

Abstract
Glial cells are a key element to the process of axonal regeneration, either promoting or inhibiting axonal growth. The study of glial derived factors induced by injury is important to understand the processes that allow or preclude regeneration, and can explain why the PNS has a remarkable ability to regenerate, while the CNS does not. In this work we focus on Apolipoprotein D (ApoD), a Lipocalin expressed by glial cells in the PNS and CNS. ApoD expression is strongly induced upon PNS injury, but its role has not been elucidated. Here we show that ApoD is required for: (1) the maintenance of peripheral nerve function and tissue homeostasis with age, and (2) an adequate and timely response to injury. We study crushed sciatic nerves at two ages using ApoD knock‐out and transgenic mice over‐expressing human ApoD. The lack of ApoD decreases motor nerve conduction velocity and the thickness of myelin sheath in intact nerves. Following injury, we analyze the functional recovery, the cellular processes, and the protein and mRNA expression profiles of a group of injury‐induced genes. ApoD helps to recover locomotor function after injury, promoting myelin clearance, and regulating the extent of angiogenesis and the number of macrophages recruited to the injury site. Axon regeneration and remyelination are delayed without ApoD and stimulated by excess ApoD. The mRNA and protein expression profiles reveal that ApoD is functionally connected in an age‐dependent manner to specific molecular programs triggered by injury. © 2010 Wiley‐Liss, Inc.
Keywords: lipocalin, sciatic nerve, myelin clearance, demyelination, axon regeneration, aging
INTRODUCTION
The role of myelinating glial cells, oligodendrocytes in the central nervous system (CNS) and Schwann cells in the peripheral nervous system (PNS), has been expanded from their effect on rapid axonal conduction to the maintenance of the axon long‐term functional integrity (reviewed by Nave and Trapp, 2008). Alterations in these glial cells lead to myelination defects and loss of support for axons, contributing to a variety of neurodegenerative diseases. We therefore need to understand the endogenous neuroprotective mechanisms that these cells provide throughout adulthood. In addition, these glial cells are responsible for the outcome in response to injury that so much differs between the CNS and PNS. A key step that allows regeneration after injury in the PNS is the clearance of myelin debris, which provides a local permissive environment for axonal regrowth (reviewed by Chen et al., 2007). This critical step is initiated by Schwann cells (SCs) and followed by macrophages recruited from the bloodstream. A complex set of signals are exchanged among the cells at the injury site before remyelination and nerve functional recovery is completed.
One of the factors secreted by SCs and strongly induced by injury is Apolipoprotein D (ApoD) (Boyles et al., 1990; Schaeren‐Wiemers et al., 1995; Verheijen et al., 2003). Together with Apolipoprotein E (ApoE) they are the only two apolipoproteins endogenously produced in nerves upon injury (Boyles et al., 1990; Spreyer et al., 1990). Both were hypothesized to help in the demyelination or remyelination processes, based on their potential for lipid transport. ApoE was initially thought to be dispensable for sciatic nerve regeneration (Fullerton et al., 1998; Goodrum et al., 1995), although ApoE‐KO mice show abnormalities in unmyelinated axons (Fullerton et al., 1998) and ApoE modulates the antigen‐presentation capacities of SCs in vitro (Duan etal., 2007). Notably, an experimental in vivo test for the function of ApoD in peripheral nerve regeneration has not been performed so far.
ApoD is a singular apolipoprotein with no evolutionary relationship to any of the other apolipoproteins. It belongs to the Lipocalins, a diverse family of extracellular proteins with functions as disparate as olfaction (Cavaggioni et al., 2006), antibacterial effects (Flo et al., 2004), or regulation of systemic insulin action (Hull‐Thompson et al., 2009). Although the nervous system is a relevant expression domain of a number of Lipocalins, information about their roles in this tissue is scarce.
In the CNS ApoD is expressed by astrocytes and oligodendrocytes (Ganfornina et al., 2005; Navarro et al., 2004) from late embryogenesis to the aged nervous system (Loerch et al., 2008; Sanchez et al., 2002). Neuronal expression has been observed in the CNS (Ganfornina et al., 2005; Ong et al., 1999), but not in peripheral axons (Boyles et al., 1990). Nervous system expression is evolutionarily conserved: neurons and glia express ApoD homologues, named Lazarillo, in fruitflies and grasshoppers (Ganfornina et al., 1995; Sanchez et al., 2000).
ApoD expression is boosted by a collection of traumatic, pathological and degenerative conditions (Rassart et al., 2000; Van Dijk et al., 2006). Using ApoD‐deficient (ApoD‐KO) and over‐expressing human ApoD (HApoD‐Tg) mice we have shown that ApoD halts lipid peroxidation in the CNS upon experimental oxidative stress (Ganfornina et al., 2008) and protects against coronavirus OC43‐induced encephalitis (Do Carmo et al., 2008). ApoD homologues in Drosophila also control oxidative stress levels and longevity (Hull‐Thompson et al., 2009; Sanchez et al., 2006), further supporting conserved ApoD neuroprotective functions.
In addition, the ApoD homologue Lazarillo promotes axon outgrowth in grasshopper embryos (Sanchez et al., 1995), and ApoD promotes neuritogenesis in dorsal root ganglion cultures (Kosacka et al., 2009).
In this state of knowledge, we set out to assay three questions: (1) Whether ApoD is one of the protecting mechanisms by which SCs maintain axonal integrity throughout normal adulthood and aging. (2) Whether ApoD, once induced after injury, contributes to the regeneration process. If so, (3) where does the primary function of ApoD lay in the well established cellular and molecular steps of Wallerian degeneration‐repair‐regeneration (WDR) processes?
We use wild‐type (WT), ApoD‐KO, and HApoD‐Tg mice to study the intact nerve and the WDR process following a controlled crush injury in the sciatic nerve. We find that ApoD is required to maintain the myelin sheath of intact peripheral nerves with age. Its absence alters the physiological properties of the nerve and provokes specific changes in the nerve transcriptome and proteome.
We also show that ApoD is required in an age‐dependent manner for a proper nerve functional recovery after injury. The transcriptional profile indicates that ApoD controls specific cellular responses during WDR. ApoD primarily affects the process of myelin removal by Schwann cells (SCs) and macrophages, the bottleneck step for the generation of a growth permissive environment. Axonal regeneration and remyelination are secondarily altered.
MATERIALS AND METHODS
Animals, Electrophysiological Recordings, Surgery, and Injury Procedures
Mice used (n = 60) were age and sex‐matched littermates of three genotypes (Wild type (WT), ApoD‐KO (KO) and Thy‐1‐HApoD (Tg)) backcrossed over 11 generations in C57BL/6 background. They were generated in our laboratories, and are genotyped by PCR as previously described (Ganfornina et al., 2008). Age groups were as follows: 112 ± 13 days (15‐week‐old sample; 15 week) and 286 ± 37 days (40‐week‐old sample, 40 week). Experimental procedures were approved by the Animal Care and Use Committees of the University of Valladolid (UVa) and Université du Québec à Montréal (UQAM) and were in accordance with the Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research (2001). Mice were maintained in positive pressure‐ventilated racks at 25 ± 1°C with a 12 h light/dark cycle, fed ad libitum with a standard rodent pellet diet (Global Diet 2014; Harlan), and allowed free access to filtered and UV‐irradiated water.
Animals subjected to surgery and injury procedures were anesthetized via intraperitoneal injection with ketamine (100 mg/kg) and xylacine (10 mg/kg) (UVa) or isofluorane (UQAM).
For sciatic nerve electrophysiology, the left sciatic nerve was exposed and stimulated with steel bipolar electrodes. Two electrodes, separated a fixed distance, were placed under the sciatic nerve (proximal electrode at the sciatic notch) and square‐wave pulses of 100 μs and 1–10 V were delivered at 0.2 Hz (CS‐14 stimulator, Cibertec, Spain). A steel needle electrode was placed in the plantar muscle of the left footpad at the base of the second digit, and a reference electrode was placed under the thigh skin. Recordings were obtained with supramaximal stimulation. Compound muscle action potentials (CMAP) were amplified (Physiorec‐2, Cibertec, Spain) and digitally recorded with a Tektronix oscilloscope and the software Origin (v7.03, OriginLab). Ten separate recordings for each mouse were analyzed. The distance between electrodes and the time lag between the responses was used for conduction velocity calculations. Mice body temperature was maintained around 30°C with a heating lamp.
Sciatic nerve crush was performed by compressing the nerve three times, 20 s each, with forceps #5. The crush location was marked with India ink, and the CMAP was measured before and after the lesion to ensure efficient axonotmesis. Subsequently, muscles and skin were sutured and the animals were allowed to recover in a warmed cage. Amoxycillin was administered in the drinking water (500 mg/L) for a week after surgery.
Sciatic nerves (from both the injured and contra‐lateral legs) were removed 14 or 48 days post‐crush injury (dPCI). A fragment of the nerve distal to the lesion was preserved in RNALater™ (Ambion) for RNA and protein expression analysis, and the remaining nerve was fixed and used in different experiments as explained below.
Behavioral Analysis
Nociceptive sensitivity
Nociceptive function was determined before surgery, and 7, 14, and 48 dPCI with a variant of the von Fray test using mechanical stimulation with a plastic filament (37 mm length, 0.6 mm diameter) fixed to a rigid handle. The filament tip was applied manually to the plantar surface of each hind paw. The number of withdrawal responses (paw lifting, shaking, or licking) in 10 trials was recorded.
