

Abstract
Oculodentodigital dysplasia (ODDD) is a rare genetic disorder associated with a characteristic craniofacial profile with variable dental, limb, eye, and ocular adnexa abnormalities. We performed an extensive literature review to highlight key eye features in patients with ODDD and report a new case of a female patient with a heterozygous missense GJA1 mutation (c.65G>A, p.G22E) and clinical features consistent with the condition. Our patient presented with multiple congenital anomalies including syndactyly, microphthalmia, microcornea, retrognathia, and a small nose with hypoplastic alae and prominent columella; in addition, an omphalocele defect was present, which has not been reported in previous cases. A systematic review of the published cases to date revealed 91 literature reports of 295 individuals with ODDD. There were 73 different GJA1 mutations associated with these cases, of which the most common were the following missense mutations: c.605G>A (p.R202H) (11%), c.389T>C (p.I130T) (10%), and c.119C>T (p.A40V) (10%). Mutations most commonly affect the extracellular-1 and cytoplasmic-1 domains of connexin-43 (gene product of GJA1), predominately manifesting in microphthalmia and microcornea. The syndrome appears with an approximately equal sex ratio. The most common eye features reported among all mutations were microcornea, microphthalmia, short palpebral fissures, and glaucoma.
1. Introduction
Oculodentodigital dysplasia (ODDD, OMIM #164200) is a rare disorder mainly characterized by abnormal craniofacial, dental, ocular, and digital development. The autosomal dominant form has been the most frequently reported inheritance pattern, although a few cases of autosomal recessive inheritance have been described [1–3]. Craniofacial abnormalities may include microcephaly, prominent columella, and underdeveloped nasal alae [2–4]. Dental abnormalities, such as hypoplastic enamel, small teeth, and premature loss of teeth, are often present [2–4]. Digit abnormalities may include syndactyly, camptodactyly, and midphalangeal hypoplasia [2–4]. Ophthalmic manifestations are common, such as microcornea and microphthalmia, and may involve a wide spectrum of eye and ocular adnexa structures, although previous analyses of prior cases show that full ocular physical exams were not performed on all patients [3, 5].
The gap junction protein alpha 1 (GJA1) gene codes for connexin-43, which is a protein that assists in the transmembrane transport of molecules through gap junctions, and mutations in the GJA1 may cause an alteration of the channel conduction properties [1–3, 6]. We report a case of an 8-month-old female patient with an identified GJA1 mutation and common clinical features associated with ODDD. This patient had an omphalocele at birth, which has not been reported in previous cases. Her eye features included microphthalmia, microcornea, narrow palpebral fissures, blonde fundus, deep anterior chambers, hyperopia, and epiphora in both eyes secondary to bilateral nasolacrimal duct obstructions. We conducted an extensive literature review to summarize the eye features in patients with ODDD reported to date.
2. Case Report
The patient, an 8-month-old female, was born to a nonconsanguineous couple from a healthy 37-year-old mother of Native American descent and a healthy 30-year-old father of German and Irish descent. Family history is notable for an older sibling with cleft palate, paternal uncle with autism, paternal second cousin with congenital heart defect, and distant paternal great-great uncle with Down syndrome and webbed/fused 4th and 5th digits of one hand. A normal pregnancy was noted until the second trimester when an omphalocele was detected on ultrasound. A subsequent ultrasound revealed possible syndactyly of the hands. The patient was born at 39 weeks by vaginal delivery with induction. The birth weight was 3.552 kg (75th percentile), birth length was 50 cm (68th percentile), and birth head circumference was 34.5 cm (70th percentile). Apgar scores were 9 at both one minute and five minutes.
Multiple congenital anomalies noted at birth included an omphalocele that measured 4 cm at base and 3.5 cm across with intestines present in the sac, but no liver. The patient had a normocephalic head with sparse wispy hair, a small nose with hypoplastic alae, a prominent columella, small-appearing palpebral fissures, a small cornea, microphthalmia, a wide anterior fontanelle, and retrognathia (Figure 1). Syndactyly of digits 4 and 5 and webbing of digits 3 and 4 of the right (Figure 2) and left hands were present. Cardiac echocardiogram on the day of birth showed the presence of a mild patent ductus arteriosus, mild patent foramen ovale, and a normal aorta. Feeding difficulties were exacerbated by the presence of the omphalocele; surgical correction was performed on day 2 of life.
Figure 1.
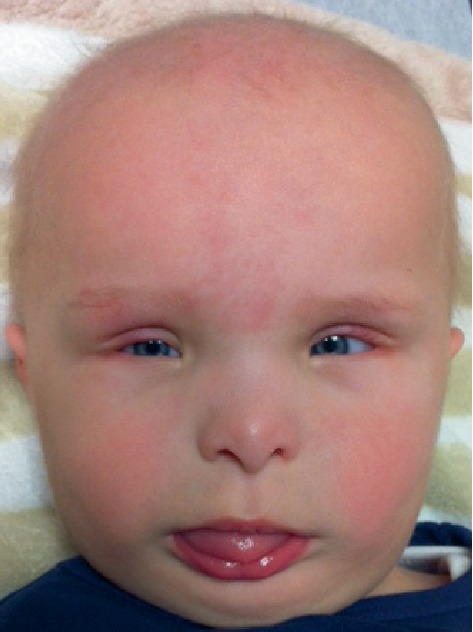
Facial photograph of a patient with oculodentodigital dysplasia; note the beaked nose with hypoplastic alae and prominent columella, microphthalmia, microcornea, small palpebral fissures, retrognathia.
Figure 2.
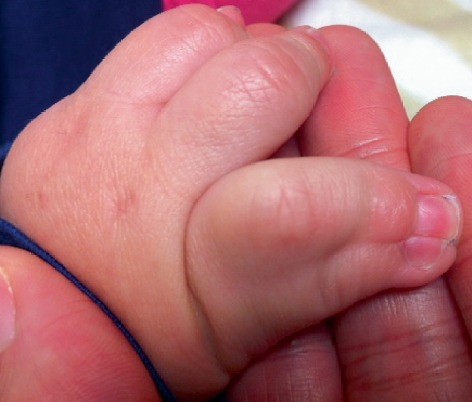
Complete syndactyly of the 4th and 5th digits of the right hand.
An ophthalmologic assessment at 4 months of age was notable for deep anterior chambers, bilateral nasolacrimal duct obstruction, microphthalmia, small 8 mm corneas, a blonde fundus, and moderate hyperopia in both eyes.
At her last examination at 8 months of age, the patient continues to have poor feeding with self-limiting volumes but has improved weight gain. The patient is at the 9th percentile for weight and 12th percentile for length. Cognitive and motor developments are delayed.
Sequencing of the GJA1 gene (transcript number: NM_000165.3) from patient genomic DNA revealed a heterozygous missense mutation in the GJA1 gene: c.65G>A (p.G22E). Deletion/duplication analysis of the GJA1 gene using the aCGH test was negative.
3. Methods
We performed a systematic review of the literature to summarize the ocular findings in individuals with ODDD. A PubMed/Medline search of “oculodentodigital syndrome” led us to find a total of 177 articles. No articles were excluded based on the year published. We reviewed the references to identify other articles that did not appear in our original search. 91 articles describing patients with a description consistent with the clinical syndrome, either with or without molecular confirmation of GJA1 pathogenic variants, were included. Within these selected articles, we identified 295 cases of ODDD with 73 different GJA1 mutations, including those that exhibited features of ODDD in the absence of molecular confirmation. Such individuals were either clinically diagnosed or were relatives of individuals with molecularly confirmed GJA1 pathogenic variants. Twelve reported that GJA1 gene coding alterations were omitted due to insufficient clinical information and data reported and are listed in Table 1 [3, 6].
Table 1.
GJA1 variants without clinical information.
| Sources | GJA1 variant | Cases | |
|---|---|---|---|
| Nucleotide | Protein | ||
| Paznekas et al. [3] | c.7G>A | p.D3N | 1 |
| Paznekas et al. [3] | c.64G>A | p.G22R | 1 |
| Paznekas et al. [3]; Richardson et al. [6] | c.79T>C | p.S27P | 1 |
| Paznekas et al. [3] | c.163A>G | p.N55D | 1 |
| Paznekas et al. [3] | c.174A>C | p.Q58H | 1 |
| Paznekas et al. [3] | c.175C>G | p.P59A | 1 |
| Paznekas et al. [3] | c.221A>T | p.H74L | 1 |
| Paznekas et al. [3] | c.428G>A | p.G143D | 1 |
| Paznekas et al. [3] | c.430A>G | p.K144E | 1 |
| Paznekas et al. [3] | c.434T>G | p.V145G | 1 |
| Paznekas et al. [3] | c.442C>G | p.R148G | 1 |
| Paznekas et al. [3] | c.578C>T | p.P193L | 1 |
4. Discussion
Oculodentodigital dysplasia (ODDD) is a rare congenital disorder manifested with developmental anomalies of the eyes, face, dentition, heart, skeletal system, and digits. The syndrome appears to be more common in Caucasian populations with an equal sex ratio [3]. Heterozygous mutation of the GJA1 gene located at chromosome 6q22.31 has been identified as the most common mutation resulting in ODDD [2, 3]. However, a compound heterozygous individual with missense mutations demonstrated mutations in the GJA1 gene (p.V41L) and the GJB2 gene (p.R127H), which encode for connexin-43 and connexin-26, respectively, and has been reported and classified as having overlapping features of Clouston syndrome and ODDD [3, 7].
