradiated cells
To analyze the effects of RT on NO secretion from fibroblasts on tumor cells and vice versa, cells were cultured with the respective conditioned medium obtained from fibroblasts for 24 h. Before use, these media was centrifuged at 10,000 rpm for 10 min.
Co-culture of irradiated and non-irradiated pancreatic tumor cells with fibroblasts
FC1245 pancreatic tumor cells (1×105 tumor cells per well) were platted into the bottom compartment of a 6-well culture plate (Corning). The fibroblasts were then seeded into the top transwell compartment (2×105 fibroblasts per well). Medium was replaced after 24 h, co-cultures were irradiated (see below for details on in vitro RT treatments), and supernatants or cell extracts were analyzed after an additional 24 h co-culture. The supernatants were centrifuged at 10,000 rpm for 10 min before use on the analysis.
Nitrite level determination by colorimetric assays
NO is a gaseous free radical with a short life and no available methods to directly measure NO levels exist. Therefore, the levels of more stable NO metabolites (nitrite, NO2−) were measured in cell culture supernatants using the NO colorimetric assay (R&D Systems) or the fluorescent probe 4,5-Diaminofluorescein (DAF-2; Cayman Chemical). See Supporting Information for details.
In vitro therapeutic treatments
Details on the different in vitro therapeutic experiments – radiation treatment, iNOS inhibition with 1400W, iNOS knockdown, iNOS amplification, NO donor treatments, glucose uptake, lactate production, cell counting, viability assessment with trypan blue exclusion, migration and invasion assays are described in Supplementary Information.
NFĸB, Cytokine, and RNA analyses
The activity of the NFĸB in FC1245 cells cultured in the presence and absence of CM from CAFs was evaluated using a NFĸB reported kit according to manufacture instructions (Bioscience, 60614). See Supporting Information for details
Cell supernatants of FC1245 PDAC cells, RT fibroblasts, non-RT fibroblasts, and FC1245 incubated with CM from fibroblasts were collected, centrifuged at 16,000 × g for 10 min, and saved at −80 °C until the day of the experiment. Cytokines were quantified using the MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead 32 Plex Panel (Millipore).
For RNA analyses, details are described in Supporting Information. mRNA expression data were processed by BioMark HD System and data analyzed using the real-time PCR analysis software (Fluidigm). mRNA levels were relative to 18S (ΔCt = Ct gene of interest – Ct 18S) and normalized versus the mean of control.
In vivo therapy studies
Bioluminescence was measured for 5 min after intraperitoneally administrating 150 mg kg−1 of potassium D-luciferin salt (Caliper Life Sciences) dissolved in PBS. Mice were deemed to have engrafted tumors (typically 7 days post orthotopic implantation of FC1245 PDAC cells) once total bioluminescence surpassed 105 photons/second/cm2/sr. Mice were randomly grouped into treatment groups (n = 10 per group): vehicle, 1400W, RT, or a combination of 1400W with RT. RT groups received three doses of 6 Gy (administrated with a 48 h interval, see sections above for details on RT protocols). Intraperitoneal administration of 1400W (1 μg g−1) was started at day 0. 1400W was administrated once daily for 7 days during the RT treatment period and every two days for two weeks after the RT treatment period.
Western blotting, Immunofluorescence, and Immunohistochemistry
Western blot, immunofluorescence, and immunohistochemistry were performed as detailed in Supporting Information.
Radiolabeling of pHLIP, PET imaging of 18F-FDG and acute biodistribution studies
The 18F-FDG was obtained from the Nuclear Pharmacy at MSKCC on the morning of injection. NO2A-variant 3 pHLIP® (>95% chemical purity) was purchased and used as received from CSBio Co. (Menlo Park, CA). For peptide radiolabeling with Gallium-67 (67Ga), 67Ga (gallium-67 citrate, Nuclear Diagnostic Products) was trapped on a silica cartridge (Sep-Pak Light, Waters), washed with water, and eluted as [67Ga]GaCl3 with 0.4 M HCl, followed by pH-adjustment to ~4.0 with 1.0 M Na2CO3. NO2A-cysVar3 was dissolved in DMSO and added to the [67Ga]GaCl3. Following addition of 200 uL of AcN, the mixture was incubated at ~75°C for 30 min, diluted with water, and loaded onto a C-18 cartridge (Sep-Pak Light, Waters). After washing the cartridge with 10 mL of water to remove unbound 67Ga, the radiolabeled peptide was eluted with 100% ethanol and reconstituted in PBS for in vivo studies. The radiochemical purity of all constructs used in animal studies exceeded 95%.
At 14 days post-therapy (see above for details on the RT protocol and 1400W administrations), mice (n = 5 per group) were administrated 20 μCi intravenous injection of 67Ga-labeled pHLIP or 200 μCi 18F-FDG. At 2 h post-intravenous administration of 18F-FDG, PET images were recorded on an Inveon PET scanner (Siemens). All images were visualized in AMIDE 1.0.4 software (http://amide.sourceforge.net).
Acute biodistribution studies were performed at 24 h after injection of 67Ga-labeled pHLIP or 2 h after intravenous injection of 18F-FDG. Mice were sacrificed and organs were harvested, weighed, and assayed in the gamma counter for biodistribution studies. Radioactivity associated with each organ was expressed as percentage of injected dose per gram of organ (% ID g−1).
RESULTS
RT increases iNOS expression in murine and human pancreatic tumors
Given that a higher iNOS expression is associated with poor survival in patients with PDAC [20] and with PDAC therapeutic resistance [29], we sought to determine if RT interferes with iNOS protein levels in pancreatic cancer. We used immunofluorescence assays to characterize iNOS protein levels in matched normal vs. tumor pancreatic samples from patient specimens obtained before treatment (treatment naïve, non-RT) and after RT (Fig. 1A, Supplementary Fig. 1). The RT-treated PDAC samples were obtained between 1 to 3 weeks after the last RT treatment fraction (Supplementary Table 1) and the number of cumulative radiation dose given to those patients decreased in the following order patient #1 > patient #2 > patient #3 > patient #4. The tumor tissues in our four different patient cohorts showed a significantly higher iNOS expression when compared to the non-tumor pancreas (Fig. 1A, Supplementary Fig. 1). These observations are consistent with previous immunohistochemical staining showing higher iNOS expression in tumor when compared with non-tumor samples in PDAC cases [20, 28]. The iNOS protein levels were higher in RT-treated tumors when compared with non-treated PDAC tumors (4.6-fold ± 0.1 patient #1, 1.6-fold ± 0.4 patient #2, 2.4-fold ± 0.9 patient #3, and 1.8-fold ± 0.5 patient #4; Fig. 1A). Immunofluorescence staining of iNOS was similar in non-RT vs. RT-treated non-tumor tissues of patients #2 and #3. Non-tumor samples of two other patients showed a two-fold increase in iNOS protein levels after RT (2.4 ± 0.3 patient #1 and 2.3 ± 0.5 patient #4). Additional immunofluorescence staining of α-smooth muscle actin (SMA) and cytokeratin in RT-treated samples obtained from patient #3, demonstrated iNOS+/α-SMA+ in 56.5% of total α-SMA+ cells and iNOS+/cytokeratin+ in 90.1% of total cytokeratin+ cells (Fig. 1B,C and Supplementary Tables 2,3).
Figure 1. RT increases iNOS expression in PDAC.

a) Quantification of immunofluorescence staining of iNOS in PDAC tumors with or without RT treatment (mean ± S.E.M, n = 3). Confocal images of iNOS staining and patient clinical history are described in Supplementary Fig. 1 and Supplementary Table 1. *P<0.05, **P<0.01 compared with corresponding non-RT tissue; &P<0.05 compared with corresponding non-tumor tissue and based on a Student’s t-test.
b,c) Immunofluorescence staining of iNOS, cytokeratin and α-SMA in PDAC samples obtained from Patient #3 after RT. DAPI was used to stain cell nuclei. Quantifications of iNOS/α-SMA and iNOS/cytokeratin staining were performed using the Halo software and data is shown in Supplementary Tables 2,3. Scale bars: 50 μm.
d,e) Immunofluorescence staining of iNOS, cytokeratin and α-SMA in FC1245 orthotopic PDAC tumors. DAPI was used to stain cell nuclei. Quantifications of iNOS/α-SMA and iNOS/cytokeratin staining were performed using the Halo software and data is shown in Supplementary Tables 4-7. Tumors were collected at 2 days after a 12 Gy RT dose. Scale bars: 2000 μm.
