

SUMMARY
The pro-longevity enzyme SIRT6 regulates various metabolic pathways. Gene expression analyses in SIRT6 heterozygotic mice identify significant decreases in PPARα signaling, known to regulate multiple metabolic pathways. SIRT6 binds PPARα and its response element within promoter regions and activates gene transcription. Sirt6+/− results in significantly reduced PPARα-induced β-oxidation and its metabolites and reduced alanine and lactate levels, while inducing pyruvate oxidation. Reciprocally, starved SIRT6 transgenic mice show increased pyruvate, acetylcarnitine, and glycerol levels and significantly induce β-oxidation genes in a PPARα-dependent manner. Furthermore, SIRT6 mediates PPARα inhibition of SREBP-dependent cholesterol and triglyceride synthesis. Mechanistically, SIRT6 binds PPARα coactivator NCOA2 and decreases liver NCOA2 K780 acetylation, which stimulates its activation of PPARα in a SIRT6-dependent manner. These coordinated SIRT6 activities lead to regulation of whole-body respiratory exchange ratio and liver fat content, revealing the interactions whereby SIRT6 synchronizes various metabolic pathways, and suggest a mechanism by which SIRT6 maintains healthy liver.
Graphical Abstract

In Brief
How the pro-longevity enzyme SIRT6 coordinates between various metabolic pathways is still obscure. Here, Naiman et al. show that SIRT6 activates PPARα to promote fatty acid beta oxidation and inhibit pyruvate oxidation during fasting. This ultimately decides the energy source under nutrient-limited conditions, promoting fat usage over other energy sources.
INTRODUCTION
Metabolic diseases such as obesity, type 2 diabetes, and fatty liver are becoming the epidemic of the twenty-first century. Their influence on society, in terms of public health and their economic cost, is enormous. The worldwide increase in lifespan is associated with a corresponding rise in obesity-related pathologies. Thus, increasing our understanding of key regulators that play a role in healthy lifespan is crucial. Sirtuins are a family of NAD+-dependent deacylases homologous to yeast SIR2 deacetylase (Imai et al., 2000), which were shown to regulate lifespan and age-related metabolic diseases (Tissenbaum and Guarente, 2001; Whitaker et al., 2013).
Of the seven mammalian sirtuins, SIRT6 was shown to regulate longevity (Kanfi et al., 2012) and various physiological pathways (Zhong et al., 2010). These include embryonic development, DNA repair, transposon stability, metabolism of carbohydrates, cholesterol and fat, inflammation, circadian rhythms, cancer, and aging (Kuang et al., 2018). SIRT6 deficiency in primates or its inactivating mutation in human causes peri- and post-embryonic lethality, respectively (Ferrer et al., 2018; Zhang et al., 2018), while transgenic (TG) mice overexpressing SIRT6 have an increased male lifespan (Kanfi et al., 2012). Furthermore, in old age these mice are less susceptible to a spectrum of aging-related dysfunctions (Roichman et al., 2017). Similarly, calorie restriction (CR), known to extend healthy lifespan in both murine and primate models, increases SIRT6 levels (Kanfi et al., 2008a). This suggests that SIRT6 might mediate the beneficial effects of CR, resulting in increased lifespan. Indeed, male SIRT6 TG mice have a liver transcription profile highly similar to male mice under CR (Kanfi et al., 2012). Thus, SIRT6 is a key regulator of healthy aging, via its effects on metabolism.
SIRT6 is involved in many facets of metabolism, regulating key metabolic pathways such as insulin-like growth factor (IGF-1) (Sundaresan et al., 2012) and AMP-activated protein kinase (AMPK) (Cui et al., 2017). Glucose metabolism is also controlled by SIRT6 via inhibition of HIF1α-dependent transcription including glucose transporter genes, subsequently reducing glycolysis (Zhong et al., 2010). Interestingly, SIRT6 was also found to negatively regulate gluconeogenesis in the liver by regulating PGC1α and FOXO1 activities (Dominy et al., 2012; Zhang et al., 2014). SIRT6 is also an important regulator of lipid homeostasis. SIRT6 inhibits cholesterol and triglyceride biosynthesis by inhibiting SREBP1/2, and controls hepatic fat metabolism by repressing miR122 (Elhanati et al., 2013, 2016). SIRT6 TG mice are protected against the physiological damages of a high-fat diet (HFD) (Kanfi et al., 2010), whereas fat-specific SIRT6 knockout (KO) increases obesity-related phenotypes (Kuang et al., 2017; Xiong et al., 2017). Liver-specific KO of SIRT6 results in increased hepatic steatosis and decreased β-oxidation (Kim et al., 2010). In contrast, SIRT6 TG mice fed an HFD have decreased micro-vesicular lipidosis and reduced body fat content compared to their wild-type (WT) littermates (Kanfi et al., 2010). Despite such extensive studies of SIRT6, our knowledge of its involvement in specific metabolic pathways, particularly β-oxidation, is still limited. Moreover, how SIRT6 coordinates between these various metabolic pathways is unknown.
PPARα, one of the three peroxisome proliferator-activated receptor (PPAR) isoforms, is a key transcription factor in hepatic β-oxidation. PPARα binds to a diverse group of compounds, such as WY 14,643 (WY) (Kersten, 2014), and to DNA as a mostly obligatory heterodimer with retinoic acid receptor RXRα, recognizing the PPAR response elements (PPREs). PPARα can be found in many large complexes and its ligand binding induces conformational changes, allowing coactivators to directly bind to PPARα, mainly via the LXXLL motif (Bugge and Mandrup, 2010). The known PPAR coactivator SRC2/NCOA2 binds PPARs and its main function is the recruitment of histone acetyltransferases (HATs), such as P300 and CBP (Powell et al., 2007). These HATs acetylate the chromatin surrounding the PPRE and activate PPARα-dependent transcription.
PPARα is expressed mainly in tissues with a high capacity for fatty acid oxidation, primarily the liver and heart (Moreno et al., 2010). Under conditions that demand fatty acids oxidation such as fasting, PPARα ensures energy availability by upregulating the expression of enzymes necessary for fat oxidation, such as CPT1α (Leone et al., 1999; Mandard et al., 2004). CR was shown to increase hepatic PPARα-mediated fatty acid oxidation and whole-body fat oxidation rates (Takemori et al., 2011). Once activated, PPARα promotes β-oxidation of fatty acids, releasing free fatty acids, thereby increasing acetyl-coA production from fats. Aside from its direct role in β-oxidation, PPARα also indirectly drives the liver toward lipid oxidation by inhibiting glycolysis-derived pyruvate oxidation via PDK4 activation, and by increasing gluconeogenic precursors such as lactate and alanine (Baes and Peeters, 2010). Additionally, PPARα was shown to inhibit SREBP-mediated cholesterol and triglyceride synthesis (König et al., 2007, 2009). Thus, PPARα also regulates β-oxidation indirectly via several modes of regulation, including activation of gluconeogenesis and inhibition of pyruvate oxidation and cholesterol/triglyceride synthesis.
PPARα shares several properties with SIRT6. Both were implicated in hepatic β-oxidation, inflammation, and circadian clock regulation (Chen and Yang, 2014; Vachharajani et al., 2016), and their activities increase under starvation and CR. SIRT6 and PPARα KO mice have shortened lifespans, and their levels decrease in aged mice (Howroyd et al., 2004; Kaluski et al., 2017; Mostoslavsky et al., 2006). Thus, these findings suggest that a coregulatory interaction between SIRT6 and PPARα may exist. Here, we underscore the interplay between SIRT6 and PPARα and demonstrate how SIRT6 coordinates different metabolic pathways.
RESULTS
SIRT6 Deficiency Inhibits PPARα Signaling Gene Expression
To identify the physiological pathways regulated by SIRT6, we performed quantitative transcriptome analysis based on RNA sequencing (RNA-seq) from livers of WT and SIRT6 heterozygotic (HZ) mice (Table S1A). The top highly differentially expressed (DE) genes (Figure 1A) were validated by quantitative real-time PCR (Figure 1B; Table S1B). Interestingly, the top three pathways differentially regulated in SIRT6 HZ mice are all directly related to β-oxidation. Ingenuity pathway analysis (IPA) (Krämer et al., 2014) identified Acyl-CoA hydrolysis, an intermediate step in β-oxidation, as the most significantly regulated pathway. The second-ranked pathway was triacylglycerol degradation, which hydrolyzes triacylglycerol into fatty acids, used in β-oxidation. The third pathway, stearate biosynthesis, involves saturated fatty acid synthesis from palmitate and carbohydrates. Other pathways include Ehprin and VEGF signaling, involved in differentiation and angiogenesis, respectively (Figure 1C). Consistent with these findings, PPARα, a key regulator of liver fatty acid β-oxidation was identified as the most significantly inhibited upstream regulator of the DE genes. Likewise, PPARγ was identified as a significantly inhibited regulator. Other regulators include TNFα, a known SIRT6 target of inflammation, and ACOX1 and EHHADH, which, similar to PPARα, regulate β-oxidation (Figure 1D). These findings strongly suggest that SIRT6 activates PPARα.
Figure 1. SIRT6 Deficiency Inhibits PPARα Signaling Gene Expression.
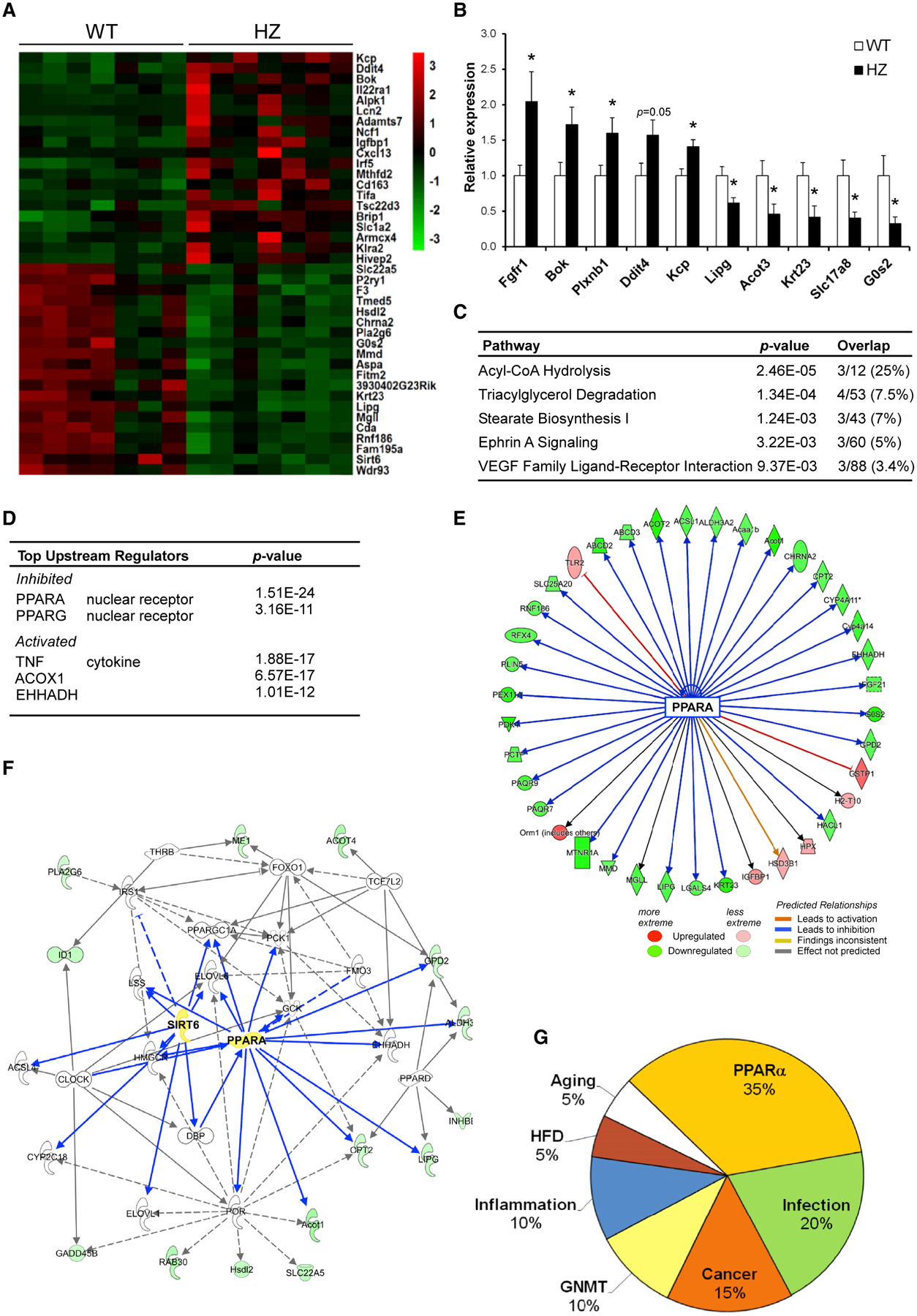
(A) Heatmap of top-20 up and down differentially expressed (DE) genes in RNA-seq of SIRT6 HZ mice.
(B) Quantitative real-time PCR analysis of mouse mRNA levels of 5 up- and down-regulated genes from the RNA-seq.
(C) Ingenuity pathway analysis (IPA).
(D) Ingenuity regulator analysis of DE genes.
(E) PPARα inhibitory gene network.
(F) Lipid metabolism network of DE genes.
(G) ExpressionBlast analysis of top-20 microarray comparison matches to SIRT6 HZ DE genes.
In (A)–(G), n = 7 mice per genotype. In (A) DESeq2 with false discovery rate (FDR) < 0.1 and a fold change cutoff > 1.5 were used to find DE genes. In (B), data are represented as means + SE; *p < 0.05, significance was calculated by two-tailed Student’s t test. In (C)–(F), IPA was used to calculate significant pathways, regulators, and networks. In (G), ExpressionBlast was used to calculate significant expression matches; *p < 0.01.
