

Abstract
Hartnup disorder (OMIM 234500) is an autosomal recessive disorder, which was first described in 1956 as an aminoaciduria of neutral amino acids accompanied by a variety of symptoms, such as a photo‐sensitive skin‐rash and cerebellar ataxia. The disorder is caused by mutations in the neutral amino acid transporter B0AT1 (SLC6A19)1. To date 21 mutations have been identified in more than twenty families. SLC6A19 requires either collectrin or angiotensin‐converting enzyme 2 for surface expression in the kidney and intestine, respectively. This ties SLC6A19 together with more complex functions such as blood‐pressure control, glomerular structure, and exocytosis. © 2009 IUBMB IUBMB Life, 61(6): 591–599, 2009
Keywords: angiotensin‐converting enzyme, epithelial cells, aminoaciduria
HARTNUP DISORDER, PHYSIOLOGICAL, AND CLINICAL ASPECTS
Hartnup disorder (OMIM 234500) is an autosomal recessive disorder occurring at a frequency of about 1:30,000 in European populations ( 1, 2). The disorder was first described in 1956 by Baron et al. (3) and the title of the publication describes the main clinical features of the disorder in a most concise way: ‘Hereditary pellagra‐like skin rash with temporary cerebellar ataxia, constant renal aminoaciduria and other bizarre biochemical features.’ Hartnup disorder received its name from the first described case Eddie Hartnup (Eddie H. in the original publication).
Although these clinical symptoms were recorded in Eddie Hartnup's case, these days most cases are asymptomatic. For instance in the Australian families described more recently, no clinical symptoms were reported other than a skin‐rash or diarrhoea in infancy ( 2, 4, 5). In a couple of cases described by Scriver et al. (6), individuals with Hartnup disorder tended to have smaller body height and weight than their siblings, but most of them were free of clinical symptoms. Nevertheless a couple of cases with all “classical” symptoms were reported from China and Japan (7, 8). In addition, acrodermatitis enterohepatica, a more severe type of skin lesions, has been reported in a recent case from Turkey (9). The clinical symptoms in the Chinese individual on admission were described as follows: “Three month ago, she felt numbness, cold, and pain below her knees. She could not walk steadily and hold a pen tightly when writing. Twenty days before admission more severe symptoms developed namely, intermittent involuntary movement appeared on her head, face and bilateral upper extremities which further developed into continuous and involuntary motion, which was aggravated during emotional upset. Her emotion was slightly instable with depression and frequent outburst of temper. She had a history of dermatitis due to photosensitivity. The rashes in the sun exposed areas such as the face, neck and dorsal surface of the hands recurred every summer and disappeared when summer was over” (7).
The renal aminoaciduria is the hallmark of the disorder, because of the variability of other symptoms, and most if not all patients were diagnosed by urine analysis. A typical example is the urine analysis of Eddie Hartnup himself as reported by Cusworth and Dent ( 10) (Fig. 1). The aminoaciduria is restricted to neutral amino acids although slightly elevated amounts of glutamate are often found as well. Particularly relevant are the increased amounts of tryptophan (not shown in Fig. 1), pointing to a lack of tryptophan reabsorption. The aminoaciduria and clinical symptoms are caused by a defect of the major renal and intestinal transporter for neutral amino acids, which has been termed B0 (Denoting a transporter for neutral amino acids (0) with broad specificity; the upper case is used to indicate Na+‐dependence) (11, 12) or neutral brush border (NBB) (13).
Figure 1.

Renal clearance of amino acids in reference subjects and in Eddie Hartnup as reported by Cusworth and Dent ( 10) (modified with permission). A renal clearance of 120 indicates a substance, which is neither reabsorbed nor excreted by the kidney tubules.
The clinical symptoms of Hartnup disorder are remarkably similar to pellagra or niacin deficiency. This vitamin deficiency is characterized by a photosensitive dermatitis. Advanced pellagra is accompanied by depressive psychosis and diarrhea ( 14). Niacin comprises nicotinic acid and nicotinamide, the two compounds having the biological activity of this vitamin. Nicotinic acid is used as a precursor for NAD(P)H biosynthesis (14). Although niacin is considered a vitamin, the body can synthesize significant amounts of NAD(P)H from tryptophan. The biochemical basis of the skin rash is ill understood, but in Hartnup disorder it appears to respond to niacin supplementation, suggesting that the reduced availability of tryptophan is the most likely cause for the skin‐rash. In addition reduced amounts of the histidine metabolite urocanic acid have been reported in the skin of pellagrins (14). The compound is important for the absorption of UV light in normal skin and histidine transport is impaired in Hartnup disorder (Fig. 1). Reduced levels of urocanic acid are also observed in hereditary zinc deficiency causing acrodermatities hepatica, which has been observed in one case of Hartnup disorder as mentioned above. In addition, mammalian skin contains the entire metabolic pathway to convert l‐tryptophan to melatonin (15). Melatonin exerts a variety of skin protective effects and is considered to be an important regulator of skin function and structure.
The ataxia might also be related to tryptophan metabolism and its conversion to serotonin (5‐hydroxytryptamine). Serotonin plays an important role as a neurotransmitter in the modulation of anger, aggression, body temperature, mood, sleep, sexuality, appetite, and metabolism. Although low levels of B0AT1 are found in the brain, its main site of expression is the brush‐border of kidney and intestinal epithelial cells. Serotonin levels in the brain, however, correspond to the levels of tryptophan in the blood because the K m of tryptophan hydroxylase, the rate limiting step of serotonin biosynthesis, is higher than tryptophan concentrations in the brain or the circulation ( 14). As a result, tryptophan has antidepressive properties and its metabolite 5‐hydroxytryptophan causes regression of various forms of cerebellar ataxia (16). Little is known whether changed plasma levels of other neurotransmitter precursor amino acids, such as tyrosine or histidine may contribute to neurological symptoms. It has been noted that clinical symptoms are more likely to occur in individuals with low plasma amino acid levels (6).
