

Abstract
We challenge the concept of idiopathic parkinsonism (IP) as inevitably progressive neurodegeneration, proposing a natural history of sequential microbial insults with predisposing host response. Proof‐of‐principle that infection can contribute to IP was provided by case studies and a placebo‐controlled efficacy study of Helicobacter eradication. “Malignant” IP appears converted to “benign”, but marked deterioration accompanies failure. Similar benefit on brady/hypokinesia from eradicating “low‐density” infection favors autoimmunity. Although a minority of UK probands are urea breath test positive for Helicobacter, the predicted probability of having the parkinsonian label depends on the serum H. pylori antibody profile, with clinically relevant gradients between this “discriminant index” and disease burden and progression. In IP, H. pylori antibodies discriminate for persistently abnormal bowel function, and specific abnormal duodenal enterocyte mitochondrial morphology is described in relation to H. pylori infection. Slow intestinal transit manifests as constipation from the prodrome. Diarrhea may flag secondary small‐intestinal bacterial overgrowth. This, coupled with genetically determined intense inflammatory response, might explain evolution from brady/hypokinetic to rigidity‐predominant parkinsonism.
Keywords: Parkinson's Disease, Helicobacter, small‐intestinal baterial overgrowth, virus, mitochrondria, aetiology, pathogenesis
In 1817, James Parkinson described the shaking palsy, a rigid brady/hypokinetic syndrome with a characteristic tremor and stooped posture [1]. Consequent therapeutic milestones are few and far between: noting the wider antiparkinsonian effect of tinctures of deadly nightshade, used to control excessive salivation (1867) [2]; describing dopamine deficiency in basal ganglia (1960), thereby instigating dopamine‐substitution therapy [3]; and discovering the antiparkinsonian effect of an antiviral agent, amantadine (1969) [4]. The way to the Helicobacter etiologic hypothesis was paved, before the discovery of H. pylori‐associated gastritis (1983) [5], by observation of an excess of previously documented peptic ulcer in Parkinson's disease (1965) [6], and speculation that an infectious agent was involved in both (1979) [7]. Explicit suggestion of a causal link followed [8].
Presence of intracytoplasmic inclusions, Lewy bodies, in a characteristic distribution in brain was, until recently [9], considered the gold standard for designation of “Parkinson's disease”. However, whittling away the population with this syndrome to a core sample, according to clinical predictors of post‐mortem brain histopathology, is not a tenable start point for unravelling causality. Were the syndrome a manifestation of systemic disease, unifying pathology would be expected outside the brain. Moreover, aged brains are frequently affected by more than one “neurodegenerative” pathology: parkinsonian and Alzheimer often coexist [10]. Polymorphisms in a single gene are associated with parkinsonism alone or with other distinctive neurologic phenotypes, and with more than one type of pathologic hallmark, even in the same neurone [11]. The same environmental insult might also result in neuronal deposition of different aberrant proteins, each having a spectrum of associated phenotypes [12].
We use Calne's broad clinical definition of idiopathic parkinsonism (IP) to describe a syndrome of unknown etiology [13]. He regards the manifestations as the net effect of “doses” of genetic predisposition and environmental risk factors [12], their balance varying between‐ and within‐proband. However, there is no convincing evidence in IP of excessive exposure to environmental chemicals [14], and studies of genes controlling their metabolism have been unrewarding [15].
Helicobacter Hypothesis: Arrival at, Pragmatic Testing, and Beyond
Our strategy and approach led to an infection hypothesis, implicating the gut (see Defining and Assembling the Jigsaw Pieces and Table 1), the initial therapeutic target being small‐intestinal bacterial overgrowth (SIBO), secondary to slow transit. Indeed, constipation features in James Parkinson's essay [1]. In IP, frequency of defecation begins to deviate from that of controls three decades before the median age of neurologic diagnosis, two before diagnosis of the first quartile of probands [16]: a finding upheld prospectively by the association of infrequent bowel movements and subsequent diagnosis of parkinsonism [17]. In IP, there is loss of, and damage to, colonic myenteric dopaminergic neurones. These, enteric plexus ganglia and physiologically related sympathetic neurones can contain Lewy bodies, as does the dorsal vagal nucleus [18, 19, 20]. Pfeiffer, thinking on similar but noninfective lines, homes in on constipation as a marker of the genesis of IP [20].
Table 1.
Generation of an “Infection Hypothesis” for idiopathic parkinsonism (IP), implicating the gut
| Year | Statistical model | Findings |
|---|---|---|
| 1992 [61] | Observational comparison nocturnal axial rotation in elderly IP probands, their spouses and control couples. | Rotation in spouses less than in controls, greater than in probands. a |
| 1993/4/6 [62, 63, 64] | Comparison measured facets of parkinsonism in elderly probands, their spouses, control couples. | Spouses significantly different from controls (toward parkinsonism) in measures of brady/hypokinesia, postural abnormality, rigidity, and frequency of seborrheic dermatitis. |
| 1997/8 [16, 65, 66] | Relationship serum immunoglobulin concentrations to presence/ absence of (i) Diagnosed IP, (ii) A parkinsonian feature in subjects without diagnosed parkinsonism, (iii) Specified medication in IP probands. | (i) No overall difference in IgM, A, or G with IP. b (ii) In controls, bradykinesia associated with higher IgA and lower M, postural abnormality with higher IgA, as if exaggerated ageing. (iii) IgA higher in probands with constipation warranting laxatives or taking antimuscarinic c . Higher IgA in ii and iii explained by A1 subclass: a systemic rather than mucosal response. |
| 1997/8 [16, 57] | (i) Comparison between IP probands and controls of current and estimated past frequency of defecation. (ii) Association serum IgA with a discriminant index for presence/ absence parkinsonism based on current bowel habit. (iii) Relative effect of tobacco smoking on bowel habit. | (i) Frequency less in probands prodromally (from fourth decade of life). (ii) IgA increased with index irrespective of subject group. (iii) Differential effect of smoking on “difficulty passing a motion”: laxative‐like effect greater in controls. |
| 1998 [56] | Comparison serum cortisol in IP probands and controls, taking into account: (i) Current or past smoking, (ii) Anti‐parkinsonian medication and laxatives in IP. | Cortisol higher in IP. In controls, the lower the cortisol, the shorter the stride and the more the deterioration over 4 years: relationship inverted in IP. (i) Smoking tended to be associated with lower cortisol, irrespective of group. (ii) Levodopa and dopaminergic agonists did not affect cortisol; selegiline and antimuscarinics d reduced it, constipation warranting laxatives tended to increase it. |
| 1999 [60] | Comparison serum IL‐6 and TNF‐α in IP probands and controls. | IL‐6 increased with age, effect of IP equivalent to more than 10 years of ageing. TNF‐α increased with age, not IP. Elevated in probands with impaired postural and psychomotor responses, suppressed with normal responses, in contrast to lack of performance relationship in controls. e |
No correlation between partners, effect independent of bed sharing.
Lymphopenia [23, 26] in IP may abrogate any overall difference in immunoglobulins present in prodrome. Higher IgA in IP with constipation likely to reflect small‐intestinal bacterial overgrowth.
Constipation is dose‐related adverse effect.
Paradoxical decrease with antimuscarinics explained by central cholingeric effects.
Differences in inflammatory response may determine predominant facets between and within individual(s). TNF‐α, tumor necrosis factor alpha; IL‐6, interleukin‐6.
As a collateral hypothesis (Table 2), we described the epidemiologic fit of Helicobacter infection to IP (including familial clusters, evidence for early acquisition, long prodrome and association with water source) and proposed an autoimmune basis [21]. By 2005, we had proof of principle that infection contributes to IP, through case studies of anti‐Helicobacter therapy in gastritis, with and without associated Helicobacter [22], and a hypothesis‐testing study [23]. In essence, “malignant” IP appeared converted to “benign” following successful eradication, but marked deterioration accompanied failure. In cases of late IP, eradicating Helicobacter produced U‐turns in both parkinsonism and cachexia [22]. In probands receiving no or only stable long‐t1/2 antiparkinsonian medication, the randomized efficacy study contrasted effect, on the time course of IP facets, of 1 week's successful anti‐H. pylori therapy against placebo, and against failure [23]. Improvement in the primary outcome, mean stride length at free‐walking speed, followed successful blinded active therapy (de‐blinding being scheduled for 1 year post‐randomization). Benefit on brady/hypokinesia did not fall off during the year after de‐blinding, and was echoed in those given open‐active anti‐H. pylori therapy subsequent to placebo [23]. Improvement was irrespective of whether patients were yet receiving background antiparkinsonian medication. Figure 1 illustrates that gait can improve dramatically following H. pylori eradication therapy where biopsies are molecular microbiology positive but culture negative (and there is no evidence of SIBO or other infection) [24]. “Low‐density” infection may be sufficient to perpetuate autoimmunity. Persistence even at this level of detection appeared detrimental [23].