In addition to the classic phenotypic features of the syndrome, a wide variety of additional physical manifestations have been observed. Ocular findings of microphthalmia and microcornea have been observed commonly in previous cases [2–4]. Craniofacial anomalies of microcephaly, poor hair growth, hypoplastic nasal alae, and prominent columella have been reported previously [2–4]. Bilateral syndactyly of the 4th and 5th digits is common [2, 3].
A systematic review of the published cases to date (ranging from 1963 to 2019) revealed 91 literature reports of 295 individuals with ODDD [1–91]. Table 2 [1–91] summarizes the sex distribution across all reviewed reports of ODDD. Patients with ODDD present with an approximately equal sex distribution (47% male and 53% female). Of the 295 individuals reported, 32 were clinically diagnosed with ODDD without molecular confirmation, 98 presented with features of ODDD and had a known relative with molecular confirmation of a GJA1 pathogenic variant, and 165 individuals had a molecularly confirmed GJA1 pathogenic variant.
Table 2.
Summary of sex distribution.
| Males | Females | Total | |||
|---|---|---|---|---|---|
| Individuals with clinical diagnosis of ODDD (with no molecular confirmation) | 14 | 45% | 18 | 56% | 32 |
|
| |||||
| Untested individuals with both ODDD phenotype and known relative with molecular confirmation | 52 | 53% | 46 | 47% | 98 |
|
| |||||
| Individuals with a molecular confirmed GJA1 pathogenic variant | 72 | 44% | 93 | 56% | 165 |
|
| |||||
| Totals | 138 | 47% | 157 | 53% | 295 |
There were 73 different GJA1 mutations identified from the 165 individuals that had a molecularly confirmed GJA1 pathogenic variant. Table 3 [1–3, 5–71, 92] summarizes the number of patients with each mutation. Patients with confirmed pathogenic variants and their relatives with no molecular confirmation but with features of ODDD were grouped separately. These two groups comprised 263 of the patients included in this study.
Table 3.
Reported GJA1 mutations and sex distribution in ODDD.
| Sources | Multiple mutations? | GJA1 mutation | Individuals with a molecular confirmed GJA1 pathogenic variant | Untested individuals with both ODDD phenotype and known relative with molecular confirmation | Total individuals with the ODDD phenotype | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Protein | Unspecified | Male | Female | Male | Female | Male | Female | Total | ||||
| Cavusoglu et al. 2019 | No | c.168_169insT | p.Q57SfsTer6 | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Aminabadi et al. 2009 & Aminabadi et al. 2010 | No | N/A | N/A | Missense mutation exon 2 (unspecified) | 1 | 0 | 2 | 1 | 3 | 75% | 1 | 25% | 4 |
| Dwarakanathan et al. 2015 & Furuta et al. 2012 | No | c.75G>T | p.W25C | N/A | 1 | 1 | 0 | 0 | 1 | 50% | 1 | 50% | 2 |
| Quick and Dobersen 2014; National Center for Biotechnology Information 2020 | Yes | c.605G>A | p.R202H | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| c.717G>A | p.R239R | ||||||||||||
| Paznekas et al. 2003 & Paznekas et al. 2009 | No | c.605G>A | p.R202H | N/A | 1 | 7 | 4 | 5 | 5 | 29% | 12 | 71% | 17 |
| Jamsheer et al. 2010 | Yes | c.301C>T | p.R101X | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| c.6delT | p.G2fsX7 | ||||||||||||
| Jamsheer et al. 2010 | No | c.301C>T | p.R101X | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Paznekas et al. 2009; Joss et al. 2008; & Richardson et al. 2006 | No | c.97C>T | p.R33X∗ | N/A | 0 | 2 | 0 | 0 | 0 | 0% | 2 | 100% | 2 |
| Paznekas et al. 2009; Richardson et al. 2004; Paznekas et al. 2003; & Gladwin et al. 1997 | No | c.93T>C | p.I31M | N/A | 0 | 0 | 4 | 4 | 4 | 50% | 4 | 50% | 8 |
| Wang et al. 2019 | No | c.91A>T | p.I311P | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Paznekas et al. 2009 & van Steensel et al. 2005 | No | c.780_781delTG | p.C260fsX306 | N/A | 1 | 2 | 0 | 0 | 1 | 33% | 2 | 67% | 3 |
| Paznekas et al. 2009; Paznekas et al. 2003; & Gorlin et al. 1963 | No | c.68A>C | p.K23T | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Dwarakanathan et al. 2015; Paznekas et al. 2009; & Vreeburg et al. 2007 | No | c.689_690delAT | p.Y230fsX236 | N/A | 0 | 3 | 1 | 0 | 1 | 25% | 3 | 75% | 4 |
| This study; Gumus 2018; Paznekas et al. 2009; Paznekas et al. 2003; & Traboulsi and Parks 1990 | No | c.65G>A | p.G22E | N/A | 0 | 3 | 0 | 0 | 0 | 0% | 3 | 100% | 3 |
| Wiest et al. 2006 | No | c.659C>A | p.S220Y | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Paznekas et al. 2009; Paznekas et al. 2003; & Norton et al. 1995 | No | c.646G>T | p.V216L | N/A | 1 | 0 | 4 | 1 | 5 | 83% | 1 | 17% | 6 |
| Park et al. 2017; Paznekas et al. 2009; & Paznekas et al. 2003 | No | c.61G>A | p.G21R | N/A | 0 | 2 | 0 | 0 | 0 | 0% | 2 | 100% | 2 |
| Brice et al. 2013 | No | c.617A>G | p.K206R | N/A | 1 | 2 | 1 | 1 | 2 | 40% | 3 | 60% | 5 |
| Paznekas et al. 2009 | No | c.602C>T | p.S201F | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Paznekas et al. 2009 & de la Parra et al. 2007 | No | c.5G>T | p.G2V | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Vitiello et al. 2005 & Vingolo et al. 1994 | No | c.581A>C | p.H194P∗ | N/A | 3 | 5 | 3 | 3 | 6 | 43% | 8 | 57% | 14 |
| Paznekas et al. 2009; Paznekas et al. 2003; & Judisch et al. 1979 | No | c.52T>C | p.S18P | N/A | 0 | 0 | 1 | 3 | 1 | 25% | 3 | 75% | 4 |
| Paznekas et al. 2009 & Paznekas et al. 2003 | No | c.50A>C | p.Y17S | N/A | 3 | 4 | 0 | 0 | 3 | 43% | 4 | 57% | 7 |
| Paznekas et al. 2009 & Debeer et al. 2005 | No | c.504_506delCTT | p.F169del | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Wiest et al. 2006 & Thomsen et al. 1998 | No | c.461C>A | p.T154N | N/A | 0 | 2 | 0 | 1 | 0 | 0% | 3 | 100% | 3 |
| Paznekas et al. 2009 & van Es et al. 2007 | No | c.460A>G | p.T154A∗ | N/A | 0 | 2 | 0 | 0 | 0 | 0% | 2 | 100% | 2 |
| Paznekas et al. 2009; Richardson et al. 2004; Paznekas et al. 2003; Gladwin et al. 1997; & Schrander-Stumpel et al. 1993 | No | c.443G>A | p.R148Q | N/A | 0 | 0 | 2 | 2 | 2 | 50% | 2 | 50% | 4 |