To further compare iNOS protein levels between non-RT and RT-treated pancreatic tumors, FC1245luc+ murine pancreatic cancer cells were orthotopically transplanted to the pancreas of C57BL/6 (B6) mice. FC1245 pancreatic cancer cells isolated from KPC (KrasLSL-G12D/+;Trp53LSL-R172H/+;Pdx1-Cre) mice in the C57BL/6 (B6) genetic background represent a valuable biological PDAC model [38] as they mimic the pathophysiological features of human PDAC, and when transplanted into mice they develop invasive metastatic PDAC characterized by extensive stroma [39]. Murine RT treatment planning utilized CT imaging with intraperitoneal (IP)-administrated contrast agent to define the orthotopic PDAC tumor area and the entire tumor mass was selectively irradiated with a single RT therapeutic absorbed dose of 12 Gy using an arc treatment [37]. The use of a 12 Gy dose to study RT-mediated iNOS production was based on previous studies demonstrating that RT at this dose accelerates the progression of PDAC invasion [11], which we hypothesize will correspondingly effectuate RT-mediated iNOS production. Western blot (Supplementary Fig. 2) and immunofluorescence (Fig. 1D) analyses of murine orthotopic PDACs obtained at 2 days after RT confirms the increase in iNOS protein levels (13.2-fold ± 0.1, mean ± S.E.M, n = 4). RT-treated PDAC exhibited a 5-fold increase in SMA when compared with non-RT PDAC (Fig. 1D, Supplementary Tables 4,5). Quantification of iNOS co-localization with α-SMA and cytokeratin in FC1245 RT-treated tumors demonstrated a two-fold increase iNOS+/α-SMA+ and a five-fold increase iNOS+/cytokeratin+ when compared with non-RT PDAC (Fig. 1D,E; Supplementary Tables 4-7). Additional immunohistochemistry studies demonstrated that under the conditions of our experiments, the percentage of collagen-positive cells is similar in non-RT (10.4 ± 3.6) and RT (11.4 ± 0.9) FC1245 PDAC tumors (Supplementary Fig. 3). Others have also demonstrated that p48Cre;LSL-KrasG12D (KC) mice irradiated at 12 Gy exhibit fibroinflammatory desmoplasia associated with an increase in collagen and α-SMA [11], suggesting pancreatic stellate cell activation. Together, these results suggest that local tumor irradiation induces iNOS expression in pancreatic tumors.
RT-activated fibroblasts induce inflammatory cytokines secretion, iNOS expression, and NO release by PDAC
Pancreatic TME largely consists of stromal fibroblasts, which are thought to contribute to PDAC resistance to treatment [40, 41]. Therefore, we examined the role of CAFs in iNOS expression and NO secretion of PDAC following RT. Previous studies have demonstrated increased invasiveness of pancreatic cancer cells after co-culture with fibroblasts irradiated at a dose of 5 Gy [7]. We performed primary cultures of fibroblasts [29] isolated from surgical resections of treatment naïve (non-RT) and RT-treated FC1245 PDACs (Supplementary Fig. 4A-B). To investigate possible paracrine iNOS/NO signaling, we analyzed conditioned media (CM) of RT vs. non-RT CAFs and in vitro co-culture models consisting of FC1245 pancreatic cancer cells and CAFs (Fig. 2A, Supplementary Fig. 5). To mimic the high PDAC desmoplastic stromal environment, we followed previous reports and used a tumor cell-CAF ratio of 1:2 in our in vitro model system [29]. Western blot analysis demonstrated similar iNOS expression in FC1245 cancer cells and CAFs (Fig. 2B). Additional studies in FC1245 cancer cells and CAFs that received an extra in vitro irradiation dose of 5 Gy demonstrated, respectively, a 2.2-fold and 3.3-fold increase in iNOS protein levels in non-RT CAFs and RT-CAFs (Fig. 2B). iNOS expression was similar in non-RT and RT-treated FC1245 cells (Fig. 2B). Additional Western blot analysis revealed a 1.4-fold ± 0.1 increase in iNOS expression when FC1245 cancer cells are incubated with CM of CAFs for 24 h (Fig. 2C). Because an extra in vitro irradiation dose increases iNOS expression in CAFs (Fig. 2B), we performed additional studies in FC1245 co-cultured with CAFs or cultured with CM of CAFs that received an extra in vitro 5 Gy. We observed a 1.8-fold ± 0.2 increase in iNOS protein levels in FC1245 cells co-cultured with CAFs or a 3.6-fold ± 0.5 increase in FC1245 cells cultured with CM of CAFs (Fig. 2C). FC1245, FC1245 cultured with CM of RT-CAFs, and FC1245 co-cultured with RT-CAFs demonstrated, respectively, a cell viability of 92.3% ± 9.42, 113.9% ± 6.07, and 64.94% ± 12.52 (Supplementary Fig. 6).
Figure 2. RT-activated CAFs enhance PDAC tumor growth.
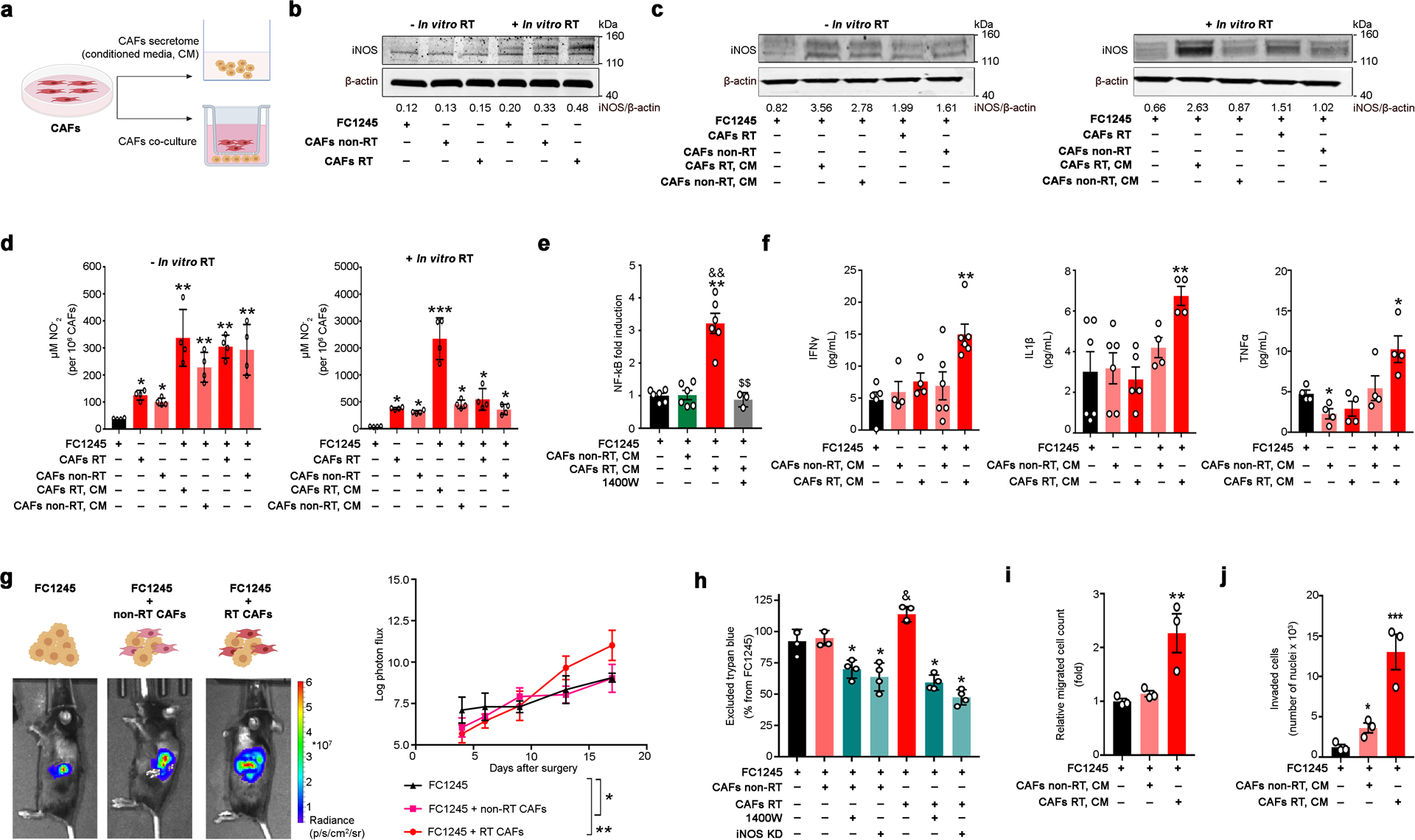
a) CAFs were isolated from non-RT treated FC1245 orthotopic PDACs (CAFs non-RT) or from mice treated with a 12 Gy RT dose (CAFs RT). FC1245 were cultured with conditioned medium from CAFs or co-cultured with CAFs using a transwell in vitro system.
