

Abstract
Summary: Host cells trigger signals for innate immune responses upon recognition of conserved structures in microbial pathogens. Nucleic acids, which are critical components for inheriting genetic information in all species including pathogens, are key structures sensed by the innate immune system. The corresponding receptors for foreign nucleic acids include members of Toll‐like receptors, RIG‐I‐like receptors, and intracellular DNA sensors. While nucleic acid recognition by these receptors is required for host defense against the pathogen, there is a potential risk to the host of self‐nucleic acids recognition, thus precipitating autoimmune and autoinflammatory diseases. In this review, we discuss the roles of nucleic acid‐sensing receptors in guarding against pathogen invasion, discriminating between self and non‐self, and contributing to autoimmunity and autoinflammatory diseases.
Keywords: TLR, RLR, DNA sensors, autoimmune disease
Introduction
Invasion of microbe to the host induces a defense response that is initiated by pattern‐recognition receptors (PRRs) (1, 2). PRRs recognize conserved structures in pathogens and are largely divided into four types based on structural homology: Toll‐like receptors (TLRs), C‐type lectin receptors (CLRs), RIG‐I‐like receptors (RLRs), and NOD‐like receptors (NLRs). In addition, intracellular DNA sensors are proposed as new types of PRRs. Each PRR induces production of proinflammatory cytokines and type I interferons (IFNs). Proinflammatory cytokines, such as interleukin‐1 (IL‐1), IL‐6, IL‐12, and tumor necrosis factor α (TNFα), induce several events including inflammation and homing and activation of adaptive immune cells, including T and B cells. Type I IFNs are composed of more than 10 members: multiple IFNα family members, IFNβ, IFNε, and IFNτ (3). Type I IFNs play a central role in antiviral responses by inducing transcription of IFN‐stimulated genes (ISGs). The ISGs include more than 1000 genes, although many remain uncharacterized. Some of these genes, such as IFN‐stimulated protein of 15 kDa (ISG15), myxovirus resistance 1 (Mx1), ribonuclease L (RNaseL), and protein kinase R (PKR), have antiviral effects, as demonstrated in mouse genetic studies (4). Antiviral states are often achieved by a combination of several ISGs, and each ISG is likely to have a specific target to suppress virus replication and budding. Recent studies have described the function of each ISG, defining its specific target pathogen, such as viperin, samhd1, or members of the gbp family (5, 6, 7).
Nucleic acids from pathogens are recognized by PRRs, including TLRs, RLRs, and cytosolic DNA sensors. RNA from RNA viruses is recognized by TLR3, TLR7, TLR8, and RLRs, and DNA from DNA viruses and microbes is recognized by TLR9 and cytosolic DNA sensors. Nucleic acids from pathogens are likely to be recognized by multiple receptors in vivo, which may cooperatively function to protect host from invasion. When infected with an RNA virus such as Sendai virus (SeV) or Newcastle disease virus (NDV), TLRs and RLRs recognize RNA in cellular compartments in different cell types and have structural preference for recognition of nucleic acids, and these receptors participated cooperatively in cytokine production (8).
Although sensing pathogenic nucleic acid is advantageous for host defense, the host species runs a risk of sensing self‐nucleic acids and producing proinflammatory cytokines and type I IFNs. Nucleic acid sensors have strict roles and mechanisms to prevent self‐nucleic acid sensing. However, recent studies have implicated that aberrant recognition of self‐nucleic acids contributes to autoimmune and autoinflammatory diseases.
Intracellular TLRs for nucleic acid recognition
TLR signaling networks
TLRs are a family of single membrane‐spanning receptors of which there are 10 in human and 12 in mouse (1, 2). TLRs have conserved homology regions, containing leucine‐rich repeats (LRRs) in the extracellular space linked by the transmembrane region to the intracellular Toll/IL‐1 receptor (TIR) domain. Specific patterns from microbial pathogens are recognized by LRRs, which are predicted to predominantly form a dimer for ligand binding. The TLRs are characterized by an ectodomain for ligand recognition (discussed in the following section), whereas the TIR domain is highly homologous, as demonstrated by structural analysis (9, 10). Activation of the TLRs by ligand binding conducts a signal from its TIR domain to the adapter protein that contains the TIR domain. The adapter protein acts mainly via two signaling pathways: the TIR domain containing the adapter inducing IFNβ (TRIF)‐dependent pathway, and the myeloid differentiation primary response protein (MyD88)‐dependent pathway. TLR3 depends on TRIF, whereas TLR7, TLR8, and TLR9 depend on MyD88 (11, 12). Both adapter proteins culminate in the activation of transcription factors, such as the nuclear factor of κ light polypeptide gene enhancer in B cells 1 (NF‐κB), IFN regulatory factor 3 (IRF3) and IRF7 for transcription of proinflammatory cytokines, and type I IFNs. Briefly, TRIF recruits TNF receptor‐associated factor 6 (TRAF6), TRAF3, TNF receptor‐associated death domain (TRADD), receptor interacting protein‐1 (RIP‐1), and transforming growth factor‐β activated kinase 1 (TAK1), whereas MyD88 recruits IRAK1, IRAK2, IRAK4, TRAF6, and TAK1. TAK1 induces activation of the canonical inhibitor of NF‐κB kinase (IKK) α/β complex, resulting in NF‐κB activation. TRAF3 leads to activation of TANK‐binding kinase 1 (TBK1) and IKKι to phosphorylate IRF3 (1, 2). In general, signaling networks are shared among the TLR family members and are utilized by the same signaling proteins. MyD88 forms the helical structure for providing signaling platform with downstream protein. Structural analysis demonstrated that MyD88 forms a well‐ordered helix with IRAK2 and IRAK4 through their death domains (13, 14) ( Fig. 1 ).
Figure 1.
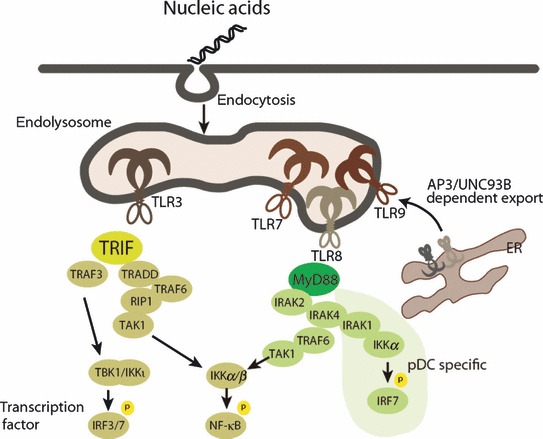
Overview of signaling pathway in nucleic acid‐sensing TLRs. Nucleic acid‐sensing TLR3, TLR7, TLR8, and TLR9 are localized in the endolysosome and recognize nucleic acids that enter the endolysosome. TLR3 signals through TRIF, whereas TLR7, TLR8, and TLR9 signal through MyD88. Each adapter molecule induces complex formation of specific downstream signaling proteins. TRIF recruits TRAF6, TRAF3, TRADD, RIP1, and TAK1, and MyD88 recruits IRAK2, IRAK4, TRAF6, and TAK1. TAK1 induces activation of canonical IKKα/β complex, resulting in NF‐κB activation. TRAF3 is recruited to TRIF and leads to activation of TBK1 and IKKι to phosphorylate IRF3.