Locomotor function
Locomotor abilities before surgery and 14, and 48 dPCI were determined using the horizontal walking pole test. Mice were placed at one end of a horizontal wooden pole (100 cm long) suspended 20 cm above soft pads and allowed to run towards an escape box located at the opposite end. Mice were first trained on 44, 27, and 18 mm wide poles successively. Three runs were performed in the test pole (18 mm wide) and the number of times each hind leg slipped out of the pole was scored.
Footprint
Footprints were analyzed prior to surgery and 14, and 48 dPCI without preliminary training. The forelimb and hind limb paws were colored red and blue respectively using water‐based dyes (Pelikan stamp pad ink). Mice were then placed at the start of a walkway (100 cm long, 10 cm wide, walls 22 cm high) and allowed to run on white paper towards a dark box. Hind limb stride width (distance between left and right foot tracks) was measured for a minimum of six steps per mouse.
Histological Procedures
Histological staining and immunohistochemistry
Nerve samples were fixed with 4% paraformaldehyde and embedded in paraffin following standard procedures. Longitudinal sections (4 μm) were performed with a rotary microtome (Microm), mounted in four series on Polysine™ slides (Menzel‐Gläser), and dried.
Sections were dewaxed and rehydrated through an ethanol series into phosphate buffered saline (PBS). Hematoxylin‐eosin staining (H&E) was carried out following standard protocols.
Before HRP immunohistochemistry, endogenous peroxidase was inactivated with H2O2 0.9% in distilled H2O for 5 min in the dark followed by washes in PBS. Sections were then blocked and permeabilized with TritonX‐100 (0.25% in PBS) and 1% normal goat serum. The following antibodies were used: Rat monoclonal anti‐Galectin‐3 (American Type Culture Collection, ATCC); Rabbit serum anti‐GAP‐43 (Abcam); Rabbit serum anti‐MBP (Abcam); Mouse monoclonal anti‐S100 (Sigma‐Aldrich); HRP‐conjugated Goat anti‐Rat IgG and Goat anti‐Rabbit IgG (Abcam). HRP development was performed with DAB (0.03%) and H2O2 (0.002% in 50 mM Tris, pH 8.0) in parallel for all sections subjected to the same antibody. Its extent was empirically established for each antibody by monitoring chromogen reaction to be kept in its lineal phase. Sections were mounted with coverslips and Eukitt (after dehydration and clearing with xylene.
For fluorescence immunohistochemistry, the following antibodies were used: Rat monoclonal anti‐Galectin‐3 (ATCC); Rabbit serum anti‐GAP‐43 (Abcam); Mouse monoclonal anti‐S100 (Sigma‐Aldrich); mouse monoclonal CD11b (DSHB); mouse monoclonal 2F11 anti‐70kD neurofilament protein (Dako); Cy3, Alexa‐594 or FITC‐conjugated Goat anti‐Mouse, Goat anti‐Rat or Goat anti‐Rabbit IgG secondary antibodies (Abcam/Molecular Probes). After washes in PBS, sections were mounted with Vectashield‐DAPI (Vector Labs).
Image acquisition and analysis
H‐E and immunohistochemistry sections were visualized with an Eclipse 90i (Nikon) fluorescence microscope equipped with a DS‐Ri1 (Nikon) digital camera.
Images were acquired and processed with the NIS‐Elements BR 3.0 software (Nikon). Acquisition was performed under the same conditions of illumination, diaphragm and condenser adjustment, exposure time, background correction and color levels for each set of WT, ApoD‐KO and HApoD‐Tg nerve sections labeled with a given antibody. For all quantifications described below a region comprising 500 μm on both sides of the crush site was analyzed in three sections spaced 50 μm from the center of the nerve.
To quantify GAP‐43 and Galectin‐3 labeling we selected a threshold for the color of a control DAB‐developed section and processed all images using the same color mask. Color integrated density was measured in four 25,000 μm2 windows randomly located in each section. MBP‐positive bodies or S‐100‐positive nuclei were counted manually. A cellularization index was obtained from H&E labeled sections after thresholding and masking the hematoxylin blue reaction. The percent area occupied by nuclei was estimated in three 20,000 μm2 random windows per section.
Semithin sections
Nerves were fixed with 4% paraformaldehyde‐2% glutaraldehyde in 0.05 M cacodylate buffer for 18 h, postfixed in 1% osmium tetroxide in the same buffer for 1 h, and included in acrylic resin (Durkupan‐ACM, Fluka) after dehydration in acetone series. Transversal 1 μm cross sections were obtained every 0.1 mm along the nerve with a Sorval MT‐5000 ultramicrotome, and stained following the first step of Tolivia et al. (1994) method (for myelin and axonal profiles) or with toluidine blue (for cell counts). For myelin and axonal profile analysis, seven sections per nerve were visualized with a Nikon Eclipse E400 microscope, digital images were taken with a Nikon DC100 camera and a morphometric analysis performed as previously described (Tolivia et al., 2006). Cells and blood vessels were counted in the injury region (five sections sampled along 500 μm of each nerve) in duplicate by two different observers.
Transcriptional Expression Profile Analysis
Quantitative real‐time RT‐PCR
Distal portions of nerves (1 mm3 fragments) were stored in RNALater™ (Ambion) at −80°C, extracted with Trizol and linearly amplified with MessageAmp™ II (Ambion). Total RNA (1 μg) was reverse‐transcribed with PrimeScript™ (Takara), and cDNA used as template for qRT‐PCR amplifications in TaqMan® Custom Arrays (Applied Biosystems) performed at the Genomic Unit of CNIC (Madrid). Rpl18 was used as the reference gene. Amplifications were performed in quadruplicate in an ABI Prism 7900HT thermal cycler. Cycling conditions: 94.5°C, 10 min; 40x(97°C, 30 s; 59.7°C, 1 min).
qRT‐PCR analysis
Changes in transcriptional expression were assayed with the ΔΔC T method (Livak and Schmittgen, 2001; Schmittgen and Livak, 2008), where 2−ΔΔCt is the fold change in gene expression relative to the experimental condition chosen as reference. We use a Log 2−ΔΔCt axis for representation of fold changes, so that up‐ and down‐regulations are symmetrically scaled around zero.
The following criteria were applied in order to extract relevant biological information from our experiments: (1) Replicates with variation coefficient >2.5% were excluded. (2) Undetermined C T values (gene expression below detection levels) were assigned C T = 40. (3) Comparisons between two experimental conditions were performed by calculating ΔΔC T values. (4) Pairwise comparisons where the gene average C T > 35 cycles in both conditions were excluded from the analysis to avoid meaningless comparisons between levels of expression close to the detection limit. (5) Low amounts of a given template in a given condition often results in highly variable C T values between replicates (ranging between 35 and indetermination) that often prevents statistical evaluation of comparisons that show otherwise clear differences. In comparisons where gene amplification shows an average C T > 35 cycles in one of the conditions only (meaning that the gene is either turned off or turned on by the experimental condition), we filtered the outlier C T values reflecting marginal expression by substitution with the average C T of the quadruplicate. This filter around the average homogenizes values, increasing the weight of the more reliable ones, and allows for on and off changes to be assessed statistically. (6) Only transcriptional changes with Log 2−ΔΔCt ≥ ±0.3 (twofold increase) were included in the analysis.
Significant differences of gene transcriptional changes were evaluated with a Mann‐Whitney U‐test (Yuan et al., 2006), using ΔC T of each replica (calculated by subtracting the average C T of the reference gene). Values are expressed as mean fold‐change ± SEM, and the level of significance was set at P < 0.05. Only statistically significant differences of expression are presented in results and discussed in the text.
Immunoblot Analysis
Intact sciatic nerves used for comparisons between genotypes were dissected and 10 mm fragments were homogenized in lysis buffer (1% Nonidet P‐40, 0.1% SDS, 0.5% sodium deoxycholate, and 10% Complete Protease Inhibitors (Roche) in PBS), centrifuged after 30 min at 4°C, and stored at −80°C. Protein concentration was determined with Micro‐BCA™ protein assay (Pierce).
For comparisons between control and experimental nerves, 1 mm3 fragments from the distal portion of each nerve were pooled by experimental condition and genotype, and proteins were obtained after Trizol extraction of RNA (see above) following the manufacturer protocol.
Immunoblot analysis was carried out with 15 μg of total protein/lane using standard procedures with the following primary antibodies: Goat anti‐human Il6 (R&D), rat anti‐Galectin‐3 (ATCC), and rabbit anti‐GAP43 (Abcam). HRP‐conjugated anti‐β actin antibody (Sigma) was used to normalize protein loads. Membranes were developed with ECL (BioRad), and the signal visualized with a digital camera (VersaDoc, BioRad). The integrated optical density of the immunoreactive protein bands was measured in images taken within the linear range of the camera (avoiding signal saturation). The mean ±SD of arbitrary density units was calculated from duplicate blots.
2D electrophoresis and MALDI‐TOF Analysis
Homogenized sciatic nerve tissues were treated with DNaseI and RNaseA. Proteins (150 μg) were separated by pI (pH gradient 3.0–10.0) and size in 16 × 14 cm 10% acrylamide gels. Silver‐stained gel images were digitized, and WT and ApoD‐KO spots were compared by densitometry. Selected spots differentially present in ApoD‐KO nerves were excised, digested with trypsin, and subjected to MALDI‐TOF. Protein identification was based in the profiles with Score values >62 found in MSDB 20060831 database.