| Taşdelen et al. 2018 | No | c.442C>T | p.R148Ter | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Paznekas et al. 2009; Debeer et al. 2005; & Spaepen et al. 1991 | No | c.440Y>C | p.M147T | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Paznekas et al. 2009; Richardson et al. 2004; & Brueton et al. 1990 | No | c.427G>A | p.G143S | N/A | 0 | 0 | 8 | 1 | 8 | 89% | 1 | 11% | 9 |
| Orosz et al. 2018 | No | c.413G>A | p.G138D | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Paznekas et al. 2009; Paznekas et al. 2003; & Shapiro et al. 1997 | No | c.412G>C | p.G138R | N/A | 1 | 2 | 2 | 2 | 3 | 43% | 4 | 57% | 7 |
| Kogame et al. 2014 | No | c.412G>A | p.G138S | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Paznekas et al. 2009; Richardson et al. 2004; Paznekas et al. 2003; & Gladwin et al. 1997 | No | c.402G>T | p.K134N | N/A | 0 | 0 | 0 | 2 | 0 | 0% | 2 | 100% | 2 |
| Paznekas et al. 2009 & Paznekas et al. 2003 | No | c.400A>G | p.K134E | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Nishat et al. 2012; Paznekas et al. 2009; Paznekas et al. 2003; & Amador et al. 2008 | No | c.389T>C | p.I130T | N/A | 7 | 4 | 5 | 1 | 12 | 71% | 5 | 29% | 17 |
| Paznekas et al. 2009; Musa et al. 2008; Wiest et al. 2006; & Loddenkemper et al. 2002 | No | c.338T>C | p.L113P | N/A | 2 | 2 | 1 | 0 | 3 | 60% | 2 | 40% | 5 |
| Paznekas et al. 2009 & Debeer et al. 2005 | No | c.330G>C | p.E110D | N/A | 2 | 3 | 1 | 2 | 3 | 38% | 5 | 63% | 8 |
| Paznekas et al. 2009 & Kelly et al. 2006 | No | c.32T>C | p.L11P | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Gabriel et al. 2011 & Jamsheer et al. 2009 | No | c.31C>T | p.L11F | N/A | 0 | 2 | 0 | 0 | 0 | 0% | 2 | 100% | 2 |
| Porntaveetus et al. 2017 | No | c.31C>A | p.L11I | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Jamsheer et al. 2014 | No | c.317T>G | p.L106R | N/A | 2 | 0 | 0 | 0 | 2 | 100% | 0 | 0% | 2 |
| Paznekas et al. 2009 & Nivelon-Chevallier et al. 1981 | No | c.317T>C | p.L106P | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Paznekas et al. 2009 & Paznekas et al. 2003 | No | c.306G>C | p.K102N | N/A | 1 | 2 | 0 | 0 | 1 | 33% | 2 | 67% | 3 |
| Paznekas et al. 2009; Paznekas et al. 2003; & Wooldridge et al. 1977 | No | c.293A>G | p.Y98C | N/A | 1 | 3 | 1 | 1 | 2 | 33% | 4 | 67% | 6 |
| Paznekas et al. 2009 | No | c.287T>C | p.V96A | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Wiest et al. 2006 | No | c.287T>A | p.V96E | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Paznekas et al. 2009 & Kjaer et al. 2004 | No | c.286G>A | p.V96M | N/A | 2 | 2 | 0 | 0 | 2 | 50% | 2 | 50% | 4 |
| Paznekas et al. 2009 & Honkaniemi et al. 2005 | No | c.284A>G | p.H95R | N/A | 0 | 1 | 0 | 1 | 0 | 0% | 2 | 100% | 2 |
| Paznekas et al. 2009; Paznekas et al. 2003; & Opjordsmoen and Nyberg-Hansen 1980 | No | c.268C>G | p.L90V | N/A | 4 | 0 | 3 | 2 | 7 | 78% | 2 | 22% | 9 |
| Jamsheer et al. 2014 | No | c.257C>A | p.S86Y | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Pizzuti et al. 2004 | No | c.227G>A | p.R76H | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Izumi et al. 2013 | No | c.226C>T | p.R76C | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Paznekas et al. 2009; Paznekas et al. 2003; & Stanislaw et al. 1998 | No | c.226C>A | p.R76S | N/A | 0 | 2 | 0 | 2 | 0 | 0% | 4 | 100% | 4 |
| Choi et al. 2018 | No | c.221A>C | p.H74P∗ | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Paznekas et al. 2009; Richardson et al. 2004; Paznekas et al. 2003; & Gladwin et al. 1997 | No | c.206C>A | p.S69Y | N/A | 0 | 0 | 2 | 5 | 2 | 29% | 5 | 71% | 7 |
| Paznekas et al. 2009 & Vasconcellos et al. 2005 | No | c.176C>A | p.P59H | N/A | 4 | 4 | 1 | 0 | 5 | 56% | 4 | 44% | 9 |
| Paznekas et al. 2009 | No | c.145_147dupCAG | p.Q49dup | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Pazenkas et al. 2009; Paznekas et al. 2003; Weintraub et al. 1975; & Gellis and Feingold 1974 | No | c.154_156dupTTT | p.F52dup | N/A | 1 | 0 | 1 | 1 | 2 | 67% | 1 | 33% | 3 |
| Hadjichristou et al. 2017 & Paznekas et al. 2009 | No | c.146A>C | p.Q49P | N/A | 1 | 1 | 0 | 0 | 1 | 50% | 1 | 50% | 2 |
| Izumi et al. 2013 | No | c.145C>G | p.Q49E | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Paznekas et al. 2009 & Paznekas et al. 2003 | No | c.145C>A | p.Q49K | N/A | 3 | 2 | 0 | 0 | 3 | 60% | 2 | 40% | 5 |
| Amano et al. 2012; Feller et al. 2008; Paznekas et al. 2009; & Itro et al. 2005 | No | c.142G>A | p.E48K | N/A | 3 | 0 | 0 | 0 | 3 | 100% | 0 | 0% | 3 |
| Jamsheer et al. 2014 | No | c.139G>C | p.D47H | N/A | 0 | 3 | 0 | 0 | 0 | 0% | 3 | 100% | 3 |
| Tumminelli et al. 2016 | No | c.125G>C | p.E42Q | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Gabriel et al. 2011 | No | c.120delGGTTGAGTCAGC | p.V41_A44del | N/A | 0 | 1 | 1 | 2 | 1 | 25% | 3 | 75% | 4 |
| Paznekas et al. 2009 & Kellermayer et al. 2005 | Yes (compound heterozygous with GJB2 mutation) | c.121G>C | p.V41L | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| N/A | p.R127H (GJB2 mutation) | ||||||||||||
| Park et al. 2019; Hayashi et al. 2014; Paznekas et al. 2009; Debeer et al. 2005; & Paznekas et al. 2003 | No | c.119C>T | p.A40V | N/A | 6 | 4 | 4 | 3 | 10 | 59% | 7 | 41% | 17 |
| Wittlieb-Weber et al. 2015 | No | c. 175C>T | p.P59S | N/A | 1 | 2 | 0 | 0 | 1 | 33% | 2 | 67% | 3 |
| Attig et al. 2016 | No | c.396_398delAAA | p.I132_K133delinsM | N/A | 3 | 2 | 0 | 0 | 3 | 60% | 2 | 40% | 5 |
| Paznekas et al. 2009 | No | c.19T>G | p.L7V | N/A | 1 | 0 | 0 | 0 | 1 | 100% | 0 | 0% | 1 |
| Himi et al. 2009 | No | c.13A>T | p.S5C | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| Pace et al. 2019 | No | c.287T>G | p.V96G | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 |
| No | c.77T>C | p.L26P | N/A | 0 | 1 | 0 | 0 | 0 | 0% | 1 | 100% | 1 | |
| Totals | 72 | 93 | 52 | 46 | 124 | 47% | 139 | 53% | 263 | ||||
∗Unknown which specific individuals tested.
The eye features of all 295 patients are summarized in Table 4 [1–91]. The most common ophthalmic manifestations reported were microcornea (n = 111), microphthalmia (n = 110), short palpebral fissures (n = 56), and glaucoma (n = 51, 4 closed-angle and 1 open-angle).
Table 4.
Eye and ocular adnexa features reported in ODDD.