Alternatively it has been proposed that toxic bacterial degradation products of tryptophan might be involved ( 17). Bacterial degradation products of tryptophan such as indol‐compounds (indoxyl sulfate, indole acetic acid, indolylacetyl glutamine) and other amino acids have been identified in the urine of individuals with Hartnup disorder, demonstrating that they are absorbed in the intestine and distributed throughout the body. The occurrence of these bacterial degradation products were indeed the first evidence that amino acid transport is impaired in the intestine (18). The involvement of indole compounds in the onset of ataxia, however, appears unlikely in view of the asymptotic cases of Hartnup disorder, in which the degradation products of tryptophan would still be produced.
IS HARTNUP DISORDER GENETICALLY COMPLEX OR SIMPLE
Hartnup disorder, judged by its hallmark aminoaciduria, is a classical autosomal recessive disorder. The variability of the clinical phenotype, however, suggests modifying factors, which reduce the penetrance of the disorder. It appears likely that in countries with a protein‐rich western diet, symptoms rarely occur because peptide uptake in the intestine –mediated by the peptide transporter PepT1 ( 19)‐ compensates for the lack of amino acid uptake. In support of this notion it has been demonstrated that peptide uptake in individuals with Hartnup disorder is unimpaired (20, 21). During growth, the demand for amino acids is increased explaining why symptoms predominantly occur in children. In areas with carbohydrate‐rich, protein‐poor nutrition such as rural China, cases with all symptoms may occur. The possible role of nutrition as a modifying factor in Hartnup disorder was observed by Scriver et al. (6), who noticed that individuals with clinical manifestations appear to have low plasma amino acid concentrations. This was further highlighted in the same study by examination of a child with undiagnosed Hartnup disorder in which cow‐milk allergy was suspected to cause diarrhea. Change of the diet to soy milk triggered the onset of the skin‐rash and a significant weight loss in this child. Surprisingly, the amino acid composition of soy protein and milk protein is similar (22), but the protein concentration of the soy milk was not reported in the study by Scriver et al. In addition to environmental factors, evidence has also been found for genetic complexity. For example purely renal cases of Hartnup disorder have been described (6, 23) as well as a purely intestinal case (24). Other conditions with general aminoaciduria are MODY3 (maturity onset diabetes of the young) (25), Fanconi syndrome (26), diabetes (27) and pregnancy (28). In some of these disorders the aminoaciduria can now be explained, demonstrating intriguing connections in the physiology of amino acid absorption. Although largely a simple Mendelian disorder, the observed complexity of Hartnup disorder points to modifying factors, some of which have now been identified at the molecular level.
IDENTIFICATION OF GENE/PROTEIN
In 2001 Nozaki et al. ( 29) reported a family with Hartnup disorder that due to a first cousin marriage allowed homozygosity mapping of regions associated with the disease. This study pointed to the tip of chromosome 5 as the region linked to the disorder. The completion of the human genome at the same time allowed a candidate approach for the identification of the causative gene. A search for genes that encode proteins with multiple transmembrane spanning domains revealed the presence of two related genes in this region (30). One of these genes had already been identified as a member of the SLC6 family of neurotransmitter transporters and named XT2, XTRP2 or ROSIT (31, 32). The other was novel and in the first drafts of the human and mouse genomes annotated as “similar to XT2”. Both genes are located next to each other on chromosome 5 spanning a region of about 50 kB. Expression of the mouse “similar to XT2” paralogue in Xenopus laevis oocytes demonstrated it to be a general transporter for neutral amino acids (33). Consequently, it was named B0AT1 (B0 amino acid transporter) and given an official gene name (SLC6A19). It facilitates Na+‐dependent uphill transport of its substrates (34, 35). The transporter prefers large aliphatic neutral amino acids. Notably it transports eight of the 10 essential amino acids, namely leucine, isoleucine, valine, methionine, phenylalanine, tryptophan, threonine, and hisitidine. Most amino acids have a similar maximum transport rate, but the K m differs, ranging from 1.1 mM (leucine), about 4 mM (alanine, phenylalanine) to 11 mM (glycine). In the intestine, B0AT1 appears to be the major transport activity for tryptophan ‐ the lack of which is key to the pathology of Hartnup disorder. Unlike other members of the SLC6 family B0AT1 is chloride independent and cotransports only 1 Na+ per amino acid. Subsequently, it was demonstrated that the human B0AT1 has the same transport activity as the mouse orthologue (4, 5) and similar affinity for its substrates. Low expression levels of the transporter in Xenopus laevis oocytes, until recently, hampered further characterization of the transporter. The transporter is mainly expressed in the S1 segment of the mouse proximal tubule, but is found in all segments of the proximal tubule in human tissue (36, 37).
MUTATIONS
The identification of B0AT1 (SLC6A19) as the gene for Hartnup disorder initiated sequencing attempts in several families with Hartnup disorder. These revealed mutations in SLC6A19, which cosegregated with the disease and inactivated transport function ( 4, 5). While initially there was some speculation about additional Hartnup disorder genes, reevaluation of the families identified so far and sequencing of new families revealed mutations in SLC6A19 in all cases (38). To date 21 different mutations have been identified in more than 20 families (Table 1 and Fig. 2). Samples of the original Hartnup case led to the identification of a splice mutation in intervening sequence 8 (4).
Table 1.