Statistical Analysis
Statistical analyses were performed with SPSS (v 11.0) or Statgraphics plus (v 5.0) softwares. Student t‐test (or Mann‐Whitney U‐Test when normality tests failed) was used to assess two‐sample comparisons with P < 0.05 as threshold for significant changes.
RESULTS
ApoD Is Required for Myelin Sheath Maintenance, and Influences the Axon Functional Properties in an Age‐Dependent Way
Compound motor action potentials of sciatic nerves were studied in WT and ApoD‐KO mice to assess whether ApoD is important for normal nerve conduction physiology. Motor nerve conduction velocity (MNCV) (Fig. 1A) and the peak‐to‐peak amplitude and duration of the compound motor action potential were measured. We obtained values similar to those determined in WT mice of similar age (Weiss et al., 2001). No differences in MNCV are observed between genotypes in the 15‐week‐old mice, but MNCV decreases significantly (29%) in the 40‐week‐old ApoD‐KO mice (Fig. 1B). Neither amplitude nor duration of the CMAP changed with genotype (not shown).
Figure 1.
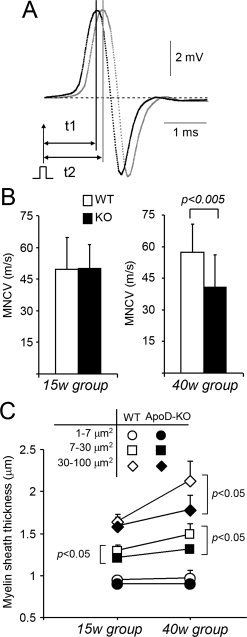
ApoD influences the myelin of intact sciatic nerves. A: Representative recordings of CMAP evoked by electrical stimulation with two bipolar electrodes. Time differences from stimulus to CMAP peak were used to estimate conduction velocity. B: MNCV from WT and ApoD‐KO mice at two ages. Conduction velocity is compromised in 40‐week‐old ApoD‐KO mice. Data shown as mean ± SEM; n = 10 recordings/mice; 13 mice/age group/genotype. C: Morphometric analysis of myelin sheath thickness in intact myelinated axons (15 weeks and 40 weeks, WT and ApoD‐KO nerves). Statistical differences assayed by Student's t‐test.
The decrease in MNCV suggests anomalies in either axon membrane physiology and/or myelin sheath. We examined myelin sheath in stained semithin sciatic nerve cross‐sections. Fibers were categorized in three groups according to their area, following the classification by Persohn et al. (2005). Myelin sheath thickness decreases significantly (Fig. 1C) in the intermediate size axons of ApoD‐KO nerves in both age groups. Large diameter axons, however, show thinner myelin only in the 40 week ApoD‐KO mice. This effect parallels, and can explain, the observed decrease in MNCV with age, where the activity of large motor axons is the main component of the recorded signal.
Our results support that ApoD participates in the myelin maintenance mechanism required to keep axon long‐term functional integrity.
ApoD Influences Functional Recovery After Sciatic Nerve Crush Injury
We chose crush injury (CI) as a well‐established paradigm to evaluate the role of ApoD in the WDR process (Chen et al., 2007) since it allows for axonal regeneration and full functional recovery. Two time points, 14 and 48 days post‐crush injury (d‐PCI), were chosen based on ApoD protein profile after sciatic CI in rats (Boyles et al., 1990).
To ensure injury completeness we recorded the CMAP before and immediately after injury (Fig. 2A). Stimulation with the proximal electrode (pointed by arrows in Fig. 2A) evoked no CMAP after crush.
Figure 2.
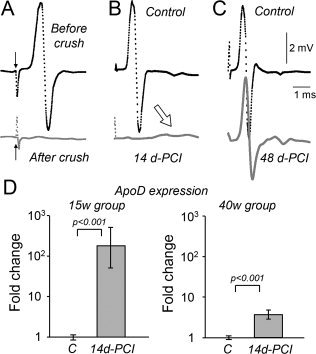
Validation of the crush injury method to study ApoD function in WDR. A: Crush effect validation through CMAP recording. CMAP disappears completely after crush. Arrows point to the stimulus artifact. B, C: Recovery of nerve conduction evaluated 14 and 48 d‐PCI. Full recovery of CMAP is achieved at 48 d‐PCI. The small deflections observed at 14 d‐PCI (arrow) are the first signs of recuperation of nerve conduction. D: qRT‐PCR levels of ApoD in intact (C = control nerves) and in the distal region of crushed nerves at 14 d‐PCI (15‐week and 40‐week mice). ApoD is up‐regulated with injury. Statistical differences assayed by Mann‐Whitney's U‐test.
We confirmed that ApoD mRNA is also up‐regulated in the mouse nerve upon CI, as reported for rats (Bosse et al., 2006; Boyles et al., 1990; Spreyer et al., 1990). ApoD transcript increases 180‐fold in the distal region of 15‐week nerves at 14 d‐PCI (Fig. 2D). In 40‐week nerves the basal expression levels of ApoD are lower (not shown), but gene expression is still significantly induced in response to injury (fourfold, Fig. 2D). A pattern of low expression and low induction by CI is also observed in many of the injury‐related genes we analyze (see below). These data support the idea that aging is accompanied by a weakening of the defense response to injury.
Recovery of locomotor and sensory function
In agreement with the existing literature, the first sign of nerve conduction recovery is seen at 14 d‐PCI as small voltage deflections following nerve stimulation (Fig. 2B, white arrow), and the conductive properties are fully recovered at 48 d‐PCI (Fig. 2C). No difference was found between WT and ApoD‐KO nerves in the recovery of CMAP parameters (latency, amplitude and duration; not shown). However, a mixture of functional differences in sensory and motor components of the recovered nerve can still be present without showing major effects in the CMAP parameters. We therefore performed a set of behavioral tests to examine different components of sensorimotor function after CI (Table 1). Locomotion was assessed by analysis of walking tracks and walking pole tests, and sensory function was assayed by the withdrawal reflex upon mechanical plantar stimulation. Footprint width showed statistically significant changes between WT and ApoD‐KO at 14 d‐PCI in 40‐week mice. In the walking pole test, WT mice in both age groups return to control values at 48 d‐PCI, while significant differences from control conditions persist in ApoD‐KO mice. Sensory performance was more altered in the 15‐week mice, with a significant reduction in the performance of ApoD‐KO at 7 d‐PCI and an enhancement of the reflex response at 14 and 48 d‐PCI. The same trend is evident in 40‐week mice.
Table 1.
Behavioral Assessment of Functional Recovery After Sciatic Nerve Crush in 15 w and 40 w WT and ApoD‐KO Mice
| 15‐week‐old group | ||||||||
|---|---|---|---|---|---|---|---|---|
| WT | ApoD‐KO | |||||||
| 0 d‐PCI | 7 d‐PCI | 14 d‐PCI | 48 d‐PCI | 0 d‐PCI | 7 d‐PCI | 14 d‐PCI | 48 d‐PCI | |
| Footprint width (cm) | 2.19 ± 0.13 | – | 1.98 ± 0.17 b | 2.14 ± 0.15 | 2.11 ± 0.17 | – | 1.91 ± 0.15 | 2.13 ± 0.18 |
| Walking pole (# sideslips) | 0.30 ± 0.48 | – | 0.67 ± 0.59 | 0.33 ± 0.5 | 0.37 ± 0.49 | – | 1.33 ± 1.48 b | 1.17 ± 1.06 b |
| Withdrawal reflex (% WT 0d) | 100 ± 27.66 | 45.82 ± 17.50 b | 66.35 ± 15.13 | 125.69 ± 70.48 | 94.89 ± 27.01 | 23.52 ± 21.73 a, b | 81.59 ± 26.55 | 152.32 ± 60.17 |
| 40‐week‐old group | ||||||||
|---|---|---|---|---|---|---|---|---|
| WT | ApoD‐KO | |||||||
| 0 d‐PCI | 7 d‐PCI | 14 d‐PCI | 48 d‐PCI | 0 d‐PCI | 7 d‐PCI | 14 d‐PCI | 48 d‐PCI | |
| Footprint width (cm) | 2.38 ± 0.15 | – | 2.18 ± 0.17 b | 2.33 ± 0.11 | 2.44 ± 0.26 | – | 1.98 ± 0.15 a, b | 2.35 ± 0.18 |
| Walking pole (# sideslips) | 0.29 ± 0.45 | – | 1.38 ± 1.25 b | 0.67 ± 0.56 | 0.31 ± 0.4 | – | 2.42 ± 2.57 b | 1.83 ± 1.31 a, b |
| Withdrawal reflex (% WT 0d) | 100 ± 31.86 | 18.57 ± 20.71 b | 24.84 ± 15.06 | 63.12 ± 26.87 | 76.47 ± 18.13 | 6.94 ± 6.94 b | 38.76 ± 26.26 | 77.53 ± 53.48 |
Mice were subjected to three behavioral tests exploring locomotor and sensory domains before injury (t = 0) and at specified days post crush (d‐PCI). Data shown as mean ± SD. Significant changes (P < 0.05) shown in bold.
Differences between genotypes (ApoD‐KO relative to WT mice) for the same parameter and time point.
Differences due to treatment (post‐crush values relative to pre‐treatment test, for a given genotype). n = 8–14/group. Statistical differences assayed by Student t‐Test (footprints analysis) and Mann‐Whitney U‐test (walking pole and withdrawal reflex tests).