Table 2.
Generation of “Helicobacter Hypothesis” for idiopathic parkinsonism (IP)
| Year | Statistical model | Findings |
|---|---|---|
| 1999 [31] | Observational comparison of facets of parkinsonism, and H. pylori anti‐urease ELISA seropositivity, in IP probands and their siblings with controls. | Siblings significantly different from controls (toward parkinsonism) in measures of brady/hypokinesia, rigidity, abnormal posture, and frequency seborrhea/seborrheic dermatitis. Odds ratio of 3 for seropositivity in probands and siblings cf. controls. a |
| 1999 [67] | Explanation of facets of syndrome by H. pylori urease antibody in subjects with and without diagnosed parkinsonism. | Seropositivity unrelated to presence/absence facets in those who have not passed diagnostic threshold, but decreased with abnormal posture in IP. b |
| 1999 [68] | Relationship of increase in serum IL‐6 and TNF‐α with age, and in IL‐6 and cortisol with parkinsonism, to H. pylori urease antibody. | These immune/inflammatory responses not explained by antibodies measured in routine ELISA. |
| 2000 [69] | Explanation of increase in serum cortisol with IP, over that in controls, by presence/absence of antibodies against VacA, CagA, and urease‐B. | Effect of antibodies independent of disease status: anti‐VacA seropositivity associated with elevated cortisol, IP with additional elevation, neither anti‐urease nor anti‐CagA adding to variance explained. |
| 2000 [59] | (i) Contrast of relationship of H. pylori urease antibody to age in subjects with and without diagnosed parkinsonism. | (i) Birth‐cohort effect in ELISA value (EV), seen in controls, obliterated in IP. Probands twice as likely to be seropositive before 72.5 years. |
| (ii) Relationship of titer to severity IP. | (ii) EV lower with greater global disease severity. b | |
| 2000 [70, 71] | Contrast of relationship of serum immunoglobulin classes to H. pylori urease antibody in subjects with and without diagnosed parkinsonism. | In controls, downward shift in IgM with anti‐urease positivity (equivalent to 25 years ageing). In IP, IgM higher than in controls in seropositive, c lower in seronegative. No seropositivity effect on IgA and IgG, either group. |
| 2000 [72] | Discrimination for seborrheic dermatitis by H. pylori serum immunoblot antibody profile in subjects without diagnosed parkinsonism. | Discriminant index for presence characteristic rash contained anti‐CagA (directly associated) and anti‐VacA (inversely). d , e |
| 2005 [22] | Contrast of relationship between being underweight and inflammatory products in subjects with and without diagnosed parkinsonism. | Association of low body mass index with serum IL‐6 concentration specific to parkinsonism, unlike that with anti‐VacA and anti‐CagA. |
| 2005 [23] | Explanation of failure of Helicobacter eradication in IP by blood lymphocyte subset counts. | Failed eradication associated with lower B‐cell count. |
| 2005/6 [23, 26] | Comparison blood counts in IP probands and their spouses with routine general practitioner requests | Total lymphocyte counts in spouses and untreated probands similar, lower than in controls. Higher counts in probands and spouses with VacA antibody. Count in IP not explained by serum B12 or folate. Probands’ CD4+, CD8+ and CD19+ counts lower than in spouses, but CD16+56+ higher. f |
| 2005 [27] | Discrimination for parkinsonism, and explanation of its severity and progression, by H. pylori serum immunoblot antibody profile. | Predicted probability of being labeled parkinsonian greatest with anti‐CagA seropositivity and anti‐VacA and ‐urease‐B negativity. e Clinically‐relevant association between index and measures of disease facets and their progression in IP, despite potentially confounding effect of anti‐parkinsonian medication. |
| 2007 [28] | Discrimination for abnormal bowel function (constipation and/or diarrhea) in IP probands and their spouses by H. pylori serum immunoblot profile. | Bowel function abnormal in 53% probands and in 36% spouses. Fourfold increase in odds for abnormal function with urease‐B band, sixfold decrement with outer‐membrane protein band, irrespective of urea breath test status, nature of abnormality, or subject group. Irritable bowel syndrome (15%) had same band associates. |
In contrast ELISA seropositivity not increased in (older) spouses (unpublished data further to [62]).
b H. pylori antigens, other than urease, are known to stimulate cytokine production [47]. They, like serum immunoglobulin classes [65], may be associated with facets of parkinsonism in prodrome/early disease. Helicobacter infection is more likely a forerunner of postural abnormality and global severity in IP, than protective.
Although CD19+ count is low [26], there may be increase in naïve B‐cells, or B1 cells, producing polyspecific IgM. Alternatively, local sequestration of IgM may fail in IP.
In IP, seborrheic dermatitis occasional flares up on treatment of H. pylori (personal observation).
There may be incomplete immune tolerance, regulatory T‐lymphocytes recognizing H. pylori but not CagA; or infection may be truncated, but cagA gene, its product or antibodies against CagA persist.
Increased natural killer cells in IP indicate preserved innate response and ongoing viral, bacterial, or parasitic infection. TNF‐α, tumor necrosis factor alpha; IL‐6, interleukin‐6.
Figure 1.

Frames, at 2 second intervals from videos over a fixed course, in an idiopathic parkinsonism (IP) patient before (top) and 6 weeks (below), 18 weeks (below again), and 1 year (bottom) after biopsy‐proven eradication of H. pylori. Initial detection by molecular microbiology on culture‐negative antral and corporal biopsies. The patient can now cycle 10 miles, and continues not to require anti‐IP medication. It took 6 frames to cover the course initially, 4 or fewer subsequently. The initial gait was tentative, stilted, wooden and doll‐like. Subsequently, it became relaxed, free‐flowing and, finally, vigorous. (Reproduced with patient's consent.)
However, we are seeing disease modification, not cure. Time trends in objective measurements of facets of parkinsonism dissociate following H. pylori eradication, with insidious increase in rigidity, in mirror image to the pattern for hypokinesia [25]. Mean torque required to extend the relaxed forearm [23] increased following successful blinded‐active therapy compared with placebo, again irrespective of antiparkinsonian medication status. This was echoed in those given open‐active. Size of effect was clinically relevant compared with baseline measurements.
Stepping back, peptic ulceration and, presumably, the zenith of gastric inflammatory response to Helicobacter precede the diagnosis of IP, often by decades [6]. In the UK, 40% of IP probands are serum H. pylori immunoblot positive, 20% urea breath test positive [23]. Immunoblot positivity is directly related to the blood lymphocyte count, lymphopenia, a feature of IP [23, 26]. Irrespective of evidence for current Helicobacter infection, the serum immunoblot antibody profile predicts not just the presence and severity of IP, but also the progression over 4 years [27].
In contrast to peptic ulceration, the clinical manifestations of slow gastrointestinal transit become more apparent post‐diagnosis [16]. The H. pylori immunoblot is predictive of abnormal bowel function within IP, irrespective of current infection [28]. Helicobacter‐associated autoimmunity might have wrought irreparable damage to the enteric nervous system. Sixty percent of our probands, without current Helicobacter infection, are lactulose‐hydrogen breath test positive for SIBO. Obvious benefit accrues from Helicobacter eradication, but loss of hypersecretion in response to antral gastritis may weaken the gastric acid barrier, making conditions for SIBO more opportune.
Proposed Stages in Pathogenesis
Staging accommodates variability between probands in manifestations of IP, and within subject change in predominant manifestations. In Stage 1 (Fig. 2), we propose that Helicobacter infection evokes autoimmunity against mitochondria, and that this mechanism underlies brady/hypokinesia predominant IP.