Indeed, many genes activated by PPARα were inhibited in HZ mice, whereas genes inhibited by PPARα were activated in HZ mice (Figure 1E). Likewise, lipid metabolism, the primary pathway of PPARα, was a top network found in the DE genes. Both SIRT6 and PPARα are centrally placed in the lipid metabolism network, indicating possible coordination between both proteins in regulating fat metabolism (Figure 1F). Other known SIRT6-regulated networks such as inflammation and carbohydrate metabolism were identified as well (Figure S1). Furthermore, comparison of HZ liver DE genes to all microarrays currently in the GEO database using the ExpressionBlast tool (Zinman et al., 2013) identified the top three highly significant matches to be PPARα KO microarrays (Figure 1G). Moreover, 7 of the top 20 matches were from PPARα KO experiments (Table S2). This shows that SIRT6 HZ mice display a gene expression profile most similar to PPARα KO mice, compared to all other existing microarray expression profiles. Aside from PPARα, SIRT6 HZ expression data were similar to known SIRT6-regulated pathways such as infection and cancer (Lerrer et al., 2016; Table S2). Altogether, these findings identified PPARα as a candidate SIRT6-regulated pathway.
SIRT6 Directly Interacts with PPARα and Binds to and Activates the PPRE In Vivo
To investigate the mechanism underlying SIRT6 activation of PPARα signaling, PPARα levels and acetylation were examined in WT and HZ mice. PPARα mRNA and protein levels were similar in WT and HZ mice (Figures S2A and S2B). In addition, in line with previous studies (Oka et al., 2011), no acetylation was detected on PPARα (Figure S2C). Next, we examined whether SIRT6 and PPARα might physically interact. Recombinant PPARα-GST tagged protein co-immunoprecipitated with recombinant SIRT6-FLAG-tagged protein, but not with FLAG-tagged BIP used as a negative control. Additionally, GST protein alone did not co-immunoprecipitate with SIRT6 (Figure 2A). These findings show that SIRT6 specifically binds PPARα in vitro. The binding affinity of SIRT6 to PPARα in comparison to other known SIRT6 interactors was then measured. A microfluidics protein binding platform (Ben-Ari et al., 2013) was used to quantify SIRT6 binding affinity to selected proteins. Binding was measured as fluorescent signals of interactors normalized to SIRT6 levels. Interestingly, in comparison to two known SIRT6 interacting proteins, HIF1α and CTIP, the binding affinity of SIRT6 to PPARα was significantly higher than to HIF1α, suggesting strong interaction between PPARα and SIRT6 (Figure 2B). A representative binding signal is shown (Figure 2B, right panel). We then examined SIRT6 binding to PPARα in HEK293T cells. As seen in Figure 2C, PPARα-GFP specifically co-immunoprecipitated with SIRT6-FLAG but not with GFP negative control. Reciprocally, endogenous SIRT6 specifically co-immunoprecipitated with PPARα-FLAG (Figure 2D). In all the binding experiments using intact cells, the binding was not DNA dependent, as ethidium bromide did not abolish the interaction (Figures 2C and 2D). Endogenous SIRT6 and GFP-tagged PPARα or endogenous PPARα and FLAG-SIRT6 interactions were shown in mouse Hepa1–6 hepatocytes as well (Figure S2D). These results show that SIRT6 and PPARα directly interact and are present in common protein complexes in cells.
Figure 2. SIRT6 Directly Interacts with PPARα and Binds to and Activates the PPRE In Vivo.

(A) In vitro pulldown assay of SIRT6-FLAG and co-immunoprecipitation of GST-PPARα from recombinant proteins.
(B) Microfluidics in vitro association assay. SIRT6-FLAG was fixed onto the chip, and Myc-tagged associated proteins were incubated and then washed. Interaction ratio was detected by fluorescence (left). Representative fluorescence binding on chip (right).
(C) Co-immunoprecipitation of FLAG-tagged SIRT6 and GFP-tagged PPARα.
(D) Co-immunoprecipitation of FLAG-tagged PPARα and endogenous SIRT6 from HEK293T cells.
(E) Microfluidics assay of SIRT6 binding to PPRE or mutant sequence in the presence/absence of PPARα and representative fluorescence binding on chip.
(F) Luciferase activity of PPRE promoter in HEK293T cells overexpressing either SIRT6 WT or dominant-negative (DN) mutant.
(G) Luciferase activity in HEK293T cells overexpressing SIRT6 and increasing amounts of PPARα.
(H) Srebp2 and Gapdh were used as positive/negative controls, respectively.
(I) ChIP-quantitative real-time PCR analysis of H3K9 acetylation on PPREs of indicated genes in WT and Sirt6+/− livers.
In (B), data are represented as means + SE and significance was calculated by one-way ANOVA followed by a Bonferroni multiple comparisons test; n = 10. In (E)–(I), data are represented as means + SE. In (E) and (I), two-way ANOVA followed by a Bonferroni multiple comparisons test were used; n = 10 for (A) and (E) and n = 3 for (I). In (H), significance was calculated by two-tailed Student’s t test. *p < 0.05, **p < 0.01.
We demonstrated that SIRT6 and PPARα associate with one another. Thus, the direct binding of SIRT6 to the PPARα-binding DNA element-PPRE and whether PPARα is required for this interaction were examined by using the quantitative protein interacting DNA (QPID) microfluidics method (Glick et al., 2016b). Recombinant SIRT6-FLAG was immobilized to the chip surface and incubated with Cy5-labeled PPREx3 or mutant DNA sequences (Oka et al., 2012) in the presence or absence of recombinant free Myc-tagged PPARα. The interaction ratio was calculated and is shown in Figure 2E. Importantly, significant and strong SIRT6 binding to PPRE was found specifically in the presence of PPARα. In contrast, SIRT6 did not bind to a mutant motif regardless of the presence of PPARα. A representative binding experiment is shown (Figure 2E). SIRT6’s interaction with the PPRE was also examined in the presence or absence of PPARα using serial DNA dilutions (Glick et al., 2016b). In the presence of PPARα, SIRT6 displayed a logarithmic binding curve that reached saturation, indicating marked affinity to the PPRE but not to mutant PPRE sequence, whereas SIRT6 alone displays a non-specific linear binding curve similar to the PPRE mutant (Glick et al., 2016b; Figures S3A and S3B). These findings show that SIRT6 binds specifically to the PPRE in a PPARα-dependent manner.
Next, we examined whether SIRT6 can activate the PPRE in vivo using a luciferase reporter assay. A construct containing the luciferase gene fused to three tandem repeats of the PPRE (Kim et al., 1998) was transfected into mouse Aml-12 hepatocyte cells along with SIRT6 or control plasmids. SIRT6 overexpression significantly induced the luciferase signal (Figure S3C). Importantly, SIRT6 does not activate negative control promoter sequences (Figure S3D). Thus, SIRT6 stimulates endogenous PPARα-dependent promoter activity in liver cells. To examine whether SIRT6 catalytic activity is required for PPARα transactivation, HEK293T cells were transfected with either SIRT6 or a catalytically inactive mutant, SIRT6 H133Y. Notably, SIRT6 but not the SIRT6 catalytic mutant activated PPRE transcriptional activity (Figure 2F). These findings suggest that SIRT6 enzymatic activity is required to activate the PPRE. Moreover, induction of the PPRE by PPARα overexpression was further increased in SIRT6 overexpressing cells (Figure 2G). Thus, the two proteins may work cooperatively to activate the PPRE. These data indicate that SIRT6 directly activates the PPRE via PPARα.
Subsequently, SIRT6 binding to the PPRE within promoters of PPARα target genes in vivo was measured using chromatin immunoprecipitation (ChIP) assay in primary hepatocytes. As shown in Figure 2H, in comparison to immunoglobulin G (IgG) control, endogenous SIRT6 significantly binds to the PPREs of several PPARα target genes. Strikingly, SIRT6 binds to the PPREs of Cpt1α, the rate-limiting enzyme of β-oxidation, as well as to the positive control Srebp2 promoter (Elhanati et al., 2013). This binding was specific, as SIRT6 does not bind to a negative control DNA sequence in the GAPDH gene promoter (Figure 2H). Srebp2 promoter was shown to be inhibited and deacetylated on H3K9 by SIRT6 (Tao et al., 2013). Indeed, in comparison to WT livers, Sirt6+/− livers showed significant increased Srebp2 promoter H3K9 acetylation levels. In contrast, specifically on the SIRT6-bound PPREs, Sirt6+/− livers showed significantly decreased H3K9 acetylation levels. (Figure 2I). Notably, SIRT6 binds not only to PPREs within promoters but also to the PPRE localized 4 Kb distal of Angptl4 (Figures 2H and S3E). These findings further indicate that SIRT6 binding is PPREs specific and not due to its proximity to other transcription elements near the promoter region. Moreover, these findings suggest that SIRT6 deacetylase activity promotes the activation of PPREs potentially via deacetylation of a PPARα cofactor and not via deacetylation of PPARα or the PPRE.
SIRT6 was shown to bind to PPARα and PPREs under normal growth conditions (Figure 2). Next, we examined whether SIRT6 binding to PPRE depends on PPARα activity. Primary hepatocytes were treated with the specific PPARα agonist, WY to induce PPARα activity. Interestingly, treatment with WY did not further increase SIRT6 binding to PPREs in comparison to untreated controls (Figure S3F). These findings imply that the in vivo association between SIRT6 and the PPRE is constant, irrespective of PPARα activation. Taken together, these results conclusively show that SIRT6 binds to and activates the PPRE in vivo in a PPARα-dependent manner.
SIRT6 Stimulates WY-Induced PPARα Transcriptional Activity In Vivo
As SIRT6 was found to bind to the PPRE (Figure 2), the effect of SIRT6 on transcription of various hepatic PPARα signaling pathways was investigated. Gene expression of PPARα-regulated pathways was examined in WT or SIRT6 HZ primary hepatocytes treated with WY. As previously demonstrated, upon WY treatment a strong induction of PPARα transcription was observed both in mouse liver and primary hepatocytes relative to controls (Szalowska et al., 2014; Figure 3). These pathways include mitochondrial β-oxidation, peroxisomal β-oxidation, acyl-CoA binding/hydrolysis, lipid storage/transport, and ketogenesis. WY does not directly bind to SIRT6, nor does it affect SIRT6 deacylase activity or RNA levels (Figures S3A–S3C). Importantly, SIRT6 HZ primary hepatocytes consistently displayed reduced PPARα activation (Figure 3A). Conversely, in comparison to WT, SIRT6 TG mice had significantly increased induction of PPARα targets (Figure 3B) in primary hepatocytes. These findings show that SIRT6 uniformly promotes PPARα transcriptional activity in multiple pathways in primary liver cells.
Figure 3. SIRT6 Stimulates WY-Induced PPARα Transcriptional Activity In Vivo.

(A–D) Quantitative real-time PCR analysis of mouse mRNA levels of PPARα target genes from specified pathways in WT and SIRT6 HZ (A), and SIRT6 TG (B) control and WY-treated primary hepatocytes. Quantitative real-time PCR analysis of mouse mRNA levels of PPARα target genes from specified pathways in WT and SIRT6 HZ (C), and SIRT6 TG (D) control and WY-gavaged mice. Genes are categorized into different pathways as labeled above.
In (A)–(D), data are represented as means + SE; *p < 0.05, calculated by two-tailed Student’s t test; n = 7 per genotype.
Next, in order to examine whether SIRT6 can activate PPARα in vivo, SIRT6 HZ and TG mice along with their appropriate WT controls were treated with WY. Consistent with the response of primary hepatocytes, SIRT6 HZ mice displayed significantly reduced PPARα activation (Figure 3C). Additionally, SIRT6 TG mice had significantly increased PPARα activation (Figure 3D). SIRT6 was previously shown to regulate β-oxidation via AMPK (Cui et al., 2017; Elhanati et al., 2016). Yet, WY is a specific PPARα activator, and no changes in phosphorylated AMPK were found following this treatment (Figure S4D). These results show that SIRT6 activates PPARα signaling in livers in vivo. Interestingly, as previously shown in mice but not in primary hepatocytes, the lipid transport gene Slc27a1 is induced by WY treatment (Rakhshandehroo et al., 2010), and SIRT6 further activated this gene in mice (Figures 3C and 3D). In addition, SIRT6 also increases the expression of Fgf21 longevity hepatokine, a critical factor for PPARα activity (Figure 3; Goto et al., 2017). This indicates that in vivo, SIRT6 may have a broader effect in regulating PPARα signaling. Altogether, these results show that SIRT6 specifically activates PPARα-dependent transcription of several metabolic pathways.
SIRT6 Deficiency Inhibits PPARα-Regulated Metabolic Pathways
SIRT6 activates the PPARα-dependent transcription of genes involved in several metabolic pathways (Figure 3). Thus, we examined the downstream effects of SIRT6 on a range of PPARα-regulated pathways. Specifically, we investigated the ability of SIRT6 to activate PPARα pathways β-oxidation, gluconeogenesis, and glycerol transport, and to inhibit glycogenolysis and pyruvate metabolism, in WY treated mice. When activated, in order to increase β-oxidation, PPARα inhibits pyruvate oxidation by upregulating PDK4 and increases fatty acid and glycerol transport. PPARα also increases precursors of gluconeogenesis such as lactate and alanine (Baes and Peeters, 2010; Figure 4A).
Figure 4. SIRT6 Deficiency Inhibits PPARα-Regulated Metabolic Pathways.

(A) Scheme depicting SIRT6 regulation of metabolic pathways via PPARα.
(B) Western blot analysis of pyruvate oxidation signaling following WY treatment (50 mg/kg) (left) in WT mice, and in WT versus HZ WY-treated mice (middle), and ImageJ quantification (right). Tubulin was used as loading control and CMC 0.1% was used as control for the WY.