b) Western blot of iNOS in total lysates of FC1245, CAFs non-RT, and CAFs RT. Density of western blot bands was quantified by scanning densitometry with ImageJ software.
c) Western blot of iNOS in total lysates of FC1245. FC1245 cells were co-cultured with CAFs (CAFs RT or CAFs non-RT) or treated with conditioned medium from CAFs (CAFs RT, CM or CAFs non-RT, CM) for 24 h. Density of western blot bands was quantified by scanning densitometry with ImageJ software.
d) NO2− levels in supernatants of FC1245, CAFs RT, CAFs non-RT, FC1245 treated with supernatants from CAFs, and FC1245 co-cultured with CAFs. *P<0.05, **P<0.01, ***P<0.001 compared with FC1245 and based on a Student’s t-test (mean ± S.E.M, n = 4).
e) Luciferase NFĸB activity assay (mean ± S.E.M, n = 3) in FC1245 cells cultured with CM non-RT CAFs, CM RT-CAFs, and CM RT-CAFs in the presence of 10 μM 1400W. **P<0.01 compared with FC1245, &&P<0.01 compared with FC1245 treated with CM from non-RT CAFs, $$P<0.01 compared with FC1245 treated with CM from RT-CAFs and based on a Student’s t-test. 1400W N-(3-(aminomethyl)benzyl)acetamidine.
f) IFNγ, IL1β, and TNFα concentrations in the supernatants of FC1245 PDAC cells (first bar on the graph), non-RT CAFs (second bar), RT-CAFs (third bar), FC1245 incubated with CM from non-RT CAFs (fourth bar), and FC1245 incubated with CM from RT CAFs (fifth bar). Cell supernatants were collected at 24 h after cell culture and cytokine activity was determined using the MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead 32 Plex Panel. Data are presented as mean ± S.E.M., n = 4 to 6 independent experiments. *P<0.05, **P<0.01 compared with FC1245 and based on a Student’s t-test. IFNγ interferon-α, IL1β interleukin 1β, TNFα tumor necrosis factor-α.
g) Bioluminescence images and tumor growth rate (mean ± S.D., n = 4) of pancreatic orthotopic tumors developed by implantation of FC1245luc+ alone, FC1245luc+ mixed with non-RT CAFs, and FC1245luc+ mixed with RT-CAFs into the pancreas of C57BL/6 (B6) mice. *P<0.05, **P<0.01 compared with FC1245.
h) Cell growth of FC1245 cultured in the presence or absence of CM of irradiated and non-irradiated CAFs. To deplete iNOS, FC1245 tumor cells were treated with 10 μM 1400W or transfected with iNOS siRNA (iNOS KD). Data are presented as mean ± S.E.M., n = 3. *P<0.05 compared with FC1245 plus CAFs non-RT or FC1245 plus CAFs RT, &P<0.05 compared with FC1245 and based on a Student’s t-test. 1400W N-(3-(aminomethyl)benzyl)acetamidine.
i,j) FC1245 migration (i) and invasion (j) after treatment with CM of CAFs using the Boyden and Matrigel invasion chambers, respectively. Data are presented as mean ± S.E.M., n = 3. *P<0.05, **P<0.01, ***P<0.001 compared with FC1245 and based on a Student’s t-test.
We next measured nitrite (NO2–), a stable byproduct of NO, in the culture medium of CAFs, FC1245 cells or FC1245 cells co-cultured with CAFs (Fig. 2D). Levels of NO2– were higher in CAFs relative to FC1245 cells in both non-irradiated and irradiated experimental conditions (Fig. 2D). These results are consistent with previous studies reporting low amounts of NO in T3M4 and PT45-P1 pancreatic cancer cells [29]. Cultures or FC1245 cells with CM of RT-CAFs displayed higher NO2– levels when compared with non-RT CAFs (Fig. 2D). NO2– secretion by FC1245 cells was strongly up-regulated by CM of RT-activated CAFs receiving an extra in vitro RT dose of 5 Gy (44.7 ± 0.1, mean ± S.E.M, n = 4; Fig. 2D). Additional studies in SUIT-2, CAPAN-2, MIA PaCa-2, and BxPC3 human pancreatic cancer cell lines cultured with CM of RT-activated human CAFs demonstrated that an in vitro RT dose of 5 Gy increases NO2– secretion by cancer cells (Supplementary Fig. 7). These results show that RT-activated CAFs produce high levels of iNOS and NO secretion, which are further induced by PDAC cells and that RT enhances iNOS/NO paracrine signaling in PDACs.
Given that inflammatory cytokines such as interferon-γ (IFNγ), interleukin 1β (IL1β), and tumor necrosis factor-α (TNFα) induce iNOS expression through nuclear factor ĸB (NFĸB) [22, 29, 30], we performed analyses of NFĸB and 32 cytokines using a MILLIPLEX kit. NFĸB activity in FC1245 cells was monitored using a NFĸB luciferase reporter assay. Culture of FC1245 with CM from RT-CAFs increased NFĸB in the tumor cells (Fig. 2E). Cytokine analyses were performed using the cell supernatant of FC1245, non-RT CAFs, RT CAFs, and FC1245 cells cultured either with medium from non-RT fibroblasts or with medium from RT-fibroblasts. IFNγ, IL1β, leukemia inhibitory factor (LIF), RANTES (regulated on activation normal T cell expressed and secreted), and interleukin-13 (IL-13) were similar in FC1245 and CAFs (Fig. 2F). Culture of FC1245 PDAC cells in the presence of CM from RT-fibroblasts increased the production of IFNγ, IL1β, and TNFα (Fig. 2F). Additionally, when FC1245 cells were cultured with medium from RT-fibroblasts, we observed an increase in the production of interleukin-6 (IL-6), LIF, and RANTES (Supplementary Fig. 8). On the other hand, IL-13 decreases when FC1245 cells were cultured with medium from non-RT fibroblasts (Supplementary Fig. 8). We next used real-time PCR to understand the major genes in RT-activated CAFs driving these transcriptional differences in FC1245 cancer cells (Supplementary Fig. 9). We observed upregulation in IFNβ, iNOS, and TNFα in RT-activated CAFs when compared with non-RT CAFs. When compared with RT-activated CAFs, non-RT CAFs demonstrated higher mRNA levels for RANTES, C-X-C motif chemokine 10 (CXCL10), IL12, IL1α, IL1β, IL1R1, IL6Rα, Interferon regulatory factor 7 (IRF7), and signal transducer and activator of transcription 1 (STAT1) (Supplementary Fig. 9). These results suggest that RT-activated fibroblasts induce increased secretion of NO and iNOS expression in the tumor cells through NFĸB, which in turn leads to an elevated release of inflammatory cytokines by the tumor cells.