Figure 2.
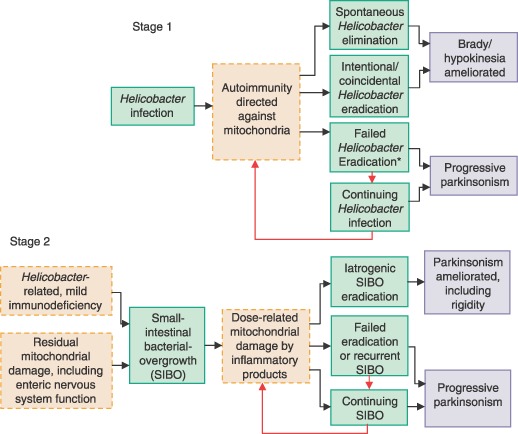
Scheme for process (green), mechanisms (pink), and outcome (blue) in idiopathic parkinsonism. Stage 1 is an autoimmune reaction to Helicobacter infection; Stage 2, a dose‐related response to inflammatory products of small‐intestinal bacterial overgrowth (SIBO). *Bolus release of antigen accompanying failed eradication may accelerate deterioration [23].
Most Helicobacter infections are transmitted where there is close contact, as between parent or sibling and infant [29]. This accords with the younger the child when a parent develops IP, the greater the risk of IP to that child [30], and with probands and their siblings sharing facets of the syndrome and increased prevalence of H. pylori seropositivity [31]. In younger people, the prevalence of urea breath test positivity is falling (associated with better hygiene/increased exposure to antimicrobials), as is incidence and age‐specific mortality of IP [32]. However, transient H. pylori infection or persistence in low density may be sufficient to trigger/perpetuate autoimmunity, thereby setting the scene for the age‐specific increase in IP in older people [32].
We propose that, in Stage 2, acquisition of SIBO causes further mitochondrial dysfunction, resulting in a rigidity‐predominant picture. Chronic infection over a large mucosal interface ratchets up dose‐related mitochondrial damage, its potency being magnified by polymorphisms for intense innate inflammatory response. Any adaptive immunodeficiency might predispose to SIBO. Our early case studies show improvement in rigidity on eradicating SIBO (lactulose‐hydrogen breath test criterion), but reinfection/recrudescence is probable. The stages may overlap: Helicobacter may be an important source of dose‐related damage, or coexistent SIBO may cause poor response to proven H. pylori eradication.
There is systemic and basal ganglia (nigro‐striatal) immune activation in IP [21, 33]. Whilst local brain inflammation does not usually signal out [34], systemic inflammation can communicate with the brain's immune system [33, 35]. Communication involving immune cells, antibody, or inflammatory products can occur where the blood brain barrier is naturally deficient, or when permeability is increased by circulating cytokines and cortisol. Peripheral afferent nerve stimulation, including vagal, can activate microglia (brain's resident macrophages) [35]. Helicobacter infection could stimulate the vagus, SIBO perhaps more so. Peripheral infection would increase traffic and signaling into brain. The substantia nigra is regarded as particularly vulnerable to everyday insults, such as products of dopamine oxidation [36]. Its homeostatic mechanisms function at full stretch. Nigral glial activation would increase stress, and recruit immune cells locally.
A Mitochondrial Disease
Mitochondrial dysfunction is described in substantia nigra in IP (complex I) and multiple‐system atrophy (complex IV) [37], and in platelets in IP [10, 38]. Indeed “cybrid” cells containing mitochondrial (platelet) and nuclear DNA, from donors with and without IP, respectively, had reduced complex I activity [39]. Our evidence for systemic, extraneuronal mitochondrial involvement in IP is of filamentous arrays encapsulated in double membranes in duodenal enterocytes (Fig. 3, upper panels) [28]. They are found among normal mitochondria in IP probands with current or recent H. pylori infection. Our electron microscopists had not previously observed similar bodies, although two examples were found subsequently in archived duodenal biopsies from patients with human immunodeficiency virus (HIV) infection. HIV can be associated with parkinsonian features [40]. There is an isolated report of similar mitochondrial inclusions in cerebral neurones in Creutzfeldt–Jakob‐like disease [41]. Prion tubulovesicular structures are larger; cluster, free within cytoplasm; and not found at prion infectivity sites outside central nervous system.
Figure 3.
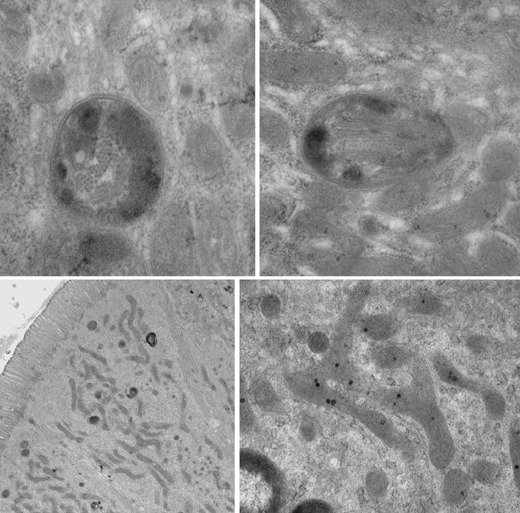
Electron micrographs of duodenal enterocytes from idiopathic parkinsonism patients. Above: double membraned, encapsulated arrays lie among normal mitochondria. Arrays are seen mainly transversely on the left (appearing tubular), longitudinally on the right. In this section every enterocyte contained 2 or 3 such bodies. Below: long thin mitochondria are seen to predominate at low magnification (left). Complex branching of a mitochondrion is shown at intermediate magnification (right).
Routine anti‐mitochondrial antibody serology has been negative in our IP patients (unpublished observation). The cytoplasmic fluorescence assay may detect only mitochondrial surface binding. Autoimmunity in IP may be directed against mitochondrial protein, neoantigen, DNA, RNA or chromatin. Matrilineal inheritance, typical of mitochondrial disorders, is rare in parkinsonism [42], but absence of mitochondrial DNA repair enzymes gives susceptibility to mutagens [43]. The modern mitochondrial proteome is widely accepted as originating, in part, from endosymbiont bacteria [44]. Helicobacter DNA might be incorporated into host mitochondria. Despite evidence for occurrence of bacteriophage in H. pylori, transduction has not been demonstrated, but there is evidence for a conjugation‐like mechanism and natural transformation between bacteria [45].
While Helicobacter infection may be the trigger, pro‐inflammatory cytokines, such as tumour necrosis factor (TNF)‐α, can intensify the mitochondrial damage [46]. Long thin mitochondria, sometimes with complex branching, become the predominant feature of probands’ duodenal enterocytes subsequent to H. pylori infection and in the presence of SIBO (Fig. 3, lower panels). This apparent hypertrophy, associated with rough endoplasmic reticulum, may compensate for hypofunction.
Acceleration, Deceleration, Perpetuation, and Attenuation
A genetically determined, intense systemic, innate inflammatory response would accelerate the natural history of IP (Fig. 4). However, an intense interleukin‐1β response could cause Helicobacter to self‐destruct, its “protective” urease continuing to produce ammonia in the face of powerful inhibition of gastric acid secretion [47]. This could explain “low‐density” infection but no gastric atrophy [22, 23] in IP, and increase susceptibility to SIBO. Certain polymorphisms increase the risk of noncardiac cancer, seropositivity for antibodies against CagA gene‐product escalates it further [48]. Variability in time course of IP may be determined analogously [49]. Exploratory studies in IP suggest greater prevalence of similar polymorphisms, and associations with early onset [50, 51, 52, 53, 54]. Here too, anti‐CagA seropositivity is a bad prognostic sign [27].
Figure 4.
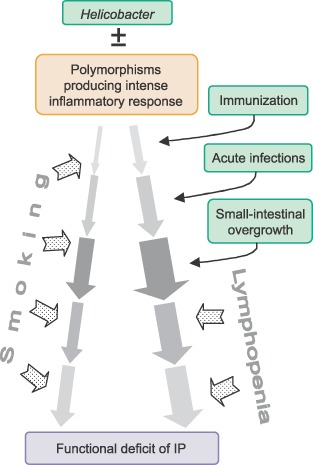
Overview of events contributing to natural history of idiopathic parkinsonism (IP).