Expression patterns of TLRs and their function in dendritic cells
TLRs are generally expressed in immune cells including monocytes, macrophages, dendritic cells (DCs), and B cells as well as non‐immune cells such as keratinocytes or epithelial cells (15). DCs play central roles that mediate innate and adaptive immune responses. TLRs involved in nucleic acid recognition are also differentially expressed in specific subsets of DCs. For example, plasmacytoid DCs (pDCs), which are a subset of DCs expressing CD11c and B220 (16, 17, 18), express high levels of TLR7 and TLR9 but not TLR3. They induce robust production of type I IFNs when activated by TLR7 or TLR9. In other types of cells, even if TLR7 and TLR9 are expressed, their stimulation generally does not cause abundant type I IFNs production but produces inflammatory cytokines. Conventional DCs (cDCs), a non‐pDC subset of DCs, also induce type I IFNs upon virus infection but mainly from RLRs (19). TLR3 and TLR8 are broadly expressed among several types of DCs and mainly participate in the production of inflammatory cytokines.
TLR7 and TLR9 stimulation induces robust production of type I IFNs by pDCs, which suggests that the signaling networks for TLR7 and TLR9 in pDCs are distinct from other TLR signaling pathways. TLR7 and TLR9 activation induces MyD88‐dependent signaling complex formation involving IRAK4, TRAF6, TRAF3, IRAK1, IKKα, and IRF7. IRF7 is phosphorylated by IRAK1 and IKKα, and translocates into the nucleus to upregulate type I IFN genes. In addition, OPNi (20), Viperin (21), PI3K‐mTOR signaling (22), and Dock2 (23) are specifically required for a robust type I IFN production in pDCs ( Fig. 1 ).
TLR activation induces DC maturation, which enhances their capacity to capture antigen and present antigen on major histocompatibility complex (MHC) to activate CD4+ or CD8+ T cells. Activation of CD4+ helper T cells by specific antigens induces the production of antibody to improve defense against the pathogen (24). Antigens that are presented on the MHC class I stimulate CD8+ T cells via a process known as cross‐presentation, which is important for elimination of virally infected cells and is facilitated by TLR3, TLR7, and TLR9 (25, 26).
Cellular location of TLRs
TLRs are divided into two subgroups according to their initial activation sites in the cells. One group, including TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11, is expressed on the plasma membrane and mainly recognizes protein, lipid, and lipoprotein in microbial membrane components. The other group includes TLR3, TLR7, TLR8, and TLR9, which recognize nucleic acids and are localized in the intracellular vesicles such as endoplasmic reticulum (ER), lysosomes, and endosome. Endosome localization of TLRs is regulated by the ER‐localized multi‐membrane spanning protein UNC93B1. DCs from mice with a single missense mutation at UNC93B1 have defects in cytokine production by TLR3, TLR7, and TLR9 ligands (26), and these mice are susceptible to the DNA virus, herpes simplex virus‐1 (HSV‐1) (27). UNC93B1 binds to the transmembrane region of TLR3, TLR7, and TLR9 and transports these TLRs from the ER to the endolysosome (28, 29). The adapter protein 3 (AP3), a well‐known trafficking protein for cargo formation, has been shown to be critical for TLR9 trafficking and type I IFN production in pDCs (30). The physiological reason for the restricted localization of TLRs is not clear, but TLR9, not TLR7, requires acidification at the endolysosome (31). Synthetic nucleotides or nucleic acids derived from dead cells are directly endocytosed and delivered to the endolysosome where they encounter with TLRs. Viruses and microbes in which the genomic nucleic acids are covered with a membrane are also endocytosed and are likely to be recognized by TLRs after the nucleic acids are exposed through mechanisms which are unknown. Viruses pretreated with ultraviolet radiation are still able to induce cytokines via nucleic acid‐sensing TLRs, suggesting that virus replication and growth of microbes are not directly linked to sensing.
Specific ligand recognition and regulation of TLRs
TLR3
TLR3 was originally identified as recognizing polyinosinic‐polycytidylic acid (polyI:C), the synthetic analog of dsRNA. TLR3 recognizes genomic dsRNA of dsRNA viruses or dsRNA that are produced during replication of ssRNA viruses or DNA viruses. TLR3‐deficient mice are susceptible to ssRNA viruses such as West Nile virus (WNV) (32), Semliki Forest virus (25), and encephalomyocarditis virus (EMCV) (33) and DNA viruses such as mouse cytomegalovirus (MCMV) (34) and HSV‐1 (35). TLR3 deficiency in humans also causes an increase in infection rates of HSV‐1 (35). The molecular mechanism of dsRNA binding to TLR3 is also revealed by structural analysis. The ectodomain is conserved through TLR2, TLR3, and TLR4. This site includes a large horseshoe‐like shape and forms a dimer with the binding pathogen, although the specific binding mechanism to the ligand differs between them. Structural analysis of the mouse TLR3 ectodomain interacting with dsRNA indicated that one end of the dsRNA of the sugar–phosphate backbone is sandwiched between two ectodomains at positively charged residues in each TLR3 (36, 37). Positively charged residues in TLR3 sites are located in two regions near the N‐ and C‐termini of the horseshoe, and the two binding sites are separated by approximately 120 Å, almost equal to approximately 45 base pairs of dsRNA in length. Supporting structural analysis, TLR3 binding to dsRNA is dependent on pH and dsRNA length. TLR3 does not bind to dsRNA at neutral pH but binds strongly at pH 6.5, which suggests that binding is mediated by a charged interaction, and requires 40–50 base pairs of dsRNA for stable binding (38).
TLR7 and TLR8
TLR7 and TLR8 recognize ssRNA from the genomes of ssRNA viruses and specific bacteria. Initial reports demonstrated that human TLR7, TLR8, and mouse TLR7 recognize imidazoquinoline derivatives such as imiquimod (R837), resiquimod (R848), and guanine analogs such as loxoribine, which have antiviral and anti‐tumor properties. TLR7 and TLR8 have sequence similarity and mostly recognize the same native pathogens, although TLR7 prefers GU‐rich RNA sequences and TLR8 prefers AU‐rich RNA sequences in human (39). In mice, sequence preference between TLR7 and TLR8 is not clearly observed. Cells derived from TLR7‐deficient mice fail to induce cytokines in response to ssRNA viruses such as influenza A virus (IAV), vesicular stomatitis virus (VSV), and human immunodeficiency virus‐1 (HIV‐1) (40, 41, 42). The preference of TLR7 for GU‐rich sequences is demonstrated by the fact that they are found in the genomes of IAV and HIV‐1 (42). In addition, TLR7 detects RNAs from bacteria such as Group B Streptococcus but not other bacteria such as Listeria monocytogenes and Group A Streptococcus. TLR7 is predominantly expressed in pDCs and involved in the robust expression of IFNα in both humans and mice, whereas TLR8 is expressed predominantly in cDCs and monocytes (43).
TLR9
TLR9 was originally discovered to recognize unmethylated 2′‐deoxyribo(cytidine‐phosphate‐guanosine) (CpG) DNA present in bacterial DNA. Later, viral DNA was also shown to be recognized by TLR9. The structure of TLR9 ectodomain with its ligand has not yet been solved. Several types of CpG DNA have been tested for their abilities to induce cytokine production by pDCs, which suggests that there is a specific recognition mechanism. TLR9 has sequence preferences but potentially recognizes a broad range of sequences, suggesting its sugar‐backbone of DNA may be the site recognized by TLR9 (44, 45). TLR9 has been shown to act as a sensor for viruses such as MCMV, HSV‐1 (46), HSV‐2 (31), and adenovirus (47) in DCs. However, the TLR9 sensing ligand in part overlaps with the cytosolic DNA sensor, as demonstrated in the response of the TLR9‐deficient mice to these viruses (46).