HApoD‐Tg mice were subjected to the same set of behavioral tests, resulting in no differences with WT (not shown). Thus, the ectopic expression of HApoD in neurons does not improve recovery. Whether the native mouse protein is more effective in this model, or the expression of ApoD from glial cells is a key factor, will need further investigations. As shown below, HApoD does produce changes in molecular and cellular events that help to fine‐tune the response to injury in a positive direction.
These results reveal that the lack of ApoD does significantly increase the functional impairment after injury and results in an imperfect recovery of locomotor function, being the defects more prominent in older mice. Therefore, the presence of ApoD is beneficial after peripheral nerve injury.
Axonal Regeneration Is Transiently Delayed in ApoD‐KO and Promoted in HApoD‐Tg Mice
Axon regeneration following CI was assessed by evaluating the presence of the regeneration‐associated protein GAP‐43, both by immunoblot of fragments distal to the crush region (Fig. 3A,B) and by immunohistochemistry of the crush region (Fig. 3C,D). The axonal marker 2F11 (anti‐70kD neurofilament protein) was also used.
Figure 3.
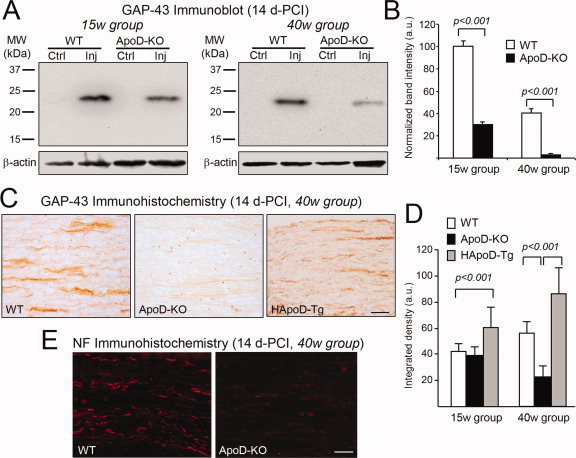
ApoD influences the expression of the regeneration marker GAP‐43 after CI. A: Immunoblot analysis of GAP‐43 in the region distal to the lesion of WT and ApoD‐KO mice at 14d‐PCI (two age groups, pooled samples, n = 6/group/genotype). Ctrl, intact nerve; Inj, injured nerve. B: Protein levels quantified by band densitometry and normalized to β‐actin signal. GAP‐43 expression decrease 69% (15‐week group) and 90% (40‐week group) in ApoD‐KO mice compared with WT nerves. C: Representative photomicrographs of GAP‐43 immunohistochemistry in sagittal sections of the lesion region at 14 d‐PCI (40‐week age group). D: Quantification of GAP‐43 immunoreactivity in the site of lesion at 14 d‐PCI (n = 4/group/genotype). Data shown as mean ± SD. Statistical differences assayed by Student's t‐test. E: 70kD neurofilament marker (2F11) immunofluorescence in sagittal sections of the lesion region at 14 d‐PCI (40‐week age group). Calibration bars in C,E: 50 μm.
GAP‐43 is not present in intact nerves (Fig. 3A) and appears in WT nerves 14 d‐PCI, as expected by the presence of newly growing axons (Fig. 3A). However, when ApoD is absent, the amount of GAP‐43 in injured nerves is reduced by 69% in 15‐week mice and by 90% in 40‐week mice (Fig. 3B).
In addition, densitometry analysis of GAP‐43 immunoreactivity in the crush region at 14 d‐PCI (Fig. 3C) shows a significant increase in nerves over‐expressing ApoD, and a reduction of GAP‐43 in ApoD‐KO nerves. These results were confirmed by immunofluorescence with a neurofilament marker (Fig. 3E). In agreement with the immunoblot analysis, the effects are more pronounced with aging (Fig. 3D). GAP‐43 immunoreactivity does not change among the three genotypes in the crush region at 48 d‐PCI (not shown), suggesting that the outgrowth phase of axonal regeneration is delayed in the absence of ApoD.
Together these data suggest that ApoD is important for establishing timely axon regeneration after injury.
Myelin Clearance Is Delayed in the Absence of ApoD
Since inhibitory molecules from myelin prevent axonal regeneration (reviewed by Chen et al., 2007) we tested whether anomalies in myelin clearance might be responsible for the delayed axonal re‐growth in the ApoD‐KO nerves. Immunodetection of myelin basic protein (MBP), present in compact myelin, was performed and quantified in the crush region (see Fig. 4), and collapsed myelin profiles were characterized in stained semi‐thin cross sections (not shown).
Figure 4.
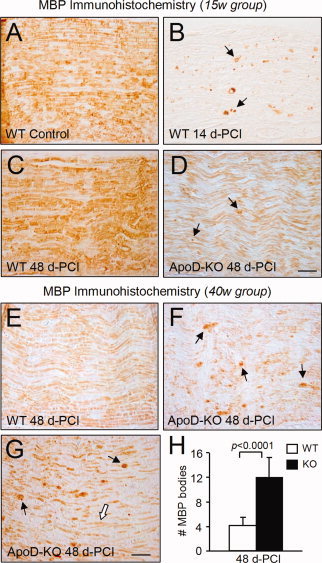
The expression of ApoD alters the duration of the myelin debris clearance phase. A–G: Representative examples of MBP immunohistochemistry in sagittal sections of the lesion region (two age groups, n = 4/group/genotype). Arrows point at MBP‐positive myelin bodies. The white arrow in G points to remyelinated axons in ApoD‐KO nerves. Calibration bars: 50 μm. H: Number of MBP‐positive myelin bodies present at 48 d‐PCI in the lesion region of WT and ApoD‐KO (40‐week age group). Statistical differences assayed by Student's t‐test.
At 14 d‐PCI, myelin clearance is underway in 15‐week‐old WT nerves. Myelin fragments were visible as MBP‐positive aggregates (Fig. 4B) or collapsed myelin profiles (not shown). By 48 d‐PCI, MBP immunoreactivity in WT nerves has returned to normal, with bundles indicating the presence of myelinated axons (compare Fig. 4A,C). A similar pattern was observed in the 40‐week age group (example of 48 d‐PCI in Fig. 4E). Neither the number of MBP‐immunopositive aggregates nor the area occupied by collapsed myelin profiles in stained semithin cross sections show differences between genotypes at 14 d‐PCI (not shown).
In contrast, myelin clearance is not completed in the ApoD‐KO nerves at 48 d‐PCI. In the 15‐week nerves some myelin debris are still present (arrows in Fig. 4D), and this effect is more noticeable at 40‐week (examples from two different mice are shown in Fig. 4F,G, arrows). A clear delay in myelin clearance is observed, though some extent of myelination has been achieved (Fig. 4G, white arrow). Quantification of MBP‐positive myelin aggregates in the lesion region of 40‐week nerves shows a significant increase in ApoD‐KO nerves (Fig. 4H).
Therefore, myelin clearance is delayed in crushed nerves when ApoD is absent, and the defect is enhanced by age.
ApoD Alters the Level of Galectin‐3 Expression by Schwann Cells and/or Macrophages
After injury, SCs are essential to determine how fast myelin and axonal debris are cleared. They are involved not only in direct phagocytosis of debris, but also in recruiting macrophages that help in the task (Chen et al., 2007; Martini et al., 2008). Galectin‐3, a galactose‐binding protein expressed by SCs and macrophages, can be used to monitor these processes. It is involved in the recruitment and activation of macrophages, and in the lectin‐dependent phagocytosis of myelin (Reichert et al., 1994; Saada et al., 1996).
Immunoblot (Fig. 5A,B) of the distal region and immunohistochemistry of the lesion region (Fig. 5C–E) were used to assay whether the time course and extent of Galectin‐3 expression was modified by ApoD.
Figure 5.
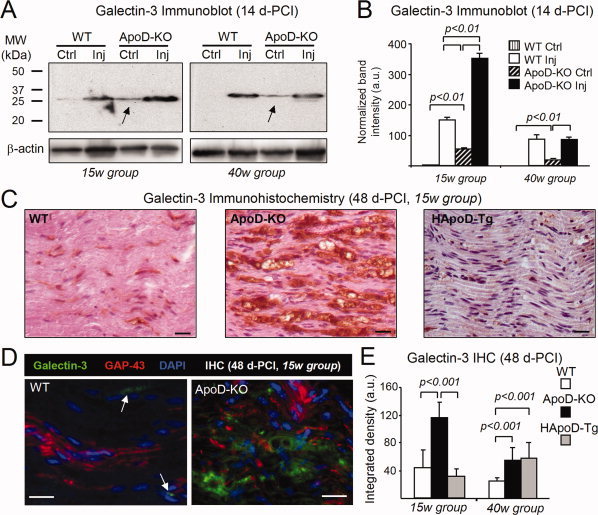
ApoD alters the timing and extent of Galectin‐3 expression in injured and intact nerves. A: Immunoblot analysis of Galectin‐3 in the region distal to the lesion of WT and ApoD‐KO mice at 14d‐PCI (two age groups, pooled samples, n = 6/group/genotype). Ctrl, intact nerve; Inj, injured nerve. B: Protein levels quantified by band densitometry normalized to β‐actin signal. In contrast to the almost null expression in WT, ApoD‐KO intact nerves show basal expression of Galectin‐3 at both ages (arrows in A). Upon injury, Galectin‐3 levels are 2.3 times higher in ApoD‐KO than in WT nerves at 15 weeks. Similar amounts are observed at 40 weeks. C: Representative photomicrographs of Galectin‐3 immunohistochemistry in sagittal sections of the lesion region (15‐week mice, 48 d‐PCI). D: Double labeling with anti‐Galectin‐3 and anti‐GAP‐43 antibodies. Scale bars in C,D: 20 μm. E: Quantification of Galectin‐3 immunoreactivity in the site of lesion at 48 d‐PCI (two age groups, n = 4/group/genotype). Data shown as mean ± SD. Statistical differences assayed by Student's t‐test.