(C) β-oxidation pathway measurements including quantitative real-time PCR analysis of mRNA levels of fatty acid transporter Cd36 (left), metabolite acetylcarnitine C2 (middle), and CO2 levels from 14C-labeled palmitate in mitochondria (right) from WY-treated control and SIRT6 HZ livers.
(D) Quantitative real-time PCR analysis of mRNA levels of glycerol transporter Aqp3.
(E) Gluconeogenic precursor metabolites lactate (left) and amino acid alanine (right).
(F) Quantitative real-time PCR analysis of mRNA levels of glycogenolysis and glycogen synthesis enzymes Pygl and Gys2, respectively. In (B)–(F), all from livers of WT and HZ mice following control (CMC) or WY treatment.
(G and H) Metabolites (G) and quantitative real-time PCR analysis of mRNA levels in livers of fasted WT and SIRT6 TG mice (H).
In (B)–(H) proteins and genes are represented as means + SE and metabolites are represented as means + max/min; *p < 0.05, **p < 0.01. In (B), (G), and (H) a two-tail Student’s t test was used; n = 6–7 per genotype. In (C)–(F), two-way ANOVA followed by a Bonferroni multiple comparisons test were used; n = 6–7 per genotype.
First, regulation of pyruvate oxidation by SIRT6 was examined. PKD4 kinase and its substrate PDC (P-PDC) are strongly induced in mouse livers following WY treatment (Figure 4B, left panel). Both PDK4 and P-PDC protein levels were significantly lower in HZ mice, while total PDC levels remained unchanged. This indicates that SIRT6 deficiency reduces PPARα-dependent inhibition of pyruvate oxidation (Figure 4B, middle and right panels). We next measured the regulation of fatty acid transport and β-oxidation. RNA levels of fatty acid transporter Cd36 were strongly induced following WY treatment and were significantly less activated in HZ mice (Figure 4C, left panel). Moreover, β-oxidation products were measured from labeled palmitate ex vivo in liver mitochondria. Acetylcarnitine metabolite levels, the product of long-chain fatty acid oxidation, were significantly reduced in SIRT6 HZ mice after WY treatment compared to controls (Figure 4C, middle panel). Likewise, significantly lower levels of the final product, palmitate-derived CO2, generated by the Krebs cycle from labeled palmitate, were found in isolated hepatic mitochondria from WY-treated HZ livers (Figure 4C, right panel). Thus, SIRT6 HZ mice have decreased β-oxidation compared to control mice upon WY treatment. These results support a role for SIRT6 in promoting PPARα-mediated β-oxidation.
PPARα is also known to increase gluconeogenic precursors glycerol, lactate, and alanine. Thus, we first measured RNA levels of the glycerol transporter Aqp3 in the liver. As can be seen in Figure 4D, in comparison to WT littermates, upon WY treatment HZ mice showed significantly reduced induction of RNA levels of Aqp3. Likewise, the metabolites lactate and alanine were also significantly less induced in HZ mice in response to WY treatment (Figure 4E). We subsequently measured RNA levels of key enzymes of glycogenolysis, Gys2 and Pygl. Similar to previous studies, WY treatment inhibited the expression of the glycogenolysis gene Pygl and activated the glycogen synthesis gene Gys2. This effect was nearly completely abolished in HZ mice (Figure 4F). These data demonstrate that SIRT6 activates a broad spectrum of PPARα signaling pathways to regulate metabolism, ultimately increasing β-oxidation.
Next, we aimed to examine whether the effects of SIRT6 on gene expression and metabolites are also found under normal physiological conditions in which PPARα is active. PPARα is known to be important for activating hepatic β-oxidation and inhibiting pyruvate oxidation in response to fasting. Thus, we examined whether SIRT6 TG mice have any defects in these fasting-regulated pathways. Indeed, as can be seen in Figure 4G, under fasting conditions, SIRT6 overexpression results in a significant increase in pyruvate levels, a sign of inhibited pyruvate oxidation, and an increase in β-oxidation metabolite acetylcarnitine (Figure 4G). In addition, a significant increase in glycerol levels was found in TG mice under starvation. These changes in metabolites were correlated with a significant increase in the transcription of the pyruvate oxidation inhibitor Pdk4 and the β-oxidation activators Cpt1a, Acot3, and Ehhadh genes and Gos2, a lipid storage gene, were found (Figure 4H). Altogether, these data show that SIRT6 activates PPARα, leading to decreased pyruvate oxidation, increased β-oxidation, and increased uptake of gluconeogenic precursors, as found under a normal fasting response.
SIRT6’s Regulation of Pyruvate and β-oxidation Is PPARα Dependent
To show a PPARα dependency in the effect of SIRT6 on PPARα signaling, the following experiment was performed: PPARα was knocked down (KD) using two different small interfering RNAs (siRNAs) in both primary and Hepa1–6 hepatocytes overexpressing SIRT6. PPARα KD was validated in both RNA and protein levels (Figures 5A and S5A–S5C). First, we examined to what extent SIRT6 can stimulate PPRE luciferase reporter when PPARα is depleted. As seen in Figure 5B, in comparison to control scrambled siRNA, SIRT6 activation of PPRE was abolished in PPARα KD Hepa1–6 cells. The importance of PPARα in mediating SIRT6’s effect on β-oxidation genes was investigated as well. Indeed, SIRT6 activation of β-oxidation genes was significantly reduced in PPARα KD primary hepatocytes (Figure 5C). Only two genes were found to be not wholly PPARα dependent (Figures S5D and S5E). Lastly, in comparison to control cells, the effect of SIRT6 on pyruvate signaling was also significantly inhibited in PPARα KD primary hepatocytes (Figures 5D and S5F). Altogether, these data conclusively show that PPARα mediates the effect of SIRT6 on multiple PPARα signaling pathways.
Figure 5. SIRT6 Regulation of Pyruvate and b-oxidation Is PPARα Dependent.

(A) PPARα protein expression levels in nuclear fractions of primary hepatocytes overexpressing either scrambled siRNA control or two different PPARα siRNAs. Histone H3 was used as loading control. ImageJ quantification of PPARα levels is shown on the right.
(B) Luciferase activity of PPRE in WT or SIRT6 overexpressing Hepa1–6 mouse hepatocytes overexpressing either scrambled siRNA control or two different PPARα siRNAs with or without WY treatment.
(C) Quantitative real-time PCR analysis of mouse mRNA levels of PPARα target genes from specified pathways in WT and SIRT6 overexpression (OE) primary hepatocytes overexpressing either scrambled siRNA control or two different PPARα siRNAs with or without WY treatment.
(D) Western blot analysis of pyruvate oxidation signaling following WY treatment in WT mice and SIRT6 OE primary hepatocytes overexpressing either scrambled siRNA control (labeled as c) or two different PPARα siRNAs (left). PDK4/Tubulin and pPDC/PDC were quantified by ImageJ (right). Tubulin was used as loading control.
In (A)–(D), data are represented as means + SE; *p < 0.05, **p < 0.01. In (A), one-way Anova was used to calculate significance followed by a Bonferroni multiple comparisons test, n = 2 per genotype. In (B) and (C), significance was calculated by two-way ANOVA followed by a Bonferroni multiple comparisons test; n = 2 per genotype. In (D), significance was calculated by two-tailed Student’s t test; n = 2 per genotype.
SIRT6 Increases Hepatic Fat Loss and Improves RER via PPARα in Response to WY
Finally, we further explored the broader effects of activation of PPARα by SIRT6. RNA-seq was performed from livers of WT and SIRT6 HZ mice treated with WY (Figure 6A; Table S1, tabs C and D). In line with the abovementioned results, PPARα was found to be one of the most strongly inhibited regulators in HZ mice (Figure 6B). Previous studies showed that induction of PPARα strongly represses SREBP1/2-mediated transcription (Zhang et al., 2015). Indeed, SREBP1/2 was among the most highly activated regulators found in HZ mice under WY treatment (Figure 6B). Importantly, this effect was not observed under normal conditions, in the absence of WY (Figure 1), suggesting that the effect of SIRT6 haploinsufficiency on the SREBP pathway is PPARα dependent. Moreover, under normal conditions, in contrast to SIRT6 KO (Tao et al., 2013), SIRT6 HZ mice have similar SREBP levels as WT mice (Figure S4D). Further pathway analysis showed that the top-five pathways differentially expressed in SIRT6 HZ mice were activation of SREBP-regulated pathways, cholesterol and sterol biosynthesis, and activation of inflammation-related pathways, B cell development, and antigen presentation (Figure 6C). Network analysis showed that the top significant network in the WY-treated hepatic transcriptome placed PPARα, SREBP, and TNFα/NFKb as central nodes (Figure S6). This indicates a broad and integrative role for SIRT6 in inflammation, cholesterol synthesis, and β-oxidation in a PPARα-dependent manner.
Figure 6. SIRT6 Increases Hepatic Fat Loss and Improved RER via PPARα.
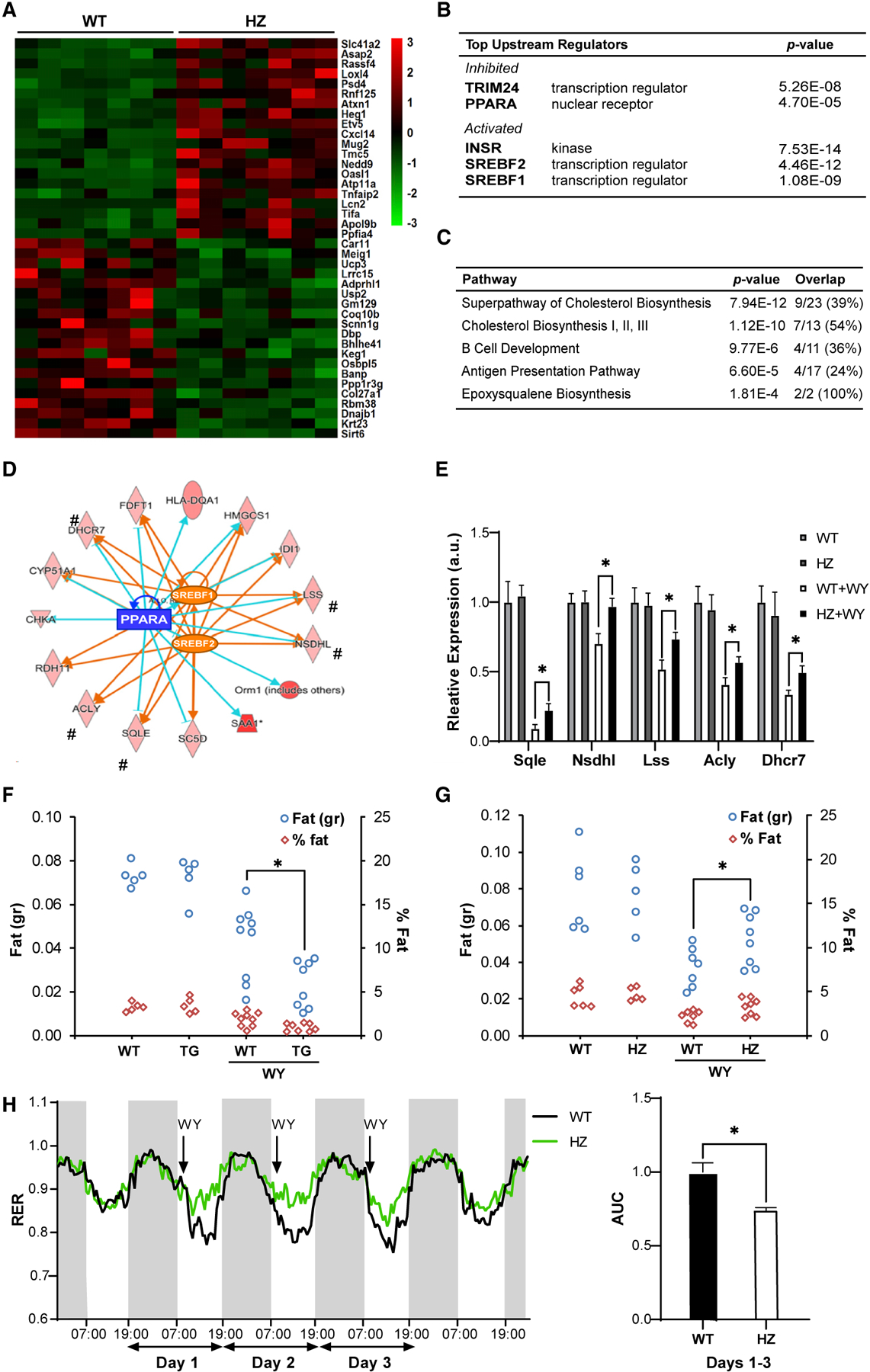
(A) Heatmap of top-20 up and down differentially expressed (DE) genes in RNA-seq of livers of WY-treated SIRT6 HZ mice.
(B and C) Ingenuity regulator analysis (B) and IPA (C) of DE genes in WY-treated SIRT6 HZ mice.
(D) Integrated PPARα-activated and SREBP1/2-inhibited overlapping gene network. The pound sign (#) indicates genes validated by quantitative real-time PCR analysis.
(E) Quantitative real-time PCR analysis of mRNA levels of PPARα-dependent SREBP cholesterol and triglyceride synthesis genes from (D).
(F and G) NMR measurements of total fat content in livers of WY-treated SIRT6 TG (F) and SIRT6 HZ (G) mice, in both grams and normalized to total liver weight.
(H) Respiratory exchange ratio (RER) of WT or HZ mice given daily WY treatment. Black arrows indicate time of WY treatment (left). AUC, area under curve for average 3 days of treatment (right); arrow indicates start of treatment.
In (A), DESeq2 with FDR < 0.1, fold change > 1.5 was used to find DE genes, n = 7. In (B)–(D), IPA was used to calculate significant pathways, regulators, and networks, n = 7. In (E)–(H), data are represented as means + SE. In (E)–(G), *p < 0.05, significance calculated by two-way ANOVA followed by a Bonferroni multiple comparisons test; n = 5–8 per genotype. In (H), AUC was calculated using Prism followed by two-tailed Student’s t test; n = 3 per genotype.