TLR9 forms a dimer and changes its conformation when it binds to CpG DNA (45). TLR9 and CpG DNA are likely to bind directly (45), although several proteins have been proposed as intermediate cofactors to initiate the activation. The high mobility group box (HMGB) is an intermediate protein. It is initially retained inside the cell cytosol, is secreted in response to cell damage or inflammation, and acts as a cytosolic DNA sensor. After binding to DNA, HMGB1 activates downstream DNA‐sensing receptors, including TLR9 and a cytosolic DNA sensor (48). Self‐DNA does not normally initiate DC activation, but self‐DNA and protein complexes induce autoimmune disease under certain conditions (as discussed in the following section). Another converter of self‐DNA to pathogenic ligand is LL37, which was found to bind to self‐DNA from a psoriasis patient. LL37‐DNA complex may promote the endocytosis pathway and sustain TLR9 activation by modification of the interaction with DNA (49). LL37 facilitates TLR9 activation of self‐DNA and synthetic CpG DNA. Another cofactor, graulin, which is constantly secreted from macrophages, was recently identified as another protein that binds to TLR9. Granulin is critical for production of IFNα from DCs by facilitating internalization of CpG DNA (50). In addition to the intermediate proteins, TLR9 is modified by proteases by cleavage of the ectodomain. TLR9 is initially localized in the ER as a precursor. N‐terminal cleavage by proteolytic enzymes such as cathepsin B, S, L, H, K (51), and asparagine endopeptidase (52) induces its localization change to the endosome where it attains its functional form.
Cytosolic RNA sensors
RNA virus infection in macrophages and fibroblast cells induces production of inflammatory cytokines and type I IFN through RLRs (RIG‐I, MDA5, and LGP2) ( Fig. 2 ). RIG‐I was initially identified by expression screening for IFNβ promoter activity, and MDA5 and LGP2 were later identified as family members (53). RIG‐I and MDA5 have similar domain structures, having N‐terminal tandem Caspase recruitment domains (CARDs) that are necessary for activating downstream signaling, and the DEAD box helicase/ATPase domain. LGP2, however, lacks the CARD. Cells deficient for RIG‐I or MDA5 fail to induce cytokine production when challenged with several RNA viruses or stimulated with synthetic RNA. LGP2 is dispensable for cytokine production after stimulation with synthetic RNA but is required for cytokine production following infection with certain RNA viruses that are recognized by RIG‐I and MDA5 (54). Normal response to synthetic RNA ligands in the absence of LGP2 suggests that LGP2 may modify viral RNA by removing proteins from viral ribonucleoprotein complexes that can access RNA to RIG‐I and MDA5 for recognition. RIG‐I and MDA5 may recognize the same synthetic polyI:C that is commonly used for mimicking viral RNA but may have different length preferences. The short polymers of polyI:C (approximately 70 base pairs‐long) are recognized by RIG‐I, whereas the long polymers of polyI:C (2 kilo base pairs‐long) are recognized by MDA5 (55). Separation of ligand preferences by sequence length is unsatisfactory, because common DNA and RNA binding domains recognize roughly one helical turn, namely a 5–8 amino acid sequence. In addition, binding analysis using several synthetic analogs such as the 5′‐phosphorylated RNA or the blunt ended dsRNA suggests that the specific structure of the 5′ end is critical for binding to the C‐terminal domain of RIG‐I and MDA5. Consistent with its ligand preference, structural analysis indicated that the acidic surface of C‐terminal binding site in RIG‐I is important for recognition of the 5′ end of the RNA sequence (56). The C‐terminal region of MDA5 has a similar overall structure to that of RIG‐I (57, 58), which does not fully explain the ligand preferences. Although the molecular mechanism for discrimination of RNA recognition between RIG‐I and MDA5 is not fully clear, the receptor responsible for recognition of each virus was identified among fibroblast cells isolated from the knockout mouse. Viruses can be separated into three groups: those specifically sensitive to RIG‐I, those specifically sensitive to MDA5, and those sensitive to both RIG‐I and MDA5. The NDV, SeV, VSV, influenza virus, and Japanese encephalitis virus are specifically recognized by RIG‐I. Piconaviruses including EMCV, Mengo virus, and Theiler’s virus are recognized by MDA5. Dengue virus and WNV are recognized by both RIG‐I and MDA5 (19, 59, 60, 61, 62).
Figure 2.
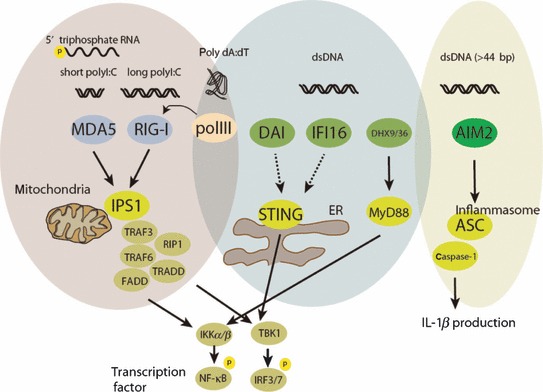
Signaling scheme of RLRs and cytosolic DNA sensors. RNA from RNA viruses is recognized by RIG‐I and MDA5. RIG‐I and MDA5 form a complex with an adapter IPS‐1, which is located in the mitochondria. IPS‐1 induces the assembly of downstream signaling proteins: TRAF3/6, caspase‐8/10, RIP1, FADD, and TRADD. This complex further induces activation of NF‐κB and IRF3 through IKKα/β and TBK1/IKKι, leading to production of inflammatory cytokines and type I IFNs, respectively. DNA from DNA viruses or bacteria causes cytokine production via several pathways. AIM2 forms an inflammasome along with ASC and Caspase‐1 and produces IL‐1β and IL‐18. Poly (dA:dT) that is transcribed to RNA via RNA pol II is recognized by RIG‐I. DAI and IFI16 may signal through STING, whereas DHX9 and DHX36 signal through MyD88. STING is localized in the ER or mitochondria and may change distribution during activation, leading to TBK1/IKKι‐dependent IRF3 activation.
RIG‐I also mediates signaling from a particular DNA sequence of poly(dA:dT) repeating adenosine and thymine residues of around approximately 100 base pairs, which have experimentally been used as a synthetic IFNβ‐inducing ligand. RNA is synthesized from DNA by RNA polymerase (pol) III that uses poly(dA:dT) as a template, which can be detected by RIG‐I (63, 64, 65). The physiological implications of the pol III‐dependent RIG‐I sensing are not clear, as DNA sensing proteins are independently proposed as sensors for natural DNA viral and bacterial pathogens (discussed in the next section).
Type I IFNs establish an antiviral state by inducing ISGs. In addition, recent genome wide screening for antiviral effector genes indicated that RIG‐I and MDA5, but not LGP2, function as potent antiviral effector genes against several viruses such as hepatitis C virus and WNV (66). Although they also have an indirect effect by inducing other ISGs through type I IFN production, RIG‐I and MDA5 may directly interfere with viral replication or budding by interaction to the viral genome RNA. As supporting mechanism of antivirus effect, it has been suggested that RIG‐I binds directly to the full length of the viral genomic RNA at the replication step of influenza and SeV (59).