Galectin‐3 increases after injury in all genotypes and age groups. However, at 15‐week the level of injury‐induced Galectin‐3 in ApoD‐KO nerves doubles that of WT, both at 14 and 48 d‐PCI (Fig. 5B,E). Galectin‐3 positive cells are still abundant in the lesion region of ApoD‐KO injured nerves at 48 d‐PCI (Fig. 5C–E), a time when expression levels have already decreased in WT, and the process of myelin clearance is close to completion. Low levels of Galectin‐3 are observed at 48 d‐PCI in 15w HApoD‐Tg nerves (Fig. 5C). Galectin‐3 expression changes after injury are ameliorated by age, and differences between genotypes become less marked.
It is important to note that ApoD‐KO mice express Galectin‐3 even in intact nerves (Fig. 5A arrows, Fig. 5B striped bars). This basal Galectin‐3 expression suggests that nerve homeostasis is already impaired in the absence of ApoD, giving rise to an injury‐like environment.
ApoD Alters the Number of Macrophages Present at the Injury Site
Elevated levels of Galectin‐3 can result from a stronger expression by the same number of cells, or from the presence of a higher number of Galectin‐3 expressing cells (SCs or macrophages). The number of cells present after injury can be modified either by changing the proliferation‐apoptosis balance of SCs (Yang et al., 2008), or by altering the process of recruitment and clearance of macrophages (Martini et al., 2008).
We first estimated a cellularization index for the lesion region by measuring the area occupied by nuclei in a series of H&E stained sections (Fig. 6A). As expected, this estimate of the number of cells increases in WT nerves at 14 d‐PCI and is maintained at 48 d‐PCI in both age groups. Aging, once again, results in a milder response to injury. Genotype‐dependent significant changes are evident at 48 d‐PCI in young mice, with a higher cellularization index in ApoD‐KO and a lower index in HApoD‐Tg. At 40‐week, the increase in ApoD‐KO nerves is also significant starting at 14 d‐PCI.
Figure 6.
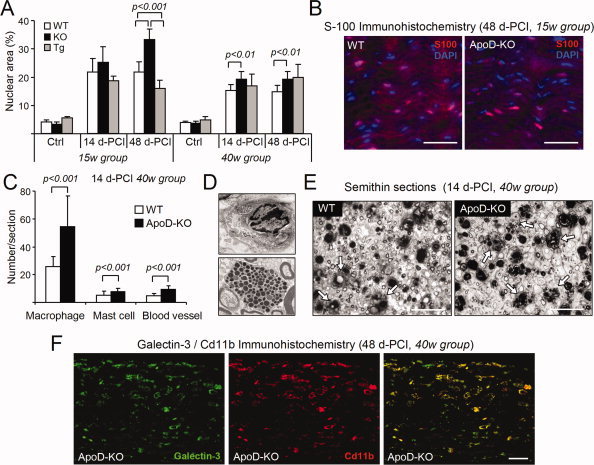
Effect of ApoD on the recruitment and persistence of cells at the injury site. A: Cellularization index (% area occupied by nuclei) in the lesion region of intact (Ctrl) and crushed nerves at two time points after crush (two age groups, n = 4/group/genotype). B: Representative examples of S100‐positive cells in the lesion region of WT and ApoD‐KO crushed nerves (15‐week mice, 48 d‐PCI). C: Number of macrophages, mast cells, and blood vessels in semithin cross‐sections of the lesion region of WT and ApoD‐KO crushed nerves. D: Examples of macrophage (top) and mast cell (bottom). E: Representative micrographs of semithin cross‐sections of the lesion regions of WT and ApoD‐KO nerves (40 week mice, 14 d‐PCI). Arrows point to macrophages engulfing myelin debris. F: Representative micrographs of CD11b and Galectin‐3 double immunofluorescence in sagittal sections of the lesion region (40 week mice, 48 d‐PCI). Merged images are shown in the third panel. Calibration bars: 50 μm in B,F; 30 μm in E. Statistical differences assayed by Student's t‐test in A,C.
The number of S100‐positive cells in the lesion region showed no genotype‐dependent differences (examples of 15‐week nerves at 48 d‐PCI are shown in Fig. 6B). Therefore, myelinating SCs are not responsible for the changes observed.
Alternatively, the higher number of cells in the ApoD‐KO injured nerves could be macrophages that enter the injured nerve and persist in the lesion region longer than in WT nerves, where they start leaving the injury site by 14 d‐PCI (Martini et al., 2008). To test this idea we first counted cells in transversal semithin sections of the lesion region at 14 d‐PCI. Macrophages, with characteristic filopodia, dark cytoplasm and myelin inclusions, and mast cells were identified by their distinct morphological appearance (Fig. 6D). ApoD‐KO nerves have a significant increase in the number of macrophages and mast cells in the lesion region, also accompanied by a significant increase in the number of blood vessels (Fig. 6C,E). In addition, we performed double immunofluorescence with Galectin‐3 and the macrophage marker CD11b to assay which proportion of Galectin‐3 expression (see Fig. 5) is due to macrophages. Most of Galectin‐3 labeling co‐localizes with the cell‐surface macrophage marker (Fig. 6F), reinforcing the idea of an elevated presence of macrophages in the injured ApoD‐KO nerves.
In summary, the presence of ApoD alters the recruitment and/or persistence of monocyte lineage cells in the injured nerves. This effect is particularly prominent in the 15‐week age group and is coherent with the Galectin‐3 observations. The process of angiogenesis during WDR is also altered. In the absence of ApoD a higher neovascularization of the injury site can in turn promote the entrance of blood born cells and the persistence of a proinflammatory environment.
ApoD Alters the Sciatic Nerve Expression Profile and the Gene Programs Triggered by Injury
So far, we have shown that ApoD contributes to the maintenance of myelination, and therefore the functional properties of nerves in normal conditions. Upon injury, ApoD controls the magnitude and duration of the inflammatory response, particularly the extent of macrophage infiltration and galectin‐3 expression. In turn, these parameters affect the speed at which myelin is cleared, and a permissive environment for axonal regeneration and remyelination is allowed.
To further study the molecular mechanisms underlying these effects of ApoD in WDR, we analyzed the transcriptional profile of a group of genes known to alter their expression in the distal region of injured nerves. We chose 14 d‐PCI as the time point for gene expression profiling, and selected genes with distinct ontology terms so that different signaling pathways and cellular events could be monitored. The final set of 46 selected genes is shown in Table 1S, and hereafter will be referred to as “injury‐regulated” genes.
The transcriptional profile was assayed for WT, ApoD‐KO and HApoD‐Tg nerves of both 15‐week and 40‐week age groups by qRT‐PCR (see Methods for details on exclusion criteria and analysis). We analyzed: (1) the effect of genotype on the transcriptional profile of intact nerves, (2) the age‐dependent changes in intact nerves, and (3) the transcriptional changes in both age groups upon crush injury, and their relationship to ApoD expression.
Figure 7 shows the genes that change their expression in intact nerves when comparing ApoD‐KO or HApoD‐Tg with WT. Only the statistically supported changes are shown (see Methods section). Seven genes are down‐regulated and five genes up‐regulated in ApoD‐KO nerves at 15‐week (Fig. 7A), with the largest increase corresponding to the cytokine Il6, also confirmed at the protein level (40% increase in intact ApoD‐KO nerves, Fig. 7C). Il6 and Vegfa are in fact turned on in the intact ApoD‐KO nerves (marked by grey boxes), since mRNA levels are below detection levels in WT nerves. Two other genes (Ptn and S100a4) show genotype‐dependent opposite patterns, being down‐regulated in the over‐expressor HApoD‐Tg and up‐regulated in ApoD‐KO nerves. A striking finding is that the presence of neuron‐expressed HApoD differentially down‐regulates several genes (Fig. 7A).
Figure 7.
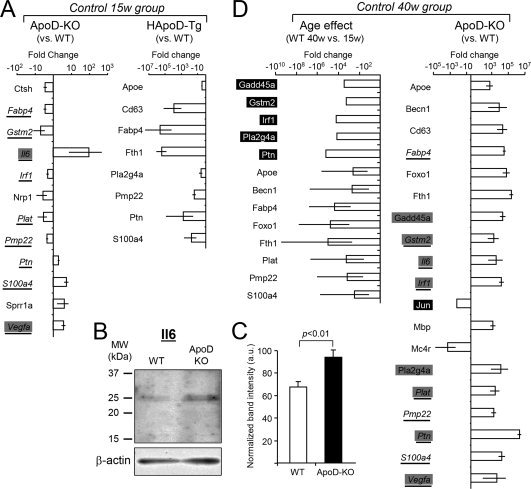
Quantitative RT‐PCR expression profiles of intact nerves of different genotypes and age groups. A: Profile of genes that change their expression in intact 15 weeks nerves of ApoD‐KO and HApoD‐Tg mice. WT intact nerve expression is used as the calibrator for each gene (n = 6/group). B, C: Immunoblot analysis of Il6 in total protein preparations of 33 weeks old intact WT and ApoD‐KO nerves. Protein levels were quantified by band densitometry normalized to β‐actin signal. Il6 expression shows a 1.4 fold increase in ApoD‐KO nerves with respect to WT. D: Gene profiles in 40‐week‐old intact nerves. Left graph shows the age effect in the profile of WT gene expression (WT 40‐week and 15‐week nerves are compared). Right graph shows the gene profile of 40‐week intact nerves from ApoD‐KO mice using the 40‐week WT intact nerve as calibrator. Gray and black boxes point to genes turned on and off respectively. Underlined genes are ApoD‐dependent in both age groups. n = 8/group. Only statistically supported changes (Mann‐Whitney's U‐test) with Log 2−ΔΔCt ≥ ±0.3 (twofold increase) are shown.