| Orbit | — | Microphthalmia (110/37%) | Hypotelorism (24/8%) | Hypertelorism (22/7%) | Short axial length (4/1%) | ||||||
| Anterior segment | Anterior chamber | Shallow anterior chamber (12/4%) | Deep anterior chambers (2/<1%) | ||||||||
| Cornea | Microcornea (111/38%) | Thick corneas (4/1%) | Corneal opacities (3/1%) | Corneal farinata (1/<1%) | Band keratopathy (1/<1%) | Corneal keratosis (1/<1%) | Abnormal Descemet's membrane (1/<1%) | Anteriorly deviated Schwalbe's line (1/<1%) | |||
| Sclera | Blue sclera (1/<1%) | ||||||||||
| Pupil | Persistent pupillary membranes (13/4%) | Eccentric pupils (3/1%) | |||||||||
| Lens | Cataracts (17/6%) | Lens opacities (2/<1%) | White retrolental masses (1/<1%) | ||||||||
| Uvea (iris, ciliary body) | Pale/atrophic irides (11/4%) | Uveitis (10/3%) | General iris abnormalities (7/2%) | Synechiae (4/1%) | Hypoplastic anterior iris stroma (3/1%) | Ciliary body cysts (2/<1%) | Flat iris (1/<1%) | Iridoschisis (1/<1%) | Inferior iris coloboma (1/<1%) | Dysplastic iris (1/<1%) | |
|
| |||||||||||
| Posterior segment | Uvea (choroid) | Thick choroid (2/<1%) | Thin choroid (1/<1%) | ||||||||
| Vitreous | Vitreous degeneration (1/<1%) | Vitreous membrane attachment to optic nerve and lens (1/<1%) | Persistent hyperplastic primary vitreous (1/<1%) | ||||||||
| Retina/fundus | Dysplastic retina/fundus (3/1%) | Pale retina/fundus (2/<1%) | Thread-like retinal vasculature (2/<1%) | Dystrophic retinal epithelium (1/<1%) | Hypoplastic macula (1/<1%) | Absent fundal glow with B-scan ultrasound (1/<1%) | |||||
| Optic disc | Pale/atrophic optic disc (3/1%) | Dysplastic optic disc (2/<1%) | Ellipsoid optic disc (1/<1%) | Optociliary vein presence (1/<1%) | Optic disc hypervascularity (1/<1%) | ||||||
|
| |||||||||||
| Ocular adnexa | Eyelid | Short/narrow palpebral fissures (56/19%) | Epicanthus (36/12%) | Telecanthus (11/4%) | Ptosis (7/2%) | Blepharophimosis (1/<1%) | Entropion (1/<1%) | Ectropion (1/<1%) | Epiblepharon (1/<1%) | Mucosal hypertrophy (1/<1%) | |
| Eyebrow/eyelash | Madarosis (19/6%) | Flared eyebrows (3/1%) (2 medially flared) | Synophyrs (1/<1%) | ||||||||
| Nasolacrimal duct | Nasolacrimal duct abnormalities (2/<1%) | Hypolacrimation (1/<1%) | |||||||||
|
| |||||||||||
| Other | Refractive errors | Myopia (16/5%) (2 anisometropic) | Hyperopia (8/3%) (2 anisometropic) | Astigmatism (1/<1%) | |||||||
| Eye movement disorders | Strabismus (27/9%) (9 esotropic, 1 exotropic) | Nystagmus (8/3%) | Amblyopia (3/1%) | Duane syndrome (2/<1%) | Brown syndrome (1/<1%) | ||||||
| Additional eye disorders | Glaucoma (51/17%) (4 closed-angle, 1 open-angle) | Paracentral scotoma (1/<1%) | |||||||||
| ERG/neurological | Abnormal ERG (2/<1%) | Delayed visual evoked responses (2/<1%) | Occipital subcortical white matter changes (1/<1%) | ||||||||
Twenty-three patients presented with refractive error, of which isolated myopia was the most frequently noted (n = 14), followed by isolated hyperopia (n = 6), anisometropia (n = 2), and astigmatism (n = 1). Forty patients presented with eye movement disorders, with strabismus (n = 27, 9 esotropic, 1 exotropic) being the most common, followed by nystagmus (n = 8), amblyopia (n = 3), Duane syndrome (n = 2), and Brown syndrome (n = 1). Note that 1 patient had both nystagmus and esotropia [71]. Other common findings included epicanthus (n = 36), hypotelorism (n = 24), hypertelorism (n = 22), madarosis (n = 19), cataracts (n = 17), persistent pupillary membranes (n = 13), shallow anterior chambers (n = 12), pale/atrophic irides (n = 11), telecanthus (n = 11), and uveitis (n = 10).
A variety of abnormal findings for the retina and optic disc were noted (n = 18), with dysplasia of the retina/fundus (n = 3) and pale/atrophic optic discs (n = 3) being the most common documented findings.
Of the individuals with molecularly confirmed mutations, the most common mutations present were c.605G>A (p.R202H) (11%; with 1 patient also having a c.717G>A synonymous mutation), c.389T>C (p.I130T) (10%), and c.119C>T (p.A40V) (10%). Table 5 [2, 3, 12, 30, 40, 41, 66, 67, 92] summarizes the eye features present in the patients with these mutations.
Table 5.
Common GJA1 mutations with associated eye features.
| Sources | Multiple mutations? | GJA1 mutation | Individuals with GJA1 mutation (confirmed and affected relatives) | Associated eye features | |
|---|---|---|---|---|---|
| Nucleotide | Protein | Total | |||
| Quick and Dobersen 2014; National Center for Biotechnology Information 2020 | Yes | c.605G>A | p.R202H | 1 | Microphthalmia (1) |
| c.717G>A | p.R239R | ||||
|
| |||||
| Paznekas et al. 2009; Paznekas et al. 2003 | No | c.605G>A | p.R202H | 17 | Microphthalmia (1), microcornea (2) |
|
| |||||
| Nishat et al. 2012; Paznekas et al. 2009; Paznekas et al. 2003; and Amador et al. 2008 | No | c.389T>C | p.I130T | 17 | Microphthalmia (4), hypotelorism (6), cataract (1), pale/atrophic optic disc (1), and short palpebral fissures (4) |
|
| |||||
| Park et al. 2019; Hayashi et al. 2014; Paznekas et al. 2009; Debeer et al. 2005; and Paznekas et al. 2003 | No | c.119C>T | p.A40V | 17 | Microphthalmia (9), hypertelorism (3), hypotelorism (4), short axial length (4), cataract (1), microcornea (8), thick cornea (4), macular hypoplasia (1), shallow anterior chamber (4), myopia (4), strabismus (6) (1 esotropic), glaucoma (6), and epicanthus (3) |
Less common features of the phenotype observed in our presented case were also reported in other cases as well. These include nasolacrimal duct abnormalities (n = 2), pale/atrophic retina/fundus (n = 2), and deep anterior chambers (n = 2). Additionally, including this study, the three patients with the p.G22E mutation have the following findings: microphthalmia (n = 3), cataracts (n = 1), microcornea (n = 2), blonde fundus (n = 1), persistent pupillary membrane (n = 1), deep anterior chamber (n = 1), hyperopia (n = 1), strabismus (n = 2, 1 esotropic), amblyopia (n = 1), glaucoma (n = 1), short palpebral fissures (n = 1), nasolacrimal duct abnormalities (n = 1), and epicanthus (n = 1) [2, 3, 21, 22].
Some unique genotype-phenotype correlations were noted upon further analysis. Three patients presented with eccentric pupils, but only 2 of these patients were reported with an associated mutation. Both mutations (p.Q49dup and p.Q49P) seem to affect the same amino acid in connexin-43 [3, 61, 72]. Additionally, uveitis was reported in 10 patients, 9 of which were associated with similar mutations. Eight of these patients were within the same study and had the p.H194P mutation, another patient had no molecular confirmation of a GJA1 mutation, and the other patient was reported with a missense mutation on exon 2 [4, 9, 10, 27, 28]. However, since the majority of these patients were reported within the same study, the apparent genotype-phenotype correlation of p.H194P and uveitis might be due to underreporting of uveitis from other sources with different pathogenic variants or may be due to other factors of the family not identified within the study.
Further analysis of the genotype-phenotype correlation was conducted by pairing the phenotypic manifestations of each mutation with the corresponding defects in the connexin-43 domains. The domains were defined by the amino acid ranges provided on UniProt (P17302–CXA1_HUMAN) [93]. Table 6 [1–3, 5–71, 92, 93] provides a summary of the phenotypes associated with mutations from each domain.
Table 6.
Mutant connexin-43 domains and associated phenotype.