Mild acquired, adaptive immunodeficiency, flagged in later IP by lymphopenia [23, 26], may attenuate inflammatory response. This may be double‐edged, guarding against “parkinsonian effects” of chronic inflammation, but leaving probands open to infection. “Quieter” disease is usual in old age. Tobacco smoking protects against parkinsonism, as it does for ulcerative colitis [55]. This may be immuno‐modulatory; a pharmacologic effect on injurious (but “physiologic”) serum cortisol elevation [56]; or just a laxative action [57]. Higher prevalence of H. pylori‐seropositivity in smokers [58], irrespective of IP [59], might keep SIBO at bay in IP.
Spectrum of Disease
Shifting IP/overlap diseases to a more appropriate nosologic grouping seems important to unravelling them. A systemic mitochondrial disorder may give a spectrum from “functional bowel disease” alone to its co‐manifestation with parkinsonism (Fig. 5). Intensity and nature of inflammatory response is primarily a between‐subject determinant [56, 60], acquired or inherited vulnerability of the basal ganglia is a susceptibility factor.
Figure 5.
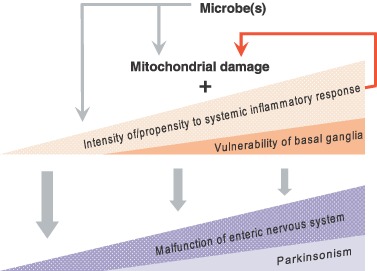
Scheme for within‐ and between‐subject determinants of position in a spectrum from isolated functional bowel disease to co‐manifestation with parkinsonism.
Defining and Assembling the Jigsaw Pieces
The above scheme, with possibilities for pragmatic testing, grew out of an exploratory strategy, approaching from different clinical clues, addressing different questions, and assessing the balance of probabilities (1, 2). The aim is a scientifically plausible etiologic fit. Oversights in interpretation of a given statistical model within the series does not jeopardize the whole. Particular hierarchical layers in the modelling are addressed below:
Is IP a systemic disease?
There are biologic gradients between measures of IP and markers of inflammation. Higher serum cortisol or TNF‐α is detrimental in IP compared with controls [56, 60]. Serum cortisol and interleukin (IL)‐6 are elevated overall in IP, TNF‐α is not. Association of a low body mass index with increased circulating IL‐6 is specific to parkinsonism, by contrast with healthy controls [22]. Cachexia in IP is usually intractable and associated with rapid deterioration. Where Helicobacter infection was found, gastritis was not severe, but successful treatment produced a turnabout [22]. Where body mass index is normal and there is no evidence of Helicobacter, ravenous appetite and night sweats are common, and may largely be attributable to SIBO.
Is it transmissible?
The classical spousal approach to environmental causality was used. Figure 6 illustrates the short but highly significant “distance down the pathway” of spouses cohabiting with IP probands for half a century. Marked, multifarious, relevant differences (physiologic/psychomotor/dermatologic) between spouses and control couples are difficult to explain by selective mating or learned/reactive behavior [61, 62, 63, 64]. These and lymphopenia, in a large group of probands and spouses [23, 26], suggest adult transmission. Moreover, half of the latter probands and a third of their spouses had chronic bowel abnormality (criteria [73]) [28]. Seven percent of probands and 14% of spouses had diarrhea (unformed stool during at least three‐quarters of past year, plus three or more bowel movements/day for half). Neuronal damage in probands may evade a diarrheal response to a shared insult.
Figure 6.
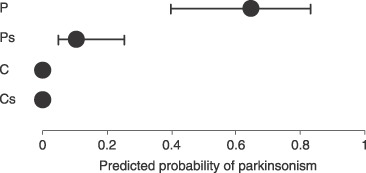
A prognostic index for parkinsonism [based on brady/hypokinesia variables in 104 subjects with idiopathic parkinsonism (IP), 144 withou] applied in 20 index controls (C), their spouses (Cs), 20 probands (P) with clinically definite (treated) IP, and their spouses (Ps). Means (95% confidence interval) are shown. P and C were matched for age and gender. p < .0001 for predicted probability in Ps cf. C + Cs combined [62].
Is Helicobacter a prerequisite?
Biologic gradients between measures of IP and serum H. pylori immunobot antibody profile strengthen the case for causality [27]. In controls without diagnosed parkinsonism, there were no such relationships, suggesting undefined bacterial pathogenicity or host susceptibility factors. Indeed, the double‐membraned bodies containing filamentous arrays in duodenal enterocytes might flag the course to parkinsonism. Association of the H. pylori immunoblot profile with abnormal bowel function within IP [28], and with seborrheic dermatitis (frequent accompaniment of IP [64]) in subjects without parkinsonism [72], further implicates Helicobacter.
Increasing prevalence of H. pylori anti‐urease seropositivity with age is typical of the general population of socioeconomically advanced countries. This “birth cohort effect” is said to reflect greater early life acquisition of persistent infection in less hygienic times. In IP, there was an increased prevalence of anti‐urease seropositivity before old age, allowing no significant age effect to be captured in anti‐urease titer, despite demonstration of the birth cohort effect in contemporaneous controls [59]. Lack of birth cohort effect is also documented for peptic ulcer and gastric carcinoma, where causal links with H. pylori are generally accepted. In IP, there is also a differential trend in total serum IgM concentration in relation to anti‐urease titer [70, 71].
H. pylori culture supernatants, whole bacteria, protein products, and H. pylori‐specific regulatory T‐lymphocytes inhibit human T‐cell proliferation [74, 75]. Since lymphopenia in IP is more marked with H. pylori immunoblot negativity, it is unlikely to be due to direct inhibition, but might relate to Helicobacter‐triggered autoimmunity. Unlike its effect on platelets in idiopathic thrombocytopenic purpura [76], H. pylori eradication has little impact on IP lymphopenia.
Does peripheral infection drive neuronal death in brain?
Mitochondrial damage in IP, and also oxidative stress, excitatory amino acid production and proteosomal damage [36], could be secondary to, or augmented by, infection. Substantia nigra microglia are activated in IP [77], and secrete TNF‐α[46]. Dopaminergic neurones express TNF‐α receptors [46] and up‐regulate nuclear factor κB in the apoptotic pathway [78]. Basal ganglia and cerebrospinal fluid (CSF) IL‐1β and IL‐6 concentrations are raised [79, 80]. Cytotoxic T‐lymphocytes are seen in relation to “degenerating” nigral dopaminergic neurones showing IgG binding [81]. These neurones appeared labelled for destruction, their proportion being less in severe/longer‐standing disease. Activated microglia persist, presumably continuing to mediate damage.
In IP, there are activation markers on circulating T‐lymphocytes and CSF monocytes [21, 33, 82, 83, 84, 85] Blood and CSF γδ + T‐cells are increased [86]. They are biased toward bacterial antigens (e.g. heat‐shock proteins), associated with autoimmune conditions, and prominent in gut‐associated lymphoid tissue. CSF contains antibodies against bacterial heat‐shock proteins [87]. Both CSF and blood contain antibodies against sympathetic and dopaminergic neurones [88]. CSF from IP probands recognizes rat nigral dopaminergic neurones [89]. Injection of purified IgG from IP serum into rat nigra selectively destroys dopaminergic neurones [90]. Purified IgG from probands’ CSF or serum is toxic to them in culture [91, 92]. Toxicity depends on complement and microglial Fcγ antibody receptors [92, 93]. Serum from IP probands also inhibits high‐affinity neuronal dopamine uptake, but not gamma‐amino butyric acid uptake [92].
Supplementary and Alternative Explanations
An underlying viral infection?