The age effect in expression profiles of intact nerves is shown in Fig. 7D. Seventeen genes are up‐regulated and two down‐regulated with age in the absence of ApoD (Fig. 7D, right panel), being eight genes turned on and one gene turned off (gray and black boxes respectively). Nine of the genes in this group are also present in the group of genes that change at 15‐week of age (overlapping genes are underlined in Fig. 7A,D). Four genes that were up‐regulated in the 15w group (Il6, Ptn, S100a4, and Vegfa), maintain their up‐regulation in the 40w intact ApoD‐KO nerves. Particularly, Il6 and Vegfa are consistently turned on in ApoD‐KO regardless of age. The genotype‐dependent expression changes of the 40w group must be interpreted in the context of an overall (13 genes) down‐regulation with age (Fig. 7D, left panel), with five genes turned off in the 40‐week group (black boxes in Fig. 7D).
Genes whose injury‐regulated expression is affected by ApoD genotype are shown in Fig. 8. Only the statistically supported changes are shown (see Methods). The effect of genotype in the injury response is age‐dependent in the subsets shown in Fig. 8A,B, while other genes show ApoD‐dependent changes irrespective of age (Fig. 8C). Alox15 and Ngfr show a higher up‐regulation by injury in ApoD‐KO 15‐week nerves (Fig. 8A, arrows), but undetectable transcript levels in all genotypes at 40‐week. The same holds for Bak1, showing an elevated up‐regulation by injury in the absence of ApoD at 40‐week (Fig. 8B, arrow). The remaining genes reduce their response to injury in the absence of ApoD. Four genes (Prx, Plat, Mcpt1, and Mapk1, Fig. 8A) do so at 15‐week, three genes (Apoe, Foxo1, and Pla2g4a, Fig. 8B) at 40‐week, and 12 genes at both ages (Fig. 8C). Six genes of the latter set (Becn1, Cd63, Fabp4, Fth1, Ptn, and S100a4) show opposite changes in the HApoD‐Tg nerves (Fig. 8C).
Figure 8.
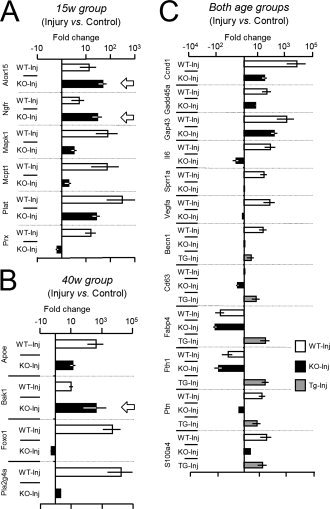
Effects of injury at 14 d‐PCI in the gene expression profile of nerves with different genotype and age. Expression was assayed by quantitative RT‐PCR using the intact nerve expression level as calibrator for each gene and genotype. A, B: Profiles of ApoD‐dependent genes at 15 week (A) and 40 week (B). C: Injury‐regulated genes with similar patterns of ApoD‐dependence at both ages. Arrows point to genes that show an increased up‐regulation with injury (compared to WT) in the absence of ApoD. n = 6/group. A threshold of Log 2−ΔΔCt ≥±0.3 (twofold increase) was used. Only genes in which the response to injury was statistically different between genotypes (Mann‐Whitney's U‐test) are shown.
Among the genes with decreased or null response to injury in the ApoD‐KO nerves we need to distinguish those that already have high basal levels of expression (e.g.: Il6, Vegfa, or S100a4), from others with basal expression levels equal to WT nerves (e.g. Mapk1 or Ccnd1). The former genes are indicators of processes restrained or inhibited by the presence of ApoD in an uninjured condition. Genes of the second category require ApoD for their injury‐dependent expression.
In the ApoD‐dependent injury‐regulated gene subsets (all genes shown in Fig. 8) there is a significant enrichment of genes associated to a stress response (gene ontology term GO:0006950) and an under representation of genes related to transcription (GO:0006351).
Finally, a complementary proteomic analysis of intact 33 weeks old ApoD‐KO and WT nerves, identified several proteins with differential expression (Fig. 1S). Four proteins, Lamin‐A, Tubulin polymerization promoting protein, Parvalbumin, and Triosephosphate isomerase, are up‐regulated, whereas 14‐3‐3 and Calpain small subunit are down‐regulated in ApoD‐KO intact nerves. The expression of all these proteins is regulated in neurodegenerative processes, therefore strengthening the conclusion that the absence of ApoD creates an injury‐like environment in the peripheral nerve.
From the transcriptional and protein expression profile analysis we conclude that (1) ApoD is needed to maintain homeostasis in peripheral nerves; (2) ApoD is differentially affecting the nerve response to injury with age; 3) ApoD influences the expression of only a subset of genes known to respond to nerve injury, thus functionally connecting ApoD to specific pathways triggered by injury.
DISCUSSION
Since the first description of ApoD as an apolipoprotein expressed in the peripheral nerve and strongly induced by damage in the PNS (Boyles et al., 1990; Spreyer et al., 1990), no proof for its function has been obtained. Several lines of evidence support that Schwann cells are ApoD main site of expression in the PNS (Schaeren‐Wiemers et al., 1995; Verheijen et al., 2003). Although it might not be the only cell type expressing it, ApoD is a relevant glia‐derived extracellular protein. Our work represents the first experimental test of the role of ApoD in mammalian peripheral nerve function and regeneration after injury.
ApoD Promotes Myelin Maintenance and Tissue Homeostasis in the PNS
Four lines of evidence place ApoD as a factor involved in myelin maintenance and nerve tissue homeostasis. First, eliminating ApoD expression alters the conductive properties of sciatic motor axons. Second, myelin sheath thickness is reduced in ApoD‐KO nerves and the effect worsens with age. Third, the transcriptional profile of injury‐regulated genes in the intact ApoD‐KO nerves resembles the profile of a damaged WT sciatic nerve, while the expression of these genes is largely decreased in HApoD‐Tg intact nerves. Finally, the proteomic analysis uncovers proteins related to elevated stress and neurodegeneration.
Our results are also consistent with the known reduced response to injury in aged WT nerves (reviewed by Verdu et al., 2000). In addition, in the absence of ApoD, 40w intact nerves show the gene expression profile of a reactive tissue, with an overall up‐regulation of injury‐related genes (25 genes expressed in 40‐week ApoD‐KO intact nerves, in contrast with only 11 and 19 genes detected in WT and HApoD‐Tg intact nerves, respectively).
In the maintenance of myelin, the ability of ApoD to bind hydrophobic ligands in vitro (Vogt and Skerra, 2001) can be exploited, since lipids management is necessary for the turnover of the specialized myelin membrane. Lipid composition in the CNS is altered in the absence of ApoD (Thomas and Yao, 2007), therefore it can be expected that lipid content of PNS myelin membranes is also modulated by ApoD. Nonetheless our data support that, in addition to lipid‐management, ApoD might be exerting signaling roles in homeostatic glia‐nerve interactions that result in the panoply of molecular effects obtained.
An altered homeostasis in ApoD‐KO nerves is revealed by the high levels of proinflammatory cytokines (Il6), genes normally up‐regulated in situations of tissue remodeling (S100a4 or Galectin‐3) and stress situations (Triosephosphate isomerase), or genes promoting vasculogenesis (Vegfa). Whether this altered homeostasis could be accompanied by an impairment of the blood‐nerve barrier, as it is the case for ApoE (Fullerton et al., 2001), is not known. In either case, the molecular changes observed are monitoring the functional consequences generated by the lack of ApoD in the tissue, since there is no evidence for a direct regulation of gene expression by ApoD.
ApoD can bind arachidonic acid, an important signaling lipid which is mobilized from membranes by A2 phospholipases (Balboa and Balsinde, 2006). Notably, the expression of phospholipase Pla2g4a is elevated in intact 40‐week ApoD‐KO nerves. In agreement with our data on ApoD‐KO injured nerves, the loss of Pla2g4a function results in delayed myelin clearance by macrophages and delayed axon regeneration (De et al., 2003; Lopez‐Vales et al., 2008). Its elevated expression in intact ApoD‐KO nerves suggests both proteins might be acting in concert, controlling the process of myelin recycling in normal conditions. The lack of ApoD might trigger compensatory up‐regulation of the enzymes producing its ligand.