Mutations associated with Hartnup disorder
| Mutation (DNA) | Mutation (protein) | Frequency | Reference |
|---|---|---|---|
| Missense | |||
| 169C>T | R57C | n.r. | ( 4) |
| 196G>A | G66R | <0.001 | ( 37) |
| 205G>A | A69T | n.r. | ( 48) |
| 277G>A | G93R | <0.001 | ( 37) |
| 517G>A | D173N | 0.004–0.007 | ( 5) |
| 532C>T | R178X | <0.001 | ( 37) |
| 719G>A | R240Q | <0.001 | ( 5) |
| 725T>C | L242P | n.r. | ( 5) |
| 794C>T | P265L | n.r. | ( 48) |
| 850G>A | G284R | <0.001 | ( 37) |
| 982C>T | R328C | <0.001 | ( 37) |
| 1213G>A | E405K | <0.001 | ( 37) |
| 1501G>A | E501K | n.r. | ( 5) |
| 1550A>G | D517G | <0.001 | ( 37) |
| 1735C>T | P579L | n.r. | ( 48) |
| Nonsense | |||
| 682‐683AC>TA | T228X | n.r. | ( 4) |
| 718C>T | R240X | 0.001 | ( 5) |
| Deletions | |||
| 340delC | L114fsX114 | n.r. | ( 4) |
| c884_885delTG | V295fsX351 | n.r. | ( 4) |
| Splice site | |||
| IVS8 + 2G | Aberrant splicing | <0.01 | ( 5) |
| IVS11 + 1A | Aberrant splicing | n.r. | ( 5) |
n.r.: not reported.
Figure 2.

Overview of Hartnup disorder associated mutations in a topological model of human SLC6A19. The arrangement of the helices reflects the consensus model of the SLC6 family based on the structure of the leucine transporter LeuT from Aquifex aeolicus ( 40). Due to this arrangement, the backbone is graphically interrupted between helix 5 and 6. The substrate and residues involved in substrate binding are shown in green. The Na+‐ion is shown in red and residues involved in Na+‐binding are decorated with a red ring. Mutants associated with Hartnup disorder are labelled and shown in orange with a blue ring. Residues thought to form the two gates of the transporter are connected by a dashed line and coloured blue (basic residues) or yellow (acidic).
The most frequent mutation in pedigrees of western European descent appears to be the D173N allele. Genetic analysis suggests that it probably originated in central Europe about 50 generations ago and was introduced into Australia and Canada by emigrants carrying the allele ( 39). Functional analysis of mutations associated with Hartnup disorder revealed further complexity. The D173N allele only partially inactivates the protein (5). Since Hartnup disorder is recessive, a partial inactivation is unlikely to cause Hartnup disorder. Similarly the R240Q mutation, if anything even increased transport activity (5). These mutations are now explained by B0AT1 associated proteins (see below). Functional analysis of SLC6A19 mutations was significantly improved by the high‐resolution structure of the bacterial SLC6 homologue LeuT, a leucine transporter isolated from Aquifex aeolicus (40). The bacterial transporter has similar substrate specificity as B0AT1. One notable exception is tryptophan, which is a competitive inhibitor of LeuT (41) but a substrate, albeit weak, of B0AT1 (34). Another difference between the two transporters is the cotransport of 2 Na+ in the case of LeuT, while only 1 Na+ is cotransported by B0AT1. There is good evidence that the Na+‐binding site 1 is the active binding site in B0AT1. Notably, the carboxyl‐group of the substrate leucine forms part of the Na+‐binding site in the bacterial transporter (42). This elegantly explains a behavior that was noted during the functional characterization of the transporter, namely that increased Na+ concentrations decrease the apparent K m for the substrate and vice versa (34). Consistent with the substrate forming part of the Na+‐binding site, Camargo et al. (35) proposed that leucine binds first in an ordered mechanism, while Bohmer proposed a more random model (34). Thus Na+ could also contribute to the leucine binding site. The role of the second Na+‐binding site in B0AT1 remains unclear. The binding site is conserved and a second Na+ may play a structural role in the transporter without being translocated.
The sequence similarity with LeuT allowed the generation of homology models, which have been instrumental in explaining the loss of function in some mutations ( 42). G284, for instance, is located in helix 6 of the transporter (Figs. 2 and 3). Together with helix 1 it lines the substrate translocation pathway and has been suggested to move during the transport cycle in a rocker‐switch mode (40). In the center of the membrane both helices are partially unwound, allowing backbone residues to contact the cotransported Na+‐ion. Due to the flexibility afforded by its lack of a side chain, residue G284 allows unwinding of the helix in this area. Mutation of R57, located in helix 1, disrupts the proposed extracellular gate of the transporter (Fig. 2). This gate is essential for the transport mechanism closing the pore by interacting with D486 (Fig. 4). Mutation of R57 to a neutral residue abolishes this interaction. A completely different mode of action was suggested for the R240Q mutation by the homology model. The residue is located at the apex of the protein protruding to the outside (37). As a result it appeared unlikely to affect the transporter itself but rather pointed to an interaction site with accessory proteins.
Figure 3.

Location of the Hartnup disorder associated mutation G284. A homology model of B0AT1 (SLC6A19) was created based on the structure of LeuT from Aquifex aeolicus. Helix 1 is shown in yellow and helix 6 is shown in blue. The cartoon indicates the nonhelical part of both helices close to the substrate binding site. Other helices are depicted in red. Two Na+‐ions are depicted in analogy to the bacterial structure. Experimental evidence suggests that Na is cotransported in B0AT1. The presence and function of Na is speculative. G284 is located in the center of helix 6, where the helical structure is interrupted. The figure was generated using Pymol (DeLano Scientific).
Figure 4.

Location of the Hartnup disorder associated mutation R57. A homology model of B0AT1 (SLC6A19) was created based on the structure of LeuT from Aquifex aeolicus. Helix 1 is shown in yellow and helix 6 is shown in blue. Other helices are depicted in red. The position of the substrate leucine and the hypothetical Na are indicated. Residue R57 is in close contact with D486 and forms an ionic bond, which closes the translocation pore toward the extracellular space. The figure was generated using Pymol (DeLano Scientific).