RT-activated CAFs increase pancreatic tumor growth, an effect that can be abolished by genetic or pharmacologic blockade of iNOS
Recent studies have demonstrated that PDAC tumor growth in orthotopic murine models is faster in tumors developed by inoculation of tumor cells plus CAFs when compared with tumor cells alone [9]. In our study, FC1245 pancreatic orthotopic tumor models were used to determine the role of non-RT and RT-activated CAFs in PDAC tumor growth in vivo. FC1245luc+ alone, FC1245luc+ mixed with non-RT CAFs, and FC1245luc+ mixed with RT-CAFs were implanted into the pancreas of C57BL/6 (B6) mice and tumor growth was evaluated using bioluminescence. As shown in Fig. 2G, the tumor growth rate was significantly higher in FC1245 cells mixed with RT-activated CAFs than that in the model developed by mixing FC1245 cells with non-RT activated CAFs or FC1245 cells alone (Fig. 2G). At 14 days after cells’ implantation in the pancreas, we observed significantly higher α-SMA+ in tumors developed by FC1245luc+ mixed with CAFs when compared with FC1245luc+ cells alone (Supplementary Fig. 10). To explain the pro-tumor effect of RT-activated CAFs, the influence of these cells on the growth of FC1245 cells was investigated in vitro using the Trypan blue assay. Enhanced FC1245 cell growth was observed after cells being cultured with CM of RT-activated CAFs (Fig. 2H). Culture of FC1245 with CM of non-RT CAFs did not interfere with PDAC cancer cell growth (Fig. 2H). Next we used the Boyden chamber (Fig. 2I) and scratch assays (Supplementary Fig. 11) to determine if RT-activated CAFs affected the migratory properties of FC1245 cells. Matrigel invasion chamber assays were also used to measure FC1245 invasion properties in the presence of CAFs (Fig. 2J). FC1245 cells in culture with CM of RT-CAFs demonstrated a significant increase in both migration and invasion properties as compared with FC1245 cells alone, which is consistent with previous studies [7]. Culture of FC1245 cells with CM of RT-CAFs showed a significantly larger number of migratory and invading cells compared with culture with CM of non-irradiated fibroblasts (Fig. 2I,J and Supplementary Fig. 11). We did not detect metastatic spread at 6 days after cells’ implantation in the pancreas (Supplementary Fig. 12). At 14 days, metastatic spread of tumor was observed in the liver, spleen and stomach in tumors developed by FC1245luc+ mixed with RT-CAFs (Supplementary Fig. 13).
Premised on our findings (Figs. 1,2), we expected that the pro-tumor effects of RT-activated CAFs are mediated by iNOS/NO signaling. Therefore, we sought to investigate FC1245 cells growth after iNOS amplification or exposure to physiologic levels of the NO donor diethylenetriamine NONOate (DETA-NO). DETA-NO has a half-life of 20 h and in aqueous solution it spontaneously decomposes to produce a long lasting NO release. Previous studies have reported that DETA-NO increases migration of lung carcinoma cells and proliferation of breast cancer cells [26, 42]. Others have reported that low concentrations of DETA-NO (20–2000 nM) induce cancer cell growth, while concentrations higher than 20 μM inhibit cell proliferation [34]. In our studies, NO induction in PDAC cells was performed using 1 μM of DETA-NO or with a clustered regularly interspaced short palindromic repeats (CRISPR) activation plasmid (Supplementary Figs. 14,15). Treatment of FC1245 pancreatic cancer cells with 1 μM of DETA-NO or with CRISPR activation plasmid increased NO levels and FC1245 cell growth, an effect that is abolished by the addition of the highly selective iNOS inhibitor 1400W [43] (Supplementary Figs. 14,15). To support the hypothesis that NO release from CAFs contributes to RT resistance of pancreatic carcinoma cells, NO induction in PDAC cells in the presence of RT-activated CAFs was blocked using small-interfering RNA (siRNA)-mediated knockdown of iNOS or pharmacologic treatments with 1400W (Fig. 2H, Supplementary Figs. 14-16). Additionally, the iNOS inhibitor 1400W decreased NFĸB induction in tumor cells cultured with CM RT-CAFs (Fig. 2E). These results indicate that CAFs can promote the growth of PDAC cells, an effect that increases when cells are cultured with CM of RT-activated CAFs, which can be abolished in vitro by iNOS depletion using genetic and pharmacologic approaches.
Pharmacologic blockade of iNOS improves therapeutic response of PDACs to RT
Previous studies demonstrated a role for iNOS in PDAC chemoresistance [20]. Our findings suggest iNOS/NO signaling as a cause of PDAC resistance to RT, with NO secretion from RT-activated CAFs preventing RT therapeutic efficacy. To translate our findings into a clinically relevant approach, we performed therapeutic studies combining iNOS pharmacologic inhibition with RT in orthotopic PDAC models. Luciferase-expressing FC1245 pancreatic cancer cells were implanted into the pancreas of C57BL/6 (B6) mice. To determine the impact of RT dose on PDAC therapy, a pilot therapeutic study was performed using 12 Gy in a single treatment fraction or 18 Gy administered in three fractions of 6 Gy at 48 h intervals. These dose schedules were selected because, per the linear quadratic model of cell kill, they have similar biologically effective dose (BED at an alpha/beta = 10, BED10). The three-fraction course of RT (BED10 = 28.8 Gy) improved survival when compared with a single fraction (BED10= 26.4 Gy) (Supplementary Fig. 17), supporting previous reports that are suggestive of the clinical benefit of RT administration in fractions [44]. In further therapeutic experiments, RT was administrated in 3 doses of 6 Gy at 48 h intervals. After an engraftment period, tumor-bearing mice as identified via bioluminescence were randomly assigned to treatment groups (non-RT, RT, 1400W, and 1400W/RT). At 14 days after the first RT dose, mice treated with 1400W or 1400W/RT demonstrated significantly decreased tumor growth and tumors of small size as measured by luminescence (Fig. 3A–C). The weight of tumors at 14 days after the first RT dose was 1.65 ± 0.60, 0.92 ± 0.04, and 0.60 ± 0.09 g in mice treated with RT, 1400W, and 1400W/RT (Supplementary Fig. 18).
Figure 3. Pharmacologic blockade of iNOS improves therapeutic response of PDACs to RT.
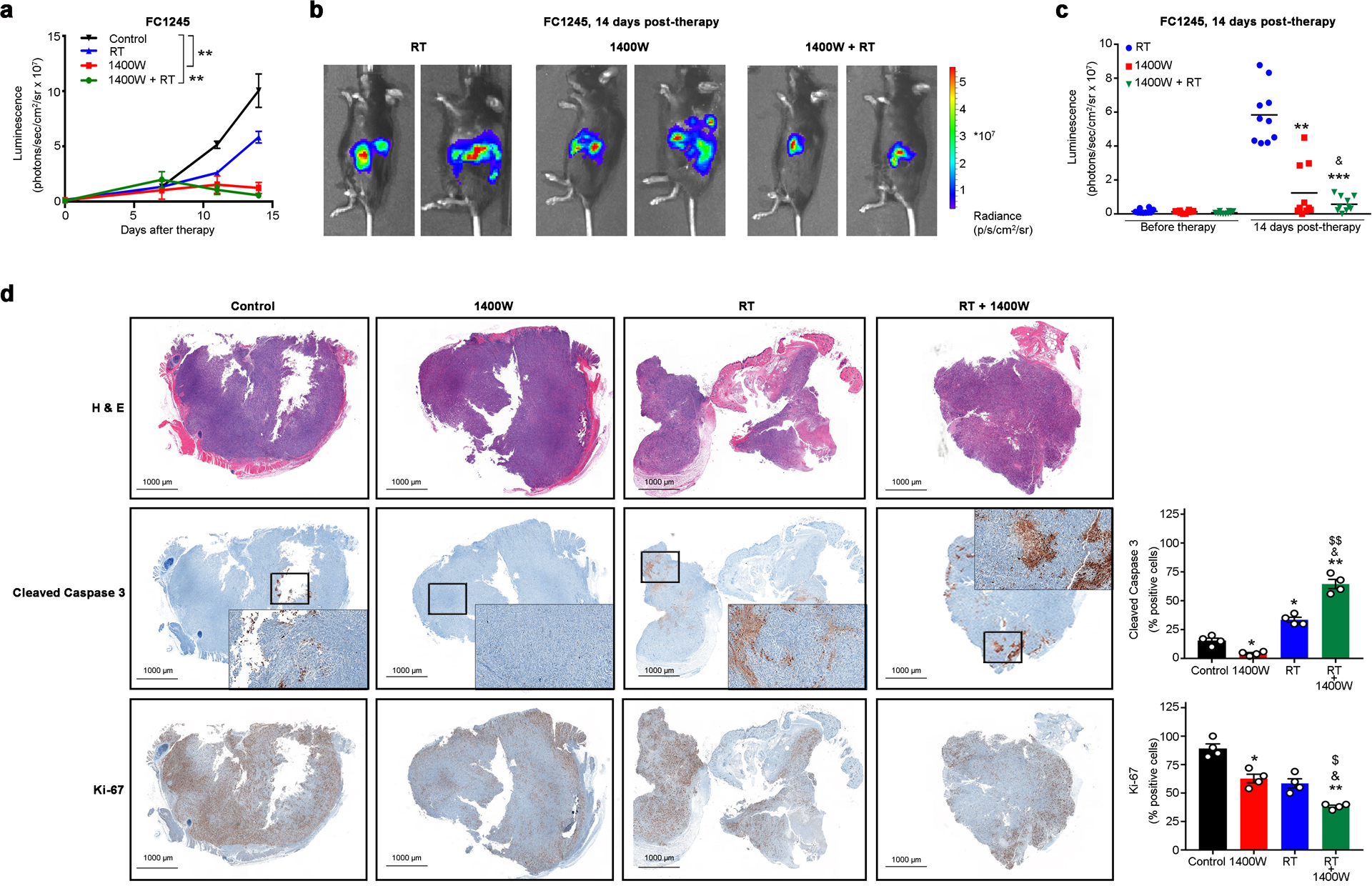
a-c) FC1245 luciferase-expressing PDAC orthotopic tumors treated with RT, 1400W or 1400W/RT (n = 10 mice per group) and tracked by bioluminescence. Mice were treated with 3 doses of 6 Gy at 48 h intervals. Intraperitoneal administration of 1400W (1 μg g−1) was started at day 0. 1400W was administrated every day for 7 days during the RT treatment period and every two days during two weeks after the RT treatment period. Real-time images from median of two animals (b) and luminescent signal (n = 10 mice per group) on day 14 post-therapy (c). **P<0.01 compared with control and based on a Student’s t-test (a). **P<0.01, ***P<0.01 compared with RT treated group and based on a Student’s t-test. &P<0.01 compared with 1400W treated group (c).