Discrimination between self and non‐self RNA is performed at the 5′ end of RNA. mRNA in mammalian cells usually has a 5′ cap structure methylated at the N7 position of the capping guanosine residue and the ribose‐2′‐O position of the 5′ penultimate residue. The capping structure in self‐mRNA is likely to be critical in distinguishing host RNA from viral RNA, but many viruses replicating in the cytoplasm develop other types of modification at the 5′ end (67). They may also contain 5′ end modification enzymes such as RNA 5′ triphosphatase, RNA guanyltranferase, RNA N7‐metyltransferase, or 2′‐O‐methyltransferase, which are homologous to the mammalian enzymes. A recent study clearly demonstrated the importance of one modification, 2′‐O‐methyltransferation in viral RNA. Coronavirus mutants lacking 2′‐O‐methyltransferase activity induce higher type I IFN production than viruses with 2′‐O‐methyltransferase activity through MDA5, suggesting that this RNA modification at the 5′ end is important for evasion of innate immune recognition (67). MDA5 is likely to recognize viral RNA at the replication step before genomic RNA is masked by the 5′ end modification enzyme. To support this speculation, RIG‐I was shown to recognize the viral genome at the replication step alongside the competing virus capping protein. Other RNA viruses such as influenza virus do not have a 5′‐phosphatase, but instead the 5′ end in genomic RNA is capped by the flu polymerase (68, 69). The RNA in the influenza genome is capped at the 5′ end and this mechanism probably protects it from RIG‐I interaction (59).
RLR signaling
Recognition of RNA induces a conformational change in the RIG‐I. E3 ubiquitin ligase tripartite motif protein 25 (TRIM25) conjugates Lys‐63‐linked ubiquitin to RIG‐I, enabling it to interact with an adapter protein IPS‐1 (also known as MAVS, Cardif, and VISA) on mitochondria (70). Formation of RIG‐I and IPS‐1 complexes induces the assembly of protein complexes to initiate downstream signaling (71, 72, 73). IPS‐1 binds to TRAF3/6, caspase‐8/10, RIP1, FAS‐associated death domain (FADD), and the TRADD (74, 75, 76). This complex induces activation of IKKα/β followed by NF‐κB activation, and TBK1/IKKι that induce IRF3 activation, leading to production of inflammatory cytokines and type I IFNs, respectively (77, 78). The physiological role of the localization of IPS‐1 in the mitochondrion is not yet clear, but several mitochondrial regulatory proteins are involved in signaling (79). NLRX1, an NLR family member localized on the mitochondrial membrane is an inhibitor of IPS‐1 signaling (80, 81). Mitofusin 2, which is a regulator of mitochondrial fusion, suppresses IPS‐1 signaling by direct interaction (82, 83). The importance of mitochondrial regulation is indirectly supported by experiments in mice that lack autophagy. It has been shown that ATG5‐ or ATG16L1‐deficient cells accumulate damaged mitochondria with IPS‐1 and induce robust type I IFN induction (84, 85) ( Fig. 2 ).
Cytosolic DNA sensors
Cytosolic DNA sensor for IL‐1β production
DNA viruses, intracellular bacteria, and parasites are recognized by DNA sensors. Absent in melanoma 2 (AIM2) is indispensable for cytoplasmic recognition of DNA and production of IL‐1β but not IFNβ (86, 87, 88, 89) ( Fig. 2 ). IL‐1β is produced following activation of inflammasome, a multiprotein complex that activates caspase‐1 with the subsequent cleavage of pro‐IL‐1β and pro‐IL‐18 and release of mature IL‐1β and IL‐18 (90). AIM2 has the HIN2000 DNA‐binding domain and the pyrin domain that interacts with an adapter protein ASC. Single‐stranded CpG DNA as short as six bases is sufficient for TLR9 recognition, whereas activation of AIM2 requires DNA at least 44 base pairs in length, suggesting that oligomer formation by binding through the HIN2000 domain onto longer DNA is necessary for AIM2 inflammasome activation (91). The preference for longer DNA for cluster formation is demonstrated by the fact that AIM2 and DNA form a puncta structure within the cells. AIM2‐deficient cells fail to produce IL‐1β in response to viral DNA and certain bacterial DNA that is released into the cytoplasm. AIM2‐deficient mice display reduced survival following infection with Gram‐negative bacteria Francisella tularensis and MCMV (92, 93).
Cytosolic DNA sensors for type I IFN production
Three DNA‐sensing proteins responsible for type I IFNs induction are proposed. DAI (DNA‐dependent activator of IFN‐regulatory factor 1), was the first candidate demonstrated as a DNA sensor (94), but type I IFN production by cytoplasmic DNA stimulation was comparable between control and DAI‐deficient fibroblast cells (95). Further analysis showed DAI knockdown in L929 cells, but not MEF cells, suppressed INFβ production after synthetic DNA stimulation, suggesting a cell type specific role of DAI (96). Secondly, IFI16, a member of HIN2000 domain‐containing protein, was also proposed as an intracellular DNA sensor (97). IFNβ production was suppressed by knockdown of IFI16 in Raw 264.7 cells after synthetic DNA transfection and HSV‐1 infection. Thirdly, DExD/H–box helicase 36 (DHX36) and DHX9 have been proposed as cytosolic CpG DNA sensors in pDCs (98). DHX36 and DHX9 have a helicase domain for recognition of DNA and have been shown to participate in MyD88‐dependent signaling. DHX36 and DHX9 have sequence preferences for binding, and knockdown of these proteins suppressed cytokine production by infection with DNA HSV‐1 virus. These studies support the suggestion that cytosolic DNA sensors are probably not single molecules and function in different cell types.
Induction of type I IFN by cytoplasmic DNA depends on TBK1‐IRF3 or STING (also known as MITA, MPYS, and ERIS) (99, 100, 101). TBK1 was found to phosphorylate IRF3 and IRF7 after virus infection (102). STING was identified as a signaling molecule for both RLRs and a DNA sensor signaling pathway, but further analysis using STING‐deficient cells demonstrated that STING predominantly participates in a DNA sensing pathway (103). STING is a multipass membrane protein localized in the ER or in the Golgi membrane, and is distributed from the ER to the perinuclear region, where it is exported to unknown organelles by forming puncta structures along with TBK1 after DNA stimulation (103, 104). Several proteins that modulate STING function, such as RNF5 (105) and TRIM56 (106), have been identified. STING deficiency causes complete abrogation of type I IFNs production in both DCs and fibroblasts, suggesting that DNA sensing pathways are converged to STING.
Endogenous ligands for activation of innate immunity
Autoimmune and autoinflammatory diseases
Innate immune signaling plays an important role in protection from pathogens but has the potential for the development of autoimmune or autoinflammatory diseases by inducing innate immune activation. Autoinflammatory patients often have high proinflammatory cytokine levels, which may come from activation of innate immunity. Involvement of TLRs in autoimmune diseases such as systemic lupus erythematosus (SLE) has been demonstrated by experimental models (107, 108). In experimental models, TLR ligands are commonly used as adjuvants to generate organ‐specific autoimmune diseases such as arthritis and encephalitis in mice. Moreover, mice with deficiency of negative regulators for TLR signals, such as SHP1 (109), A20 (110), TANK (111), and Zc3h12a (112), spontaneously develop autoimmune diseases by aberrant production of inflammatory cytokines and type I IFNs.