ASSOCIATED PROTEINS
A major breakthrough in our understanding of B0AT1 function was the discovery that mice deficient for the protein collectrin excreted large amounts of neutral amino acids in the urine, similar to Hartnup disorder ( 43, 44). These amino acids were visible as a precipitate when the urine was chilled. Subsequent analysis of renal brush‐border membrane vesicles revealed an almost complete absence of B0AT1 in the brush border. Collectrin is a type I membrane protein of 27 kDa (also known as TMEM27). The protein has been implicated in vesicular traffic, insulin release in pancreatic beta cells and in the function of ciliated cells in the collecting duct (hence the name collectrin) (45). However, the only noticeable phenotype of the collectrin deficient mice so far appears to be the general aminoaciduria. Subsequent experiments showed that coexpression of B0AT1 together with collectrin in Xenopus oocytes caused a five to tenfold increase of transport activity (43). Thus B0AT1 appears to require heterodimerization with collectrin for stabilization and surface expression. Collectrin is known to bind to the SNARE complex by interacting with snapin (46) (Fig. 5) and also appears to interact with elements of the cytoskeleton such as myosin heavy chain II‐A and γ‐actin (47). It is tempting to speculate that it is involved in the fusion of B0AT1 containing vesicles with the plasma membrane. The requirement for collectrin also explains the poor expression, particularly of human B0AT1 in Xenopus oocytes. Interestingly, collectrin is expressed in the kidney but not in the intestine, where B0AT1 is abundant. Subsequently, it was reported that in the intestine, the collectrin related peptidase angiotensin‐converting enzyme 2 (ACE2) functions as the partner of B0AT1 (37, 48). ACE2 is a carboxypeptidase with several physiological functions. It inactivates the blood pressure regulating peptide angiotensin (II), by converting it to angiotensin 1–7 (Fig. 5). In the intestine it is involved in peptide digestion, feeding neutral amino acids to the B0AT1 transporter (37). ACE2 has also been identified as the SARS virus receptor (49) and lack of ACE2 causes glomerulosclerosis (50). Reevaluation of a number of mutations in oocytes coexpressing B0AT1 and ACE2 (or collectrin), revealed that mutation D173N completely inhibits transport activity and not partially as observed when analysed in oocytes expressing B0AT1 alone (37). The R240Q mutation in the combined expression system also resulted in a significantly reduced transport activity. Thus interaction of B0AT1 with ACE2/collectrin helps to explain all disease causing alleles discovered so far. The interaction with two different proteins in kidney and intestine also suggests an explanation for cases of purely renal or intestinal Hartnup disorder. These might be caused either by mutations affecting interaction with one but not the other protein. Alternatively, mutations in ACE2 or collectrin could abolish interaction with B0AT1, but those have not yet been identified in Hartnup disorder. Two pieces of evidence support this theory. First, the collectrin‐deficient mouse presents with renal Hartnup disorder (43), while the ACE2‐deficient mouse presents with intestinal Hartnup disorder and ACE2 related pathology (48). Secondly, some of the mutations associated with Hartnup disorder demonstrate a slightly differential interaction with ACE2 or collectrin (48). It has not been investigated whether mutations in B0AT1 also affect surface expression of ACE2 and may thus affect blood pressure regulation. A recent study reported the association of a specific microsatellite within the SLC6A19 gene (SLC6A19‐MS7) with hypertension (51), but It remains unclear why the other microsatellites located in the gene lack this association.
Figure 5.
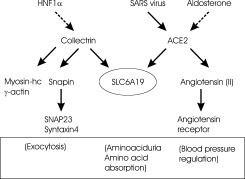
Overview of SLC6A19 interacting proteins. Protein‐Protein interactions are depicted by full arrows. Transcriptional regulation is indicated by dashed arrows. Physiological responses are shown in brackets.
The interaction of B0AT1 with collectrin also explains the aminoaciduria in cases of MODY3, which is caused by mutations in the transcription factor HNF1α (hepatic nuclear factor 1 alpha) ( 52). HNF1α is one of the major transcription factors controlling expression of collectrin in the kidney. Inactivation of HNF1α will cause a lack of expression of collectrin, which in turn will cause reduced expression of B0AT1 and the proline transporter IMINO (Fig. 5). Inactivation of HNF1α in mice results in hepatic dysfunction, phenylketonuria and renal Fanconi syndrome. The aminoaciduria observed in Fanconi syndrome thus might be related to a loss of collectrin or down‐regulation of HNF1α expression. In agreement with this notion, HNF1α has also been shown to control expression of the apical glucose transporter SGLT1 (53) and possibly of the cationic amino acid transporter rbAT/b0,+AT (54). The aminoaciduria observed in diabetes or pregnancy remains unexplained, but may also involve reduced levels of collectrin.
As pointed out above, peptide transport is likely to compensate for amino acid depletion in Hartnup disorder. So far largely synonymous single nucleotide polymorphisms (SNPs) have been identified in the peptide transporter PepT1 ( 55). Only one mutation (F28Y) was detected that reduced transport activity. It has not been investigated whether SNPs in promoter or intronic regions may alter expression of the transporter and could therefore modulate the phenotype of Hartnup disorder. Neither has it been investigated whether the recent cases of classical Hartnup disorder might carry the F28Y mutation in PepT1 thereby aggravating symptoms.
PATRIARCH OF A NEW FAMILY
As outlined above, B0AT1 is a member of the SLC6 family of neurotransmitter transporters. When this family was discovered, homology cloning revealed a subfamily of related transporters the function of which could not be identified. These transporters were named XT1 (alias NTT4) ( 56, 57), XT2 (alias ROSIT) (31, 32), XT3 (58), NTT5 (59), and v7‐3 (60). This branch of the SLC6 family was hence called the orphan transporter branch. The discovery of B0AT1 placed it in the orphan transporter branch suggesting that the “orphans” might in fact be amino acid transporters (61) (Table 2). This was subsequently confirmed for XT3, showing it to be the apical system IMINO transporter (66, 67), a transporter specific for proline, hydroxyproline, betaine, and MeAIB. Next v7‐3 was identified as B0AT2 a transporter with similar properties as B0AT1, but with a more narrow substrate specificity preferring branched‐chain amino acids and methionine (62, 63). More recently NTT4 was shown to be a neutral amino acid transporter (64, 65), which is either Na+ or H+‐dependent and was aptly named B0AT3. XT2 appears to prefer glycine and alanine (manuscript in preparation). The orphan transporters are thus forming a large family of Na+‐dependent neutral amino acid transporters.