Next, we examined the overlap between PPARα-dependent genes and known SREBP targets in HZ DE genes. Notably, SIRT6 DE genes activated by SREBP1/2 are inhibited by PPARα, showing an opposite regulation (Figure 6D). These genes are involved in triglyceride and cholesterol synthesis. Importantly, as seen in Figure 6F, in the absence of PPARα activation, SIRT6 did not affect these genes in HZ mice. Yet, upon WY treatment, SIRT6 haploinsufficiency significantly blunted PPARα inhibition of SREBP-dependent genes (Figure 6E). Thus, these findings show that SIRT6 regulates PPARα inhibition of SREBP-dependent cholesterol synthesis.
We then examined the physiological effect of the SIRT6 regulation of PPARα-mediated inhibition of SREBP. Hepatic lipid content is composed of triglycerides and cholesterol, regulated by SREBP-1 and −2, respectively (Xu et al., 2013). Therefore, we treated mice with WY and analyzed whole-liver fat content using NMR. WY treatment significantly decreased fat content in liver by almost 50%. Strikingly, in SIRT6 TG mice, this fat reduction was significantly enhanced (Figure 6F). Likewise, in HZ mice this fat reduction was significantly inhibited (Figure 6G). In addition, we examined the cumulative physiological outcome of these metabolic changes. Respiratory exchange ratio (RER) was quantified in WY treatment in WT and HZ mice. The RER ratio under a normal diet was similar between WT and HZ mice. Daily WY administration caused a decrease in RER shortly after WY supplementation in WT mice (Figure 6H). Strikingly, the HZ mice had a significantly weaker response to WY treatment, indicating reduced β-oxidation. In addition, once the WY treatment was stopped, the RER returned to normal, and the changes previously seen between WT and HZ disappeared (Figure 6H). These findings suggest that SIRT6 directs the energy source utilization toward fat under broader conditions requiring PPARα activation.
SIRT6 Regulates PPARα Activity via NCOA2 K780 Deacetylation
SIRT6 deacetylase activates PPARα, yet no acetylation was detected on PPARα (Figure S2C). In addition, previously SIRT6 was reported as a histone deacetylase, which usually results in reduced levels of transcription. Intriguingly, here, SIRT6 induces PPRE H3 K9 acetylation and activity (Figure 2). Therefore, we examined whether the mechanism underlying SIRT6’s activation of PPARα is potentially via the deacetylation of one of its coactivators or corepressors. The acetylation levels of 12 PPARα coactivators or corepressors were compared between WT and SIRT6 TG livers using stable isotope labeling by amino acids in cell culture (SILAC) mass spectrometry (MS) analyses. Within these, only one protein, NCOA2, a known PPARα coactivator, showed significantly lower differentially acetylated levels on K780 in TG mice (Figure 7A). Therefore, SIRT6 and NCOA2 interaction was examined. As seen in Figure 7B, endogenous SIRT6 interacts with FLAG-tagged NCOA2. Conversely, endogenous NCOA2 interacts with FLAG-tagged SIRT6. This suggests that NCOA2 might be the PPARα coactivator through which SIRT6 mediates the induction of PPARα activity.
Figure 7. SIRT6 Regulates PPARα Activity via NCOA2 K780 Deacetylation.
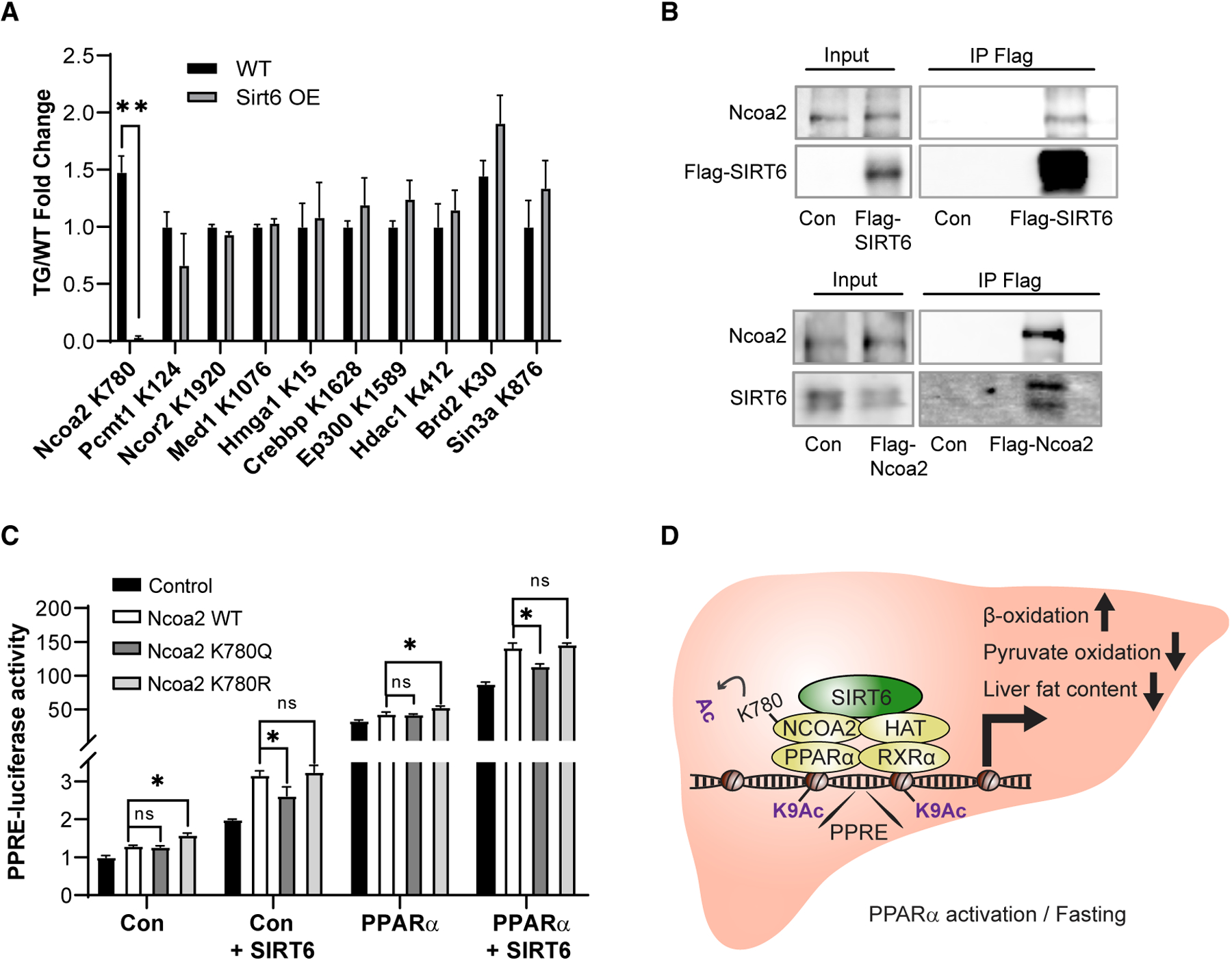
(A) MS quantification of acetylated levels of various PPARα co-activators/repressors in WT and SIRT6 OE mouse liver.
(B) Luciferase activity of PPRE in WT- or SIRT6-overexpressing HEK293T cells overexpressing either empty plasmid control or NCOA2 WT, K780Q, and K780R with or without PPARα overexpression.
(C) Co-immunoprecipitation of FLAG-tagged SIRT6 and endogenous NCOA2 (top) or FLAG-tagged NCOA2 and endogenous SIRT6 (bottom).
(D) Schematic representation of SIRT6 regulation of PPARα via NCOA2 deacetylation under conditions in which is PPARα activated.
In (A) and (B), data are represented as means + SE. In (A), FDR q < 0.1 was deemed significant, n = 3 per genotype. In (B), significance was calculated by two-way ANOVA followed by a Bonferroni multiple comparisons test; *p < 0.05, n = 7 per genotype.
To explore the role of NCOA2 acetylation in PPARα activation, NCOA2 K780 was mutated into arginine (K780R) or glutamine (K780Q), mimicking constitutive deacetylated or acetylated lysine, respectively. PPRE-luciferase activity was measured in HEK293T cells overexpressing WT, K780R, or K780Q with or without overexpression of SIRT6, PPARα, or both. As previously published, NCOA2 significantly induced PPRE activity (Røst et al., 2009). Notably, in comparison to WT or K780Q, the K780R mutant significantly further increases PPRE activation (Figures 7C and S7A). WT and K780Q showed similar levels of PPRE activation, suggesting that NCOA is highly acetylated and its deacetylation is required for further PPRE activation. Strikingly, indeed, SIRT6 overexpression significantly induced WT NCOA2-dependent PPRE activation to the same levels as K780R, the deacetylated NCOA2 mimicker. Moreover, SIRT6 overexpression had no effect on either K780Q- or K780R-induced PPRE activity or on NCOA2-PPARα association (Figures 7C and S7B). These findings suggest a model in which SIRT6 promotes PPARα activity via NCOA2 K780 deacetylation, which leads to increased HAT acetylation of PPRE chromatin and subsequent activation of PPARα-dependent signaling (Figure 7D).
DISCUSSION
Here, we show that SIRT6 regulates hepatic β-oxidation by activating PPARα via NCOA2 deacetylation. This regulation culminates in a PPARα-dependent maintenance of normal whole-body RER and improved liver fat content. These findings significantly contribute to our understanding of how SIRT6 coordinates central metabolic pathways in response to energy fluctuations.
Previously, we showed that SIRT6 overexpression mimics key aspects of the rodent CR response (Kanfi et al., 2012). β-oxidation is a known hallmark of CR in both rodents and monkeys (Bruss et al., 2010; Rhoads et al., 2018). Remarkably, SIRT6 activation of PPARα also increases fatty acid degradation and peroxisomal β-oxidation, revealing an additional similarity to the CR response. Moreover, both SIRT6 and PPARα levels increase under starvation and CR (Masternak and Bartke, 2007). Altogether, in parallel to increased PPARα levels, we suggest here a pathway of PPARα regulation under CR by induction of its activity via SIRT6.
What are the physiological outcomes of this PPARα induction by SIRT6? A significant amount of data show a role for SIRT6 as a guardian of healthy liver (Kim et al., 2010). HFD or liver-specific deletion of SIRT6 in mice causes fatty liver formation while SIRT6 overexpression significantly reduced fatty liver incidence (Kanfi et al., 2010). Thus, potentially SIRT6 protects against hepatic steatosis through PPARα activation and the increase in fatty acid β-oxidation. In addition, liver-specific SIRT6 KO impairs hepatic ketogenesis via induced Fsp27β. When fed a ketogenic diet, these mice also showed exacerbated hepatic steatosis and inflammation (Chen et al., 2019). This adds another interesting layer for SIRT6’s protective effect on liver pathologies. Yet, it is possible that SIRT6 expression in other tissues also contributes to this and other liver phenotypes observed in the current study due to the use of a whole-body HZ model. However, samples from human patients with fatty liver or liver fibrosis exhibited significantly lower levels of SIRT6 than did normal controls (Ka et al., 2017; Kim et al., 2010). Thus, SIRT6 activation has great therapeutic potential in treating fatty liver diseases.
SIRT6 possesses histone deacetylase activity that is usually associated with repressed transcription. Here, we show an example of direct activation of gene transcription by SIRT6. SIRT6 binds to PPARα and its PPREs in a PPARα-dependent manner, inducing PPARα-mediated transcription. Importantly, SIRT6 enzymatic activity is required for this induction. Therefore, here, we aimed to identify the precise target for SIRT6 within the PPARα complex on the PPRE. There are at least three possible options. First, SIRT6 might directly affect PPARα, either by deac(et)ylation, or mono-ADP-ribosylation (monoADPR). Yet, we and others were unable to detect acetylation on PPARα (Figure S2C; Oka et al., 2011). However, this possibility cannot be rigorously excluded at this stage. A second possibility is that SIRT6 binding to PPARα results in a conformational change, allowing increased binding or recruitment of PPARα coactivators. In support of this model, SIRT6 contains the well-known LXXLL motif (Figure S7C) in its coactivator domain, a motif commonly found in PPARα-binding coactivators. Yet, given that SIRT6 enzymatic activity is required for PPARα activation, such a model does not fully explain the activity of SIRT6 on PPARα. Thus, a final, more comprehensive model suggests that SIRT6 activates PPARα via deacetylation and activation of a member of the PPARα complex, such as a coactivator acetyltransferase. Indeed, we found that SIRT6 regulates PPARα activity via NCOA2 K780 deacetylation. In analogy, SIRT1 was shown to activate PPARα indirectly via deacetylation and activation of its coactivator, PCG1α (Purushotham et al., 2009). Furthermore, in contrast, SIRT4 was shown to inhibit PPARα signaling and β-oxidation indirectly by inhibiting SIRT1 activity by NAD+ substrate competition (Laurent et al., 2013). While SIRT6 and SIRT1 increase in starvation, SIRT4 decreases in starvation, which may potentially explain their opposite roles in β-oxidation (Laurent et al., 2013; Kanfi et al., 2008a, 2008b).
Deacetylation of NCOA2 K780 promotes its activation of PPARα. Likewise, NCOA2 Ser736 phosphorylation is also known to augment PPAR activity via increased binding and recruitment of P300 acetyltransferase to NCOA2 and PPREs (Frigo et al., 2006). Thus, it would be interesting to further investigate the interaction between these posttranslational modifications on PPARα activation potentially by increased P300 recruitment. In support of this model, indeed, we found an inverse correlation between NCOA2 and H3K9 acetylation along with increased PPARα activation.