| GJA1 mutation | Protein domain (amino acid range) (obtained from UniProt-P17302) | Associated phenotype (no. of individuals) |
|---|---|---|
| p.G2fsX7 (with p.R101X) p.G2V p.L11P p.L11F p.L11I p.L7V p.S5C |
Cytoplasmic N-terminus(1-13) | Microcornea (7), microphthalmia (5), epicanthus (4), strabismus (3) (1 esotropic), short palpebral fissures (2), telecanthus (2), amblyopia (1), dysplastic fundus (1), optociliary vein (1), dysplastic optic disc (1), pale/atrophic optic disc (1), persistent pupillary membrane (1), myopia (3), hyperopia (1) (anisometropic), glaucoma (1), ptosis (1), entropion (1), madarosis (1), hypertelorism (1), and cataract (1) |
|
| ||
| p.W25C p. R33X p.I31M p.K23T p.G22E p.G21R p.S18P p.Y17S p.L26P |
Transmembrane-1 (14-36) | Microcornea (21), microphthalmia (14), short palpebral fissures (11), persistent pupillary membrane (6), madarosis (6), epicanthus (6), glaucoma (5), anterior iris stroma hypoplasia (3), hypertelorism (2), cataract (2), iris abnormalities (2), blonde fundus (1), iridoschisis (1), deep anterior chamber (1), hyperopia (2), strabismus (7) (3 esotropic), amblyopia (1), nystagmus (1), ptosis (1), epiblepharon (1), nasolacrimal duct obstruction (1), and flared eyebrows (1) (medially flared) |
|
| ||
| p.Q57SfsTer6 p.R76H p.R76C p.R76S p.H74P p.S69Y p.P59H p.Q49dup p.F52dup p.Q49P p.Q49E p.Q49K p.E48K p.D47H p.E42Q p.V41_A44del p.V41L (with p.R127H (GJB2 mutation)) p.A40V p.P59S |
Extracellular-1 (37-76) | Microphthalmia (32), microcornea (30), glaucoma (15) (2 closed-angle, 1 open-angle), hypertelorism (11), epicanthus (10), strabismus (9) (3 esotropic), short palpebral fissures (9), iris atrophy (peripupillary) (8), cataract (6), shallow anterior chamber (6), hypotelorism (5), short axial length (4), myopia (4), corneal farinata (4), telecanthus (3), iris abnormalities (2), eccentric pupils (2), persistent pupillary membrane (2), dysplastic fundus (1), dysplastic optic (1), macular hypoplasia (1), synechiae (1), ciliary body cysts (1), deep anterior chamber (1), hyperopia (1), ptosis (1), blepharophimosis (1), madarosis (1), nasolacrimal duct abnormalities (1), and low-voltage ERG (1) |
|
| ||
| p.Y98C p.V96A p.V96E p.V96M p.H95R p.L90V p.S86Y p.V96G |
Transmembrane-2 (77-99) | Hypertelorism (5), microcornea (2), microphthalmia (3), glaucoma (3), strabismus (2) (1 esotropic), short palpebral fissures (2), eyelid mucosal hypertrophy (1), telecanthus (1), epicanthus (1), optic disc atrophy (1), hyperopia (1), myopia (1), strabismus (1), paracentral scotoma (1), madarosis (1), and delayed visual evoked potentials (1) |
|
| ||
| p.R101X (with p.G2fsX7) p.R101X p.T154N p.T154A p.R148Q p.R148Ter p.M147T p.G143S p.G138D p.G138R p.G138S p.K134N p.K134E p.I130T p.L113P p.E110D p.L106R p.L106P p.K102N p.I132_K133delinsM |
Cytoplasmic-1 (100-154) | Microphthalmia (20), microcornea (18), short palpebral fissures (14), hypotelorism (14), glaucoma (9), myopia (7), epicanthus (5), cataract (3), strabismus (3), shallow anterior chamber (3), hypertelorism (2), opaque lens (1), optic disc hypervascularity (1), pale/atrophic optic disc (1), pale irides (1), iris abnormalities (2), astigmatism (1), Duane syndrome (1), ptosis (1), occipital subcortical white matter changes (1), and delayed visual evoked responses (1) |
|
| ||
| p.F169del | Transmembrane-3 (155-177) | Short palpebral fissures (1) |
|
| ||
| p.R202H (with p.R239R) p.R202H p.K206R p.S201F p.H194P |
Extracellular-2 (178-208) | Microphthalmia (18), uveitis (8), glaucoma (8), microcornea (4), opaque cornea (2), thick choroid (2), cataract (1), shallow anterior chamber (1), nystagmus (2), and ptosis (1) |
|
| ||
| p.S220Y p.V216L |
Transmembrane-4 (209-231) | Microphthalmia (1), glaucoma (1), microcornea (1), and persistent pupillary membrane (1) |
|
| ||
| p.Y230fsX236 | Transmembrane-4 & cytoplasmic C-terminus (209-382) | Hypertelorism (2), hypotelorism (1), and flared eyebrows (2) (1 medially flared) |
|
| ||
| p.R239R (with p.R202H) p.I311P p.C260fsX306 |
Cytoplasmic C-terminus(232-382) | Short palpebral fissures (3), epicanthus (2), hypotelorism (2), microcornea (2), pale irides (2), myopia (2), hyperopia (1) (1 anisometropic), corneal opacity (1), microphthalmia (1), retinal dysplasia (1), choroid thinning (1), glaucoma (1), madarosis (1), and loss of flash ERG (1) |
|
| ||
| Missense mutation exon 2 (unspecified) | Unknown | Microphthalmia (1), cataract (1), microcornea (1), uveitis (1), glaucoma (1), epicanthus (1), telecanthus (1), short palpebral fissures (1), and ptosis (1) |
The domains most commonly affected by GJA1 mutations are the extracellular-1 loop and the cytoplasmic-1 loop of connexin-43, accounting for 19 and 20 mutations, respectively. Disruptions in the extracellular-1 loop presented primarily as microphthalmia (n = 32) and microcornea (n = 30). A similar pattern can be seen in the cytoplasmic-1 loop, as the most common presentations were microphthalmia (n = 20) and microcornea (n = 18). Other clinical findings, however, may be able to distinguish mutations resulting from these domains. The next most common findings associated with mutations in the extracellular-1 loop were glaucoma (n = 15) and hypertelorism (n = 11), as opposed to short palpebral fissures (n = 14) and hypotelorism (n = 14) for the cytoplasmic-1 loop.
Mutations affecting the cytoplasmic N-terminus and the transmembrane-1 domain shared similar features to the ones in the extracellular-1 and cytoplasmic-1 domains, as microphthalmia and microcornea were the most common clinical findings. However, the mutations in the cytoplasmic N-terminus and transmembrane-1 domain presented with microcornea (n = 17 and n = 21, respectively) more frequently than microphthalmia (n = 5 and n = 14, respectively). The opposite pattern is true for the extracellular-1 and cytoplasmic-1 domains.
The mutations in the extracellular-2 loop demonstrate a different phenotypic pattern, as microphthalmia (n = 14) occurs the most frequently, while microcornea is less frequent (n = 4). Mutations in the transmembrane-2 domain also display a unique pattern, with hypertelorism (n = 5) being the most frequent clinical finding. Other domains listed in Table 6 also demonstrate some unique clinical patterns, but this may be due to variability from the small number of samples. The patterns mentioned previously, however, still provide insight into the role of different connexin-43 domains in providing phenotypic variability among patients with ODDD.
In conclusion, this report provides a comprehensive review of the eye and ocular adnexa abnormalities that are currently known to be associated with the ODDD phenotype. Limitations of this report include the possibility of an incomplete ophthalmologic evaluation and/or lack of reporting of eye features in all of the evaluated case reports or misdiagnosis in the individuals with the ODDD phenotype without molecular confirmation. As such, it is possible that the reported common eye features within this summary may be over or underrepresented. Ophthalmic manifestations are commonly associated within the phenotype, and a wide spectrum of eye and ocular adnexa structures may be affected. The rarity of this condition provides further incentive to further investigate the phenotype.
Consent
Consent has been obtained.
Conflicts of Interest
Virang Kumar and Arti Pandya declare that they have no conflicts of interest. Natario L. Couser, MD, MS, is a principal investigator at the Virginia Commonwealth University site of Retrophin, Inc., and book editor in Elsevier.
Supplementary Materials
Supplementary Material 1: all GJA1 mutations with associated eye and ocular adnexa features. This dataset groups patients with ODDD by GJA1 mutation and reports the associated eye and ocular adnexa features.