The pandemic of encephalitis lethargica (1917) sparked the viral hypothesis. Parkinsonism occurs in uncomplicated HIV infection, and when precipitated by opportunistic infections (e.g. toxoplasmosis) in acquired immunodeficiency syndrome [40, 94, 95]. HIV sensitizes to anti‐dopaminergic medication. Parkinsonian features may be associated with HIV dementia. Motor dysfunction compatible with basal ganglia damage is found in early HIV, and basal ganglia dopaminergic cell loss is seen without clinical parkinsonism. In simian immunodeficiency virus‐infected monkeys, nigrostriatal dopamine is halved within 2 months [40]. Seborrheic dermatitis is associated with both IP [31, 64] and HIV [94]. Although the epidemiology of HIV is distinct, a relatively benign retrovirus could be pathogenic in IP. (Lewy bodies are not reported in HIV [40]). As well as lymphopenia, damage to the enteric nervous system in IP might be viral in origin. Jejunal autonomic denervation is described with HIV infection [96]. Enteroviruses infect via the gastrointestinal tract and are associated with neurologic syndromes. A combined virologic approach is needed: current standard‐of‐care diagnostic assays to address the clinical features of IP (Table 3), pathogen discovery for uncharacterized viruses.
Table 3.
Classification of viral and other intracellular microbial targets by clinical clues
| Clinical feature compatible with: (description) | Target |
|---|---|
| Upper respiratory tract symptoms (rhinitis, post‐nasal catarrh, eye injection) a | Adenovirus |
| Human meta‐pneumovirus | |
| Influenza A virus | |
| Influenza B virus | |
| Parainfluenza viruses 1, 2, and 3 | |
| Respiratory syncytial virus subtypes A and B | |
| Rhinovirus | |
| Hot sweats (especially at night) | Epstein–Barr virus b |
| Duodenitis | Adenovirus |
| Cytomegalovirus | |
| Enterovirus | |
| Mycoplasma [97] | |
| Diarrhea | Aichi virus c |
| Astrovirus | |
| Bocavirus c | |
| Coronavirus c , d | |
| Enteric adenovirus d | |
| Norovirus d | |
| Parvovirus c , d | |
| Rotavirus | |
| Sapovirus d | |
| Torovirus c | |
| Bloating and constipation related to slow transit (consequent on damage to enteric nervous system) | Enterovirus e |
| Lymphopenia f | Cytomegalovirus g |
| Epstein–Barr virus g | |
| Hepatitis C virus | |
| Human immunodeficiency virus 1 and 2 g | |
| Human T‐lymphotropic virus 1 and 2 | |
| Parvovirus B19 | |
| Herpes simplex virus I and II | |
| Varicella zoster virus | |
| Human herpes virus 6 | |
| Toxoplasma gondii g | |
| Urinary frequency | Chlamydia trachomatis |
Includes viruses associated with laryngitis. Rhinitis sometimes coincides with diarrheal attack. Risk of developing IP over 20 years is 2.9 times greater in those with hayfever/allergic rhinitis [98]. Droplet‐spread infection proposed reason for higher risk IP in teachers and health workers [99].
Also relevant to isolated pharyngitis.
Associated with gastroenteritis, but causality in humans unproven (except SARS coronavirus).
Belongs to family group causing wide spectrum of systemic illnesses in humans/other mammals.
Infect via gastrointestinal tract. Not primarily a cause of gastroenteritis but may cause gastrointestinal upset. Cause illness ranging from mild non‐specific fevers/myalgia to meningitis/poliomyelitis.
Potentially persistent viruses that can cause insidious human disease and lymphopenia (or pancytopenia).
Associated with atypical lymphocytes, rather than lymphopenia.
Effect of antimicrobials on host?
Antimicrobial medication could produce benefit independent of eliminating target organism(s). In rats, minocycline reduces inflammation, blood‐brain‐barrier permeability, and dopaminergic neurone damage, produced by intranigral injection of bacterial lipopolysaccharide [100].
Antithesis of One‐Step Pragmatism
Table 4 outlines our strategy and approach to etiology and pathogenesis. Investigational medicine yields clinical clues to elucidating processes and mechanisms, and pinpointing potential causal agents. Consequent observational and experimental studies provide raw data for statistical modeling to generate a cumulative hypothesis.
Table 4.
Strategy and approach to etiology/pathogenesis of an idiopathic parkinsonism
| Strategy | Approach |
|---|---|
| Step back | |
| Consider whole entity | Reject dividing syndrome by clinical minutiae and nature of cellular protein aggregates |
| Define clinical syndrome and be aware of overlap diseases | Assemble all raw clinical clues, without selectivity |
| Avoid focusing on tip of an iceberg in a disease with a long prodrome | Early disease may hold clues masked in later stages. Indeed, solution may be untenable without acknowledging preclinical state. |
| Question adequacy of measurement methodology | Reject subjective global scores. Embrace valid, sensitive/specific, reliable measures (objective where possible) of disease facets, which can identify clinically relevant changes with time or intervention. a |
| Adopt exploratory statistical methodology for hypothesis generation; defer any pragmatic testing | Seek to explain clues by associations. Measure potential biologic effect in small well‐defined subject groups, taking into account candidate confounders and effect modifiers. Carefully select control groups, avoiding convenient family (including spouses) and close contacts. |
| Define jigsaw pieces | |
| Collect observational data to generate statistical models, each focusing on different clue(s) or addressing a different question | Identify associations and effects. Adjustments for multiple comparisons are inappropriate: false positives are not anathema at this stage, but failing to notice (or falsely rejecting) leads is. |
| Retain “odd‐ball” results, pending future insights | Avoid peer pressures to conform, and demands for repetition. |
| Assemble features in jigsaw | |
| Seek coherent explanation of associations with each clinical clue | Look for biologically plausible explanations and connections |
| Identify pieces which appear not to fit | Present conundrum to diverse experts to gain fresh insights. |
| Conduct pragmatic studies when testable cause/effect hypothesis generated | Obtain case study evidence for appropriateness of interventions licensed for treating suspected etiologic agent (i.e. explore a “new indication”). Proceed to per‐protocol analysis of randomized efficacy study of effect on facets of syndrome. |
| Where novel intervention is needed, seek pharmaceutical collaboration | Apply stages of pharmaceutical testing to candidate compounds with a view to licensing |
| Fit features into composition | |
| Define chain of events in natural history | Elucidate process mechanistically |
| Define hierarchical ordering of interventions in established cases | Tailor optimal treatment for an individual by screening tests |
| Plan for prophylaxis in early clinical syndrome and preclinical state | Identify core event(s) and perpetuating circumstance(s) |
| Proceed to pivotal multicentre studies b | |
| Perform effectiveness studies on available interventions. | Use environmentally/genetically heterogeneous subject groups, with large sample size and outcome criteria appropriate to an effectiveness study. Use intention‐to‐treat analysis to give generalizable results on benefit. Continue to explore differential effects of intervention in per‐protocol analyses to challenge robustness of hypothesis and suggest refinements. Document adverse events and their predictors. |
| Translate to service clinic | |
| Control introduction into clinical practice | Initiate surveillance program. b Be alert to unmasking further etiologic insights and unanticipated adverse events in long term. |
They allow economy in sample size; give clarity in defining differential time trends between disease facets; and allow pre‐ and post‐ presentational states to be considered as a continuum, in a disease widely accepted as having a long prodrome. Global scores, and even relevant subscores, are relatively insensitive to intervention [101]: adding in the immutable can mask important changes.
Required for “evidence beyond reasonable doubt” of clinical importance, as with discovery of H. pylori.
In contrast, neuropsychiatric disease is classified, a priori, as non‐communicable by the World Health Organization. The systemic nature of IP has been ignored as a template for intervention: the dogma of a “cold” neurodegeneration demands that constitutional illness be attributed to other causes. A one‐step pragmatic rather than an exploratory strategy is the mode. (e.g. An elegant pathologic study sees IP as evolving from the gastrointestinal tract [9], but readers seize upon just one mechanism, migration along neural pathways.) Assessment of IP by “global subjective scores” is ubiquitous. This is analogous to studying diabetes mellitus without blood glucose or cardiovascular risk factors.
Financial constraints on the risks associated with innovation mean that the same fields are reploughed. Traditional funding has favored the laboratory‐to‐clinic approach of arbitrary selection of putative pathogenic mechanisms for detailed examination. It has been directed to patching up damage (e.g. by seeking to modify components of cellular mechanisms, and replace/restore dopaminergic neurones by surgical implants/intracerebral infusion of neurotrophins), rather than getting to grips with what might be driving the process. Such dissociation is unwise: ongoing inflammation may jeopardize patching‐up.