d) H&E, cleaved caspase 3, and Ki-67 immunohistochemistry and quantification (mean ± S.E.M, n = 3) in tumors from control, RT, 1400W, and 1400W/RT groups collected on day 14 post-therapy. *P<0.05 and **P<0.01 compared with control; &P<0.05 compared with RT; $P<0.05 and $$P <0.01 compared with 1400W and based on a Student’s t-test.
One of the downstream effects of NO in cancer includes evasion to apoptosis [24, 45] and therefore, tumor sections of the different therapeutic groups were immunostained to assess proliferation and apoptosis. The 1400W/RT treated mice displayed diminished proliferation of tumor cells based on the reduced number of Ki67+ tumor cells compared to control, 1400W, and RT groups (Fig. 3D). Immunohistochemical staining of cleaved caspase-3 indicated significantly increased apoptosis in pancreatic tumors treated with RT and 1400W/RT (Fig. 3D). Collectively, these results demonstrate that RT/iNOS inhibition slows pancreatic tumor growth.
NO secretion from RT-activated CAFs changes PDAC TME extracellular pH
One of the mechanisms by which NO modulates tumor cell proliferation is through upregulation of glycolysis [34]. NO release into tumor cells reduces mitochondrial oxidative phosphorylation at complexes III and IV of the electron transport chain and increases activity of the 6-phosphofructokinase 1 (PFK-1), which results in the activation of glycolysis promoting a metabolic switch from an aerobic to a glycolytic phenotype (Warburg effect) [34, 46–51]. We attempted to study changes in pancreatic cancer cells glycolysis after FC1245 cells incubation with the NO-donor DETA-NO or culture with CM of RT-activated CAFs. Our results show that NO production in FC1245 cells is increased through exposure to DETA-NO (Supplementary Fig. 14). Glucose consumption and lactate production (Supplementary Fig. 19) are increased compared with without DETA-NO addition. This effect was further inhibited by the addition of the iNOS inhibitor 1400W (Supplementary Fig. 19).
Since our results showed that DETA-NO increased the glycolytic rate when compared with untreated FC1245 cells, we hypothesized that NO release from CAFs could increase glucose consumption and lactate secretion in cancer cells. Cultures of FC1245 with CM of CAFs demonstrated that RT-activated CAFs led to a significant increase in glucose uptake (1.3 ± 0.1, mean ± S.E.M, n = 4; Fig. 4A) and lactate production (2.1 ± 0.6, mean ± S.E.M, n = 4; Fig. 4A). At 14 days post-RT therapy, we observed an increase in GLUT1 expression of PDAC tumors (1.7 ± 0.2, mean ± S.E.M, n = 3; Fig. 4B). Additional immunofluorescence staining of α-SMA, cytokeratin, and GLUT1 in RT-treated PDAC samples demonstrated GLUT1+/α-SMA+ in 8.4% of total α-SMA+ cells and GLUT1+/cytokeratin+ in 34.3% of total cytokeratin+ cells (Fig. 4C,D; Supplementary Tables 8-11). The percentage of GLUT1+/cytokeratin+ cells was 7.6-fold higher in RT-treated samples when compared with control (Fig. 4C,D; Supplementary Tables 8-11). We next used fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) to validate in vivo changes in GLUT1 and glucose consumption observed in Fig. 4A–D. Our biodistribution and PET imaging results at 14 days post first RT dose in mice bearing FC1245 orthotopic PDAC tumors demonstrated a significant increase in 18F-FDG PDAC uptake, an effect that decreases in mice treated with RT/1400W (Fig. 4E,F; Supplementary Fig. 20). As glycolysis promotes intracellular H+ generation contributing to the membrane dynamics of Na+/H+ flux, as mediated by the sodium/hydrogen exchanger isoform 1 (NHE1), an increase in glycolysis as mediated by NO release from RT-activated CAFs could activate NHE1. Our results revealed that RT increases NHE1 expression in FC1245 PDACs, an effect that was inhibited with the iNOS inhibitor 1400W (Fig. 4G). In sum, our findings indicate that an increase in iNOS/NO signaling in RT-activated fibroblasts upregulates PDAC glycolysis.
Figure 4. RT-activated CAFs change PDAC-TME extracellular pH allowing detection with pH-targeted pHLIP.

a) Glucose uptake and lactate production of FC1245 PDAC cells treated with CM from CAFs. Data are presented as mean ± S.E.M., n = 4. *P <0.05 compared with FC1245 and based on a Student’s t-test. NBDG 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose.
b-d) Western blot of GLUT1 (b) and immunofluorescence of GLUT1/α-SMA (c) and GLUT1/cytokeratin (d) in total lysates of FC1245 orthotopic PDAC tumors obtained at 14 days post-RT therapy. Density of western blot bands was quantified by scanning densitometry with ImageJ software.
e,f) PDAC tumor uptake (e) and MIPs PET images (f) at 2 h after injection of 18F-FDG in mice bearing FC1245 orthotopic tumors. At 14 days post-therapy, mice were administrated 200 μCi intravenous injection of 18F-FDG. Bars, n = 4 mice per group, mean±S.E.M. MIPs maximum intensity projection. %ID, percentage of injected dose.
g) Confocal images and quantification (mean ± S.E.M, n = 3) of immunofluorescence staining of NHE1 (green) in FC1245 orthotopic PDAC tumors treated with or without 5Gy RT. DAPI (blue) was used to stain cell nuclei. Scale bars: 50 μm.
h,i) PDAC tumor uptake as determined by acute biodistribution studies at 24 h after injection of 67Ga-labeled pHLIP® in mice bearing FC1245 orthotopic tumors. At 14 days post-RT, mice were intravenously given 20 μCi of 67Ga-labeled pHLIP®. Bars, n = 4 mice per group, mean±S.E.M. %ID g−1, percentage of injected dose per gram.
j) An increase in glycolysis as mediated by NO release from RT-activated CAFs activates NHE1. Activation of NHE1 causes a reversal of the plasma membrane pH gradient resulting in a more alkaline intracellular pH and a more acidic extracellular pH that can be detected using pH-targeted molecular imaging with the pH (low) insertion peptide (pHLIP®) technology.
pH-targeted pHLIP® allows assessment of RT/1400W response within PDAC
Activation of NHE1 causes a reversal of the plasma membrane pH gradient [52], resulting in a more alkaline intracellular pH and a more acidic extracellular pH (Fig. 4H–J). As we observed an increase in GLUT1 and NHE1 in RT-treated tumors (Fig. 4A–G), we attempted to use a pH-targeted imaging probe to detect changes in the extracellular microenvironment pH, as mediated by NO secretion from RT-activated CAFs. pH (Low) Insertion Peptide (pHLIP®) allows detection of tumor cell surface acidity (Fig. 4H) [53]. We performed in vivo biodistribution studies with the radiometallated pHLIP variant [67Ga]Ga-NO2A-cysVar3 in mice bearing FC1245 orthotopic PDAC tumors. Based on our studies demonstrating that at 14 days post-therapy the tumor growth was slowed in mice treated with 1400W/RT when compared with RT alone (Fig. 3A–C), mice were administrated with [67Ga]Ga-NO2A-cysVar3 at 14 days post first RT dose. At 24 h post-injection of [67Ga]Ga-NO2A-cysVar3, PDAC RT-treated tumors had an uptake of 8.6 ± 0.7 percentage injected dose per gram tissue (%ID/g, mean ± S.E.M, n = 5), while control tumors had an uptake of 3.6 ± 1.9 % ID/g, mean ± S.E.M, n = 5 (Fig. 4I and Supplementary Fig. 21). The increase in [67Ga]Ga-NO2A-cysVar3 PDAC tumor uptake, due to an increase in extracellular acidity, was further decreased in mice treated with RT plus 1400W (3.1 ID/g ± 1.0, mean ± S.E.M, n = 5, Fig. 4I). Taken together, these results suggest that NO signaling activation in RT-treated tumors will result in changes of the PDAC extracellular pH (Fig. 4J) that can be detected using radiolabeled pHLIP®, suggesting a clinical potential of pHLIP® to assess RT response within pancreatic cancer, and suggesting ultimately that the benefit of RT intervention in PDAC can be improved by blocking NO signaling.