References
- 1.Dwarakanathan A., Bhat M., GN S., Shetty S. Missense and deletion mutations in GJA1 causing oculodentodigital dysplasia in two Indian families. Clinical Dysmorphology. 2015;24(4):159–162. doi: 10.1097/MCD.0000000000000094. [DOI] [PubMed] [Google Scholar]
- 2.Paznekas W. A., Boyadjiev S. A., Shapiro R. E., et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. American Journal of Human Genetics. 2003;72(2):408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paznekas W. A., Karczeski B., Vermeer S., et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Human Mutation. 2009;30(5):724–733. doi: 10.1002/humu.20958. [DOI] [PubMed] [Google Scholar]
- 4.Kayalvizhi G., Subramaniyan B., Suganya G. Clinical manifestations of oculodentodigital dysplasia. Journal of the Indian Society of Pedodontics and Preventive Dentistry. 2014;32(4):350–352. doi: 10.4103/0970-4388.140973. [DOI] [PubMed] [Google Scholar]
- 5.de la Parra D. R., Zenteno J. C. A new GJA1 (connexin 43) mutation causing oculodentodigital dysplasia associated to uncommon features. Ophthalmic Genetics. 2007;28(4):198–202. doi: 10.1080/13816810701538620. [DOI] [PubMed] [Google Scholar]
- 6.Richardson R., Donnai D., Meire F., Dixon M. J. Expression of Gja1 correlates with the phenotype observed in oculodentodigital syndrome/type III syndactyly. Journal of Medical Genetics. 2004;41(1):60–67. doi: 10.1136/jmg.2003.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kellermayer R., Keller M., Ratajczak P., et al. Bigenic connexin mutations in a patient with hidrotic ectodermal dysplasia. European Journal of Dermatology. 2005;15(2):75–79. [PubMed] [Google Scholar]
- 8.Cavusoglu D., Dundar N. O., Arican P., Ozyilmaz B., Gencpinar P. A hypomyelinating leukodystrophy with calcification: oculodentodigital dysplasia. Acta Neurologica Belgica. 2019 doi: 10.1007/s13760-019-01178-4. [DOI] [PubMed] [Google Scholar]
- 9.Aminabadi N. A., Ganji A. T., Vafaei A., Pourkazemi M., Oskouei S. G. Oculodentodigital dysplasia: disease spectrum in an eight-year-old boy, his parents and a sibling. The Journal of Clinical Pediatric Dentistry. 2009;33(4):337–341. doi: 10.17796/jcpd.33.4.0r02810u1533h168. [DOI] [PubMed] [Google Scholar]
- 10.Aminabadi N. A., Pourkazemi M., Oskouei S. G., Jamali Z. Dental management of oculodentodigital dysplasia: a case report. Journal of Oral Science. 2010;52(2):337–342. doi: 10.2334/josnusd.52.337. [DOI] [PubMed] [Google Scholar]
- 11.Furuta N., Ikeda M., Hirayanagi K., Fujita Y., Amanuma M., Okamoto K. A novel GJA1 mutation in oculodentodigital dysplasia with progressive spastic paraplegia and sensory deficits. Internal Medicine. 2012;51(1):93–98. doi: 10.2169/internalmedicine.51.5770. [DOI] [PubMed] [Google Scholar]
- 12.Quick J. S., Dobersen M. Cardiac arrhythmia and death of teenager linked to rare genetic disorder diagnosed at autopsy. The American Journal of Forensic Medicine and Pathology. 2014;35(2):103–105. doi: 10.1097/PAF.0000000000000092. [DOI] [PubMed] [Google Scholar]
- 13.Jamsheer A., Badura-Stronka M., Sowińska A., Debicki S., Kiryluk K., Latos-Bieleńska A. A severe progressive oculodentodigital dysplasia due to compound heterozygous GJA1 mutation. Clinical Genetics. 2010;78(1):94–97. doi: 10.1111/j.1399-0004.2010.01412.x. [DOI] [PubMed] [Google Scholar]
- 14.Joss S. K., Ghazawy S., Tomkins S., Ahmed M., Bradbury J., Sheridan E. Variable expression of neurological phenotype in autosomal recessive oculodentodigital dysplasia of two sibs and review of the literature. European Journal of Pediatrics. 2008;167(3):341–345. doi: 10.1007/s00431-007-0468-1. [DOI] [PubMed] [Google Scholar]
- 15.Richardson R. J., Joss S., Tomkin S., Ahmed M., Sheridan E., Dixon M. J. A nonsense mutation in the first transmembrane domain of connexin 43 underlies autosomal recessive oculodentodigital syndrome. Journal of Medical Genetics. 2006;43(7, article e37) doi: 10.1136/jmg.2005.037655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gladwin A., Donnai D., Metcalfe K., et al. Localization of a gene for oculodentodigital syndrome to human chromosome 6q22-q24. Human Molecular Genetics. 1997;6(1):123–127. doi: 10.1093/hmg/6.1.123. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z., Sun L., Wang P., et al. Novel ocular findings in oculodentodigital dysplasia (ODDD): a case report and literature review. Ophthalmic Genetics. 2019;40(1):54–59. doi: 10.1080/13816810.2019.1571616. [DOI] [PubMed] [Google Scholar]
- 18.van Steensel M. A. M., Spruijt L., van der Burgt I., et al. A 2-bp deletion in theGJA1 gene is associated with oculo-dento-digital dysplasia with palmoplantar keratoderma. American Journal of Medical Genetics Part A. 2005;132a(2):171–174. doi: 10.1002/ajmg.a.30412. [DOI] [PubMed] [Google Scholar]
- 19.Gorlin R. J., Meskin L. H., Geme J. W. S. Oculodentodigital Dysplasia. The Journal of Pediatrics. 1963;63(1):69–75. doi: 10.1016/s0022-3476(63)80304-2. [DOI] [PubMed] [Google Scholar]
- 20.Vreeburg M., de Zwart-Storm E. A., Schouten M. I., et al. Skin changes in oculo-dento-digital dysplasia are correlated with C-terminal truncations of connexin 43. American Journal of Medical Genetics. Part A. 2007;143(4):360–363. doi: 10.1002/ajmg.a.31558. [DOI] [PubMed] [Google Scholar]
- 21.Gumus E. A rare symptom of a very rare disease: a case report of a oculodentodigital dysplasia with lymphedema. Clinical Dysmorphology. 2018;27(3):91–93. doi: 10.1097/MCD.0000000000000221. [DOI] [PubMed] [Google Scholar]
- 22.Traboulsi E. I., Parks M. M. Glaucoma in oculo-dento-osseous dysplasia. American Journal of Ophthalmology. 1990;109(3):310–313. doi: 10.1016/s0002-9394(14)74556-8. [DOI] [PubMed] [Google Scholar]
- 23.Wiest T., Herrmann O., Stögbauer F., et al. Clinical and genetic variability of oculodentodigital dysplasia. Clinical Genetics. 2006;70(1):71–72. doi: 10.1111/j.1399-0004.2006.00631.x. [DOI] [PubMed] [Google Scholar]
- 24.Norton K. K., Carey J. C., Gutmann D. H. Oculodentodigital dysplasia with cerebral white matter abnormalities in a two-generation family. American Journal of Medical Genetics. 1995;57(3):458–461. doi: 10.1002/ajmg.1320570320. [DOI] [PubMed] [Google Scholar]
- 25.Park K. W., Ryu H. S., Kim J., Chung S. J. Oculodentodigital dysplasia presenting as spastic paraparesis: the first genetically confirmed Korean case and a literature review. Journal of Movement Disorders. 2017;10(3):149–153. doi: 10.14802/jmd.17050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brice G., Ostergaard P., Jeffery S., Gordon K., Mortimer P. S., Mansour S. A novel mutation in GJA1 causing oculodentodigital syndrome and primary lymphoedema in a three generation family. Clinical Genetics. 2013;84(4):378–381. doi: 10.1111/cge.12158. [DOI] [PubMed] [Google Scholar]
- 27.Vitiello C., D'Adamo P., Gentile F., Vingolo E. M., Gasparini P., Banfi S. A novel GJA1 mutation causes oculodentodigital dysplasia without syndactyly. American Journal of Medical Genetics Part A. 2005;133a(1):58–60. doi: 10.1002/ajmg.a.30554. [DOI] [PubMed] [Google Scholar]
- 28.Vingolo E. M., Steindl K., Forte R., et al. Autosomal dominant simple microphthalmos. Journal of Medical Genetics. 1994;31(9):721–725. doi: 10.1136/jmg.31.9.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Judisch G. F., Martin-Casals A., Hanson J. W., Olin W. H. Oculodentodigital dysplasia. Four new reports and a literature review. Archives of Ophthalmology. 1979;97(5):878–884. doi: 10.1001/archopht.1979.01020010436007. [DOI] [PubMed] [Google Scholar]