The work was funded initially by the Medical Research Council, and subsequently by the Psychiatry Research Trust which received grants from the Hayward Foundation, the Cyril Corden Trust and Cecil Pilkington Charitable Trust, and donations from Abbott Laboratories and AstraZeneca. Barclay's Corporate Social Responsibility Ambassador, Nicholas Smith, coordinated a fundraising program. Malcolm Plant coordinated the network of support from patients and carers. Dr Ron Hutton helped with the bibliography. The review is dedicated to the memory of Anthony Dawson Paul.
References
- 1. Parkinson J. An Essay on the Shaking Palsy. London: Sherwood, Neely and Jones; 1817. [Google Scholar]
- 2. Ordenstein L. Sur la Paralysie Agitante et la Sclérose en Plaque Géneralisée. Paris: E Martinet, 1867. [Google Scholar]
- 3. Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3‐hydroxytyramine) in human brain and its relation to diseases of the extrapyramidal system. Wien Klin Wschr 1960;38:1236–1239. [DOI] [PubMed] [Google Scholar]
- 4. Schwab RS, England AC Jr, Poskanzer C, et al Amantadine in the treatment of Parkinson's disease. JAMA 1969;208:1168–70. [PubMed] [Google Scholar]
- 5. Warren JR, Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983;1:1273–5. [PubMed] [Google Scholar]
- 6. Strang RR. The association of gastro‐duodenal ulceration with Parkinson's disease. Med J Austr 1965;52:842–3. [DOI] [PubMed] [Google Scholar]
- 7. Szabo S. Dopamine disorder in duodenal ulceration. Lancet 1979;ii:880–2. [DOI] [PubMed] [Google Scholar]
- 8. Altschuler E. Gastric Helicobacter pylori infection as a cause of idiopathic Parkinson's disease and non‐arteric anterior optic ischaemic neuropathy. Med Hypotheses 1996;47:413–4. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Del Tredici K, Rüb U, De Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 10. Lang AE, Lozano AM. Parkinson's disease. New Eng J Med 1998;339:1044–53. [DOI] [PubMed] [Google Scholar]
- 11. Zimprich A, Biskup S, Lietner P, et al Mutations in the LRRK2 gene cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601–7. [DOI] [PubMed] [Google Scholar]
- 12. Calne DB. Parkinson's disease is not one disease. Parkinsonism Related Disord 2001;7:3–7. [DOI] [PubMed] [Google Scholar]
- 13. Calne DB, Snow BJ, Lee C. Criteria for diagnosing Parkinson's disease. Ann Neurol 1992;32 (Suppl):125–7. [DOI] [PubMed] [Google Scholar]
- 14. Liu B, Gao H‐M, Hong J‐S. Parkinson's disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: Role of neuroinflammation. Environ Health Perspect 2003;111:1065–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Markopoulou K, Langston JW. Candidate genes and Parkinson's disease. Where to next? Neurology 1999;53:1382–3. [DOI] [PubMed] [Google Scholar]
- 16. Charlett A, Dobbs RJ, Weller C, Dobbs SM. Stasis in the gut: The source of xenobiotic in idiopathic parkinsonism. Eur J Clin Pharmacol 1997;52 (Suppl):168. [Google Scholar]
- 17. Abbott RD, Petrovitch H, White LR, et al Frequency of bowel movements and the future risk of Parkinson's disease. Neurology 2001;57:456–62. [DOI] [PubMed] [Google Scholar]
- 18. Singaram C, Ashraf W, Gaumnitz EA, et al Dopaminergic defect of enteric nervous system in Parkinson's disease patients with chronic constipation. Lancet 1995;346:861–4. [DOI] [PubMed] [Google Scholar]
- 19. Kupsky WJ, Grimes MM, Sweeting J, Bectsch R, Cote LJ. Parkinson's disease and megacolon: Hyaline inclusions (Lewy bodies) in enteric ganglion cells. Neurology 1987;37:1253–5. [DOI] [PubMed] [Google Scholar]
- 20. Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease. Lancet Neurol 2003;2:107–16. [DOI] [PubMed] [Google Scholar]
- 21. Dobbs SM, Dobbs RJ, Weller C, Charlett A. Link between Helicobacter pylori infection and idiopathic parkinsonism. Med Hypotheses 2000;55:93–8. [DOI] [PubMed] [Google Scholar]
- 22. Dobbs RJ, Dobbs SM, Weller C, et al Role of chronic infection and inflammation in the gastrointestinal tract in the aetiology and pathogenesis of idiopathic parkinsonism. Part 1: Eradication of Helicobacter in the cachexia of idiopathic parkinsonism. Helicobacter 2005;10:267–75. [DOI] [PubMed] [Google Scholar]
- 23. Bjarnason IT, Charlett A, Dobbs RJ, et al Role of chronic infection and inflammation in the gastrointestinal tract in the aetiology and pathogenesis of idiopathic parkinsonism. Part 2: Response of facets of clinical idiopathic parkinsonism to Helicobacter pylori eradication. A randomised, double‐blind, placebo‐controlled efficacy study. Helicobacter 2005;10:276–87. [DOI] [PubMed] [Google Scholar]
- 24. Dobbs SM, Dobbs RJ, Charlett A, et al Helicobacter in idiopathic parkinsonism: A template for intervention in the role of inflammation in neuropsychiatric disease. Helicobacter 2006;11:371. [Google Scholar]
- 25. Dobbs SM, Dobbs RJ, Charlett A, Weller C, Bjarnason IT. Differential effect of Helicobacter eradication on facets of idiopathic parkinsonism: Explanation for predominantly hypokinetic or rigid syndromes. Helicobacter 2007;12:423. [Google Scholar]
- 26. Dobbs RJ, Dobbs SM, Charlett A, et al Lymphopenia in idiopathic parkinsonism and spouses of probands as a clue to an infectious environmental insult. J Neuroimmunol 2006;178 (Suppl. 1):198–9. [Google Scholar]
- 27. Weller C, Oxlade NL, Dobbs SM, Dobbs RJ, Peterson DW, Bjarnason IT. Role of chronic infection and inflammation in the gastrointestinal tract in the aetiology and pathogenesis of idiopathic parkinsonism. Part 3: Predicted probability and gradients of severity of idiopathic parkinsonism based on H. pylori antibody profile. Helicobacter 2005;10:288–97. [DOI] [PubMed] [Google Scholar]
- 28. Ellis D, Curry A, Dobbs RJ, et al Duodenal enterocyte mitochondrial involvement and abnormal bowel function in idiopathic parkinsonism In: Hanin I, Windisch M, Poewe W, Fisher A: New Trends in Alzheimer and Parkinson Related Disorders: ADPD 2007. Bologna: Medimond S.r.l., 2007; 269–72. [Google Scholar]
- 29. Drumm B, Perez Perez GI, Blaser MJ, Sherman PM. Intrafamilial clustering of Helicobacter pylori infection. N Engl J Med 1990;322:358–63. [DOI] [PubMed] [Google Scholar]
- 30. De La Fuente‐Fernandez R, Calne DB. Evidence for environmental causation of Parkinson's disease. Parkinsonism Related Disorders 2002;8:235–41. [DOI] [PubMed] [Google Scholar]
- 31. Charlett A, Dobbs RJ, Dobbs SM, Weller C, Brady P, Peterson DW. Parkinsonism: Siblings share Helicobacter pylori seropositivity and facets of syndrome. Acta Neurol Scand 1999;99:26–35. [DOI] [PubMed] [Google Scholar]
- 32. Ben‐Shlomo Y. The epidemiology of Parkinson's disease. Bailliere's Clin Neurol 1997;6:55–67. [PubMed] [Google Scholar]
- 33. Weller C, Oxlade NL, Charlett A, et al Role of inflammation in gastrointestinal tract in aetiology and pathogenesis of idiopathic parkinsonism. FEMS Immunol Med Microbiol 2005;7:129–35. [DOI] [PubMed] [Google Scholar]
- 34. Perry VH. Persistent pathogens in the parenchyma of the brain. J Neurovirol 2000;6 (Suppl. 1):86–9. [PubMed] [Google Scholar]