DISCUSSION
The mechanisms of iNOS/NO signaling and their actions on PDAC response to RT remain to be completely understood. Here, we demonstrate that RT enhances iNOS expression and subsequent NO secretion in PDAC. Mechanistic studies demonstrated that the CM of irradiated CAFs drives the production of iNOS in PDACs. Cultures of PDAC cells with CM of RT-activated CAFs demonstrate that NO secretion and iNOS expression by RT-CAFs are further induced by pancreatic tumor cells through NFĸB, which in turn leads to the release of inflammatory cytokines by the tumor cells. In addition, we found that NO increases after RT in PDAC, results in acidification of the microenvironment, which can be detected by molecular imaging using pHLIP®. These data support that NO has a pro-tumor function in irradiated PDACs by promoting a glycolytic phenotype as detected in vivo by 18F-FDG PET imaging, which leads to extracellular acidosis. iNOS pharmacological inhibition or knockdown improved RT efficacy and decreased PDAC tumor growth, suggesting an integral role for NO and RT-mediated iNOS activity in PDAC therapy. Until our study, a role for RT-mediated iNOS/NO signaling in PDAC remained unexplored.
NOS exist as three isoforms: inducible NOS (iNOS or NOS2), neuronal NOS (nNOS or NOS1), and endothelial NOS (eNOS or NOS3). nNOS and eNOS produce low amounts of NO and are constitutively expressed by neuronal and vascular endothelial cells. iNOS produce a higher and sustained level of NO and are transcriptionally regulated and induced by inflammatory cytokines [21, 22, 29]. The expression of iNOS and production of NO have been detected in several tumor types and are comprehensively reviewed elsewhere [22]. Our findings show that, in contrast to PDAC samples, non-tumor tissues exhibit lower iNOS expression levels (Figs. 1,2) [20, 28]. Previous studies have demonstrated that iNOS expression is found not only in the tumor, but also in CAFs, inflammatory, and endothelial cells [29]. Further studies are necessary to evaluate the consequences of RT on NOS expression in other non-cancer cells that contribute to the stromal compartment of the PDAC TME.
While some studies have demonstrated that tumor-cell-derived NO inhibits tumor progression [22, 23, 26, 42, 54, 55], others have demonstrated its role in the development of tumor growth [22, 56, 57]. Similar to what has been observed for other tumor types, the role NO plays in PDAC is also not clearly defined. PDAC tumor cells containing low levels of NO exhibit an aggressive metastatic phenotype when compared with tumor cells containing high NO levels [21] and treatment with a NO-donor inhibited proliferation and invasion of PDAC [55]. Other studies have reported an association between high iNOS expression and poorer survival in PDAC patients [20] and a role for iNOS/NO signaling in PDAC chemoresistance [29]. Conversely, low iNOS levels are associated with enhanced survival in KPC mice containing orthotopic PDAC [20]. Our studies clearly establish that RT increases iNOS expression and NO signaling in PDAC contributing to tumor resistance to therapy.
The contribution of NO to tumor progression depends on its source: tumor cells, tumor-associated stromal fibroblasts, and immune cells. RT-mediated cellular damage of surviving CAFs [6, 16] alters the paracrine interactions between tumor and stroma through alterations in inflammatory secretome [6]. To determine the cellular sources responsible for iNOS/NO increase in PDAC after RT, we performed in vitro studies using pancreatic tumor cells in culture with CAFs in a transwell co-culture system or with CM of these fibroblasts. Our data show that, although undetectable in RT-activated tumor cells, RT-CAFs showed significant iNOS expression and NO secretion (Fig. 2B–D). In our experiments, an extra in vitro RT dose was necessary to study the mechanisms by which RT-CAFs induce iNOS/NO secretion in the tumors cells (Fig. 2). When compared with non-RT CAFs, RT-CAFs demonstrated upregulation in IFNβ, iNOS, and TNFα (Supplementary Fig. 9). Co-culture of PDAC cells with RT-CAFs and culture with CM of CAFs increased iNOS expression/NO secretion in the tumors cells (Fig. 2C,D). The increase in iNOS expression and NO secretion by the tumor cells was higher when cells were cultured with CAFs-conditioned media when compared with cells co-cultured with CAFs (Fig. 2C,D). These results suggest that the secretome of RT-CAFs is the major driving force in the production of iNOS in PDACs. The differences in iNOS/NO between FC1245 co-cultures and cultures with CAFs-conditioned media can be explained by changes in PDAC cells viability (Supplementary Fig. 6) and future studies are necessary to determine the temporal dynamics of iNOS/NO signaling in these in vitro culture systems.
RT-activated CAFs increased PDAC cell growth, migration, invasion, and metastatic spread of tumor (Fig. 2G–J, Supplementary Fig. 13). Blocking of NO release, by iNOS inhibition with 1400W or using knockdown via siRNA, abolished the pro-tumor effect of RT-activated CAFs. Admittedly, while we successfully demonstrate that CAFs play a role in the RT-mediated increase of iNOS/NO signaling in PDAC in vitro, this system does not fully reflect the complexity and cellular diversity of the PDAC microenvironment in vivo.
Since NO regulates several biochemical pathways, the molecular mechanisms by which NO increases in CAFs after RT, and how iNOS/NO signaling can be further induced by the tumor cells facilitating tumor growth (Fig. 2E) are likely broad. At the transcriptional level, TNFα, IFN-γ, IL-6, and IL-1β are all known to activate NFĸB, which induces iNOS expression [22, 29, 30]. Here, we show that the pro-tumor inducing effect of RT-activated fibroblasts occurs through NFĸB and involves increased secretion of IFNγ, IL1β, TNFα, IL-6, and LIF (an IL-6 class cytokine) by pancreatic tumor cells. Additionally, our studies demonstrate that the iNOS inhibitor 1400W decreases NFĸB induction in PDACs cultured with secretome from RT-CAFs.
Preclinical and clinical studies have shown low toxicity of iNOS inhibitors [33, 58–60], suggesting their potential as therapeutic agents. For instance, iNOS inhibitors have been show to block colon adenocarcinoma [61] and glioma tumor stem cell tumor growth [33]. Our findings that RT increases iNOS expression, suggests iNOS inhibition as a promising strategy that could be applied in the treatment of PDAC. In our studies, the iNOS inhibitor 1400W delayed tumor growth, an effect that was enhanced with the 1400W/RT combination therapy (Fig. 3A–C). Importantly, the 1400W/RT therapeutic approach shows potential to increase preservation of healthy tissues during surgical PDAC interventions due to the observation of increasingly localized tumors with less diffuse margins (Fig. 3B). The 1400W/RT combination therapy was unable to completely eliminate tumor growth and further studies are necessary to determine whether other iNOS inhibitors or combination of iNOS inhibition with other treatment regimens may further augment efficacy when compared with 1400W/RT. Previous reports in orthotopic intracranial tumors demonstrated higher therapeutic efficacy for the iNOS inhibitor BYK191023 when compared with 1400W [33] plausibly due to its improved pharmacokinetic and bioavailability properties.