Intermediate self‐nucleic acid recognition by TLR7 and TLR9
Nucleic acid‐sensing TLRs do not have strong mechanisms for preventing self‐nucleic acid recognition. However, structural differences, such as the high levels of unmethylated CpG motifs in viral DNA for TLR9 and clusters of U or GU‐rich sequences in viral RNA for TLR7, have been considered key factors in the discrimination between self and non‐self nucleic acids. In addition to sequence preference, all nucleic acid‐sensing TLRs are localized in the endosome and the specific localization in the cells clearly protects them from access to self‐nucleic acid (113, 114). Supporting these findings, several DNA‐binding proteins are proposed to facilitate the development of autoimmune and autoinflammatory diseases. LL37 was implicated in mediating TLR9 and TLR7 responses to self‐nucleic acids in psoriasis, a common chronic inflammatory disease in the skin (49, 115). LL37 was isolated from psoriasis patient samples accompanied by high level of IFNα in pDCs. LL37 binds to self‐DNA and ‐RNA, and protects it from degradation by DNase and facilitates internalization. HMBG1 is another protein that facilitates entry of self‐nucleic acids into cells by forming complexes. Initial reports have demonstrated that extracellular DNA released from dying cells in these complexes is recognized by TLR9 (116, 117). Several mechanisms for internalization process are proposed. One report demonstrated that HMGB1 associates with receptor for advanced glycosylation end products (RAGE), inducing endocytosis, and these binding complexes activate TLR9 in the endosome (117). Another report demonstrated that HMGB1 and the related family members HMGB2 and HMGB3 bind to DNA or RNA and activate TLRs and cytosolic nucleic sensors, capturing both self and non‐self nucleic acids to present them to sensors (48). In systematic autoimmune diseases such as SLE, scleroderma, and Sjögren’s syndrome, antibodies against self‐DNA complexed with DNA from dead cells are internalized by FcγRII on B cells and DCs (118). The antibody complexes activate TLRs or cytosolic DNA sensors to produce an excess of type I IFNs, which precipitates the development and progression of systemic autoimmune diseases ( Fig. 3 ).
Figure 3.
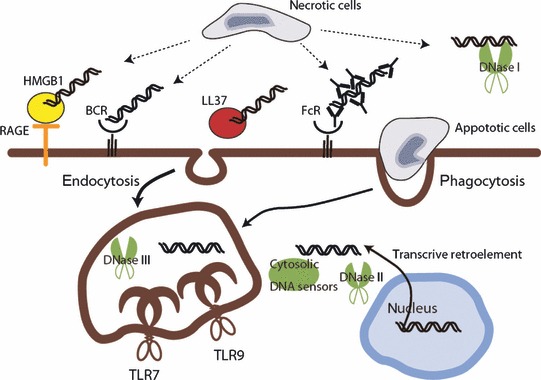
Self‐nucleic acid recognition mechanisms and degradation of DNA. Self‐nucleic acids, which are produced from necrotic or apoptotic cells, are internalized via several pathways and induce innate immune responses. Extracellular DNA is degraded by DNase I, but some of this escapes from degradation by binding to intermediate proteins, such as autoantibodies, LL37, or HMGB1. DNA–protein complexes facilitate internalization through the endocytosis pathway by binding to specific mediators. DNase II plays a role similar to DNase I after self‐DNA internalization or degradation of DNA derived from phagocytosed apoptotic cells in the macrophage. DNase III degrades intracellular DNA derived from the retro element from genomic DNA, which is likely to prevent activation of the cytosolic DNA sensor.
SLE and nucleic acid‐sensing TLRs
SLE is a chronic inflammatory systemic autoimmune disease which affects the skin, joints, kidneys, lungs, nervous system, and other organs. Mice which have the homozygous LPR mutation (lpr/lpr) does not express the functional death receptor (FAS) and develop an SLE‐like disease (118). The lpr/lpr mice produce antibodies against self‐DNA or nucleic acid‐associated protein, and self‐antibody production is commonly used for evaluating the degree of the SLE phenotype. Initial study demonstrated a critical link between innate immunity and systemic autoimmunity in B cells (107). Consistent with the initial finding, lpr/lpr mice deficient in MyD88 do not produce autoantibody (108), but the contribution of TLR7 or TLR9 is controversial. TLR7 deficiency in lpr/lpr mice reduced the level of autoantibodies against RNA and RNA‐binding proteins and increased mouse survival. However, TLR9 deficiency in lpr/lpr mice also decreased autoantibody production against self‐DNA, facilitating disease level and increasing survival (119). Another SLE‐like disease mouse model, involving mutation of Ali5 that increases the function of phospholipase‐Cγ2, also aggravated disease and increased production of self‐antibody, when crossed with TLR9‐deficient mice (120). The reason for unexpected difference in TLR9 is not clear, but one possible explanation is that the cytosolic DNA sensor is redundantly contributing to the production of self‐antibody. Recently DHX36 and DHX9 have been proposed as cytosolic DNA sensors that mediate a signal though MyD88 in the helicase domain (98). This function is supported by the demonstration of MyD88 dependency for self‐antibody production in lpr/lpr mice. The TLR7‐dependent signal might also be contributing to TLR9 activation in the lpr/lpr mice model (121). More direct evidence for the contribution of the TLR signal in SLE has been revealed by the discovery of a Y chromosome‐linked autoimmune accelerator (Yaa) mutation, causing an SLE‐like disease in mice. The Yaa mutation results from translocation of a 4 megabase translocation of the X chromosome to the Y chromosome, leading to a twofold increase in genes in the region containing TLR7 gene (122, 123). A later study shows that overexpression of TLR7 is sufficient for inducing an SLE‐like disease (124).
To stimulate adaptive immunity by self‐DNA‐antibody, it is necessary for the antibody complexes to be internalized for activation of TLR7 or TLR9. Aberrant expression of type I IFNs is correlated with disease severity in SLE (118). Several processes for internalization of DNA–antibody complexes have been proposed for the activation of TLRs. One major activation process is internalization through FcγRIII or FcγRIIα on the DCs, which are the main source of type I IFNs to develop disease in lpr/lpr mice (125, 126). The Fc region of DNA–antibody complexes is recognized by FcγR and internalized DNA is recognized by TLR9 in the endosomes. Blocking peptides specific to the FcγR did not stimulate pDCs by immune complex from an SLE patient (127). The alternative process is internalization through the BCR‐mediated endocytosis pathway. B cells expressing an antigen receptor specific for self‐IgG internalize DNA–IgG2α–chromatin immune complexes by synergistic engagement of the antigen receptor (107) ( Fig. 3 ).
SLE and other autoimmune diseases can be treated with glucocorticoids administrated orally on a daily basis. Glucocorticoids have strong anti‐inflammatory effects by inhibiting NF‐κB activity (128), but it is not clear why SLE patients commonly need higher doses of glucocorticoids than patients with other autoimmune disease. Furthermore, oral administration of glucocorticoids does not affect IFNα production. Recent findings show that nucleic acid‐containing immune complexes activate NF‐κB through TLR7 or TLR9, and activation of NF‐κB is suppressed by glucocorticoid treatment (129). This finding supports the importance of TLR activation in development of SLE. Inhibition of TLR signaling may provide an effective control for SLE in the future.