Regarding the involvement of PPARα in SIRT6-dependent phenotypes, an interesting question is the potential contribution of PPARα to the gender-specific effects found in SIRT6 TG mice. In mice, SIRT6 regulation of body and fat mass is more pronounced in males. In addition, the reduced lifespan in SIRT6-deficient mice is more severe in males (Peshti et al., 2017). Similarly, SIRT6 overexpression extends lifespan, mimics a DR-like transcription profile, and reduces IGF-1 signaling only in males (Kanfi et al., 2012). This gender-specific effect is maintained over the course of evolution, in both monkeys and humans (Naiman and Cohen, 2018). In SIRT6 KO monkeys, only females survive to birth, and in humans, SIRT6 deficiency results in sex reversal in the male fetus (Ferrer et al., 2018; Zhang et al., 2018). Interestingly, the effects of PPARα are also gender-specific. PPARα activators decrease adiposity in male but not female animals fed HFD, and reduce total cholesterol and triglycerides only in males in normal diet (Yoon et al., 2002). Moreover, the response of β-oxidation gene expression to PPARα agonist is much stronger in male mice and ovariectomized females than in WT females, indicating the involvement of gonadal sex steroids (Yoon, 2010). Similarly, in humans, males but not females carrying a PPARα polymorphism demonstrated improved triglyceride levels in response to statin treatment (Khan et al., 2004). Therefore, PPARα and SIRT6 share several gender-specific effects, particularly in protecting against obesity and its physiological consequences. It would be of great interest in future research to explore whether PPARα is involved in other male-specific SIRT6-regulated pathways such as lifespan extension.
Interestingly, in comparison to their WT littermates, no significant physiological effects were observed in SIRT6 HZ mice under normal growth conditions. For example, no changes in body weight and composition, daily activity, RER, or food intake were detected. This suggests that SIRT6 haploinsufficiency retains sufficient activity under unstressed conditions, at least to adulthood. Yet, under conditions in which PPARα is activated, such as increased β-oxidation (i.e., under food limitation), both copies of active SIRT6 are required. Notably, gene transcription changes in PPARα genes were found in SIRT6 HZ even under normal conditions, but these were not phenotypically evident until PPARα was activated. This may indicate that SIRT6 primes transcription to respond quickly to energy fluctuations such as changes in nutrient availability. We suggest that a certain minimum level of SIRT6 is necessary for long-term survival under fluctuating or chronic mild metabolic stress, such as that found in obese or old animals. Thus, SIRT6 may have therapeutic potential under chronic long-term stress conditions, for example, those that can be found in age-related diseases.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
This study generated new unique plasmids. All unique plasmids generated in this study are available from the Lead Contact without restriction. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Haim Cohen (Haim.Cohen@biu.ac.il)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Subjects
All animal procedures and use of mice were approved by the Institutional Animal Care and Use Committee of Bar Ilan University. Mice were housed on a 12hr light/dark cycle at room temperature (22–24C) in the Bar Ilan animal facilities. Mice were maintained on standard rodent chow diet with ad libitum access to food and water. All mice used in these experiments were in good health. Mice were kept under specific pathogen free conditions in IVC cages that are routinely examined and found negative for viral serology and both endo and ectoparasites. Male littermates were used for all experiments. Mice ranging from 12–15 months in age were randomly assigned to experimental groups. SIRT6 whole-body overexpressing (TG) mice were produced in CB6F1 strain, and maintained on a segregating stock containing equal contributions of C57BL/6J and BALB/cOlaHsd (Harlan laboratories) backgrounds as previously described (Kanfi et al., 2010). SIRT6 whole-body heterozygote (HZ) 129SVJ mice were a kind gift from Mostoslavsky’s lab (Mostoslavsky et al., 2006).
Cell Culture
HEK293T, Hepa1–6 and Aml12 cells were obtained from ATCC. All cells were grown at 37C with 95% humidity and 5% CO2. Cells were tested for mycoplasma contamination (Mycoplasma PCR Detection Kit, Hylabs) and found negative. Cells were maintained in Dulbecco’s modified Eagle’s medium (GIBCO) supplemented with 10% FBS, 1% penicillin, 1% streptomycin and 1% glutamine (Biological Industries).
Primary Cell Culture
Primary mouse hepatocytes were prepared as previously published (Lustig et al., 2011). Mice were sacrificed by CO2 and livers were immediately perfused for 7 minutes with warm (42°C) perfusion buffer (HBSSx1 [CellGro], NaHCO3 2.1g/L, EDTA 0.2g/L, KCl 0.4g/L, Glucose 1g/L, pH = 7.4) at a constant rate of 4.5ml/min with a 25 g needle. Needles were threaded via the inferior vena cava and the portal vein was severed to allow drainage. This was followed by 10 minutes perfusion with warm (42°C) liver digest media (Invitrogen #17703–034). Livers were then extracted into 10cm sterile tissue culture plates in 10ml plating media (DMEM with FBS 10%, Sodium pyruvate 2mM, Pen/Strep 2%, Dexamethasone 1uM and Insulin 0.1uM). Livers were manually disrupted with scalpels and passed through a 10ml electronic pipette several times until a homogeneous mix was formed. Samples were then passed through a 70uM cell strainer (Corning) into a 50ml Falcon and centrifuged in swinging buckets at RT 50 g for 10min. Pelleted cells were then mixed with 10ml plating media and 10ml Percoll (GE Healthcare GE17-0891-01), and centrifuged again to separate hepatocytes. Hepatocytes were washed twice more with plating media and stained with Trypan Blue. 500,000 live cells were plated into collagen coated 6 well plates or 3 million live cells were plated on a 10cm plate in plating media for 6 hours. Media was then switched to maintenance media (10% FBS 4.5 g/L glucose, 2 mM sodium pyruvate, 0.1 mM dexamethasone, and 1% penicillin/streptomycin) overnight. All experiments were performed in maintenance media after overnight incubation, and cells were harvested within 24 hours of extraction.
METHOD DETAILS
WY Administration
In Vivo
Mice were gavaged with Wy 14,643 (AbMole) at a concentration of 50mg/kg dissolved in 0.5% Carboxymethylcellulose (Sigma). Control littermates were gavaged with only 0.5% Carboxymethylcellulose. WY mixture was sonicated briefly 10 s twice at low speed before use, and mixtures were prepared fresh before each daily gavage. Approximately 0.2ml solution was given to each mouse, depending on their specific weight (50mg/kg). Gavage was performed in the evening at 9 pm, and mice were sacrificed 9am for the 12 hour WY treatment. Mice were gavaged with water for one week prior to treatment to accustom them to the procedure and monitored for signs of stress; food consumption, weight and overall health remained constant prior to the WY treatment.
In Vitro
Primary hepatocytes incubated overnight in maintenance media were treated the morning after hepatocyte extraction with 50uM WY freshly dissolved in DMSO for 12 hours, with corresponding DMSO controls.
Cell Transfections
Aml12 and Hepa1–6 Cells
One day before transfection 300,000 cells were seeded onto 6 well plates (Corning). Lipofectamene 2000 (Invitrogen) was used according to manufacturer’s protocol. 4ug total DNA was mixed with 10ul Lipofectamene in antibiotic-free medium, incubated for 10 minutes, and dripped on cells. Media was changed 6 hours post-transfection. Cells were harvested 48 hours post transfection.
293T Cells
One day before transfection 200,000 or 2 million cells were seeded onto either 6 well or 10cm plates (Corning), respectively. Calcium phosphate transfection protocol was used, with Hebsx2 buffer (NaCl 280mM, HEPES 50mM, 1.42 mM Na2HPO4·7H2O, pH = 7.05). 14ug of total DNA for a 10cm, and 4ug total DNA for 6well plate were used. DNA was mixed with CaCl2 250mM, and gently dripped onto the Hebsx2 buffer over a vortex to produce bubbles. DNA and Hebs was incubated for 20 minutes. This mixture was then dripped onto cells. Medium was changed 6 hours post-transfection. Cells were harvested 48 hours post-transfection.
Primary Hepatocytes
400,000 cells were extracted and incubated overnight in maintenance media prior to transfection. Transfection was performed with RNAiMax and TriFECTA RNAi Kit according to manufacturer’s protocol. Medium was changed 24 hours post-transfection and cells was harvested 48 hours post-transfection.
DNA Constructs
For in vitro immunoprecipitation: SIRT6-Flag, PPARα-GFP and PPARα -FLAG were cloned from cDNA of 293T cells or mouse liver, for human or mouse constructs, respectively. pcDNA 3.1+or pCAGGS plasmids were used for mammalian cell expression. GFP was cloned from pEGFP and inserted into pcDNA- PPARα. For recombinant protein constructs: pET28a plasmid was used to add an N-terminal 6xHis tag to C-tagged Flag SIRT6 and BIP, and pGEX4t3 was used to add an N-terminal GST tag to GST- PPARα. For luciferase assays: SIRT6 H133Y mutant and Ncoa2 K780R and K780Q mutants were created using mutagenesis protocol using PFU Ultra II Fusion (Agilent) according to manufacturer’s protocol from pcDNA-SIRT6 or pcDNA-Ncoa2-Flag. PPREx3 TK luciferase plasmid (Kim et al., 1998) was purchased from Addgene. pRL-TK renilla and pGL4 (hygro) luciferase was used (Promega).
Recombinant Protein Purification
Proteins were purified as previously published (Gil et al., 2013). pGEX4t3-GST-PPARα and pET28a-SIRT6/BIP-FLAG plasmids were transformed into bacteria BL21 (DE3) competent cells. A single positive colony from each plate was grown overnight at 37°C, 200RPM in 10ml LB, then diluted in the morning to 400ml LB and incubated until O.D.600nm = 0.8. Bacteria were induced with 1mM IPTG for 3–4 hours at 37°C then washed with PBSx1 and resuspended in buffer A (50mM Sodium Phosphate Buffer pH 7.2, 50mM NaCl, 1mM β-mercaptoethanol) with mini EDTA-free protease inhibitor cocktail (Roche). Bacteria was homogenized using Dounce homogenizer and strained, then lysed with a microfluidizer (Microfluidics). 6xHis-tagged proteins were then incubated with Talon metal affinity resin (Clontech), and GST-conjugated proteins were incubated with GST-binding resin (Novagen) for 2 hours in 4°C with rotation, then resin was separated and washed. Elution for His tagged proteins was performed with 500mM imidazole, and 50mM reduced glutathione (Sigma) was used to elute GST-conjugated proteins. PD10 desalting columns (Amersham) were used for buffer exchange to purify recombinant proteins, which were then kept at −70°C in storage buffer ((30 mM Tris-HCl, pH 7.4, 50 mM NaCl, 1 mM DTT, 10% Glycerol).
Quantitative Real-Time PCR
Total RNA was extracted using Trizol (Sigma) according to manufacturer’s protocol. cDNA was generated using RevertAid First Strand cDNA Synthesis Kit (Fermentas). PCR reaction was performed using ABsoluteblue SYBR Green (Thermo Scientific) in StepOnePlus (Applied Biosystem) or CFX96 (Biorad) instruments. Beta-Actin gene was used as an endogenous control and the expression data was analyzed using the relative quantification method 2-ΔΔCt. Primer sequences are detailed in Table S3.
Western Blot
Livers were collected and snap frozen in liquid nitrogen. Tissue was lysed in tissue lysis buffer with phosphatase inhibitors (7M Urea, 0.5% Triton X-100, 150 mM NaCl, 10mM TrisHCl pH7.4, 1 mM EDTA, 1mM EGTA, 2mM Vanadate, 10mM NaF, 2.5mM Sodium Pyrophosphate, proteinase inhibitor cocktail pill (Roshe Diagnostics). For PPARα immunoblots an additional step of nuclear extraction was first performed using the REAP method (Suzuki et al., 2010). Lysates were centrifuged and protein concentration was determined by Bradford protein assay (Biorad). 15ug protein were denatured at 100°C for 10 minutes in Laemmli sample buffer then loaded onto a 10% Tris-glycine polyacrylamide gel and subjected to SDS-PAGE electrophoresis. Gels were transferred to nitrocellulose membranes (Biorad) using the Bio-Rad semidry transfer and blocked for 1 hour in filtered 5% BSA. Immunoblotting was carried out with the following antibodies in 5% filtered BSA: SIRT6 (Cell Signaling), αTubulin (Sigma-Aldrich), Flag (Sigma), PDK4 (kindly donated by Robert Harris), PDH-E1a(pSer293) (Millipore), PDH-E1α (sc-377092), GFP and GST (Santa Cruz), and HRP-conjugated secondary antibodies mouse or rabbit (Jackson). Results were quantified with ImageJ.
ChIP
This protocol was adapted from Ram et al. (2011). 100mg frozen liver were homogenized using plastic disposable homogenizing pestles in cold PBS, the final volumen filtered through a 70uM filter with a 20 g needle to create a liver cell suspension in 10ml PBS. Cells were then crosslinked in 1% formaldehyde for 10 minutes at 37 degrees with gentle agitation then quenched with 5% glycine for 5 minutes at 37 degrees with gentle agitation. Cells were then washed x2 in PBS then lysed in nuclear extraction buffer (0.1% NP40-PBS) using REAP method (Suzuki et al., 2010). Nuclei were then lysed in chip lysis buffer (1% SDS, 50mM Tris-HCL pH 8.0, 10mM EDTA) containing protease inhibitors (Roche). Chromatin was sonicated using covaris S220 at 4°C (165 peak power, 10 duty factor, 200 cycles/burst, 6 minutes) to obtain 150–300bp DNA fragments. Chromatin lysate was then precleared using prewashed 40ul protein A Dynabeads for 4 hours; in parallel 5ug of SIRT6 (AB-ab62739) or IGG (CS-#2729) antibody were conjugated to 50ul protein A Dynabeads for 4 hours according to manufacturer’s instructions using 250ul blocking buffer (PBS with 0.5%Tween and 0.5% BSA). Beads were then washed and added to chromatin lysate overnight. After incubation beads were washed x6 with RIPA, x2 with RIPA-NaCl (500mM), x2 with LiCl (250mM LiCl, 10mM TE, 0.5% DOC, 0.5% NP-40), x2 with TE, and then eluted overnight at 65 degrees in elution buffer (0.5% SDS, 5mM EDTA, 300mM NaCl, 10mM Tris-Hcl pH 8.0). Following elution samples were treated with proteinase K at 55 degrees for 2 hours. DNA was then purified using Agencourt AMPure XP beads (Beckman Coulter) according to manufacturer’s instructions and eluted in TE. DNA concentration was quantified by Qubit, and 3ul of each chip was used as template. Data was normalized to inputs.