- 35. Watkins AD. Perceptions, emotions and immunity: An integrated homeostatic network. Q J Med 1995;88:283–94. [PubMed] [Google Scholar]
- 36. Gerlach M, Riederer P, Youdim MBH. Molecular mechanisms for neurodegeneration. Synergism between reactive oxygen species, calcium, and excitotoxic amino acids. Adv Neurol 1996;69:177–94. [PubMed] [Google Scholar]
- 37. Schapria AHV, Mann VM, Cooper JM, et al Anatomic and disease specificity of NADH CoQ1 reductase (Complex I) deficiency in Parkinson's disease. J Neurochem 1990;55:2142–5. [DOI] [PubMed] [Google Scholar]
- 38. Oeretel WH, Bandmann O, Eichhorn T, Gasser T. Peripheral markers in Parkinson's disease. An overview. Adv Neurol, 1996;69:283–91. [PubMed] [Google Scholar]
- 39. Swerdlow RH, Parks JK, Miller SW, et al Origin and functional consequences of the complex I defect in Parkinson's disease. Ann Neurol 1996;40:663–71. [DOI] [PubMed] [Google Scholar]
- 40. Koutsilieri E, Sopper S, Scheller C, Ter Meulen V, Reiderer P. Parkinsonism in HIV dementia. J Neural Transm 2002;109:767–75. [DOI] [PubMed] [Google Scholar]
- 41. Lewin PK, Edwards V. Mitochondrial inclusions in neurons of Creutzfeldt–Jakob‐like disease. Lancet 1991;337:236–7. [DOI] [PubMed] [Google Scholar]
- 42. Wooten GE, Currie LJ, Bennett JP, Harrison MB, Trugman JM, Parker WD. Maternal inheritance in Parkinson's disease. Ann Neurol 1997;41:265–8. [DOI] [PubMed] [Google Scholar]
- 43. Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci USA 1988;85:6465–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andersson SG, Karlberg O, Canbäck B, Kurland CG. On the origin of mitochondria: A genomics perspective. Philos Trans R Soc London B Biol Sci 2003;358:165–77; discussion 177–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marshall DG, Dundon WG, Beesley SM, Smyth CJ. Helicobacter pylori– A conundrum of genetic diversity. Microbiology 1998;144:2925–39. [DOI] [PubMed] [Google Scholar]
- 46. Boka G, Anglade P, Wallach D, Javoy‐Agid F, Agid Y, Hirsch EC. Immunocytochemical analysis of tumour necrosis factor and its receptors in Parkinson's disease. Neurosci Lett 1994;172:151–4. [DOI] [PubMed] [Google Scholar]
- 47. El‐Omar EM. The importance of interleukin 1β in Helicobacter pylori associated disease. Gut 2001;48:743–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. El‐Omar EM, Rabkin CS, Gammon MD, et al Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 2003;124:1193–201. [DOI] [PubMed] [Google Scholar]
- 49. Dobbs SM, Dobbs RJ, Charlett A, et al A direct, or surrogate, not necessarily unique, role for Helicobacter infection in pathogenesis of idiopathic parkinsonism: Analogy and contrast with gastric cancer In: Hanin I, Windisch M, Poewe W, Fisher A: New Trends in Alzheimer and Parkinson Related Disorders: ADPD 2007. Bologna: Medimond S.r.l., 273–6. [Google Scholar]
- 50. Krüger R, Hardt C, Tschentscher F, et al Genetic analysis of immunomodulating factors in sporadic Parkinson's disease. J Neural Transm 2000;107:553–62. [DOI] [PubMed] [Google Scholar]
- 51. Schulte T, Schöls L, Müller T, Woitalla D, Berer K, Krüger R. Polymorphisms in the interleukin‐1 alpha and beta genes and the risk for Parkinson's disease. Neurosci Lett 2002;326:70–2. [DOI] [PubMed] [Google Scholar]
- 52. McGeer PL, Yasojima K, McGeer EG. Association of interleukin‐1β polymorphisms with idiopathic Parkinson's disease. Neurosci Lett 2002;326:67–9. [DOI] [PubMed] [Google Scholar]
- 53. Nishimura M, Mizuta I, Mizuta E, Yamasaki S, Ohta M, Kuno S. Influence of interleukin‐1β gene polymorphisms on age‐at‐onset of sporadic Parkinson's disease. Neurosci Lett 2000;284:73–6. [DOI] [PubMed] [Google Scholar]
- 54. Nishimura M, Mizuta I, Mizuta E, et al Tumour necrosis factor gene polymorphisms in patients with sporadic Parkinson's disease. Neurosci Lett 2001;311:1–4. [DOI] [PubMed] [Google Scholar]
- 55. Doll R, Peto R. Mortality in relation to smoking: 20 years’ observations on male British doctors. Br Med J 1976;2:1525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Charlett A, Dobbs RJ, Purkiss AG, et al Cortisol is higher in parkinsonism and associated with gait deficit. Acta Neurol Scand 1998;97:77–85. [DOI] [PubMed] [Google Scholar]
- 57. Dobbs SM, Charlett A, Dobbs RJ, Weller C. Does the protective effect of tobacco smoking in idiopathic parkinsonism reside in its laxative action. Naunyn Schmiedebergs Arch Pharmacol 1998;358 (Suppl. 2):479. [Google Scholar]
- 58. Murray LJ, McCrum EE, Evans AE, Bamford KB. Epidemiology of Helicobacter pylori infection amongst 4742 randomly selected subjects from Northern Ireland. Int J Epidemiol 1997;26:880–7. [DOI] [PubMed] [Google Scholar]
- 59. Charlett A, Dobbs RJ, Dobbs SM, Weller C, Peterson DW. Parkinsonism: Differential age‐trend in Helicobacter pylori antibody. Alimentary Pharmacol Therapeutics 2000;14:1199–205. [DOI] [PubMed] [Google Scholar]
- 60. Charlett A, Purkiss AG, Dobbs SM, Dobbs RJ, Weller C, Peterson DW. Association of circulating TNF‐α and IL‐6 with ageing and parkinsonism. Acta Neurol Scand 1999;100:34–41. [DOI] [PubMed] [Google Scholar]
- 61. Weller C, Nicholson PW, Dobbs SM, Bowes SG, Purkiss A, Dobbs RJ. Reduced axial rotation in the spouses of sufferers from idiopathic Parkinsonism. Age Ageing 1992;21:189–94. [DOI] [PubMed] [Google Scholar]
- 62. Kirollos C, ’Neill O. CJA, Dobbs RJ, et al Quantification of the cardinal signs of parkinsonism and of associated disability in spouses of sufferers. Age Ageing 1993;22:20–6. [DOI] [PubMed] [Google Scholar]
- 63. Kirollos C, Charlett A, ’Neill O. CJA, et al Objective measurement of activation of rigidity: Diagnostic, pathogenetic and therapeutic implications in parkinsonism. Br J Clin Pharmacol 1996;41:557–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. O.’Neill CJA, Richardson MD, Charlett A, et al Could seborrhoeic dermatitis be implicated in the pathogenesis of parkinsonism? Acta Neurol Scand 1994;89:252–7. [DOI] [PubMed] [Google Scholar]
- 65. Purkiss AG, Charlett A, Wright DJ, et al Associations of immunoglobulins, IgG, IgA and IgM, with treatment in those with diagnosed parkinsonism, and with parkinsonian‐like postural abnormality in those without. Br J Clin Pharmacol 1998;41:457–8. [Google Scholar]
- 66. Purkiss AG, Charlett A, Weller C, Dobbs SM, Dobbs RJ. Association of serum immunoglobulin subclasses IgA1 and IgA2 with facets of parkinsonism. Br J Clin Pharmacol 1998;45:207. [Google Scholar]
- 67. Charlett A, Dobbs SM, Dobbs RJ, Peterson DW, Weller C. Could antibodies to Helicobacter pylori urease explain the facets of parkinsonism quantified in subjects who have and have not passed the diagnostic threshold? Br J Clin Pharmacol 1999;48:888. [Google Scholar]
- 68. Charlett A, Dobbs RJ, Dobbs SM, Weller C, Peterson DW. Does Helicobacter pylori account for increase in serum interleukin‐6 and tumour necrosis factor‐α with age and interleukin‐6 and cortisol with parkinsonism? Br J Pharmacol 1999;127 (Suppl):79. [Google Scholar]