Table 2.
Transport function of orphan transporters of the SLC6 family
| Solute carrier | Name | Alias | Substrates (References) |
|---|---|---|---|
| SLC6A15 | B0AT2 | V7‐3, NTT7‐3, SBAT1 | Leu, Ile, Val, Met, Pro ( 62, 63) |
| SLC6A16 | NTT5 | Unknown | |
| SLC6A17 | B0AT3 | NTT4, RXT1 | Ala, Pro, Gly, Leu ( 64) |
| Leu, Met, Pro, Cys, Ala, Gln, Ser, His, Gly ( 65) | |||
| SLC6A18 | XT2, | XTRP2, ROSIT | Ala, Gly > other neutral AA |
| SLC6A19 | B0AT1 | Similar to XT2 | All neutral ( 34, 35) |
| SLC6A20 | IMINO | SIT1, XT3 | Pro, OH‐Pro ( 66, 67) |
CONCLUSION
Although initially described as a simple autosomal recessive disorder, molecular identification of the underlying gene SLC6A19 reveals complex interactions between the transporter and its associated proteins collectrin and ACE2. The interactions of SLC6A19 (Fig. 5) possibly connects it to complex disorders such as diabetes, dysfunction of blood pressure regulation and glomerular sclerosis.
Footnotes
The acronym B0AT1 refers to a Na+‐dependent transport activity for neutral (0) amino acids of broad (B) specificity. In mammalian genomes it is referred to as solute carrier family 6 member 19 (SLC6A19).
REFERENCES
- 1. Lemieux,B. , Auray‐Blais,C. , Giguere,R. , Shapcott,D. , and Scriver,C. R. ( 1988) Newborn urine screening experience with over one million infants in the Quebec Network of Genetic Medicine. J. Inherit. Metab. Dis. 11, 45–55. [DOI] [PubMed] [Google Scholar]
- 2. Wilcken,B. , Yu,J. S. , and Brown,D. A. ( 1977) Natural history of Hartnup disease. Arch. Dis. Child. 52, 38–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baron,D. N. , Dent,C. E. , Harris,H. , Hart,E. W. , and Jepson,J. B. ( 1956) Hereditary pellagra‐like skin rash with temporary cerebellar ataxia, constant renal aminoaciduria and other bizarre biochemical features. Lancet 2, 421–428. [DOI] [PubMed] [Google Scholar]
- 4. Kleta,R. , Romeo,E. , Ristic,Z. , Ohura,T. , Stuart,C. ,Arcos‐Burgos M., Dave,M. H. , Wagner,C. A. , Camargo,S. R. , Inoue,S. , Matsuura,N. ,Helip‐Wooley A., Bockenhauer,D. , Warth,R. , Bernardini,I. , Visser,G. , Eggermann,T. , Lee,P. , Chairoungdua,A. , Jutabha,P. , Babu,E. , Nilwarangkoon,S. , Anzai,N. , Kanai,Y. , Verrey,F. , Gahl,W. A. , and Koizumi,A. ( 2004) Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat. Genet. 36, 999–1002. [DOI] [PubMed] [Google Scholar]
- 5. Seow,H. F. , Broer,S. , Broer,A. , Bailey,C. G. , Potter,S. J. , Cavanaugh,J. A. , and Rasko,J. E. ( 2004) Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat. Genet. 36, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 6. Scriver,C. R. , Mahon,B. , Levy,H. L. , Clow,C. L. , Reade,T. M. , Kronick,J. , Lemieux,B. , and Laberge,C. ( 1987) The Hartnup phenotype: Mendelian transport disorder, multifactorial disease. Am. J. Hum. Genet. 40, 401–412. [PMC free article] [PubMed] [Google Scholar]
- 7. Zheng,Y. , Zhou,C. , Huang,Y. , Bu,D. , Zhu,X. , Jiang,W. ( 2009) A novel missense mutation in the SLC6A19 gene in a Chinese family with Hartnup disorder, Int J Dermatol. 48, 388–392. [DOI] [PubMed] [Google Scholar]
- 8. Hosoyamada,T. ( 2006) Clinical studies of pediatric malabsorption syndromes. Fukuoka Igaku Zasshi 97, 322–350. [PubMed] [Google Scholar]
- 9. Seyhan,M. E. , Selimoglu,M. A. , Ertekin,V. , Fidanoglu,O. , and Altinkaynak,S. ( 2006) Acrodermatitis enteropathica‐like eruptions in a child with Hartnup disease. Pediatr. Dermatol. 23, 262–265. [DOI] [PubMed] [Google Scholar]
- 10. Cusworth,D. C. and Dent,C. E. ( 1960) Renal clearances of amino acids in normal adults and in patients with aminoaciduria. Biochem. J. 74, 550–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doyle,F. A. and McGivan,J. D. ( 1992) The bovine renal epithelial cell line NBL‐1 expresses a broad specificity Na(+)‐dependent neutral amino acid transport system (System Bo) similar to that in bovine renal brush border membrane vesicles. Biochim. Biophys. Acta. 1104, 55–62. [DOI] [PubMed] [Google Scholar]
- 12. Maenz,D. D. and Patience,J. F. ( 1992) l‐threonine transport in pig jejunal brush border membrane vesicles. Functional characterization of the unique system B in the intestinal epithelium. J. Biol. Chem. 267, 22079–22086. [PubMed] [Google Scholar]
- 13. Stevens,B. R. , Kaunitz,J. D. , and Wright,E. M. ( 1984) Intestinal transport of amino acids and sugars: advances using membrane vesicles. Annu. Rev. Physiol. 46, 417–433. [DOI] [PubMed] [Google Scholar]
- 14. Bender,D. A. ( 1983) Biochemistry of tryptophan in health and disease. Mol. Aspects Med. 6, 101–197. [DOI] [PubMed] [Google Scholar]