Interestingly, Pmp22, a Schwann cell‐specific gene causing Charcot‐Marie‐Tooth (CMT) disease type 1, is up‐regulated in the 40‐week ApoD‐KO. In keeping with our current findings, Pmp22 over‐expression leads to defects in myelination that affect mainly large diameter axons (Robaglia‐Schlupp et al., 2002). The decreased MNCV and myelin thickness in 40w ApoD‐KO nerves can therefore be mediated by the induction of demyelinating genes. This suggests that in normal conditions ApoD is antagonizing demyelination.
In summary, the absence of ApoD causes a reactive environment in intact peripheral nerves that is aggravated with age. ApoD is revealed as a key node in the gene network responsible for the correct function of peripheral nerves, including the turnover and/or repair of myelin that is required throughout life.
ApoD and the Response to Axonotmesis
Axon regeneration
The expression pattern of GAP‐43 in ApoD‐KO and HApoD‐Tg nerves suggests that ApoD promotes axonal growth in the regeneration phase after injury, which is compatible with ApoD promoting neuritogenesis in cultured dorsal root ganglion cells (Kosacka et al., 2009). However, GAP‐43 expression is ApoD‐independent at late regeneration stages, and nerve regeneration is finally attained in ApoD‐KO nerves. This suggests that ApoD is dispensable for the intrinsic process of axonal growth in vivo. Besides, our gene expression data support that ApoD intervenes in a specific set of the glia‐neuron interactions that modulate regeneration. The lack of ApoD modify the gene network after injury so that growth‐inhibiting factors (Ngfr and Alox15) increase their expression, and neurotrophic factors (Ptn) decrease their expression in response to injury.
Wallerian degeneration and preparation of a permissive environment for axonal regrowth
Four lines of evidence locate ApoD primary function in a phase earlier than axonal growth itself, particularly in the preparation of a permissive environment for growth. First, myelin clearance, monitored by MBP labeling, is delayed in the ApoD‐KO. Second, genes involved in phagocytosis and autophagy (Cd63 and Becn1), as well as extracellular proteases (Plat and Mcp1) require ApoD for a full response to injury. Third, Galectin‐3 expression is elevated in the injured ApoD‐KO nerves and decreased in the HApoD‐Tg nerves. Finally, in the absence of ApoD more macrophages and mast cells colonize the injury site.
In addition, the basal high levels of pro‐inflammatory IL6 and Vegfa in the ApoD‐KO are factors that, once the nerve is injured, contribute to a more efficient recruitment of blood born phagocytic cells. One could expect that this would lead to a faster preparation of a good environment for axonal growth. However, if this effect is accompanied by a slower than normal myelin phagocytosis, it would result in an excessive number of macrophages and a longer permanence at the injury site (reviewed by Martini et al., 2008). The fact that Galectin‐3‐KO mice show an accelerated functional recovery and faster regeneration than wild‐type animals (Narciso et al., 2009), is in agreement with our data.
On the contrary, an earlier event, the initiation of compact myelin breakdown, is not impaired by the absence of ApoD. The extent of myelin aggregates do not differ from WT at 14 d‐PCI, and the basal high levels of Pla2g4a are expected to provide the necessary levels of lysophosphatidylcholine to initiate the breakdown (De et al., 2003).
Lipid management and remyelination process
As mentioned above, after the first analysis of apolipoproteins in regenerating nerves (Boyles et al., 1990), these proteins were hypothesized to work in the process of remyelination by transporting cholesterol or cholesteryl esters for myelin synthesis. However, two apolipoproteins (ApoE and ApoA‐I) seem to be dispensable in WDR (Fullerton et al., 1998; Goodrum et al., 1995), and the possibility of functional compensation by ApoD was then proposed. Our data argue against both hypotheses: (1) No ApoD‐dependent changes are observed in lecithin‐cholesterol acyltransferase (Lcat) expression upon injury, making a mechanism through cholesterol binding unlikely. (2) ApoE induction by injury is in fact decreased in ApoD‐KO nerves, thus precluding gene compensation.
Age effects in the response to injury
WDR is influenced by age through changes in the neuronal, axonal, SC and macrophage responses (Verdu et al., 2000). Here we show that, at 40 weeks of age (representing approximately one third of the mice lifespan), the basal nerve homeostasis is altered and dependent on ApoD. Our results also show age‐dependent gene expression differences in the response to injury, which results in physiological constraints to successful nerve regeneration.
ApoD is the most consistently up‐regulated gene in the brain of old rodents and primates (Loerch et al., 2008), and it is therefore expected to be an important part of the response of the nervous system to the aging process. The present work uncovers ApoD as an important factor modulating the changes with age in the regeneration potential of the PNS. Further exploration of the requirements of ApoD in aged peripheral nerves is part of our research program.
ApoD position in the Molecular Networks Participating in WDR
Our study supports that myelin clearance is the main factor curtailed in the absence of ApoD, while axon regeneration and remyelination are delayed because of the inhibitory effects of non‐cleared myelin.
Our data also suggest novel implications for ApoD in the modulation of signaling processes that coordinate the events needed for functional recovery after PNS injury. By influencing the expression of only a subset of injury‐related genes, ApoD is exerting control over a set of specific processes. ApoD is an agonist in the signaling pathways promoting: (1) myelin removal by SCs and macrophages, phagocytosis, and autophagy, (2) extracellular matrix remodeling by proteases, and (3) induction of axonal regeneration genes. ApoD is an antagonist in the signaling pathways that control: (1) the extent and timing of proinflammatory signals, and (2) the induction of growth inhibitory and axon collapse factors.
These effects can in turn delay axonal regeneration and remyelination, and are therefore sufficient to account for all the alterations observed in our experiments.
The findings reported here support that ApoD antagonizes demyelination in normal conditions, and that it is beneficial after injury, controlling the time course of regeneration, and functional recovery. These results have important implications for many peripheral demyelinating diseases, as well as for the prognosis after traumatic injury. In addition, we have uncovered interesting gene interactions dependent on ApoD that are modulated by injury and age in peripheral nerves. Understanding these gene networks should lead to more efficient therapeutic interventions.
Acknowledgements
The authors thank J.R. Acebes, E. Martin, and A. de Campos for technical assistance, our colleagues in the lab R. Bajo‐Grañeras, M. Ruiz, A. Pérez‐Castellanos, N. García‐Mateo, and M. del Caño for their stimulating discussions, and Dr. X. Navarro for his critical comments on the manuscript. 2‐D and MALDI‐TOF analysis were performed by R.M. Dégano at the Proteomic Unit of CIC (Salamanca), funded by PROTEORED.
REFERENCES
- Al‐Shahrour F,Minguez P,Tarraga J,Medina I,Alloza E,Montaner D,Dopazo J. 2007. FatiGO +: A functional profiling tool for genomic data. Integration of functional annotation, regulatory motifs and interaction data with microarray experiments. Nucleic Acids Res 35(Web Server issue): W91–W96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balboa MA,Balsinde J. 2006. Oxidative stress and arachidonic acid mobilization. Biochim Biophys Acta 1761: 385–391. [DOI] [PubMed] [Google Scholar]
- Bosse F,Hasenpusch‐Theil K,Kury P,Muller HW. 2006. Gene expression profiling reveals that peripheral nerve regeneration is a consequence of both novel injury‐dependent and reactivated developmental processes. J Neurochem 96: 1441–1457. [DOI] [PubMed] [Google Scholar]
- Boyles JK,Notterpek LM,Anderson LJ. 1990. Accumulation of apolipoproteins in the regenerating and remyelinating mammalian peripheral nerve. Identification of apolipoprotein D, apolipoprotein A‐IV, apolipoprotein E, apolipoprotein A‐I. J Biol Chem 265: 17805–17815. [PubMed] [Google Scholar]
- Cavaggioni A,Pelosi P,Edwards S,Sawyer L. 2006. Functional aspects of β‐lactoglobulin, major urinary protein and odorant binding protein. In: Akerstrom B,Borregaard N,Flower D,Salier J, editors. Lipocalins. Georgetown, Texas: Landes Bioscience. pp 131–139. [Google Scholar]
- Chen ZL,Yu WM,Strickland S. 2007. Peripheral regeneration. Annu Rev Neurosci 30: 209–233. [DOI] [PubMed] [Google Scholar]
- De S,Trigueros MA,Kalyvas A,David S. 2003. Phospholipase A2 plays an important role in myelin breakdown and phagocytosis during Wallerian degeneration. Mol Cell Neurosci 24: 753–765. [DOI] [PubMed] [Google Scholar]