- 30.Debeer P., van Esch H., Huysmans C., et al. Novel GJA1 mutations in patients with oculo-dento-digital dysplasia (ODDD) European Journal of Medical Genetics. 2005;48(4):377–387. doi: 10.1016/j.ejmg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 31.Thomsen M., Schneider U., Weber M., Niethard F. U. The different appearance of the oculodentodigital dysplasia syndrome. Journal of Pediatric Orthopaedics. Part B. 1998;7(1):23–26. doi: 10.1097/01202412-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 32.van Es R. J. J., Wittebol-Post D., Beemer F. A. Oculodentodigital dysplasia with mandibular retrognathism and absence of syndactyly: a case report with a novel mutation in the connexin 43 gene. International Journal of Oral and Maxillofacial Surgery. 2007;36(9):858–860. doi: 10.1016/j.ijom.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Schrander-Stumpel C. T., de Groot-Wijnands J. B., de Die-Smulders C., Fryns J. P. Type III syndactyly and oculodentodigital dysplasia: a clinical spectrum. Genetic Counseling. 1993;4(4):271–276. [PubMed] [Google Scholar]
- 34.Tasdelen E., Durmaz C. D., Karabulut H. G. Autosomal recessive oculodentodigital dysplasia: a case report and review of the literature. Cytogenetic and Genome Research. 2018;154(4):181–186. doi: 10.1159/000489000. [DOI] [PubMed] [Google Scholar]
- 35.Spaepen A., Schrander-Stumpel C., Fryns J. P., de Die-Smulders C., Borghgraef M., van den Berghe H. Hallermann-Streiff syndrome: clinical and psychological findings in children. Nosologic overlap with oculodentodigital dysplasia? American Journal of Medical Genetics. 1991;41(4):517–520. doi: 10.1002/ajmg.1320410428. [DOI] [PubMed] [Google Scholar]
- 36.Brueton L. A., Huson S. M., Farren B., Winter R. M. Oculodentodigital dysplasia and type III syndactyly: separate genetic entities or disease spectrum? Journal of Medical Genetics. 1990;27(3):169–175. doi: 10.1136/jmg.27.3.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orosz O., Fodor M., Balogh I., Losonczy G. Relative anterior microphthalmos in oculodentodigital dysplasia. Indian Journal of Ophthalmology. 2018;66(2):334–336. doi: 10.4103/ijo.IJO_756_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shapiro R. E., Griffin J. W., Stine O. C. Evidence for genetic anticipation in the oculodentodigital syndrome. American Journal of Medical Genetics. 1997;71(1):36–41. [PubMed] [Google Scholar]
- 39.Kogame T., Dainichi T., Shimomura Y., Tanioka M., Kabashima K., Miyachi Y. Palmoplantar keratosis in oculodentodigital dysplasia with a GJA1 point mutation out of the C-terminal region of connexin 43. The Journal of Dermatology. 2014;41(12):1095–1097. doi: 10.1111/1346-8138.12682. [DOI] [PubMed] [Google Scholar]
- 40.Nishat S., Mansoor Q., Javaid A., Ismail M. Oculodentodigital syndrome with syndactyly type III in a Pakistani consanguineous family. Journal of Dermatological Case Reports. 2012;6(2):43–48. doi: 10.3315/jdcr.2012.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amador C., Mathews A. M., del Carmen Montoya M., Laughridge M. E., Everman D. B., Holden K. R. Expanding the neurologic phenotype of oculodentodigital dysplasia in a 4-generation Hispanic family. Journal of Child Neurology. 2008;23(8):901–905. doi: 10.1177/0883073808317730. [DOI] [PubMed] [Google Scholar]
- 42.Musa F. U., Ratajczak P., Sahu J., et al. Ocular manifestations in oculodentodigital dysplasia resulting from a heterozygous missense mutation (L113P) in GJA1 (connexin 43) Eye (London, England) 2009;23(3):549–555. doi: 10.1038/eye.2008.77. [DOI] [PubMed] [Google Scholar]
- 43.Loddenkemper T., Grote K., Evers S., Oelerich M., Stögbauer F. Neurological manifestations of the oculodentodigital dysplasia syndrome. Journal of Neurology. 2002;249(5):584–595. doi: 10.1007/s004150200068. [DOI] [PubMed] [Google Scholar]
- 44.Kelly S. C., Ratajczak P., Keller M., Purcell S. M., Griffin T., Richard G. A novel GJA 1 mutation in oculo-dento-digital dysplasia with curly hair and hyperkeratosis. European Journal of Dermatology. 2006;16(3):241–245. [PubMed] [Google Scholar]
- 45.Gabriel L. A., Sachdeva R., Marcotty A., Rockwood E. J., Traboulsi E. I. Oculodentodigital dysplasia: new ocular findings and a novel connexin 43 mutation. Archives of Ophthalmology. 2011;129(6):781–784. doi: 10.1001/archophthalmol.2011.113. [DOI] [PubMed] [Google Scholar]
- 46.Jamsheer A., Wisniewska M., Szpak A., et al. A novel GJA1 missense mutation in a Polish child with oculodentodigital dysplasia. Journal of Applied Genetics. 2009;50(3):297–299. doi: 10.1007/BF03195687. [DOI] [PubMed] [Google Scholar]
- 47.Porntaveetus T., Srichomthong C., Ohazama A., Suphapeetiporn K., Shotelersuk V. A novel GJA1 mutation in oculodentodigital dysplasia with extensive loss of enamel. Oral Diseases. 2017;23(6):795–800. doi: 10.1111/odi.12663. [DOI] [PubMed] [Google Scholar]
- 48.Jamsheer A., Sowińska-Seidler A., Socha M., Stembalska A., Kiraly-Borri C., Latos-Bieleńska A. Three novel GJA1 missense substitutions resulting in oculo-dento-digital dysplasia (ODDD) - further extension of the mutational spectrum. Gene. 2014;539(1):157–161. doi: 10.1016/j.gene.2014.01.066. [DOI] [PubMed] [Google Scholar]
- 49.Nivelon-Chevallier A., Audry D., Audry F., Dumas R. Oculo-dental-digital dysplasia: report of a case with spastic paraplegia. Journal de Génétique Humaine. 1981;29(2):171–179. [PubMed] [Google Scholar]
- 50.Wooldridge W. E., Anthony D. D., Olson E. R., Bates G. P., Sammon T. J. Oculodentodigital dysplasia. Missouri Medicine. 1977;74(8):379–80, 383. 383. [PubMed] [Google Scholar]
- 51.Kjaer K. W., Hansen L., Eiberg H., Leicht P., Opitz J. M., Tommerup N. Novel connexin 43 (GJA1) mutation causes oculo-dento-digital dysplasia with curly hair. American Journal of Medical Genetics. 2004;127a(2):152–157. doi: 10.1002/ajmg.a.20614. [DOI] [PubMed] [Google Scholar]
- 52.Honkaniemi J., Kalkkila J. P., Koivisto P., Kähärä V., Latvala T., Simola K. Letter to the editor: novel GJA1 mutation in oculodentodigital dysplasia. American Journal of Medical Genetics. Part A. 2005;139(1):48–49. doi: 10.1002/ajmg.a.30925. [DOI] [PubMed] [Google Scholar]
- 53.Opjordsmoen S., Nyberg-Hansen R. Hereditary spastic paraplegia with neurogenic bladder disturbances and syndactylia. Acta Neurologica Scandinavica. 1980;61(1):35–41. doi: 10.1111/j.1600-0404.1980.tb02993.x. [DOI] [PubMed] [Google Scholar]
- 54.Pizzuti A., Flex E., Mingarelli R., Salpietro C., Zelante L., Dallapiccola B. A homozygous GJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum phenotype. Human Mutation. 2004;23(3):p. 286. doi: 10.1002/humu.9220. [DOI] [PubMed] [Google Scholar]
- 55.Izumi K., Lippa A. M., Wilkens A., Feret H. A., McDonald-McGinn D., Zackai E. H. Congenital heart defects in oculodentodigital dysplasia: report of two cases. American Journal of Medical Genetics Part A. 2013;161a(12):3150–3154. doi: 10.1002/ajmg.a.36159. [DOI] [PubMed] [Google Scholar]
- 56.Stanislaw C. L., Narvaez C., Rogers R. G., Woodard C. S. Oculodentodigital dysplasia with cerebral white matter abnormalities: an additional case. Proceedings of the Greenwood Genetic Cente. 1998;17(1):20–24. [Google Scholar]
- 57.Choi J., Yang A., Song A., et al. Oculodentodigital dysplasia with a novel mutation in GJA1 diagnosed by targeted gene panel sequencing: a case report and literature review. Annals of Clinical and Laboratory Science. 2018;48(6):776–781. [PubMed] [Google Scholar]
- 58.Vasconcellos J. P., Melo M. B., Schimiti R. B., Bressanim N. C., Costa F. F., Costa V. P. A novel mutation in the GJA1 gene in a family with oculodentodigital dysplasia. Archives of Ophthalmology. 2005;123(10):1422–1426. doi: 10.1001/archopht.123.10.1422. [DOI] [PubMed] [Google Scholar]
- 59.Weintraub D. M., Baum J. L., Pashayan H. M. A family with oculodentodigital dysplasia. The Cleft Palate Journal. 1975;12:323–329. [PubMed] [Google Scholar]
- 60.Gellis S. S., Feingold M. Oculodentodigital dysplasia. Picture of the month. American Journal of Diseases of Children. 1974;128(1):81–82. doi: 10.1001/archpedi.1974.02110260083015. [DOI] [PubMed] [Google Scholar]
- 61.Hadjichristou C., Christophidou-Anastasiadou V., Bakopoulou A., et al. Oculo-dento-digital dysplasia (ODDD) due to a GJA1 mutation: report of a case with emphasis on dental manifestations. The International Journal of Prosthodontics. 2017;30(3):280–285. doi: 10.11607/ijp.5130. [DOI] [PubMed] [Google Scholar]