Luciferase Reporter Assay
HEK293T cells were cotransfected using calcium phosphate transfection protocol in 6-well plates with SIRT6/SIRT6 H133Y and PPARα /empty plasmid, along with firefly luciferase reporter vector containing PPREx3, and Renilla as transfection control. 48hr post-transfection cells were harvested and luciferase activities were measured using Dual Luciferase Reporter Assay System (Promega).
Immunoprecipitation
In Vitro
4ug of SIRT6-Flag or BIP-Flag (negative control) were incubated with either PPARα-GST or GST (negative control) overnight at 4°C in IP binding buffer (buffer A) with gentle agitation with 50ul anti-Flag M2 affinity gel (Sigma), to immunoprecipitate the Flag-tagged proteins. Flag beads were then briefly centrifuged then washed x4 with wash buffer (50mM Tris pH 8.0, 1% NP40, 150mM NaCl, 1mM MgCl2, 10% glycerol, and protease inhibitor (Roche)), followed by elution with 5ul of diluted Flagx3 peptide (Sigma) in 100ul lysis buffer peptide with strong agitation for 30 minutes, according to manufacturer’s instructions. Elutions were then boiled in sample buffer then run on SDS-page with 0.4ug (10%) total input and probed for both Flag and GST.
In Vivo
293T cells overexpressing 7ug Flag-tagged plasmids in a 10cm plate were first washed with PBS then lysed in 700ul cell lysis buffer. Lysates were sonicated using Branson sonicator at 60% for 40 s twice, and protein concentration was measured using Bradford reagent (Biorad). 1–4mg of protein per IP was then treated with 300 mg/ml ethidium bromide (EtBr) for 30 min on ice, followed by 10 min centrifugation at 17,000 g twice. Supernatants were subjected to immunoprecipitation with 50ul anti-Flag M2 affinity gel (Sigma) overnight at 4°C with gentle agitation to immunoprecipitate Flag-tagged proteins. Flag beads were then briefly centrifuged then washed 4 times with cell lysis buffer, followed by elution with 5ul of diluted flag peptide /in 100ul lysis buffer Flagx3 peptide (Sigma) with strong agitation for 30 minutes according to manufacturer’s instructions. Elutions were then boiled in sample buffer then run on SDS-page with 50ug (5%) total input and probed for both Flag and GFP or endogenous SIRT6.
ExpressionBlast Microarray Comparison Analysis
DE genes from RNA-seq WT versus HZ control treatment (0.5% carboxymethylcellulose) from Table S1B were submitted into ExpressionBlast (https://bio.tools/expressionblast) with parameters: Input/output species Mus musculus, list of genes provided as gene symbol with fold change, distance function Euclidian, p val cutoff 0.01, count cutoff 20, % genes required to match: 65%, GO p val cutoff: 0.05. Euclidian function parameters were 3, 0.4, and 1.5 for trim expression values, magnitude weight, and sign penalty, respectively. Results were downloaded on 09/13/18 and are shown in Table S2. Categories for genes such as PPARα and GNMT had to include the word “PPARα” or “GNMT” in the GSM description or comparison (highlighted in red or blue in Table S2), and were manually examined to ensure the experiment was indeed a matching PPARα comparison. Other categories such (i.e., infection/cancer) were curated and manually validated in a similar manner. All key words for category determination are highlighted in blue in Table S2.
Respiratory Exchange Ratio (RER)
Mice were individually housed in metabolic chambers at room temperature (23°C) and allowed to acclimate for 3 days. Spontaneous movement, food and water consumption were continuously measured using automated indirect calorimetry (TSE Systems GmbH). After acclimation mice were gavaged daily with WY (50mg/kg) for 3 days, and then measured for an additional day without treatment. Oxygen consumption (VO2), carbon dioxide production (VCO2), and respiratory exchange ratio (RER) were measured every half hour.
Body Composition
Body composition was measured using quantitative nuclear magnetic resonance (NMR) using the Minispec LF90 (Bruker Optics, Billerica, MA). For liver composition, whole livers were dissected from mice and measured in the machine. Fat and Lean mass were recorded and percentages were calculated accordingly.
Metabolomics
Wy-Treated Mice
Mice were orally gavaged with WY or vehicle control (0.5% carboxymethylcellulose). Twelve hours later, mice were sacrificed by CO2 and livers were harvested and flash frozen. 50 mg of frozen liver tissue was pulverized and then homogenized in 50% aqueous acetonitrile containing 0.3% formic acid. Pyruvate, lactate, and alanine were measured as described (Jensen et al., 2006). Acetylcarnitine (C2) was measured as previously reported (Kusama et al., 2006; Wu et al., 2004).
Starved Mice
To maximize accuracy and to take into account tissue inhomogeneity, metabolites were extracted from 3 separate liver pieces for each mouse. Precellys Lysing Kits comprising prefilled tubes with beads (CK14 homogenizing tubes for soft tissue containing 1.4 mm ceramic beads) were prefilled with 1000ul of cold (−20°C) metabolites extraction solvent (MeOH:ACN:H20, 5:3:2) and kept on ice. Frozen tissue was cut, weighed and approximately 30 mg sample was added to the prefilled homogenization tube. Samples were homogenized at 4°C in a Precellys 24 homogenizer. (Homogenization at 3*20 s, 6000 rpm with a 30 s gap between each of the three cycles). Homogenized samples were centrifuged in the Precellys tubes at 18,000 g for 15 min at 0–4°C. The supernatant was collected in a microcentrifuge tube and centrifuged again. The supernatants were transferred to glass HPLC vials and kept at −75°C prior to LC-MS analysis.
LC-MS metabolomics analysis was performed as described previously (Mackay et al., 2015). Briefly, Thermo Ultimate 3000 high-performance liquid chromatography (HPLC) system coupled to Q- Exactive Orbitrap Mass Spectrometer (Thermo Fisher Scientific) was used with a resolution of 35,000 at 200 mass/charge ratio (m/z), electrospray ionization, and polarity switching mode to enable both positive and negative ions across a mass range of 67 to 1000 m/z. HPLC setup consisted ZIC-pHILIC column (SeQuant; 150 mm × 2.1 mm, 5 μm; Merck), with a ZIC-pHILIC guard column (SeQuant; 20 mm × 2.1 mm). 5 ul of Biological extracts were injected and the compounds were separated with mobile phase gradient of 15 min, starting at 20% aqueous (20 mM ammonium carbonate adjusted to pH 9.2 with 0.1% of 25% ammonium hydroxide) and 80% organic (acetonitrile) and terminated with 20% acetonitrile. Flow rate and column temperature were maintained at 0.2 ml/min and 45°C, respectively, for a total run time of 27 min. All metabolites were detected using mass accuracy below 5 ppm. Thermo Xcalibur was used for data acquisition
TraceFinder 4.1 was used for analysis. Peak areas of metabolites were determined by using the exact mass of the singly charged ions. The retention time of metabolites was predetermined on the pHILIC column by analyzing an in-house mass spectrometry metabolite library that was built by running commercially available standards. Data from each sample was normalized to its corresponding tissue weight.
Fatty Acid Oxidation
Mice were orally gavaged with WY or vehicle control (0.5% carboxymethylcellulose). Twelve hours later, mice were sacrificed by CO2 and livers were harvested for fatty acid oxidation measurements. Freshly harvested liver samples were homogenized in ice cold 0.25 M sucrose, 10 mM Tris-HCl, 1 mM EDTA, at a pH of 7.4 using a Dounce homogenizer. The homogenates were then centrifuged at 420 g for 10 minutes to remove nuclear proteins and cell debris. The supernatant was then subjected to fatty acid oxidation measurements using 14C-palmitate as described in detail by Huynh et al. (2014).
RNA-Seq and Analysis
RNA was isolated with Trizol (Sigma). RNA quality was evaluated by Tapestation RNA Assay (Agilent Technologies, CA, USA). Libraries were constructed with the NEBNext Ultra RNA library prep Kit (#E7530, NEB) using manufacturer’s instructions. Final quality was evaluated by Tapestation DNA HS Assay (Agilent Technologies, CA, USA). Equimolar pooling of libraries were performed based on Qubit values and loaded onto an Illumina Hiseq 2500 platform (Illumina, CA, USA). These libraries were sequenced in multiplex as single-end non-strand specific 56-bp/61-bp reads on 2 lanes of an Illumina HiSeq 2000, resulting in on average 28 million reads per sample.
The RNA sequencing data was aligned using STAR (v 2.5.0a) (Dobin et al., 2013) with default parameters. The UCSC mm10 iGenome build and its corresponding gtf were used to build the STAR reference database. The number of reads per gene were quantified using HTSeq-Count (v0.6.1p1) (Anders et al., 2015). The R Bioconductor package, DESeq2 (v1.18.1) (Love et al., 2014), was used to classify genes as differentially expressed (Benjamini-Hochberg adjusted p value < 0.1). Reference genome and index used:ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/Mus_musculus/UCSC/mm10/Mus_musculus_UCSC_mm10.tar.gz.
Ingenuity Analysis
The networks, pathways, and regulators functional analyses were generated through the use of IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis). Networks were customized in Ingenuity by modifying font and text size, and bolding genes of interest, but were otherwise left intact.
Microfluidics Protein-DNA Interaction
Protein-DNA interactions were carried out as previously described (Glick et al., 2016b). In each experiment, 25 mL of extract (50 ng of protein) was loaded into the device. Introduction of 3′-HIS&5′-cMyc or Flag-tagged TFs complex into the DNA chambers solubilize spotted DNA, allowing TFs and DNA to interact. TF-DNA complexes were then captured on the chip surface beneath the ‘button’ valve during a 1 hour incubation period at 32°C. Next, MITOMI was performed by closing the ‘button’ valve to trap the interactions. Protein complexes and DNA not trapped by MITOMI were then washed away. TFs were labeled with α-FLAG cy3 (Sigma). Proteins expression levels and interacting DNA signals were measured with a microarray scanner (LS Reloaded, Tecan) using a 488 nm laser and 535 filter for Cy3, and a 633 nm laser and 695 nm filter for Cy5. Cy3 intensities under the ‘button’ valve reflect the number of surface-bound protein molecules; Cy5 intensities under the ‘button’ valve reflect the number of DNA molecules bound by surface-immobilized protein. The ratio of Cy5 to Cy3 fluorescence is proportional to the number of DNA molecules bound per protein, namely, protein fractional occupancy. Cy5 intensities within the DNA chamber reflect the amount of soluble DNA available for binding.
PPRE and Mutant Sequence Mini-library Preparation
PPRE\mutant elements from Oka et al. (2012) were prepared as previously reported (Glick et al., 2016a). DNA primers were synthesized (IDT), hybridized to a Cy5-labeled primer and extended using Klenow fragment (exo-) (New England Biolabs) to produce Cy5-labeled dsDNA. Cy5-labeled dsDNA oligonucleotides were diluted to a final concentration of 2 μM then serially diluted in 32 dilutions ranging from 2 μM down to 0.0156 μM. Each sample contained 0.125% Poly ethylene glycol (Peg, Sigma-Aldrich) and 1.25 mg/ml D-trehalose dihydrate (Sigma-Aldrich) in dH2O, preventing irreversible binding of the DNA to the printed slide as well as for visualization during alignment of the device to the DNA array. A negative control sample with no DNA was included. The oligonucleotides were spotted onto epoxy coated glass substrates (CEL Associates) with a MicroGrid 610 (Bio Robotics) microarrayer using SMT-S75 silicone pins (Parallel Synthesis, USA). Column and row pitch corresponded to the specific device. The microfluidic device that was used contains 64 columns and 64 rows with a pitch of 280 μm by 560 μm, respectively.
Microfluidics Protein-Protein Interaction
Protein-protein interactions on chip were carried out as previously described (Gerber et al., 2009). Human proteins (C-TIP, PPARα and HIF1α) with N terminus c-Myc and C terminus His tags were expressed by using rabbit reticulocyte quick-coupled TNT reaction (Promega). Surface chemistry was prepared and α-HIS biotinylated antibodies (QIAGEN) were bound under buttons on chip. Next, each expressed protein was attached to a specific part of the device and bound to its corresponding antibody. Finally, Flag-tagged SIRT6 was applied to the device. By closing the sandwich valves, each unit cell separated from its environment. SIRT6 were allowed to incubate with the proteins for 30 min at 32°C. Next, the button valves opened, exposing proteins, and unbound protein was washed away. Bound proteins were then labeled with α-c-Myc Alexa Fluor 647 (Abcam) and α-FLAG cy3 (Sigma) antibodies. Protein interactions were determined with a microarray scanner (LS Reloaded; Tecan) using a 633-nm laser and 695-nm filter for Alexa Fluor 647 and a 535-nm laser and 595-nm filter for Cy3.
Microscale Thermophoresis Binding Assay (MST)
Fluorescent labeled HIS-tagged human SIRT6 was incubated with increasing concentrations of ADP-ribose or WY-14643 for 30 min at room temperature (RT) in MST assay buffer ((50 [mM] Tris-HCl pH = 8.0, 137 [mM] NaCl, 2.7 [mM] KCl, 1[mM] DTT and 1[mM] MgCl2 0.1% Tween).)prior to MST measurements in order to achieve thermodynamic equilibrium. The samples loaded into premium coated capillaries (NanoTemper Technologies). Measurements were conducted with 20% LED power and 20% of MST power (IR laser intensity) for ADP-ribose or 40% for WY-14643. Initial florescence was measured in order to dismiss fluorescence quenching and protein adhesion to the capillary surface. Data was fitted and analyzed using the MST analysis software.