- Do Carmo S,Jacomy H,Talbot PJ,Rassart E. 2008. Neuroprotective effect of apolipoprotein D against human coronavirus OC43‐induced encephalitis in mice. J Neurosci 28: 10330–10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan RS,Jin T,Yang X,Mix E,Adem A,Zhu J. 2007. Apolipoprotein E deficiency enhances the antigen‐presenting capacity of Schwann cells. Glia 55: 772–776. [DOI] [PubMed] [Google Scholar]
- Flo TH,Smith KD,Sato S,Rodriguez DJ,Holmes MA,Strong RK,Akira S,Aderem A. 2004. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432: 917–921. [DOI] [PubMed] [Google Scholar]
- Fullerton SM,Shirman GA,Strittmatter WJ,Matthew WD. 2001. Impairment of the blood‐nerve and blood‐brain barriers in apolipoprotein E knockout mice. Exp Neurol 169: 13–22. [DOI] [PubMed] [Google Scholar]
- Fullerton SM,Strittmatter WJ,Matthew WD. 1998. Peripheral sensory nerve defects in apolipoprotein E knockout mice. Exp Neurol 153: 156–163. [DOI] [PubMed] [Google Scholar]
- Ganfornina MD,Do Carmo S,Lora JM,Torres‐Schumann S,Vogel M,Allhorn M,Gonzalez C,Bastiani MJ,Rassart E,Sanchez D. 2008. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell 7: 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina MD,Sanchez D,Bastiani MJ. 1995. Lazarillo, a new GPI‐linked surface lipocalin, is restricted to a subset of neurons in the grasshopper embryo. Development 121: 123–134. [DOI] [PubMed] [Google Scholar]
- Ganfornina MD,Sanchez D,Pagano A,Tonachini L,Descalzi‐Cancedda F,Martinez S. 2005. Molecular characterization and developmental expression pattern of the chicken apolipoprotein D gene: Implications for the evolution of vertebrate lipocalins. Dev Dyn 232: 191–199. [DOI] [PubMed] [Google Scholar]
- Goodrum JF,Bouldin TW,Zhang SH,Maeda N,Popko B. 1995. Nerve regeneration and cholesterol reutilization occur in the absence of apolipoproteins E, A‐I in mice. J Neurochem 64: 408–416. [DOI] [PubMed] [Google Scholar]
- Hull‐Thompson J,Muffat J,Sanchez D,Walker DW,Benzer S,Ganfornina MD,Jasper H. 2009. Control of metabolic homeostasis by stress signaling is mediated by the lipocalin NLaz. PLoS Genet 5: e1000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosacka J,Gericke M,Nowicki M,Kacza J,Borlak J,Spanel‐Borowski K. 2009. Apolipoproteins D, E3 exert neurotrophic and synaptogenic effects in dorsal root ganglion cell cultures. Neuroscience 162: 282–291. [DOI] [PubMed] [Google Scholar]
- Livak KJ,Schmittgen TD. 2001. Analysis of relative gene expression data using real‐time quantitative PCR, the 2(‐Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Loerch PM,Lu T,Dakin KA,Vann JM,Isaacs A,Geula C,Wang J,Pan Y,Gabuzda DH,Li C, Prolla TA, Yanker BA. 2008. Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE 3: e3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Vales R,Navarro X,Shimizu T,Baskakis C,Kokotos G,Constantinou‐Kokotou V,Stephens D,Dennis EA,David S. 2008. Intracellular phospholipase A(2) group IVA, group VIA play important roles in Wallerian degeneration and axon regeneration after peripheral nerve injury. Brain 131(Part 10): 2620–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R,Fischer S,López‐Vales R,David S. 2008. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia 56: 1566–1577. [DOI] [PubMed] [Google Scholar]
- Narciso MS,Mietto BdS,Marques SA,Soares CP,Mermelstein CdS,El‐Cheikh MC,Martinez AMB. 2009. Sciatic nerve regeneration is accelerated in galectin‐3 knockout mice. Exp Neurol 217: 7–15. [DOI] [PubMed] [Google Scholar]
- Navarro A,Del Valle E,Tolivia J. 2004. Differential expression of apolipoprotein d in human astroglial and oligodendroglial cells. J Histochem Cytochem 52: 1031–1036. [DOI] [PubMed] [Google Scholar]
- Nave KA,Trapp BD. 2008. Axon‐glial signaling and the glial support of axon function. Annu Rev Neurosci 31: 535–561. [DOI] [PubMed] [Google Scholar]
- Ong WY,Lau CP,Leong SK,Kumar U,Suresh S,Patel SC. 1999. Apolipoprotein D gene expression in the rat brain and light and electron microscopic immunocytochemistry of apolipoprotein D expression in the cerebellum of neonatal, immature and adult rats. Neuroscience 90: 913–922. [DOI] [PubMed] [Google Scholar]
- Persohn E,Canta A,Schoepfer S,Traebert M,Mueller L,Gilardini A,Galbiati S,Nicolini G,Scuteri A,Lanzani F, Giussani G, Cavaletti G. 2005. Morphological and morphometric analysis of paclitaxel and docetaxel‐induced peripheral neuropathy in rats. Eur J Cancer 41: 1460–1466. [DOI] [PubMed] [Google Scholar]
- Rassart E,Bedirian A,Do Carmo S,Guinard O,Sirois J,Terrisse L,Milne R. 2000. Apolipoprotein D. Biochim Biophys Acta 1482: 185–198. [DOI] [PubMed] [Google Scholar]
- Reichert F,Saada A,Rotshenker S. 1994. Peripheral nerve injury induces Schwann cells to express two macrophage phenotypes: Phagocytosis and the galactose‐specific lectin MAC‐2. J Neurosci 14(Part 2): 3231–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robaglia‐Schlupp A,Pizant J,Norreel JC,Passage E,Saberan‐Djoneidi D,Ansaldi JL,Vinay L,Figarella‐Branger D,Levy N,Clarac F, Cau P, Pellissier JF, Fontés M. 2002. PMP22 overexpression causes dysmyelination in mice. Brain 125(Part 10): 2213–2221. [DOI] [PubMed] [Google Scholar]
- Saada A,Reichert F,Rotshenker S. 1996. Granulocyte macrophage colony stimulating factor produced in lesioned peripheral nerves induces the up‐regulation of cell surface expression of MAC‐2 by macrophages and Schwann cells. J Cell Biol 133: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez D,Ganfornina MD,Bastiani MJ. 1995. Developmental expression of the lipocalin Lazarillo and its role in axonal pathfinding in the grasshopper embryo. Development 121: 135–147. [DOI] [PubMed] [Google Scholar]
- Sanchez D,Ganfornina MD,Martinez S. 2002. Expression pattern of the lipocalin apolipoprotein D during mouse embryogenesis. Mech Dev 110(1–2): 225–229. [DOI] [PubMed] [Google Scholar]
- Sanchez D,Ganfornina MD,Torres‐Schumann S,Speese SD,Lora JM,Bastiani MJ. 2000. Characterization of two novel lipocalins expressed in the Drosophila embryonic nervous system. Int J Dev Biol 44: 349–359. [PubMed] [Google Scholar]
- Sanchez D,Lopez‐Arias B,Torroja L,Canal I,Wang X,Bastiani MJ,Ganfornina MD. 2006. Loss of glial lazarillo, a homolog of apolipoprotein D, reduces lifespan and stress resistance in Drosophila. Curr Biol 16: 680–686. [DOI] [PubMed] [Google Scholar]
- Schaeren‐Wiemers N,Schaefer C,Valenzuela DM,Yancopoulos GD,Schwab ME. 1995. Identification of new oligodendrocyte‐ and myelin‐specific genes by a differential screening approach. J Neurochem 65: 10–22. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD,Livak KJ. 2008. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Spreyer P,Schaal H,Kuhn G,Rothe T,Unterbeck A,Olek K,Muller HW. 1990. Regeneration‐associated high level expression of apolipoprotein D mRNA in endoneurial fibroblasts of peripheral nerve. EMBO J 9: 2479–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA,Yao JK. 2007. Clozapine specifically alters the arachidonic acid pathway in mice lacking apolipoprotein D. Schizophr Res 89(1–3): 147–153. [DOI] [PubMed] [Google Scholar]
- Tolivia J,Navarro A,del Valle E,Perez C,Ordoñez C,Martinez‐Pinilla E. 2006. Application of photoshop and scion image analysis to quantification of signals in histochemistry, immunocytochemistry and hybridocytochemistry. Anal Quant Cytol Histol 28: 43–53. [PubMed] [Google Scholar]
- Tolivia J,Navarro A,Tolivia D. 1994. Polychromatic staining of epoxy semithin sections: A new and simple method. Histochemistry 101: 51–55. [DOI] [PubMed] [Google Scholar]
- Van Dijk W,Do Carmo S,Rassart E,Dalhlback B,Sodetz J. 2006. The plasma lipocalins α1‐acid glycoprotein, apolipoprotein D, apolipoprotein M, complement C8γ. In: Akerstrom B,Borregaard N,Flower D,Salier J, editors. Lipocalins. Georgetown, Texas: Landes Bioscience. pp 140–166. [Google Scholar]
- Verdu E,Ceballos D,Vilches JJ,Navarro X. 2000. Influence of aging on peripheral nerve function and regeneration. J Peripher Nerv Syst 5: 191–208. [DOI] [PubMed] [Google Scholar]
- Verheijen MHG,Chrast R,Burrola P,Lemke G. 2003. Local regulation of fat metabolism in peripheral nerves. Genes Dev 17: 2450–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt M,Skerra A. 2001. Bacterially produced apolipoprotein D binds progesterone and arachidonic acid, but not bilirubin or E‐3M2H. J Mol Recognit 14: 79–86. [DOI] [PubMed] [Google Scholar]
- Weiss MD,Luciano CA,Quarles RH. 2001. Nerve conduction abnormalities in aging mice deficient for myelin‐associated glycoprotein. Muscle Nerve 24: 1380–1387. [DOI] [PubMed] [Google Scholar]
- Yang DP,Zhang DP,Mak KS,Bonder DE,Pomeroy SL,Kim HA. 2008. Schwann cell proliferation during Wallerian degeneration is not necessary for regeneration and remyelination of the peripheral nerves: Axon‐dependent removal of newly generated Schwann cells by apoptosis. Mol Cell Neurosci 38: 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JS,Reed A,Chen F,Stewart CN Jr. 2006. Statistical analysis of real‐time PCR data. BMC Bioinformatics 7: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]