In ovarian cancer cells, NO decreases mitochondrial respiration, which results in the activation of glycolysis promoting a metabolic switch from an aerobic to a glycolytic phenotype [34]. Glycolysis promotes H+ generation, which is removed from the intracellular to the extracellular compartments in exchange with Na+, by NHE1 [52]. In our studies, we observed that NO increases glycolytic rate and NHE1 expression in PDAC (Fig. 4A–G). These membrane transport mechanisms contribute to the acidification of the extracellular space. Equipped with this information, we used a radiolabeled peptide, [67Ga]Ga-NO2A-cysVar3 pHLIP® to probe changes in the TME extracellular pH arising from RT-mediated NO secretion (Fig. 4H,I). The var3 pHLIP® was used in our studies due to its remarkable ability to target acidity in vivo [53, 62–64] and a radiolabeled version of var3 pHLIP® will be entering in the clinic in the current year of 2020. The radiolabeled var3 pHLIP® demonstrated higher targeting accumulation in RT PDAC tumors when compared to unirradiated PDAC tumors (Fig. 4I). This increase in acidity, due to NO secretion from RT-activated PDACs, was further decreased when RT is combined with the iNOS inhibitor 1400W.
In conclusion, our data support the notion that the secretome of RT-CAFs initiates a paracrine activation loop increasing iNOS/NO signaling in PDAC and additional in vitro radiation is necessary for robust iNOS expression by the tumor cells. Our preclinical results demonstrate the potential of iNOS inhibition with fractionated RT regimen for improved response, and additionally that that response can be assessed via radiolabeled pHLIP, highlighting the mechanisms by which NO in the secretome of RT-activated CAFs contributes to PDAC resistance. Our study supports previous reports demonstrating that the secretome of therapy-activated CAFs contribute to PDAC pathology and that targeting iNOS/NO signaling may be a valid therapeutic approach for improving RT outcomes in PDAC.
Supplementary Material
SIGNIFICANCE.
A radiolabeled pH-targeted peptide can be used as a PET imaging tool to assess therapy response within PDAC, and blocking iNOS/NO signaling may improve radiation therapy outcomes.
Acknowledgements
The authors acknowledge the Radiochemistry and Molecular Imaging Probe Core, the Anti-tumor Assessment Core and Molecular Cytology Core Facility, which were supported by NIH, grant P30 CA08748. This study was supported in part by the Geoffrey Beene Cancer Research Center of MSKCC (JSL) and NIH NCI R35 CA232130 (JSL). We gratefully acknowledge Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Center for Experimental Therapeutics of MSKCC. PMRP acknowledges the Tow Foundation Postdoctoral Fellowship from the MSKCC Center for Molecular Imaging and Nanotechnology and the Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center of MSKCC. LMC acknowledges support from the Ruth L. Kirschstein National Research Service Award postdoctoral fellowship (NIH F32-EB025050). TM and LFC are supported by Swim Across America, Ludwig Institute for Cancer Research, Ludwig Center for Cancer Immunotherapy at Memorial Sloan Kettering, Cancer Research Institute, Parker Institute for Cancer Immunotherapy and Breast Cancer Research Foundation. Figures 2A, 4H, and 4J were created with Biorender. We thank Dr. Russel and Dr. Monette for their help with the Arc Therapy and image analysis, respectively. We thank Egger for cutting the human samples used in this work. We thank Carl DeSelm in the Michel Sadelain lab for generating the FC1245luc+ PDAC cells.
Disclosure
Lewis J. is founder of pHLIP, Inc. He has shares in the company, but the company did not fund any part of the work reported herein, which was done in his academic laboratory.
T.M. is a cofounder and holds equity in IMVAQ Therapeutics. He is a consultant of Immunos Therapeutics and Pfizer. He has research support from Bristol-Myers Squibb; Surface Oncology; Kyn Therapeutics; Infinity Pharmaceuticals, Inc.; Peregrine Pharmaceuticals, Inc.; Adaptive Biotechnologies; Leap Therapeutics, Inc.; and Aprea. He has patents on applications related to work on oncolytic viral therapy, alpha virus-based vaccine, neo antigen modeling, CD40, GITR, OX40, PD-1, and CTLA-4.
REFERENCES
- 1.Siegel RL; Miller KD; Jemal A Cancer statistics, 2019. CA Cancer J Clin 2019, 69, 7–34. [DOI] [PubMed] [Google Scholar]
- 2.Kleeff J; Korc M; Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016, 2. [DOI] [PubMed] [Google Scholar]
- 3.Conlon KC; Klimstra DS; Brennan MF Long-term survival after curative resection for pancreatic ductal adenocarcinoma. Clinicopathologic analysis of 5-year survivors. Ann Surg. 1996, 223, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neoptolemos JP; Stocken DD; Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004, 350, 1200–1210. [DOI] [PubMed] [Google Scholar]
- 5.Bhide SA; Nutting CM Recent advances in radiotherapy. Bmc Med. 2010, 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tommelein J; De Vlieghere E; Verset L, et al. Radiotherapy-activated cancer-associated fibroblasts promote tumor progression through paracrine IGF1R activation. Cancer Res. 2018, 78, 659–670. [DOI] [PubMed] [Google Scholar]
- 7.Ohuchida K; Mizumoto K; Murakami M, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004, 64, 3215–3222. [DOI] [PubMed] [Google Scholar]
- 8.Hellevik T; Pettersen I; Berg V, et al. Changes in the secretory profile of NSCLC-associated fibroblasts after ablative radiotherapy: potential impact on angiogenesis and tumor growth. Transl Oncol. 2013, 6, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ligorio M; Sil S; Malagon-Lopez J, et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell. 2019, 178, 160–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gukovsky I; Li N; Todoric J; Gukovskaya A; Karin M Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013, 144, 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seifert L; Werba G; Tiwari S, et al. Radiation therapy induces macrophages to suppress T-cell responses against pancreatic tumors in mice. Gastroenterology. 2016, 150, 1659–1672 e1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hallahan DE; Spriggs DR; Beckett MA; Kufe DW; Weichselbaum RR Increased tumor necrosis factor-alpha mRNA after cellular exposure to ionizing radiation. PNAS. 1989, 86, 10104–10107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burnette BC; Liang H; Lee Y, et al. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lugade AA; Sorensen EW; Gerber SA; Moran JP; Frelinger JG; Lord EM Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008, 180, 3132–3139. [DOI] [PubMed] [Google Scholar]
- 15.Mantoni TS; Lunardi S; Al-Assar O; Masamune A; Brunner TB Pancreatic stellate cells radioprotect pancreatic cancer cells through β1-integrin signaling. Cancer Res. 2011, 71, 3453–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hellevik T; Martinez-Zubiaurre I Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front Oncol. 2014, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagares-Garcia JA; Moore RA; Collier B; Heggere M; Diaz F; Qian F Nitric oxide synthase as a marker in colorectal carcinoma. Am Surg. 2001, 67, 709–713. [PubMed] [Google Scholar]
- 18.Ekmekcioglu S; Ellerhorst JA; Prieto VG; Johnson MM; Broemeling LD; Grimm EA Tumor iNOS predicts poor survival for stage III melanoma patients. Int J Cancer. 2006, 119, 861–866. [DOI] [PubMed] [Google Scholar]
- 19.Glynn SA; Boersma BJ; Dorsey TH, et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J Clin Invest. 2010, 120, 3843–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J; He PJ; Gaida M, et al. Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer. Oncotarget. 2016, 7, 52993–53004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang B; Wei DY; Crum VE, et al. A novel model system for studying the double-edged roles of nitric oxide production in pancreatic cancer growth and metastasis. Oncogene. 2003, 22, 1771–1782. [DOI] [PubMed] [Google Scholar]
- 22.Fukumura D; Kashiwagi S; Jain RK The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006, 6, 521–534. [DOI] [PubMed] [Google Scholar]
- 23.Bailey P; Chang DK; Forget MA, et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci Rep. 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engels K; Knauer SK; Loibl S, et al. NO signaling confers cytoprotectivity through the survivin network in ovarian carcinomas. Cancer Res. 2008, 68, 5159–5166. [DOI] [PubMed] [Google Scholar]