Clearance of self‐ligand
Autoimmune and autoinflammatory diseases are potentially induced by inappropriate clearance of self‐nucleic acids ( Fig. 3 ). Although the relationship between self‐nucleic acid recognition and disease onset in each autoimmune disease is difficult to establish from clinical evidence, several mouse models that display defects in self‐nucleic acid clearance show an autoimmune disease‐like phenotype. There are three types of mammalian DNases that mediate the degradation of self/non‐self DNA: DNase I, DNase II, and DNase III (TREX). DNase I is mainly present in serum and degrades extracellular dsDNA into tri‐ or tetra‐oligonucleotides (130). DNase I‐deficient mice develop glomerulonephritis, a feature of SLE. In humans, mutations in the dnase I gene are associated with SLE, and low DNase I activity is correlated with glomerulonephritis in patients (131). DNase II is present in the lysosomes of macrophages and is important for degradation of DNA derived from phagocytosed apoptotic cells. DNase II‐deficient mice induce accumulation of undigested DNA in macrophages, and the resulting production of IFNβ and TNF causes embryonic death (132). Embryonic lethality is rescued by type I IFN receptor deficiency (133), but the mouse still suffers arthritis by production of other proinflammatory cytokines such as TNFα, IL‐1β, and IL‐6. DNase III (Trex1), which is a 3′ repair exonuclease, is localized in the cytosol, and is involved in clearance of cytosolic DNA from the retro element from self‐genome DNA (134). DNase III‐deficient mice increase production of inflammatory cytokines and type I IFNs probably through activation of an intracellular DNA sensor. DNA intrinsically accumulated in the cells by dysregulation of clearance is related to autoimmunity and Aicardi‐Goutieres syndrome in humans (135, 136). However, a sensor for accumulated DNA by dysregulation of the relevant DNases has not been demonstrated. DNA derived from DNase II‐deficient mice produce cytokines independently on MyD88 and TRIF (137). As with self‐DNA clearance, dysregulation of self‐RNA clearance by mutations in SAMHD1, an RNase H2 subunit, is also associated with Aicardi‐Goutieres syndrome (138). The receptors responsible for self‐RNA recognition in this situation remain unidentified.
Link between nucleic sensing and other autoimmune disease
The contribution of TLR7 and TLR9 to autoimmune skin inflammation has been studied recently in a mouse model in which cutaneous injury by tape stripping leads to rapid inflammation and activation of pDCs (139, 140). pDCs infiltrate the injured skin and produce type I IFNs through MyD88‐dependent signaling.
Conclusion and perspective
In this review, we summarized the outcome of recognition of nucleic acids by TLRs, RLRs, and cytosolic DNA sensors. Nucleic acids from pathogens cause production of cytokines and type I IFNs to protect from invasion. Aberrant recognition of self‐nucleic acid also causes similar responses against pathogen but may induce autoimmune diseases. TLR7 and TLR9 are involved in autoimmune diseases, but contributions of cytosolic sensors such as RLRs and DNA sensors to the development of autoimmune diseases remains unclear. After the discovery of TLRs, knowledge about sensing mechanisms, signaling schemes, types of sensors, or target pathogens has greatly accumulated, but we still do not know mechanisms by which TLR‐mediated signaling pathways are aberrantly activated in other immune diseases. A combination of clinical and experimental approaches will be important for resolving many of the current questions, and we expect that accumulation of knowledge in innate immunity will help in the treatment and management of immune disease in the future.
Acknowledgement
The authors have no conflicts of interest to declare.
References
- 1. Kawai T, Akira S. Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011;34:637–650. [DOI] [PubMed] [Google Scholar]
- 2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010;140:805–820. [DOI] [PubMed] [Google Scholar]
- 3. Roberts RM, Liu L, Guo Q, Leaman D, Bixby J. The evolution of the type I interferons. J Interferon Cytokine Res 1998;18:805–816. [DOI] [PubMed] [Google Scholar]
- 4. Sadler AJ, Williams BR. Interferon‐inducible antiviral effectors. Nat Rev Immunol 2008;8:559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seo JY, Yaneva R, Hinson ER, Cresswell P. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 2011;332:1093–1097. [DOI] [PubMed] [Google Scholar]
- 6. Laguette N, et al. SAMHD1 is the dendritic‐ and myeloid‐cell‐specific HIV‐1 restriction factor counteracted by Vpx. Nature 2011;474:654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN‐gamma‐inducible 65‐kD GTPases protects against bacterial infection. Science 2011;332:717–721. [DOI] [PubMed] [Google Scholar]
- 8. Kumagai Y, et al. Alveolar macrophages are the primary interferon‐alpha producer in pulmonary infection with RNA viruses. Immunity 2007;27:240–252. [DOI] [PubMed] [Google Scholar]
- 9. Xu Y, et al. Structural basis for signal transduction by the Toll/interleukin‐1 receptor domains. Nature 2000;408:111–115. [DOI] [PubMed] [Google Scholar]
- 10. Nyman T, Stenmark P, Flodin S, Johansson I, Hammarstrom M, Nordlund P. The crystal structure of the human toll‐like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem 2008;283: 11861–11865. [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto M, et al. Role of adaptor TRIF in the MyD88‐independent toll‐like receptor signaling pathway. Science 2003;301:640–643. [DOI] [PubMed] [Google Scholar]
- 12. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783–801. [DOI] [PubMed] [Google Scholar]
- 13. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88‐IRAK4‐IRAK2 complex in TLR/IL‐1R signalling. Nature 2010; 465:885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Motshwene PG, et al. An oligomeric signaling platform formed by the Toll‐like receptor signal transducers MyD88 and IRAK‐4. J Biol Chem 2009;284:25404–25411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Novak N, Koch S, Allam JP, Bieber T. Dendritic cells: bridging innate and adaptive immunity in atopic dermatitis. J Allergy Clin Immunol 2010;125:50–59. [DOI] [PubMed] [Google Scholar]
- 16. Asselin‐Paturel C, et al. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med 2005;201:1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bjorck P. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte‐macrophage colony‐stimulating factor‐treated mice. Blood 2001;98:3520–3526. [DOI] [PubMed] [Google Scholar]
- 18. Nakano H, Yanagita M, Gunn MD. CD11c(+)B220(+)Gr‐1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med 2001;194:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato H, et al. Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature 2006;441:101–105. [DOI] [PubMed] [Google Scholar]
- 20. Shinohara ML, et al. Osteopontin expression is essential for interferon‐alpha production by plasmacytoid dendritic cells. Nat Immunol 2006;7:498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saitoh T, et al. Antiviral protein Viperin promotes Toll‐like receptor 7‐ and Toll‐like receptor 9‐mediated type I interferon production in plasmacytoid dendritic cells. Immunity 2011;34:352–363. [DOI] [PubMed] [Google Scholar]
- 22. Cao W, et al. Toll‐like receptor‐mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin‐sensitive PI(3)K‐mTOR‐p70S6K pathway. Nat Immunol 2008;9:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gotoh K, et al. Selective control of type I IFN induction by the Rac activator DOCK2 during TLR‐mediated plasmacytoid dendritic cell activation. J Exp Med 2010;207:721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurts C, Robinson BW, Knolle PA. Cross‐priming in health and disease. Nat Rev Immunol 2010;10:403–414. [DOI] [PubMed] [Google Scholar]
- 25. Schulz O, et al. Toll‐like receptor 3 promotes cross‐priming to virus‐infected cells. Nature 2005;433:887–892. [DOI] [PubMed] [Google Scholar]
- 26. Tabeta K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll‐like receptors 3, 7 and 9. Nat Immunol 2006;7:156–164. [DOI] [PubMed] [Google Scholar]
- 27. Casrouge A, et al. Herpes simplex virus encephalitis in human UNC‐93B deficiency. Science 2006;314:308–312. [DOI] [PubMed] [Google Scholar]
- 28. Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide‐sensing toll‐like receptors to endolysosomes. Nature 2008;452:234–238. [DOI] [PubMed] [Google Scholar]