Fluorescent Deacylation Assay (F.D.)
The assay is based on 7-amino-4-methylcoumarin (AMC) fluorophore, attached to a myristoylated peptide substrate (H3-K(Myristoyl)-AMC) (Peptron Inc). The procedure requires 3 steps in F.D. assay buffer (50 mM Tris-HCl pH = 8.0, 137 mM NaCl, 2.7 mM KCl, 1[mM] DTT and 1mM MgCl2). In the first step, 50 μM WY-14643 or DMSO control were incubated with 0.5 μM human recombinant SIRT6 along with NAD+ (100μM) and H3-K(Myristoyl)-AMC (50μM) at 37°C for 60 min in a reaction tube. Upon demyristylation, the peptide becomes a trypsin substrate. In the next step, samples at time 0 and 60 min were taken and quenched with SIRT6 inhibitor nicotinamide (NAM, 20mM) in a 96-well black plate (Perkin Elmer). Next, treatment with trypsin from bovine pancreas (sigma) at 25° for 30 min released the fluorophore and fluorescence intensity was measured (excitation: 370nm and emission 440nm). The plate containing the incubations was read using SYNERGY4 microplate reader. Increase in fluorescent intensity indicates reaction progression. For analysis, fluorescent intensities at time 0 were subtracted from time 60 min.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are expressed as mean ± SEM. For single comparisons with one condition, significance was calculated between groups using a two-tailed unpaired Student’s t test. For experiments with 2 factors (i.e., treatment and genotype) a 2-way ANOVA followed by Tukey post hoc was used. For experiments with multiple comparisons with one factor a one-way ANOVA with Bonferroni post hoc was used. Area under curve (AUC) for RER was measured from 7AM to 7PM for each day. In all experiments p < 0.05 was considered significant. For RNA-Seq experiments FDR q < 0.1 was deemed significant. All analyses were performed using Graphpad Prism 8 (GraphPad, San Diego, CA). Statistical parameters and information on biological replicates (n) are indicated in the figure legends. Statistically significant differences are indicated as * p < 0.05, **p < 0.01. Ns indicates not significant differences. Animals were randomized to treatment groups and treated blinded to genotype. Measurements were performed blinded to treatment or genotype.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-SIRT6 (clone D8D12) | Cell Signaling Technology | Cat# 12486; RRID: AB_2636969 |
| Mouse monoclonal anti-Flag (clone M2) | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| Mouse monoclonal anti-GST (clone B-14) | Santa Cruz Biotechnology | Cat# sc-138; RRID: AB_627677 |
| Mouse monoclonal anti-GFP (clones 7.1 and 13.1) | Roche | Cat# 11814460001; RRID: AB_390913 |
| Mouse monoclonal anti-PPARA (clone H-2) | Santa Cruz Biotechnology | Cat# sc-398394 |
| Mouse monoclonal anti-Flag-Cy3 (clone M2) | Sigma-Aldrich | Cat# A9594; RRID: AB_439700 |
| Rabbit polyclonal anti-c-Myc-Cy5 | Bioss | Cat# bs-0842R-Cy5; RRID: AB_11049406 |
| Mouse monoclonal anti-Cy5 (clone CY5–15) | Sigma-Aldrich | Cat# C1117; RRID: AB_477654 |
| Rabbit polyclonal anti-PDK4 | Jeoung et al., 2006 | N/A (Custom-produced by Sigma-Genosys) |
| Rabbit polyclonal anti-phospho-PDH-E1α (Ser293) | Millipore | Cat# AP1062; RRID: AB_10616069 |
| Mouse monoclonal anti-PDH-E1α (clone D-6) | Santa Cruz Biotechnology | Cat# sc-377092; RRID: AB_2716767 |
| Mouse monoclonal anti-alpha-Tubulin (clone B512) | Sigma-Aldrich | Cat# T5168; RRID: AB_477579 |
| Rabbit polyclonal anti-PPARA | Santa Cruz Biotechnology | Cat# sc-9000; RRID: AB_2165737 |
| Rabbit polyclonal anti-phospho-AMPk (Thr172) | Cell Signaling Technology | Cat# 2531; RRID: AB_330330 |
| Rabbit monoclonal anti-AMPK (clone D5A2) | Cell Signaling Technology | Cat# 5831; RRID: AB_10622186 |
| Rabbit polyclonal anti-SREBP2 | Abcam | Cat# ab30682; RRID: AB_779079 |
| Rabbit polyclonal anti-H3 | Abcam | Cat# ab39655; RRID:AB_732921 |
| Rabbit polyclonal anti-H3K9 | Abcam | Cat# ab4441; RRID:AB_2118292 |
| Bacterial Strains | ||
| Escherichia coli Bl21 (De3) strain | New England Biolabs | Cat# C2527 |
| Escherichia coli Dh5a strain | Invitrogen | Cat# 18263012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| WY 14643 | AbMole | Cat# M1880 |
| Ethidium Bromide | Sigma-Aldrich | Cat# E1510 |
| Carboxymethylcellulose sodium salt | Sigma-Aldrich | Cat# C4888 |
| Formaldehyde 37% | Sigma-Aldrich | Cat# F1635 |
| Collagen | Santa Cruz | Cat# SC-136157 |
| Liver digest medium | Thermo Fisher Scientific | Cat# 17703034 |
| Human Insulin solution | Santa Cruz | Cat# SC-360248 |
| Dexamethasone | Sigma-Aldrich | Cat# D2915 |
| DMSO | Sigma-Aldrich | Cat# D4540 |
| HBSSx10 | GIBCO | Cat# 14185045 |
| Sodium Pyruvate | Invitrogen | Cat# 11360–070 |
| Percoll | Sigma-Aldrich | Cat# P1644 |
| EDTA | Santa Cruz | Cat# SC-29092 |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10003D |
| Medium199 | Invitrogen | Cat# 11150–059 |
| Proteinase K | Roche | Cat# 3115879001 |
| TriReagent | Sigma-Aldrich | Cat# 93289 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668027 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | Cat# 13778075 |
| Flag×3 Peptide | Sigma-Aldrich | Cat# F4799 |
| Critical Commercial Assays | ||
| ANTI-FLAG® M2 Affinity Gel | Sigma-Aldrich | Cat# A2220 |
| GFP-Trap Agarose | Chromotek | Cat# gta200 |
| RevertAid First Strand cDNA Synthesis Kit | Thermo Fisher Scientific | Cat# K1621 |
| Agencourt AMPure XP kit | Beckman Coulter | Cat# A63881 |
| NEBNext® Multiplex Oligos for Illumina | New England Biolabs | Cat# E7335 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| NEBNext® Ultra DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7370 |
| NextSeq 500/550 High Output Kit v2.5 | Ilumina | Cat# 20024906 |
| Deposited Data | ||
| Raw and analyzed RNA sequencing data | This paper | GEO: GSE140063 |
| Mus musculus genome build 38 GRCm38.p6 | Ensembl | Mus_musculus.GRCm38.dna.primary_assembly.fa |
| Experimental Models: Cell Lines | ||
| Human HEK293T | ATCC | Cat. # CRL-11268; RRID:CVCL_0063 |
| Mouse Hepa1–6 | ATCC | Cat# CRL-1830, RRID:CVCL_0327 |
| Mouse AML12 | ATCC | Cat# CRL-2254, RRID:CVCL_0140 |
| Experimental Models: Organisms/Strains | ||
| C57B-SIRT6-TG (SIRT6 Overexpression) | Kanfi et. al., 2012 | Cohen Lab |
| CB6-SIRT6-TG (SIRT6 Overexpression) | Kanfi et. al., 2012 | Cohen Lab |
| 129-Sirt6tm1Fwa/J (SIRT6 Heterozygote) | The Jackson Laboratory | Kind gift from Dr. Raul Mostoslavsky |
| Oligonucleotides | ||
| Oligonucleotides for Chip | This paper | Table S3, tab A |
| Oligonucleotides for qRT-PCR | This paper | Table S3, tab B |
| TriFECTA RNAi Kit for PPARA mouse | IDT | mm.Ri.Ppara.13 |
| Recombinant DNA | ||
| pSG5-Ncoa2-Flag | Ogawa et. al., 2004 | Kind gift from Dr. Hidesato Ogawa |
| pSG5-Ncoa2 | Leers et. al., 1998 | Kind gift from Dr. Hinrich Gronemeyer |
| pCDNA3.1+-Ncoa2-Flag | This paper | N/A |
| pCDNA3.1+-Ncoa2K780Q-Flag | This paper | N/A |
| pCDNA3.1+-Ncoa2K780R-Flag | This paper | N/A |
| pCAGGS-SIRT6 | This paper | N/A |
| pCAGGS-SIRT6-H133Y | This paper | N/A |
| pCDNA3.1+-SIRT6-Flag | Gil et. al, 2013 | N/A |
| pCDNA-PPARA-GFP | This paper | N/A |
| pGEX4t3-GST-PPARA | This paper | N/A |
| pET28a-His-PPARA | This paper | N/A |
| pET28a-SIRT6-Flag | Gil et. al, 2013 | N/A |
| Software and Algorithms | ||
| ImageJ Version 1.8 | Schneider et al., 2012 | https://imagej.nih.gov/ij |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.Shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| STAR 2.6 | Dobin and Gingeras, 2015 | https://github.com/alexdobin/STAR |
| MarkDuplicates tool in picard-tools-1.8.4 | Broad Institute | http://broadinstitute.github.io/picard/ |
| R (version 3.2.3) | The R Foundation | https://www.r-project.org/ |
| R Studio | The R Foundation | https://rstudio.com/ |
| DESeq2 (version 1.10) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GraphPad Prism (version 8.0 for Windows) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Ggplot2 | Hadley Wickham | https://ggplot2.tidyverse.org/ |
| Ingenuity Pathway Analysis | QIAGEN | https://www.ingenuity.com |
| Other | ||
| Multi channel cassette pump | Watson Marlow | Cat# 205CA |
| C96 MicroWellTM Plates | Thermo Fisher Scientific | Cat# 437796 |
| 70 μm cell strainer | Corning | Cat# CLS431751 |
Highlights.
PPARα mediates various SIRT6-regulated metabolic pathways
PPARα binds to and is activated by SIRT6 to promote fatty acid beta oxidation
SIRT6 decreases NCOA2 acetylation and induces its coactivation of PPARα
Coordinated SIRT6-PPARα activities control energy production under limited nutrients
ACKNOWLEDGMENTS
We thank members of the Cohen lab for their helpful input and their comments on the manuscript. We also thank Raul Mostoslavsky (HMS) for his gift of the SIRT6 HZ mice. We thank Tamar Geiger and Michal Harel (TAU) for help with SILAC experiments and Avia Cohen for preparing the graphical abstract. This work was supported by the following funding sources: the Israel Science Foundation (621/13 and 777/16) (H.Y.C.), I-Core Foundation (41/11) (H.Y.C. and S.N.), the Economic and Social Fund for Development (ESFD) (H.Y.C.), D-Cure (H.Y.C.), Israel Cancer Association (2016-0103) (H.Y.C.), the Sagol Healthy Longevity Center (HEALS) (H.Y.C.), Israel Cancer Research Fund (ICRF) (H.Y.C.) and the U.S.-Israel Binational Science Foundation (BSF) (H.Y.C.), the Laura and Isaac Perlmutter Fund for supporting the Technion’s metabolomics facility (E.G.), the National Institutes of Health (NIH), and NIA grant R01AG045351 (M.D.H.). F.K.H. was supported by an American Diabetes Association/Canadian Diabetes Association post-doctoral fellowship (PF-3-13-4342-FH).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.067.
DECLARATION OF INTERESTS
H.C. advises SirTLab Ltd.
DATA AND CODE AVAILABILITY
Raw and processed RNA-seq data described in this manuscript are deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible using GEO: GSE140063 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140063.