- 25.Rieder J; Jahnke R; Schloesser M, et al. Nitric oxide-dependent apoptosis in ovarian carcinoma cell lines. Gynecol Oncol. 2001, 82, 172–176. [DOI] [PubMed] [Google Scholar]
- 26.Pervin S; Singh R; Hernandez E; Wu GY; Chaudhuri G Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: Involvement of mammalian target of Rapamycin/eIF4E pathway. Cancer Res. 2007, 67, 289–299. [DOI] [PubMed] [Google Scholar]
- 27.Kisley LR; Barrett BS; Bauer AK, et al. Genetic ablation of inducible nitric oxide synthase decreases mouse lung tumorigenesis. Cancer Res. 2002, 62, 6850–6856. [PubMed] [Google Scholar]
- 28.Vickers SM; MacMillan-Crow LA; Green M; Ellis C; Thompson JA Association of increased immunostaining for inducible nitric oxide synthase and nitrotyrosine with fibroblast growth factor transformation in pancreatic cancer. Arch Surg. 1999, 134, 245–251. [DOI] [PubMed] [Google Scholar]
- 29.Müerköster S; Wegehenkel K; Arlt A, et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1β. Cancer Res. 2004, 64, 1331–1337. [DOI] [PubMed] [Google Scholar]
- 30.Arlt A; Vorndamm J; Muerkoster S, et al. Autocrine production of interleukin-1-β confers constitutive NFκB activity and chemoresistance in pancreatic carcinoma cell lines. Cancer Res. 2002, 62, 910–916. [PubMed] [Google Scholar]
- 31.Khalili JS; Liu SJ; Rodriguez-Cruz TG, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012, 18, 5329–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du Q; Zhang X; Liu Q; Zhang X; Bartels CE; Geller DA Nitric oxide production upregulates Wnt/β-catenin signaling by inhibiting Dickkopf-1. Cancer Res. 2013, 73, 6526–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eyler CE; Wu QL; Yan K, et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011, 146, 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caneba CA; Yang L; Baddour J, et al. Nitric oxide is a positive regulator of the Warburg effect in ovarian cancer cells. Cell Death Dis. 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeSelm C; Palomba ML; Yahalom J, et al. Low-Dose radiation conditioning enables CAR T cells to mitigate antigen escape. Mol Ther. 2018, 26, 2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Escorcia FE; Houghton JL; Abdel-Atti D, et al. ImmunoPET predicts response to Met-targeted radioligand therapy in models of pancreatic cancer resistant to Met kinase inhibitors. Theranostics. 2019, 10, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorek DLJ; Kramer RM; Chen Q, et al. Reverse-contrast imaging and targeted radiation therapy of advanced pancreatic cancer models. Int J Radiat Oncol Biol Phys. 2015, 93, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hruban RH; Adsay NV; Albores-Saavedra J, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: Consensus report and recommendations. Cancer Res. 2006, 66, 95–106. [DOI] [PubMed] [Google Scholar]
- 39.Hingorani SR; Wang L; Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005, 7, 469–483. [DOI] [PubMed] [Google Scholar]
- 40.Erkan M; Michalski CW; Rieder S, et al. The Activated Stroma Index Is a Novel and Independent Prognostic Marker in Pancreatic Ductal Adenocarcinoma. Clin Gastroenterol Hepatol. 2008, 6, 1155–1161. [DOI] [PubMed] [Google Scholar]
- 41.Erkan M; Hausmann S; Michalski CW, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012, 9, 454–467. [DOI] [PubMed] [Google Scholar]
- 42.Sanuphan A; Chunhacha P; Pongrakhananon V; Chanvorachote P Long-term nitric oxide exposure enhances lung cancer cell migration. Biomed Res Int. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garvey EP; Oplinger JA; Furfine ES, et al. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997, 272, 4959–4963. [DOI] [PubMed] [Google Scholar]
- 44.Seifert L; Werba G; Tiwari S, et al. Radiation therapy induces macrophages to suppress T-cell responses against pancreatic tumors in mice. Gastroenterology. 2016, 150, 1659–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levesque MC; Misukonis MA; O’Loughlin CW, et al. IL-4 and interferon gamma regulate expression of inducible nitric oxide synthase in chronic lymphocytic leukemia cells. Leukemia. 2003, 17, 442–450. [DOI] [PubMed] [Google Scholar]
- 46.Sanhueza C; Araos J; Naranjo L, et al. NHE1 promote cell proliferation in ovarian cancer: a role of hypoxia-inducible factors. Int J Gynecol Cancer. 2015, 25, 55–56. [DOI] [PubMed] [Google Scholar]
- 47.Sanhueza C; Araos J; Naranjo L, et al. Modulation of intracellular pH in human ovarian cancer. Curr Mol Med. 2016, 16, 23–32. [DOI] [PubMed] [Google Scholar]
- 48.Spugnini EP; Sonveaux P; Stock C, et al. Proton channels and exchangers in cancer. Bba-Biomembranes. 2015, 1848, 2715–2726. [DOI] [PubMed] [Google Scholar]
- 49.Granados-Principal S; Liu Y; Guevara ML, et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harhaji L; Popadic D; Miljkovic D; Cvetkovic I; Isakovic A; Trajkovic V Acidosis affects tumor cell survival through modulation of nitric oxide release. Free Radic Biol Med. 2006, 40, 226–235. [DOI] [PubMed] [Google Scholar]
- 51.Riemann A; Ihling A; Thomas J; Schneider B; Thews O; Gekle M Acidic environment activates inflammatory programs in fibroblasts via a cAMP-MAPK pathway. Bba-Mol Cell Res. 2015, 1853, 299–307. [DOI] [PubMed] [Google Scholar]
- 52.Orlowski J; Grinstein S Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflügers Arch Eur J Physiol. 2004, 447, 549–565. [DOI] [PubMed] [Google Scholar]
- 53.Demoin DW; Wyatt LC; Edwards KJ, et al. PET Imaging of extracellular pH in tumors with 64Cu- and 18F-labeled pHLIP peptides: a structure-activity optimization study. Bioconjug Chem. 2016, 27, 2014–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arcos MLB; Gorelik G; Klecha A; Goren N; Cerquetti C; Cremaschi GA Inducible nitric oxide synthase-mediated proliferation of a T lymphoma cell line. Nitric Oxide. 2003, 8, 111–118. [DOI] [PubMed] [Google Scholar]
- 55.Sugita H; Kaneki M; Furuhashi S; Hirota M; Takamori H; Baba H Nitric oxide inhibits the proliferation and invasion of pancreatic cancer cells through degradation of insulin receptor substrate-1 protein. Mol Cancer Res. 2010, 8, 1152–1163. [DOI] [PubMed] [Google Scholar]
- 56.Le XD; Wei DY; Huang SY; Lancaster JR; Xie KP Nitric oxide synthase II suppresses the growth and metastasis of human cancer regardless of its up-regulation of protumor factors. Proc Natl Acad Sci USA. 2005, 102, 8758–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nunokawa Y; Tanaka S Interferon-γ inhibits proliferation of rat vascular smooth-muscle cells by nitric-oxide generation. Biochem Bioph Res Commun. 1992, 188, 409–415. [DOI] [PubMed] [Google Scholar]
- 58.Brindicci C; Ito K; Torre O; Barnes PJ; Kharitonov SA Effects of aminoguanidine, an inhibitor of inducible nitric oxide synthase, on nitric oxide production and its metabolites in healthy control subjects, healthy smokers, and COPD patients. Chest 2009, 135, 353–367. [DOI] [PubMed] [Google Scholar]
- 59.Dover AR; Chia S; Ferguson JW; Megson IL; Fox KAA; Newby DE Inducible nitric oxide synthase activity does not contribute to the maintenance of peripheral vascular tone in patients with heart failure. Clin Sci. 2006, 111, 275–280. [DOI] [PubMed] [Google Scholar]
- 60.Singh D; Richards D; Knowles RG, et al. Selective inducible nitric oxide synthase inhibition has no effect on allergen challenge in asthma. Am J Resp Crit Care Med. 2007, 176, 988–993. [DOI] [PubMed] [Google Scholar]
- 61.Thomsen LL; Scott JMJ; Topley P; Knowles RG; Keerie AJ; Frend AJ Selective inhibition of inducible nitric oxide synthase inhibits tumor growth in vivo: Studies with 1400W, a novel inhibitor. Cancer Res. 1997, 57, 3300–3304. [PubMed] [Google Scholar]
- 62.Weerakkody D; Moshnikova A; Thakur MS, et al. Family of pH (low) insertion peptides for tumor targeting. P Natl Acad Sci USA. 2013, 110, 5834–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tapmeier TT; Moshnikova A; Beech J, et al. The pH low insertion peptide pHLIP Variant 3 as a novel marker of acidic malignant lesions. P Natl Acad Sci USA. 2015, 112, 9710–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roberts S; Strome A; Choi C, et al. Acid specific dark quencher QC1 pHLIP for multi-spectral optoacoustic diagnoses of breast cancer. Sci Rep. 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.