- 69. Charlett A, Weller C, Oxlade N, Dobbs SM, Dobbs RJ. Systemic cortisol response to Helicobacter pylori vacuolating toxin in parkinsonism and controls. Br J Clin Pharmacol 2000;131:220. [Google Scholar]
- 70. Dobbs RJ, Dobbs SM, Charlett A, Weller C. Downward shift in serum IgM with Helicobacter pylori seropositivity. J Infection 2000;41:240–4. [DOI] [PubMed] [Google Scholar]
- 71. Charlett A, Dobbs RJ, Weller C, Dobbs SM, Peterson DW. Evidence for autoimmunity triggered by Helicobacter pylori in idiopathic parkinsonism. Br J Clin Pharmacol 2000;49:506–7. [Google Scholar]
- 72. Oxlade N, Charlett A, Weller C, Dobbs RJ, Dobbs SM. Discriminant index for seborrhoeic dermatitis based on antibody profile against Helicobacter pylori . Brit J Clin Pharmacol 2000;49:506. [Google Scholar]
- 73. Drossman DA, ed. The Functional Gastrointestinal Disorders. Diagnosis, Pathology, and Treatment – A Multinational Consensus. Boston, MA: Little, Brown and Co, 1994. [Google Scholar]
- 74. Lundgren A, Suri‐Payer E, Enarsson K, Svennerholm A‐M, Lundin BS. Helicobacter pylori‐specific CD4+ CD25high regulatory T‐cells suppress memory T‐cell responses to Helicobacter pylori in infected individuals. Infect Immun 2003;71:1755–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schmees C, Prinz C, Treptau T, et al Inhibition of T‐cell proliferation by Helicobacter pylori gamma‐glutamyl transpeptidase. Gastroenterology 2007;132:1820–33. [DOI] [PubMed] [Google Scholar]
- 76. Nilsson H‐O, Pietroiusti A, Gabrielli M, Assunta Zocco M, Gasbarrini G, Gasbarrini A. Helicobacter pylori and extragastric diseases – Other Helicobacters . Helicobacter 2005;10 (Suppl. 1):54–65. [DOI] [PubMed] [Google Scholar]
- 77. McGeer P, et al Reactive microglia are positive for HLA‐DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 1998;38:1285–9. [DOI] [PubMed] [Google Scholar]
- 78. Hunot S, Brugg B, Ricard D, et al Nuclear translocation of NF‐kappaB is increased in dopaminergic neurons of patients with Parkinson's disease. Proc Natl Acad Sci USA 1997;94:7531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mogi M, et al Interleukin‐1‐beta, interleukin‐6, epidermal growth‐factor‐alpha and transforming growth‐factor‐alpha are elevated in the brain from parkinsonian patients. Neurosci Letts 1994;180:147–50. [DOI] [PubMed] [Google Scholar]
- 80. Mogi M, et al Interleukin (IL)‐1‐beta, IL‐2, IL‐4, Il‐6 and transforming growth‐factor‐alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson's disease. Neurosci Letts 1996;211:13–6. [DOI] [PubMed] [Google Scholar]
- 81. Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson's disease. Brain 2005;28:2665–74. [DOI] [PubMed] [Google Scholar]
- 82. Chiba S, et al A correlation study between serum adenosine‐deaminase activities and peripheral lymphocyte subsets in Parkinson's disease. J Neurol Sci 1995;132:170–3. [DOI] [PubMed] [Google Scholar]
- 83. Fiszer U, et al Parkinson's disease and immunological abnormalities: Increase of HLA‐DR expression on monocytes in cerebrospinal fluid and of CD45RO+ T cells in peripheral blood. Acta Neurol Scand 1994;90:160–6. [DOI] [PubMed] [Google Scholar]
- 84. Bas J, Calopa M, Mestre M, et al Lymphocyte populations in Parkinson's disease and in rat models of Parkinsonism. J Neuroimmunol 2001;113:146–52. [DOI] [PubMed] [Google Scholar]
- 85. Hisanaga K, Asagi M, Itoyama Y, Iwasaki Y. Increase in peripheral CD4 bright+ CD8 dull+ T cells in Parkinson disease. Arch Neurol 2001;58:1580–3. [DOI] [PubMed] [Google Scholar]
- 86. Fiszer U, et al γδ+ T cells are increased in patients with Parkinson's disease. J Neurol Sci 1994;121:39–45. [DOI] [PubMed] [Google Scholar]
- 87. Fiszer U, et al Humoral response to hsp 65 and hsp 70 in cerebrospinal fluid in Parkinson's disease. J Neurol Sci 1996;139:66–70. [PubMed] [Google Scholar]
- 88. Poupard A, Emile J. Autoimmunity in Parkinson's disease In: Hassler RG. Christ JF, eds. Parkinson‐Specific Motor and Mental Disorders. Role of the Pallidum: Pathophysiological Biochemical and Therapeutic Aspects. Adv Neurol 40. New York: Raven Press, 1984;307–14. [Google Scholar]
- 89. McRae A, Degueurce A, Gottfries C‐G, Karlsson I, Svennerholm L, Dahlström A. Antibodies in the CSF of a Parkinson patient recognizes neurons in rat mesencephalic regions. Acta Physiol Scand 1986;126:313–5. [DOI] [PubMed] [Google Scholar]
- 90. Chen S, Le WD, Xie WJ, et al Experimental destruction of substantia nigra initiated by Parkinson's disease immunoglobulins. Arch Neurol 1998;55:1075–80. [DOI] [PubMed] [Google Scholar]
- 91. Dahlström A, Wigander A, Lundmark K, Gottfries C‐G, Carvey PM, McRae A. Investigations on auto‐antibodies in Alzheimer's and Parkinson's diseases using defined neuronal cultures. J Neural Transm 1990;29 (Suppl):195–206. [DOI] [PubMed] [Google Scholar]
- 92. Defazio G, Dal Toso R, Benvegnù D, Minozzi MC, Cananzi AR, Leon A. Parkinson serum carries complement‐dependent toxicity for rat mesencephalic dopaminergic neurons in culture. Brain Res 1994;633:206–12. [DOI] [PubMed] [Google Scholar]
- 93. He Y, Le W‐D, Appel SH. Role of Fcγ receptors in nigral cell injury induced by Parkinson disease immunoglobulin injection into mouse substantia nigra. Exp Neurol 2002;176:322–7. [DOI] [PubMed] [Google Scholar]
- 94. Karlsen NR, Reinvang I, Frøland SS. Slowed reaction time in asymptomatic HIV‐positive patients. Acta Neurol Scand 1992;86:242–6. [DOI] [PubMed] [Google Scholar]
- 95. Berger JR, Arendt G. HIV dementia: The role of the basal ganglia and dopaminergic systems. J Psychopharmacol 2000;14:214–21. [DOI] [PubMed] [Google Scholar]
- 96. Bateman PA, Miller AR, Sedgwick PM, Griffin GE. Autonomic denervation in jejunal mucosa of homosexual men infected with HIV. AIDS 1991;5:1247–52. [DOI] [PubMed] [Google Scholar]
- 97. Kwon H‐J, Kang J‐O, Cho S‐H, et al Presence of human mycoplasma DNA in gastric tissue samples from Korean chronic gastritis patients. Cancer Sci 2004;95:311–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bower JH, Ahlskog JE, Maraganone DM, Rocca WA, Peterson BJ. Immunologic diseases, anti‐inflammatory drugs and Parkinson's disease: A case control study. Neurology 2006;67:494–6. [DOI] [PubMed] [Google Scholar]
- 99. Tsui JK, Calne DB, Wang Y, Schulzer M, Marion SA. Occupational risk factors in Parkinson's disease. Can J Public Health 1999;90:334–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tomas‐Camardiel M, Rite I, Herrera AJ, et al Minocycline reduces the lipopolysaccharide‐induced inflammatory reaction, peroxynitrite‐mediated nitration of proteins, disruption of the blood‐brain barrier, and damage in the nigral dopaminergic system. Neurobiol Dis 2004;16:190–201. [DOI] [PubMed] [Google Scholar]
- 101. Bowes S, Dobbs RJ, Henley M, et al Objective evidence for tolerance, against a back‐ground of improvement during maintenance therapy with controlled release levodopa/carbidopa. Eur J Clin Pharmacol 1992;43:483–9. [DOI] [PubMed] [Google Scholar]