All R scripts written for data processing used for analyses are available upon request. All software used are available on-line from a commercial supplier and are summarized in the Key Resources Table.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baes M, and Peeters A (2010). Role of PPARα in hepatic carbohydrate metabolism. PPAR Res. 2010, 572405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Glick Y, Kipper S, Schwartz N, Avrahami D, Barbiro-Michaely E, and Gerber D (2013). Microfluidic large scale integration of viral-host interaction analysis. Lab Chip 13, 2202–2209. [DOI] [PubMed] [Google Scholar]
- Bruss MD, Khambatta CF, Ruby MA, Aggarwal I, and Hellerstein MK (2010). Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am. J. Physiol. Endocrinol. Metab 298, E108–E116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge A, and Mandrup S (2010). Molecular mechanisms and genome-wide aspects of PPAR subtype specific transactivation. PPAR Res. 2010, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Yang G (2014). PPARs integrate the mammalian clock and energy metabolism. PPAR Res. 2014, 653017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu Q, Tang Q, Kuang J, Li H, Pu S, Wu T, Yang X, Li R, Zhang J, et al. (2019). Hepatocyte-specific Sirt6 deficiency impairs ketogenesis. J. Biol. Chem 294, 1579–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Yao L, Yang X, Gao Y, Fang F, Zhang J, Wang Q, and Chang Y (2017). SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. Am. J. Physiol. Endocrinol. Metab 313, E493–E505. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, and Gingeras TR (2015). Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinforma 51, 11.14.1–11.14.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy JE Jr., Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan HB, Feldman J, Pierce K, Mostoslavsky R, et al. (2012). The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 48, 900–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhanati S, Kanfi Y, Varvak A, Roichman A, Carmel-Gross I, Barth S, Gibor G, and Cohen HY (2013). Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep. 4, 905–912. [DOI] [PubMed] [Google Scholar]
- Elhanati S, Ben-Hamo R, Kanfi Y, Varvak A, Glazz R, Lerrer B, Efroni S, and Cohen HY (2016). Reciprocal Regulation between SIRT6 and miR-122 Controls Liver Metabolism and Predicts Hepatocarcinoma Prognosis. Cell Rep. 14, 234–242. [DOI] [PubMed] [Google Scholar]
- Ferrer CM, Alders M, Postma AV, Park S, Klein MA, Cetinbas M, Pajkrt E, Glas A, van Koningsbruggen S, Christoffels VM, et al. (2018). An inactivating mutation in the histone deacetylase SIRT6 causes human perinatal lethality. Genes Dev. 32, 373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigo DE, Basu A, Nierth-Simpson EN, Weldon CB, Dugan CM, Elliott S, Collins-Burow BM, Salvo VA, Zhu Y, Melnik LI, et al. (2006). p38 mitogen-activated protein kinase stimulates estrogen-mediated transcription and proliferation through the phosphorylation and potentiation of the p160 coactivator glucocorticoid receptor-interacting protein 1. Mol. Endocrinol 20, 971–983. [DOI] [PubMed] [Google Scholar]
- Gerber D, Maerkl SJ, and Quake SR (2009). An in vitro microfluidic approach to generating protein-interaction networks. Nat. Methods 6, 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil R, Barth S, Kanfi Y, and Cohen HY (2013). SIRT6 exhibits nucleo-some-dependent deacetylase activity. Nucleic Acids Res. 41, 8537–8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick Y, Ben-Ari Y, Drayman N, Pellach M, Neveu G, Boonyaratanakornkit J, Avrahami D, Einav S, Oppenheim A, and Gerber D (2016a). Pathogen receptor discovery with a microfluidic human membrane protein array. Proc. Natl. Acad. Sci. USA 113, 4344–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick Y, Orenstein Y, Chen D, Avrahami D, Zor T, Shamir R, and Gerber D (2016b). Integrated microfluidic approach for quantitative high-throughput measurements of transcription factor binding affinities. Nucleic Acids Res. 44, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto T, Hirata M, Aoki Y, Iwase M, Takahashi H, Kim M, Li Y, Jheng HF, Nomura W, Takahashi N, et al. (2017). The hepatokine FGF21 is crucial for peroxisome proliferator-activated receptor-α agonist-induced amelioration of metabolic disorders in obese mice. J. Biol. Chem 292, 9175–9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howroyd P, Swanson C, Dunn C, Cattley RC, and Corton JC (2004). Decreased longevity and enhancement of age-dependent lesions in mice lacking the nuclear receptor peroxisome proliferator-activated receptor α (PPARalpha). Toxicol. Pathol 32, 591–599. [DOI] [PubMed] [Google Scholar]
- Huynh FK, Green MF, Koves TR, and Hirschey MD (2014). Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol. 542, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, and Guarente L (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. [DOI] [PubMed] [Google Scholar]
- Jeoung NH, Wu P, Joshi MA, Jaskiewicz J, Bock CB, DePaoli-Roach AA, and Harris RA (2006). Role of pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) in glucose homoeostasis during starvation. Biochem. J 397, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MV, Joseph JW, Ilkayeva O, Burgess S, Lu D, Ronnebaum SM, Odegaard M, Becker TC, Sherry AD, and Newgard CB (2006). Compensatory responses to pyruvate carboxylase suppression in islet β-cells. Preservation of glucose-stimulated insulin secretion. J. Biol. Chem 281, 22342–22351. [DOI] [PubMed] [Google Scholar]
- Ka SO, Bang IH, Bae EJ, and Park BH (2017). Hepatocyte-specific sirtuin 6 deletion predisposes to nonalcoholic steatohepatitis by up-regulation of Bach1, an Nrf2 repressor. FASEB J. 31, 3999–4010. [DOI] [PubMed] [Google Scholar]
- Kaluski S, Portillo M, Besnard A, Stein D, Einav M, Zhong L, Ueberham U, Arendt T, Mostoslavsky R, Sahay A, and Toiber D (2017). Neuroprotective Functions for the Histone Deacetylase SIRT6. Cell Rep. 18, 3052–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanfi Y, Peshti V, Gozlan YM, Rathaus M, Gil R, and Cohen HY (2008a). Regulation of SIRT1 protein levels by nutrient availability. FEBS Lett. 582, 2417–2423. [DOI] [PubMed] [Google Scholar]
- Kanfi Y, Shalman R, Peshti V, Pilosof SN, Gozlan YM, Pearson KJ, Lerrer B, Moazed D, Marine J-C, de Cabo R, and Cohen HY (2008b). Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett. 582, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, Kronfeld-Schor N, and Cohen HY (2010). SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 9, 162–173. [DOI] [PubMed] [Google Scholar]
- Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, and Cohen HY (2012). The sirtuin SIRT6 regulates lifespan in male mice. Nature 483, 218–221. [DOI] [PubMed] [Google Scholar]
- Kersten S (2014). Integrated physiology and systems biology of PPARα. Mol. Metab 3, 354–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan QH, Pontefract DE, Iyengar S, and Ye S (2004). Evidence of differing genotypic effects of PPARalpha in women and men. J. Med. Genet 41, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, et al. (2010). Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 12, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Wright HM, Wright M, and Spiegelman BM (1998). ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA 95, 4333–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König B, Koch A, Spielmann J, Hilgenfeld C, Stangl GI, and Eder K (2007). Activation of PPARalpha lowers synthesis and concentration of cholesterol by reduction of nuclear SREBP-2. Biochem. Pharmacol 73, 574–585. [DOI] [PubMed] [Google Scholar]
- König B, Koch A, Spielmann J, Hilgenfeld C, Hirche F, Stangl GI, and Eder K (2009). Activation of PPARalpha and PPARgamma reduces triacylglycerol synthesis in rat hepatoma cells by reduction of nuclear SREBP-1. Eur. J. Pharmacol 605, 23–30. [DOI] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang J, Zhang Y, Liu Q, Shen J, Pu S, Cheng S, Chen L, Li H, Wu T, Li R, et al. (2017). Fat-specific Sirt6 ablation sensitizes mice to high-fat diet-induced obesity and insulin resistance by inhibiting lipolysis. Diabetes 66, 1159–1171. [DOI] [PubMed] [Google Scholar]
- Kuang J, Chen L, Tang Q, Zhang J, Li Y, and He J (2018). The role of Sirt6 in obesity and diabetes. Front. Physiol 9, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusama T, Mukai M, Tatsuta M, Nakamura H, and Inoue M (2006). Inhibition of transendothelial migration and invasion of human breast cancer cells by preventing geranylgeranylation of Rho. Int. J. Oncol 29, 217–223. [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent G, de Boer VC, Finley LW, Sweeney M, Lu H, Schug TT, Cen Y, Jeong SM, Li X, Sauve AA, and Haigis MC (2013). SIRT4 represses peroxisome proliferator-activated receptor α activity to suppress hepatic fat oxidation. Mol. Cell. Biol 33, 4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leers J, Treuter E, and Gustafsson J-Å (1998). Mechanistic Principles in NR Box-Dependent Interaction between Nuclear Hormone Receptors and the Coactivator TIF2. Mol. Cell. Biol 18, 6001–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Weinheimer CJ, and Kelly DP (1999). A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 96, 7473–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerrer B, Gertler AA, and Cohen HY (2016). The complex role of SIRT6 in carcinogenesis. Carcinogenesis 37, 108–118. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig Y, Ruas JL, Estall JL, Lo JC, Devarakonda S, Laznik D, Choi JH, Ono H, Olsen JV, and Spiegelman BM (2011). Separation of the gluconeogenic and mitochondrial functions of PGC-1alpha through S6 kinase. Genes Dev. 25, 1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay GM, Zheng L, van den Broek NJF, and Gottlieb E (2015). Analysis of Cell Metabolism Using LC-MS and Isotope Tracers. Methods Enzymol. 561, 171–196. [DOI] [PubMed] [Google Scholar]
- Mandard S, Müller M, and Kersten S (2004). Peroxisome proliferator-activated receptor α target genes. Cell. Mol. Life Sci 61, 393–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masternak MM, and Bartke A (2007). PPARs in calorie restricted and genetically long-lived mice. PPAR Res. 2007, 28436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno M, Lombardi A, Silvestri E, Senese R, Cioffi F, Goglia F, Lanni A, and de Lange P (2010). PPARs: Nuclear receptors controlled by, and controlling, nutrient handling through nuclear and cytosolic signaling. PPAR Res. 2010, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329. [DOI] [PubMed] [Google Scholar]
- Naiman S, and Cohen HY (2018). Role for the longevity protein SIRT6 in primate development. Nature 560, 559–560. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Yu RT, Haraguchi T, Hiraoka Y, Nakatani Y, Morohashi KI, and Umesono K (2004). Nuclear structure-associated TIF2 recruits glucocorticoid receptor and its target DNA. Biochem. Biophys. Res. Commun 320, 218–225. [DOI] [PubMed] [Google Scholar]
- Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, and Sadoshima J (2011). PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 14, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka S, Zhai P, Alcendor R, Park JY, Tian B, and Sadoshima J (2012). Suppression of ERR targets by a PPARα/Sirt1 complex in the failing heart. Cell Cycle 11, 856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshti V, Obolensky A, Nahum L, Kanfi Y, Rathaus M, Avraham M, Tinman S, Alt FW, Banin E, and Cohen HY (2017). Characterization of physiological defects in adult SIRT6−/− mice. PLoS ONE 12, e0176371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell E, Kuhn P, and Xu W (2007). Nuclear receptor cofactors in PPAR-gamma-mediated adipogenesis and adipocyte energy metabolism. PPAR Res. 2007, 53843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, and Li X (2009). Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhshandehroo M, Knoch B, Müller M, and Kersten S (2010). Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. (2011). Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147, 1628–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads TW, Burhans MS, Chen VB, Hutchins PD, Rush MJP, Clark JP, Stark JL, McIlwain SJ, Eghbalnia HR, Pavelec DM, et al. (2018). Caloric Restriction Engages Hepatic RNA Processing Mechanisms in Rhesus Monkeys. Cell Metab. 27, 677–688.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roichman A, Kanfi Y, Glazz R, Naiman S, Amit U, Landa N, Tinman S, Stein I, Pikarsky E, Leor J, and Cohen HY (2017). SIRT6 Overexpression Improves Various Aspects of Mouse Healthspan. J. Gerontol. A Biol. Sci. Med. Sci 72, 603–615. [DOI] [PubMed] [Google Scholar]
- Røst TH, Haugan Moi LL, Berge K, Staels B, Mellgren G, and Berge RK (2009). A pan-PPAR ligand induces hepatic fatty acid oxidation in PPARα−/− mice possibly through PGC-1 mediated PPARδ coactivation. Cell Biol. Lipids 1791, 1076–1083. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, et al. (2012). The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med 18, 1643–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, and Riabowol K (2010). REAP: A two minute cell fractionation method. BMC Res. Notes 3, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalowska E, Tesfay HA, van Hijum SAFT, and Kersten S (2014). Transcriptomic signatures of peroxisome proliferator-activated receptor α (PPAR α) in different mouse liver models identify novel aspects of its biology. BMC Genomics 15, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemori K, Kimura T, Shirasaka N, Inoue T, Masuno K, and Ito H (2011). Food restriction improves glucose and lipid metabolism through Sirt1 expression: a study using a new rat model with obesity and severe hypertension. Life Sci. 88, 1088–1094. [DOI] [PubMed] [Google Scholar]
- Tao R, Xiong X, DePinho RA, Deng C-XX, and Dong XC (2013). Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J. Lipid Res 54, 2745–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, and Guarente L (2001). Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410, 227–230. [DOI] [PubMed] [Google Scholar]
- Vachharajani VT, Liu T, Wang X, Hoth JJ, Yoza BK, and McCall CE (2016). Sirtuins Link Inflammation and Metabolism. J. Immunol. Res 2016, 8167273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker R, Faulkner S, Miyokawa R, Burhenn L, Henriksen M, Wood JG, and Helfand SL (2013). Increased expression of Drosophila Sir2 extends life span in a dose-dependent manner. Aging (Albany N.Y.) 5, 682–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Kao HJ, Li SC, Stevens R, Hillman S, Millington D, and Chen YT (2004). ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J. Clin. Invest 113, 434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Zhang C, Zhang Y, Fan R, Qian X, and Dong XC (2017). Fabp4-Cre-mediated Sirt6 deletion impairs adipose tissue function and metabolic homeostasis in mice. J. Endocrinol 233, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, So JS, Park JG, and Lee AH (2013). Transcriptional control of hepatic lipid metabolism by SREBP and ChREBP. Semin. Liver Dis 33, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M (2010). PPARα in Obesity: Sex Difference and Estrogen Involvement. PPAR Res. 2010, 584296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M, Jeong S, Nicol CJ, Lee H, Han M, Kim JJ, Seo YJ, Ryu C, and Oh GT (2002). Fenofibrate regulates obesity and lipid metabolism with sexual dimorphism. Exp. Mol. Med 34, 481–488. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li C, Wang F, Zhou S, Shangguan M, Xue L, Zhang B, Ding F, Hui D, Liang A, et al. (2015). Treatment with PPAR α agonist clofibrate inhibits the transcription and activation of srebps and reduces triglyceride and cholesterol levels in liver of broiler chickens. PPAR Res. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Tu B, Wang H, Cao Z, Tang M, Zhang C, Gu B, Li Z, Wang L, Yang Y, et al. (2014). Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. USA 111, 10684–10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wan H, Feng G, Qu J, Wang J, Jing Y, Ren R, Liu Z, Zhang L, Chen Z, et al. (2018). SIRT6 deficiency results in developmental retardation in cynomolgus monkeys. Nature 560, 661–665. [DOI] [PubMed] [Google Scholar]
- Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, et al. (2010). The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell 140, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinman GE, Naiman S, Kanfi Y, Cohen H, and Bar-Joseph Z (2013). ExpressionBlast: mining large, unstructured expression databases. Nat. Methods 10, 925–926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.