- 29. Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol 2007;177:265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll‐like receptor 9 signaling by adaptor protein 3. Science 2010;329:1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll‐like receptor 9‐mediated recognition of Herpes simplex virus‐2 by plasmacytoid dendritic cells. J Exp Med 2003;198:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Daffis S, Samuel MA, Suthar MS, Gale M Jr, Diamond MS. Toll‐like receptor 3 has a protective role against West Nile virus infection. J Virol 2008;82:10349–13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hardarson HS, et al. Toll‐like receptor 3 is an essential component of the innate stress response in virus‐induced cardiac injury. Am J Physiol Heart Circ Physiol 2007;292: H251–H258. [DOI] [PubMed] [Google Scholar]
- 34. Tabeta K, et al. Toll‐like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA 2004;101:3516–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang SY, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007;317:1522–1527. [DOI] [PubMed] [Google Scholar]
- 36. Liu L, et al. Structural basis of toll‐like receptor 3 signaling with double‐stranded RNA. Science 2008;320:379–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Choe J, Kelker MS, Wilson IA. Crystal structure of human toll‐like receptor 3 (TLR3) ectodomain. Science 2005;309:581–585. [DOI] [PubMed] [Google Scholar]
- 38. Botos I, Liu L, Wang Y, Segal DM, Davies DR. The toll‐like receptor 3: dsRNA signaling complex. Biochim Biophys Acta 2009;1789:667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forsbach A, et al. Identification of RNA sequence motifs stimulating sequence‐specific TLR8‐dependent immune responses. J Immunol 2008;180:3729–3738. [DOI] [PubMed] [Google Scholar]
- 40. Lund JM, et al. Recognition of single‐stranded RNA viruses by Toll‐like receptor 7. Proc Natl Acad Sci USA 2004;101:5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hemmi H, et al. Small anti‐viral compounds activate immune cells via the TLR7 MyD88‐dependent signaling pathway. Nat Immunol 2002;3:196–200. [DOI] [PubMed] [Google Scholar]
- 42. Heil F, et al. Species‐specific recognition of single‐stranded RNA via toll‐like receptor 7 and 8. Science 2004;303:1526–1529. [DOI] [PubMed] [Google Scholar]
- 43. Mancuso G, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol 2009;10:587–594. [DOI] [PubMed] [Google Scholar]
- 44. Vollmer J, et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol 2004;34:251–262. [DOI] [PubMed] [Google Scholar]
- 45. Latz E, et al. Ligand‐induced conformational changes allosterically activate Toll‐like receptor 9. Nat Immunol 2007;8:772–779. [DOI] [PubMed] [Google Scholar]
- 46. Hochrein H, et al. Herpes simplex virus type‐1 induces IFN‐alpha production via Toll‐like receptor 9‐dependent and ‐independent pathways. Proc Natl Acad Sci USA 2004;101:11416–11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu J, Huang X, Yang Y. Innate immune response to adenoviral vectors is mediated by both Toll‐like receptor‐dependent and ‐independent pathways. J Virol 2007;81:3170–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yanai H, et al. HMGB proteins function as universal sentinels for nucleic‐acid‐mediated innate immune responses. Nature 2009;462:99–103. [DOI] [PubMed] [Google Scholar]
- 49. Lande R, et al. Plasmacytoid dendritic cells sense self‐DNA coupled with antimicrobial peptide. Nature 2007;449:564–569. [DOI] [PubMed] [Google Scholar]
- 50. Park B, et al. Granulin is a soluble cofactor for Toll‐like receptor 9 signaling. Immunity 2011;34:505–513. [DOI] [PubMed] [Google Scholar]
- 51. Ewald SE, et al. The ectodomain of Toll‐like receptor 9 is cleaved to generate a functional receptor. Nature 2008;456:658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sepulveda FE, et al. Critical role for asparagine endopeptidase in endocytic Toll‐like receptor signaling in dendritic cells. Immunity 2009;31:737–748. [DOI] [PubMed] [Google Scholar]
- 53. Yoneyama M, et al. The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 2004;5:730–737. [DOI] [PubMed] [Google Scholar]
- 54. Satoh T, et al. LGP2 is a positive regulator of RIG‐I‐ and MDA5‐mediated antiviral responses. Proc Natl Acad Sci USA 2010;107:1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kato H, et al. Length‐dependent recognition of double‐stranded ribonucleic acids by retinoic acid‐inducible gene‐I and melanoma differentiation‐associated gene 5. J Exp Med 2008;205:1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu C, et al. The structural basis of 5′ triphosphate double‐stranded RNA recognition by RIG‐I C‐terminal domain. Structure 2010;18:1032–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Takahasi K, et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C‐terminal domains: identification of the RNA recognition loop in RIG‐I‐like receptors. J Biol Chem 2009;284:17465–17474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takahasi K, et al. Nonself RNA‐sensing mechanism of RIG‐I helicase and activation of antiviral immune responses. Mol Cell 2008;29:428–440. [DOI] [PubMed] [Google Scholar]
- 59. Rehwinkel J, et al. RIG‐I detects viral genomic RNA during negative‐strand RNA virus infection. Cell 2010;140:397–408. [DOI] [PubMed] [Google Scholar]
- 60. Gitlin L, et al. Essential role of mda‐5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA 2006;103:8459–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M Jr. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG‐I and MDA5 signaling through IPS‐1. J Virol 2008;82:609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loo YM, et al. Distinct RIG‐I and MDA5 signaling by RNA viruses in innate immunity. J Virol 2008;82:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG‐I‐dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III‐transcribed RNA intermediate. Nat Immunol 2009;10:1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Choi MK, et al. A selective contribution of the RIG‐I‐like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc Natl Acad Sci USA 2009;106:17870–17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG‐I pathway. Cell 2009;138:576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011;472:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zust R, et al. Ribose 2′‐O‐methylation provides a molecular signature for the distinction of self and non‐self mRNA dependent on the RNA sensor Mda5. Nat Immunol 2011;12:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fodor E, et al. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol 2002;76:8989–9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tiley LS, Hagen M, Matthews JT, Krystal M. Sequence‐specific binding of the influenza virus RNA polymerase to sequences located at the 5′ ends of the viral RNAs. J Virol 1994;68:5108–5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gack MU, et al. TRIM25 RING‐finger E3 ubiquitin ligase is essential for RIG‐I‐mediated antiviral activity. Nature 2007;446:916–920. [DOI] [PubMed] [Google Scholar]
- 71. Schmidt A, et al. 5′‐triphosphate RNA requires base‐paired structures to activate antiviral signaling via RIG‐I. Proc Natl Acad Sci USA 2009;106:12067–12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cui S, et al. The C‐terminal regulatory domain is the RNA 5′‐triphosphate sensor of RIG‐I. Mol Cell 2008;29:169–179. [DOI] [PubMed] [Google Scholar]
- 73. Myong S, et al. Cytosolic viral sensor RIG‐I is a 5′‐triphosphate‐dependent translocase on double‐stranded RNA. Science 2009;323:1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus‐triggered IFN‐beta signaling. Mol Cell 2005;19:727–740. [DOI] [PubMed] [Google Scholar]
- 75. Balachandran S, Thomas E, Barber GN. A FADD‐dependent innate immune mechanism in mammalian cells. Nature 2004;432:401–405. [DOI] [PubMed] [Google Scholar]
- 76. Michallet MC, et al. TRADD protein is an essential component of the RIG‐like helicase antiviral pathway. Immunity 2008;28:651–661. [DOI] [PubMed] [Google Scholar]
- 77. Hemmi H, et al. The roles of two IkappaB kinase‐related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 2004;199:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN‐regulatory factor 3‐dependent gene expression is defective in Tbk1‐deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA 2004;101:233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell 2005;122:669–682. [DOI] [PubMed] [Google Scholar]
- 80. Moore CB, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008;451:573–577. [DOI] [PubMed] [Google Scholar]
- 81. Tattoli I, et al. NLRX1 is a mitochondrial NOD‐like receptor that amplifies NF‐kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep 2008;9:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG‐I‐like receptor antiviral pathway. EMBO Rep 2010;11:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yasukawa K, et al. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal 2009;2:ra47. [DOI] [PubMed] [Google Scholar]
- 84. Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature 2008;456:264–268. [DOI] [PubMed] [Google Scholar]