- 62.Amano K., Ishiguchi M., Aikawa T., et al. Cleft lip in oculodentodigital dysplasia suggests novel roles for connexin43. Journal of Dental Research. 2012;91(7_suppl):S38–S44. doi: 10.1177/0022034512447952. [DOI] [PubMed] [Google Scholar]
- 63.Feller L., Wood N. H., Sluiter M. D., et al. Report of a black South African child with oculodentodigital dysplasia and a novel GJA1 gene mutation. American Journal of Medical Genetics Part A. 2008;146a(10):1350–1353. doi: 10.1002/ajmg.a.32272. [DOI] [PubMed] [Google Scholar]
- 64.Itro A., Marra A., Urciuolo V., Difalco P., Amodio A. Oculodentodigital dysplasia. A case report. Minerva Stomatologica. 2005;54(7-8):453–459. [PubMed] [Google Scholar]
- 65.Tumminelli G., di Donato I., Guida V., Rufa A., de Luca A., Federico A. Oculodentodigital dysplasia with massive brain calcification and a new mutation of GJA1 gene. Journal of Alzheimer's Disease. 2016;49(1):27–30. doi: 10.3233/JAD-150424. [DOI] [PubMed] [Google Scholar]
- 66.Park D. Y., Cho S. Y., Jin D. K., Kee C. Clinical characteristics of autosomal dominant GJA1 missense mutation linked to oculodentodigital dysplasia in a Korean family. Journal of Glaucoma. 2019;28(4):357–362. doi: 10.1097/IJG.0000000000001190. [DOI] [PubMed] [Google Scholar]
- 67.Hayashi R., Bito T., Taniguchi-Ikeda M., Farooq M., Ito M., Shimomura Y. Japanese case of oculodentodigital dysplasia caused by a mutation in the GJA1 gene. The Journal of Dermatology. 2014;41(12):1109–1110. doi: 10.1111/1346-8138.12656. [DOI] [PubMed] [Google Scholar]
- 68.Wittlieb-Weber C. A., Haude K. M., Fong C. T., Vinocur J. M. A novel GJA1 mutation causing familial oculodentodigital dysplasia with dilated cardiomyopathy and arrhythmia. HeartRhythm Case Reports. 2016;2(1):32–35. doi: 10.1016/j.hrcr.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Attig A., Trabelsi M., Hizem S., et al. Oculo-dento-digital dysplasia in a Tunisian family with a novel GJA1 mutation. Genetic Counseling. 2016;27(3):433–439. [PubMed] [Google Scholar]
- 70.Himi M., Fujimaki T., Yokoyama T., Fujiki K., Takizawa T., Murakami A. A case of oculodentodigital dysplasia syndrome with novel GJA1 gene mutation. Japanese Journal of Ophthalmology. 2009;53(5):541–545. doi: 10.1007/s10384-009-0711-6. [DOI] [PubMed] [Google Scholar]
- 71.Pace N. P., Benoit V., Agius D., et al. Two novel GJA1 variants in oculodentodigital dysplasia. Molecular Genetics & Genomic Medicine. 2019;7(9, article e882) doi: 10.1002/mgg3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parashari U. C., Khanduri S., Bhadury S., Qayyum F. A. Radiographic diagnosis of a rare case of oculo-dento-digital dysplasia. South African Journal of Radiology. 2011;15(4):p. 134. doi: 10.4102/sajr.v15i4.359. [DOI] [Google Scholar]
- 73.Beighton P., Hamersma H., Raad M. Oculodento-osseous dysplasia: heterogeneity or variable expression? Clinical Genetics. 1979;16(3):169–177. doi: 10.1111/j.1399-0004.1979.tb00987.x. [DOI] [PubMed] [Google Scholar]
- 74.Doshi D. C., Limdi P. K., Parekh N. V., Gohil N. R. Oculodentodigital dysplasia. Indian Journal of Ophthalmology. 2016;64(3):227–230. doi: 10.4103/0301-4738.180191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frasson M., Calixto N., Cronemberger S., Pessoa de Aguiar R. A. L., Leão L. L., Burle de Aguiar M. J. Oculodentodigital dysplasia: study of ophthalmological and clinical manifestations in three boys with probably autosomal recessive inheritance. Ophthalmic Genetics. 2004;25(3):227–236. doi: 10.1080/13816810490513424. [DOI] [PubMed] [Google Scholar]
- 76.Gillespie F. D. A hereditary syndrome: dysplasia oculodentodigitalis. Archives of Ophthalmology. 1964;71(2):187–192. doi: 10.1001/archopht.1964.00970010203009. [DOI] [PubMed] [Google Scholar]
- 77.Gutmann D. H., Zackai E. H., McDonald-McGinn D., Fischbeck K. H., Kamholz J. Oculodentodigital dysplasia syndrome associated with abnormal cerebral white matter. American Journal of Medical Genetics. 1991;41(1):18–20. doi: 10.1002/ajmg.1320410106. [DOI] [PubMed] [Google Scholar]
- 78.Kurlander G. J., Lavy N. W., Campbell J. A. Roentgen differentiation of the oculodentodigital syndrome and the Hallermann-Streiff syndrome in infancy. Radiology. 1966;86(1):77–86. doi: 10.1148/86.1.77. [DOI] [PubMed] [Google Scholar]
- 79.Levine D. S. Delayed gastric emptying and chronic diarrhea in a patient with oculodentodigital dysplasia syndrome. Journal of Pediatric Gastroenterology and Nutrition. 1986;5(2):329–333. [PubMed] [Google Scholar]
- 80.Martínez-García M., Bustamante-Aragonés A., Lorda I., Trujillo-Tiebas M. J. Displasia oculodentodigital: consejo genético, opciones reproductivas y estudio molecular de un caso clínico referido para diagnóstico preimplantacional. Medicina Clínica. 2012;138(13):592–593. doi: 10.1016/j.medcli.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 81.Mills J. K., Wheeler L., Oishi S. N. A case of familial syndactyly associated with eye and dental abnormalities. Jaapa. 2015;28(12):40–43. doi: 10.1097/01.JAA.0000471611.99902.fe. [DOI] [PubMed] [Google Scholar]
- 82.Mosaed S., Jacobsen B. H., Lin K. Y. Case report: imaging and treatment of ophthalmic manifestations in oculodentodigital dysplasia. BMC Ophthalmology. 2016;16(1) doi: 10.1186/s12886-015-0173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Owlia F., Akhavan Karbassi M. H., Hakimian R., Alemrajabi M. S. A highlighted case for emphasizing on clinical diagnosis for rare syndrome in third world. Iranian Journal of Child Neurology. 2017;11(4):77–80. [PMC free article] [PubMed] [Google Scholar]
- 84.Scheutzel P. Oculodentodigital syndrome: report of a case. Dento Maxillo Facial Radiology. 1991;20(3):175–178. doi: 10.1259/dmfr.20.3.1808004. [DOI] [PubMed] [Google Scholar]
- 85.Schneider J. A., Shaw G. G., van Reken D. Congenital heart disease in oculodentodigital dysplasia. Virginia Medical. 1977;104(4):262–263. [PubMed] [Google Scholar]
- 86.Schuller M. G., Barnett M. L., Strassburger K., Friedman D. L., Sonnenberg E. M. Oculodentodigital dysplasia. Oral Surgery, Oral Medicine, and Oral Pathology. 1986;61(4):418–421. doi: 10.1016/0030-4220(86)90431-7. [DOI] [PubMed] [Google Scholar]
- 87.Sharma N. L., Sharma R. C., Goyal A., Goyal B. K., Lakhanpal K. R. Oculodentodlgital dysplasia with cutaneous keratotic papules. Indian Journal of Dermatology, Venereology and Leprology. 1982;48(5):271–273. [PubMed] [Google Scholar]
- 88.Sugar H. S. Oculodentodigital dysplasia syndrome with angle-closure glaucoma. American Journal of Ophthalmology. 1978;86(1):36–38. doi: 10.1016/0002-9394(78)90011-9. [DOI] [PubMed] [Google Scholar]
- 89.Tejada P., Eduardo Y. W., Gutiérrez E., Barceló A., Sánchez J. Glaucoma hereditario asociado a displasia oculodentodigital. Archivos de la Sociedad Española de Oftalmología. 2011;86(9):292–294. doi: 10.1016/j.oftal.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 90.Thoden C. J., Ryoppy S., Kuitunen P. Oculodentodigital dysplasia syndrome. Report of four cases. Acta Paediatrica Scandinavica. 1977;66(5):635–638. doi: 10.1111/j.1651-2227.1977.tb07960.x. [DOI] [PubMed] [Google Scholar]
- 91.Traboulsi E. I., Faris B. M., Kaloustian V. M. D., Opitz J. M., Reynolds J. F. Persistent hyperplastic primary vitreous and recessive oculo-dento- osseous dysplasia. American Journal of Medical Genetics. 1986;24(1):95–100. doi: 10.1002/ajmg.1320240111. [DOI] [PubMed] [Google Scholar]
- 92.National Center for Biotechnology Information. ClinVar. March 2020, https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000137482.1.
- 93.Consortium T. U. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research. 2018;47(D1):D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1: all GJA1 mutations with associated eye and ocular adnexa features. This dataset groups patients with ODDD by GJA1 mutation and reports the associated eye and ocular adnexa features.