- 15. Slominski,A. , Tobin,D. J. , Zmijewski,M. A. , Wortsman,J. , and Paus,R. ( 2008) Melatonin in the skin: synthesis, metabolism and functions. Trends Endocrinol. Metab. 19, 17–24. [DOI] [PubMed] [Google Scholar]
- 16. Takei,A. , Hamada,T. , Yabe,I. , and Sasaki,H. ( 2005) Treatment of cerebellar ataxia with 5‐HT1A agonist. Cerebellum 4, 211–215. [DOI] [PubMed] [Google Scholar]
- 17. Levy,L. L. ( 2001) Hartnup disorder. InThe Metabolic and Molecular Bases of Inherited Diseases, 8 edn., Vol. 3, ( Scriver,C. R. , Beaudet,A. L. , Sly,W. S. , and Valle,D. , ed.). pp. 4957–4969, McGraw‐Hill, New York. [Google Scholar]
- 18. Milne,M. D. ( 1967) Hereditary abnormalities of intestinal absorption. Br. Med. Bull. 23, 279–284. [DOI] [PubMed] [Google Scholar]
- 19. Daniel,H. ( 2004) Molecular and integrative physiology of intestinal peptide transport. Annu. Rev. Physiol. 66, 361–384. [DOI] [PubMed] [Google Scholar]
- 20. Asatoor,A. M. , Cheng,B. , Edwards,K. D. , Lant,A. F. , Matthews,D. M. , Milne,M. D. , Navab,F. , and Richards,A. J. ( 1970) Intestinal absorption of dipeptides and corresponding free amino acids in Hartnup disease. Clin. Sci. 39, 1P. [DOI] [PubMed] [Google Scholar]
- 21. Milne,M. D. ( 1971) Transport of amino acids and peptides in the gut and the kidney. Sci. Basis. Med. Annu. Rev., 1971, 161–177. [PubMed] [Google Scholar]
- 22. Rutherfurd,S. M. and Moughan,P. J. ( 1998) The digestible amino acid composition of several milk proteins: application of a new bioassay. J. Dairy Sci. 81, 909–917. [DOI] [PubMed] [Google Scholar]
- 23. Srikantia,S. G. , Venkatachalam,P. S. , and Reddy,V. ( 1964) Clinical and biochemical features of a case of hartnup disease. Br. Med. J. 1, 282–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hillman,R. E. , Stewart,A. , and Miles,J. H. ( 1986) Amino‐acid‐transport defect in intestine not affecting kidney. Pediatr. Res. 20, A265–A265. [Google Scholar]
- 25. Fajans,S. S. , Bell,G. I. , and Polonsky,K. S. ( 2001) Molecular mechanisms and clinical pathophysiology of maturity‐onset diabetes of the young. N. Engl. J. Med. 345, 971–980. [DOI] [PubMed] [Google Scholar]
- 26. Milne,M. D. ( 1967) The prognosis and management of renal tubular disorders. Proc. R. Soc. Med. 60, 1149–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bingham,C. , Ellard,S. , Nicholls,A. J. , Pennock,C. A. , Allen,J. , James,A. J. , Satchell,S. C. , Salzmann,M. B. , and Hattersley,A. T. ( 2001) The generalized aminoaciduria seen in patients with hepatocyte nuclear factor‐1alpha mutations is a feature of all patients with diabetes and is associated with glucosuria. Diabetes 50, 2047–2052. [DOI] [PubMed] [Google Scholar]
- 28. Hytten,F. E. and Cheyne,G. A. ( 1972) The aminoaciduria of pregnancy. J. Obstet. Gynaecol. Br. Common. 79, 424–432. [DOI] [PubMed] [Google Scholar]
- 29. Nozaki,J. , Dakeishi,M. , Ohura,T. , Inoue,K. , Manabe,M. , Wada,Y. , and Koizumi,A. ( 2001) Homozygosity mapping to chromosome 5p15 of a gene responsible for Hartnup disorder. Biochem. Biophys. Res. Commun. 284, 255–260. [DOI] [PubMed] [Google Scholar]
- 30. Broer,S. , Cavanaugh,J. A. , and Rasko,J. E. ( 2005) Neutral amino acid transport in epithelial cells and its malfunction in Hartnup disorder. Biochem. Soc. Trans. 33, 233–236. [DOI] [PubMed] [Google Scholar]
- 31. Nash,S. R. , Giros,B. , Kingsmore,S. F. , Kim,K. M. , el‐Mestikawy,S. , Dong,Q. , Fumagalli,F. , Seldin,M. F. , and Caron,M. G. ( 1998) Cloning, gene structure and genomic localization of an orphan transporter from mouse kidney with six alternatively‐spliced isoforms. Receptors Channels 6, 113–128. [PubMed] [Google Scholar]
- 32. Wasserman,J. C. , Delpire,E. , Tonidandel,W. , Kojima,R. , and Gullans,S. R. ( 1994) Molecular characterization of ROSIT, a renal osmotic stress‐induced Na(+)‐Cl(−)‐organic solute cotransporter. Am. J. Physiol. 267, F688–F694. [DOI] [PubMed] [Google Scholar]
- 33. Broer,A. , Klingel,K. , Kowalczuk,S. , Rasko,J. E. , Cavanaugh,J. , and Broer,S. ( 2004) Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J. Biol. Chem. 279, 24467–24476. [DOI] [PubMed] [Google Scholar]
- 34. Bohmer,C. , Broer,A. , Munzinger,M. , Kowalczuk,S. , Rasko,J. E. , Lang,F. , and Broer,S. ( 2005) Characterization of mouse amino acid transporter B0AT1 (slc6a19). Biochem. J. 389, 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Camargo,S. M. , Makrides,V. , Virkki,L. V. , Forster,I. C. , and Verrey,F. ( 2005) Steady‐state kinetic characterization of the mouse B(0)AT1 sodium‐dependent neutral amino acid transporter. Pflugers Arch. 451, 338–348. [DOI] [PubMed] [Google Scholar]