- 85. Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008;456:259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Burckstummer T, et al. An orthogonal proteomic‐genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 2009;10:266–272. [DOI] [PubMed] [Google Scholar]
- 87. Fernandes‐Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009;458:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hornung V, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 2009;458:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Roberts TL, et al. HIN‐200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009;323:1057–1060. [DOI] [PubMed] [Google Scholar]
- 90. Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 2007;7:31–40. [DOI] [PubMed] [Google Scholar]
- 91. Muruve DA, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008;452:103–107. [DOI] [PubMed] [Google Scholar]
- 92. Fernandes‐Alnemri T, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis . Nat Immunol 2010;11:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rathinam VA, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 2010;11:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Takaoka A, et al. DAI (DLM‐1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007;448:501–505. [DOI] [PubMed] [Google Scholar]
- 95. Ishii KJ, et al. TANK‐binding kinase‐1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008;451:725–729. [DOI] [PubMed] [Google Scholar]
- 96. Wang Z, et al. Regulation of innate immune responses by DAI (DLM‐1/ZBP1) and other DNA‐sensing molecules. Proc Natl Acad Sci USA 2008;105:5477–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Unterholzner L, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 2010;11:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kim T, et al. Aspartate‐glutamate‐alanine‐histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc Natl Acad Sci USA 2010;107:15181–15186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008;455:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sun W, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 2009;106:8653–8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhong B, et al. The adaptor protein MITA links virus‐sensing receptors to IRF3 transcription factor activation. Immunity 2008;29:538–550. [DOI] [PubMed] [Google Scholar]
- 102. Ishii KJ, et al. A Toll‐like receptor‐independent antiviral response induced by double‐stranded B‐form DNA. Nat Immunol 2006;7:40–48. [DOI] [PubMed] [Google Scholar]
- 103. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA‐mediated, type I interferon‐dependent innate immunity. Nature 2009;461:788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Saitoh T, et al. Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 2009;106:20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhong B, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 2009;30:397–407. [DOI] [PubMed] [Google Scholar]
- 106. Tsuchida T, et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double‐stranded DNA. Immunity 2010;33:765–776. [DOI] [PubMed] [Google Scholar]
- 107. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak‐Rothstein A. Chromatin‐IgG complexes activate B cells by dual engagement of IgM and Toll‐like receptors. Nature 2002;416:603–607. [DOI] [PubMed] [Google Scholar]
- 108. Lau CM, et al. RNA‐associated autoantigens activate B cells by combined B cell antigen receptor/Toll‐like receptor 7 engagement. J Exp Med 2005;202:1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Croker BA, et al. Inflammation and autoimmunity caused by a SHP1 mutation depend on IL‐1, MyD88, and a microbial trigger. Proc Natl Acad Sci USA 2008;105:15028–15033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Turer EE, et al. Homeostatic MyD88‐dependent signals cause lethal inflamMation in the absence of A20. J Exp Med 2008;205:451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kawagoe T, et al. TANK is a negative regulator of Toll‐like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol 2009;10:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Matsushita K, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009;458:1185–1190. [DOI] [PubMed] [Google Scholar]
- 113. Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll‐like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol 2006;7:49–56. [DOI] [PubMed] [Google Scholar]
- 114. Yasuda K, et al. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9‐dependent and ‐independent pathways. J Immunol 2005;174:6129–6136. [DOI] [PubMed] [Google Scholar]
- 115. Ganguly D, et al. Self‐RNA‐antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 2009;206:1983–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ivanov S, et al. A novel role for HMGB1 in TLR9‐mediated inflammatory responses to CpG‐DNA. Blood 2007;110:1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Tian J, et al. Toll‐like receptor 9‐dependent activation by DNA‐containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 2007;8:487–496. [DOI] [PubMed] [Google Scholar]
- 118. Marshak‐Rothstein A. Toll‐like receptors in systemic autoimmune disease. Nat Rev Immunol 2006;6:823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll‐like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006;25:417–428. [DOI] [PubMed] [Google Scholar]
- 120. Yu P, et al. Toll‐like receptor 9‐independent aggravation of glomerulonephritis in a novel model of SLE. Int Immunol 2006;18:1211–1219. [DOI] [PubMed] [Google Scholar]
- 121. Nickerson KM, et al. TLR9 regulates TLR7‐ and MyD88‐dependent autoantibody production and disease in a murine model of lupus. J Immunol 2010;184:1840–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA‐related antigens due to TLR7 gene duplication. Science 2006;312:1669–1672. [DOI] [PubMed] [Google Scholar]
- 123. Subramanian S, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA 2006;103:9970–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Deane JA, et al. Control of toll‐like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007;27:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Boule MW, Broughton C, Mackay F, Akira S, Marshak‐Rothstein A, Rifkin IR. Toll‐like receptor 9‐dependent and ‐independent dendritic cell activation by chromatin‐immunoglobulin G complexes. J Exp Med 2004;199:1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody‐DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 2005;115:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bave U, Magnusson M, Eloranta ML, Perers A, Alm GV, Ronnblom L. Fc gamma RIIa is expressed on natural IFN‐alpha‐producing cells (plasmacytoid dendritic cells) and is required for the IFN‐alpha production induced by apoptotic cells combined with lupus IgG. J Immunol 2003;171:3296–3302. [DOI] [PubMed] [Google Scholar]
- 128. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor‐kappaB or activator protein‐1: molecular mechanisms for gene repression. Endocr Rev 2003;24:488–522. [DOI] [PubMed] [Google Scholar]
- 129. Guiducci C, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 2010;465:937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Martinez Valle F, Balada E, Ordi‐Ros J, Vilardell‐Tarres M. DNase 1 and systemic lupus erythematosus. Autoimmun Rev 2008;7:359–363. [DOI] [PubMed] [Google Scholar]
- 131. Yasutomo K, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet 2001;28:313–314. [DOI] [PubMed] [Google Scholar]
- 132. Kawane K, et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 2001;292:1546–1549. [DOI] [PubMed] [Google Scholar]
- 133. Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon‐beta produced in mouse embryos carrying undigested DNA. Nat Immunol 2005;6:49–56. [DOI] [PubMed] [Google Scholar]
- 134. Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell‐intrinsic initiation of autoimmunity. Cell 2008;134:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Crow YJ, et al. Mutations in the gene encoding the 3′‐5′ DNA exonuclease TREX1 cause Aicardi‐Goutieres syndrome at the AGS1 locus. Nat Genet 2006;38:917–920. [DOI] [PubMed] [Google Scholar]
- 136. Lee‐Kirsch MA, et al. Mutations in the gene encoding the 3′‐5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet 2007;39:1065–1067. [DOI] [PubMed] [Google Scholar]
- 137. Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll‐like receptor‐independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med 2005;202:1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Rice GI, et al. Mutations involved in Aicardi‐Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 2009;41:829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Guiducci C, et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med 2010;207:2931–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gregorio J, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med 2010;207:2921–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]