- 36. Romeo,E. , Dave,M. H. , Bacic,D. , Ristic,Z. , Camargo,S. M. , Loffing,J. , Wagner,C. A. , and Verrey,F. ( 2006) Luminal kidney and intestine SLC6 amino acid transporters of B0AT‐cluster and their tissue distribution in Mus musculus . Am. J. Physiol. Renal Physiol. 290, F376–F383. [DOI] [PubMed] [Google Scholar]
- 37. Kowalczuk,S. , Broer,A. , Tietze,N. , Vanslambrouck,J. M. , Rasko,J. E. , and Broer,S. ( 2008) A protein complex in the brush‐border membrane explains a Hartnup disorder allele. Faseb. J. 22, 2880–2887. [DOI] [PubMed] [Google Scholar]
- 38. Azmanov,D. N. , Kowalczuk,S. , Rodgers,H. , Auray‐Blais,C. , Giguere,R. , Rasko,J. E. , Broer,S. , Cavanaugh,J. A. ( 2008) Further evidence for allelic heterogeneity in Hartnup disorder. Hum. Mutat. 29, 1217–1221. [DOI] [PubMed] [Google Scholar]
- 39. Azmanov,D. N. , Rodgers,H. ,Auray‐Blais C., Giguere,R. , Bailey,C. , Broer,S. , Rasko,J. E. , and Cavanaugh,J. A. ( 2007) Persistence of the common Hartnup disease D173N allele in populations of European origin. Ann. Hum. Genet. 71, 755–761. [DOI] [PubMed] [Google Scholar]
- 40. Yamashita,A. , Singh,S. K. , Kawate,T. , Jin,Y. , and Gouaux,E. ( 2005) Crystal structure of a bacterial homologue of Na(+)/Cl(−)‐dependent neurotransmitter transporters. Nature 437, 215–223. [DOI] [PubMed] [Google Scholar]
- 41. Singh,S. K. , Piscitelli,C. L. , Yamashita,A. , and Gouaux,E. ( 2008) A competitive inhibitor traps LeuT in an open‐to‐out conformation. Science 322, 1655–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Mara,M. , Oakley,A. , and Broer,S. ( 2006) Mechanism and putative structure of B(0)‐like neutral amino acid transporters. J. Membr. Biol. 213, 111–118. [DOI] [PubMed] [Google Scholar]
- 43. Danilczyk,U. , Sarao,R. , Remy,C. , Benabbas,C. , Stange,G. , Richter,A. , Arya,S. , Pospisilik,J. A. , Singer,D. , Camargo,S. M. , Makrides,V. , Ramadan,T. , Verrey,F. , Wagner,C. A. , and Penninger,J. M. ( 2006) Essential role for collectrin in renal amino acid transport. Nature 444, 1088–1091. [DOI] [PubMed] [Google Scholar]
- 44. Malakauskas,S. M. , Quan,H. , Fields,T. A. , McCall,S. J. , Yu,M. J. , Kourany,W. M. , Frey,C. W. , and Le,T. H. ( 2007) Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am. J. Physiol. Renal. Physiol. 292, F533–F544. [DOI] [PubMed] [Google Scholar]
- 45. Zhang,Y. and Wada,J. ( 2007) Collectrin, a homologue of ACE2, its transcriptional control and functional perspectives. Biochem. Biophys. Res. Commun. 363, 1–5. [DOI] [PubMed] [Google Scholar]
- 46. Fukui,K. , Yang,Q. , Cao,Y. , Takahashi,N. , Hatakeyama,H. , Wang,H. , Wada,J. , Zhang,Y. , Marselli,L. , Nammo,T. , Yoneda,K. , Onishi,M. , Higashiyama,S. , Matsuzawa,Y. , Gonzalez,F. J. , Weir,G. C. , Kasai,H. , Shimomura,I. , Miyagawa,J. , Wollheim,C. B. , and Yamagata,K. ( 2005) The HNF‐1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab. 2, 373–384. [DOI] [PubMed] [Google Scholar]
- 47. Zhang,Y. , Wada,J. , Yasuhara,A. , Iseda,I. , Eguchi,J. , Fukui,K. , Yang,Q. , Yamagata,K. , Hiesberger,T. , Igarashi,P. , Zhang,H. , Wang,H. , Akagi,S. , Kanwar,Y. S. , and Makino,H. ( 2007) The role for HNF‐1beta‐targeted collectrin in maintenance of primary cilia and cell polarity in collecting duct cells. PLoS ONE 2, e414. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Camargo,S. M. , Singer,D. , Makrides,V. , Huggel,K. , Pos,K. M. , Wagner,C. A. , Kuba,K. , Danilczyk,U. , Skovby,F. , Kleta,R. , Penninger,J. M. , and Verrey,F. ( 2009) Tissue‐specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 136, 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lambert,D. W. , Hooper,N. M. , and Turner,A. J. ( 2008) Angiotensin‐converting enzyme 2 and new insights into the renin‐angiotensin system. Biochem. Pharmacol. 75, 781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oudit,G. Y. , Herzenberg,A. M. , Kassiri,Z. , Wong,D. , Reich,H. , Khokha,R. , Crackower,M. A. , Backx,P. H. , Penninger,J. M. , and Scholey,J. W. ( 2006) Loss of angiotensin‐converting enzyme‐2 leads to the late development of angiotensin II‐dependent glomerulosclerosis. Am. J. Pathol. 168, 1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seol,S. Y. , Lee,S. Y. , Kim,Y. D. , Do,E. J. , Kwon,J. A. , Kim,S. I. , Chu,I. S. , and Leem,S. H. ( 2008) Minisatellite polymorphisms of the SLC6A19: susceptibility in hypertension. Biochem. Biophys. Res. Commun. 374, 714–719. [DOI] [PubMed] [Google Scholar]
- 52. Yamagata,K. , Oda,N. , Kaisaki,P. J. , Menzel,S. , Furuta,H. , Vaxillaire,M. , Southam,L. , Cox,R. D. , Lathrop,G. M. , Boriraj,V. V. , Chen,X. , Cox,N. J. , Oda,Y. , Yano,H. , Le Beau,M. M. , Yamada,S. , Nishigori,H. , Takeda,J. , Fajans,S. S. , Hattersley,A. T. , Iwasaki,N. , Hansen,T. , Pedersen,O. , Polonsky,K. S. , Turner,R.C. , Velho,G. , Chevre,J. C. , Froguel,P. , and Bell,G. I. ( 1996) Mutations in the hepatocyte nuclear factor‐1alpha gene in maturity‐onset diabetes of the young (MODY3). Nature 384, 455–458. [DOI] [PubMed] [Google Scholar]
- 53. Martin,M. G. , Wang,J. , Solorzano‐Vargas,R.S. , Lam,J. T. , Turk,E. , and Wright,E. M. ( 2000) Regulation of the human Na(+)‐glucose cotransporter gene, SGLT1, by HNF‐1 and Sp1. Am. J. Physiol. Gastrointest. Liver Physiol. 278, G591–G603. [DOI] [PubMed] [Google Scholar]
- 54. Lockwood,C. R. , Bingham,C. , and Frayling,T. M. ( 2003) In silico searching of human and mouse genome data identifies known and unknown HNF1 binding sites upstream of beta‐cell genes. Mol. Genet. Metab. 78, 145–151. [DOI] [PubMed] [Google Scholar]
- 55. Anderle,P. , Nielsen,C. U. , Pinsonneault,J. , Krog,P. L. , Brodin,B. , and Sadee,W. ( 2006) Genetic variants of the human dipeptide transporter PEPT1. J. Pharmacol. Exp. Ther. 316, 636–646. [DOI] [PubMed] [Google Scholar]
- 56. Liu,Q. R. , Mandiyan,S. ,Lopez‐Corcuera B., Nelson,H. , and Nelson,N. ( 1993) A rat brain cDNA encoding the neurotransmitter transporter with an unusual structure. FEBS Lett. 315, 114–118. [DOI] [PubMed] [Google Scholar]
- 57. el Mestikawy S., Giros,B. , Pohl,M. , Hamon,M. , Kingsmore,S. F. , Seldin,M. F. , and Caron,M. G. ( 1994) Characterization of an atypical member of the Na+/Cl(−)‐dependent transporter family: chromosomal localization and distribution in GABAergic and glutamatergic neurons in the rat brain. J. Neurochem. 62, 445–455. [DOI] [PubMed] [Google Scholar]
- 58. Smith,K. E. , Fried,S. G. , Durkin,M. M. , Gustafson,E. L. , Borden,L. A. , Branchek,T. A. , and Weinshank,R. L. ( 1995) Molecular cloning of an orphan transporter. A new member of the neurotransmitter transporter family. FEBS Lett. 357, 86–92. [DOI] [PubMed] [Google Scholar]
- 59. Farmer,M. K. , Robbins,M. J. , Medhurst,A. D. , Campbell,D. A. , Ellington,K. , Duckworth,M. , Brown,A. M. , Middlemiss,D. N. , Price,G. W. , and Pangalos,M. N. ( 2000) Cloning and characterization of human NTT5 and v7‐3: two orphan transporters of the Na+/Cl− ‐dependent neurotransmitter transporter gene family. Genomics 70, 241–252. [DOI] [PubMed] [Google Scholar]
- 60. Uhl,G. R. , Kitayama,S. , Gregor,P. , Nanthakumar,E. , Persico,A. , and Shimada,S. ( 1992) Neurotransmitter transporter family cDNAs in a rat midbrain library: orphan transporters suggest sizable structural variations. Brain. Res. Mol. Brain. Res. 16, 353–359. [DOI] [PubMed] [Google Scholar]
- 61. Broer,S. ( 2006) The SLC6 orphans are forming a family of amino acid transporters. Neurochem. Int. 48, 559–567. [DOI] [PubMed] [Google Scholar]
- 62. Broer,A. , Tietze,N. , Kowalczuk,S. , Chubb,S. , Munzinger,M. , Bak,L. K. , and Broer,S. ( 2006) The orphan transporter v7‐3 (slc6a15) is a Na+‐dependent neutral amino acid transporter (B0AT2). Biochem. J. 393, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Takanaga,H. , Mackenzie,B. , Peng,J. B. , and Hediger,M. A. ( 2005) Characterization of a branched‐chain amino‐acid transporter SBAT1 (SLC6A15) that is expressed in human brain. Biochem. Biophys. Res. Commun. 337, 892–900. [DOI] [PubMed] [Google Scholar]
- 64. Parra,L. A. , Baust,T. , El Mestikawy,S. , Quiroz,M. , Hoffman,B. , Haflett,J. M. , Yao,J. K. , and Torres,G. E. ( 2008) The orphan transporter Rxt1/NTT4 (SLC6A17) functions as a synaptic vesicle amino acid transporter selective for proline, glycine, leucine, and alanine. Mol. Pharmacol. 74, 1521–1532. [DOI] [PubMed] [Google Scholar]
- 65. Zaia,K. A. and Reimer,R. J. ( 2009) Synaptic vesicle protein NTT4/XT1 (SLC6A17) catalyzes Na+‐coupled neutral amino acid transport. J. Biol. Chem. 284, 455–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kowalczuk,S. , Broer,A. , Munzinger,M. , Tietze,N. , Klingel,K. , and Broer,S. ( 2005) Molecular cloning of the mouse IMINO system: an Na+‐ and Cl−‐dependent proline transporter. Biochem. J. 386, 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takanaga,H. , Mackenzie,B. , Suzuki,Y. , and Hediger,M. A. ( 2005) Identification of mammalian proline transporter SIT1 (SLC6A20) with characteristics of classical system imino. J. Biol. Chem. 280, 8974–8984. [DOI] [PubMed] [Google Scholar]