

Abstract
Hepatitis C virus (HCV) is a major cause of chronic liver disease. Despite recent success in improving anti‐HCV therapy, additional progress is still needed to develop cheaper and interferon (IFN)‐free treatments. Here, we report that ferroquine (FQ), an antimalarial ferrocenic analog of chloroquine, is a novel inhibitor of HCV. FQ potently inhibited HCV infection of hepatoma cell lines by affecting an early step of the viral life cycle. The antiviral activity of FQ on HCV entry was confirmed with pseudoparticles expressing HCV envelope glycoproteins E1 and E2 from six different genotypes. In addition to its effect on HCV entry, FQ also inhibited HCV RNA replication, albeit at a higher concentration. We also showed that FQ has no effect on viral assembly and virion secretion. Using a binding assay at 4°C, we showed that FQ does not prevent attachment of the virus to the cell surface. Furthermore, virus internalization was not affected by FQ, whereas the fusion process was impaired in the presence of FQ as shown in a cell‐cell fusion assay. Finally, virus with resistance to FQ was selected by sequential passage in the presence of the drug, and resistance was shown to be conferred by a single mutation in E1 glycoprotein (S327A). By inhibiting cell‐free virus transmission using a neutralizing antibody, we also showed that FQ inhibits HCV cell‐to‐cell spread between neighboring cells. Combinations of FQ with IFN, or an inhibitor of HCV NS3/4A protease, also resulted in additive to synergistic activity. Conclusion: FQ is a novel, interesting anti‐HCV molecule that could be used in combination with other direct‐acting antivirals. (Hepatology 2013)
Hepatitis C virus (HCV) is a major cause of chronic liver disease (CLD). Approximately 160 million individuals suffer from chronic hepatitis C, putting them at risk to develop cirrhosis and hepatocellular carcinoma.1 Recent improvements in the standard‐of‐care therapy, currently a combination of pegylated interferon (IFN) alpha (Peg‐IFN‐α), ribavirin (RBV), and one of two drugs interfering with the virally encoded NS3/4A protease, have raised the hope that HCV infection can be managed efficiently in countries with adequate medical infrastructure.2 However, the current mode of treatment is not equally effective for all HCV genotypes and significant side effects are still observed. Efforts are currently being made to develop a combination of new direct‐acting antivirals (DAAs) not leading to the emergence of escape mutations and, if possible, free of IFN. First proofs of concept recently emerged from clinical trials demonstrating that combinations of DAAs can result in the cure of chronic HCV infection.3, 4 However, combinations of drugs targeting different steps of the viral life cycle, including virus entry, will likely improve viral response rates and therapeutic success.
HCV is a small enveloped virus with a positive stranded RNA genome belonging to the Hepacivirus genus in the Flaviviridae family.5 Its genome encodes two envelope glycoproteins (E1 and E2), which play a key role in virus entry into the hepatocyte. However, as a result of its association with low‐ or very‐low‐density lipoproteins,6 the lipoprotein moiety can also play a role in the entry process of HCV particle. HCV entry is currently viewed as a complex multistep process, because a series of specific cellular entry factors have been shown to be essential in the early steps of the HCV life cycle.7 These molecules include the scavenger receptor class B type 1 (SRB1), the tetraspanin CD81, tight‐junction proteins claudin 1 (CLDN1) and occludin (OCLN), and receptor tyrosine kinase‐like epidermal growth factor receptor. After its interaction with entry factors at the cell surface, HCV particle is internalized by clathrin‐mediated endocytosis.8 Importantly, as for several other viruses, HCV can also spread by direct cell‐to‐cell transfer.9, 10
Abbreviations.
3D, three‐dimensional; Ab, antibody; BVDV, bovine viral diarrhea virus; CC50, 50% cytotoxic concentration; CI, combination index; CLD, chronic liver disease; CLDN1, claudin 1; CMFDA, 5‐chloromethylfluorescein diacetate; CQ, chloroquine; DAAs, direct‐acting antivirals; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethyl sulfoxide; FCS, fetal calf serum; ffu, focus forming unit; FQ, ferroquine; gRNA, genomic RNA; HCV, hepatitis C virus; HCVcc, hepatitis C virus produced in cell culture; HCVpp, hepatitis C virus pseudoparticle; IC50, half‐maximal inhibitory concentration; IC90, 90% inhibitory concentration; IF, immunofluorescence; IFN, interferon; JFH‐1, Japanese fulminant hepatitis type 1; LT, liver transplantation; mAb, monoclonal Ab; OCLN, occludin; PE, phycoerythrin; Peg‐IFN‐α, pegylated interferon alpha; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; RBV, ribavirin; SRB1, scavenger receptor class B type 1; YFV, yellow fever virus.
Ferroquine (FQ; SSR97193) is a ferrocenic analog of chloroquine (CQ) that has been developed as a new antimalarial drug (Fig. 1A).11 This bioorganometallic compound, which has a mechanism of action different from CQ,12 is currently one of the most promising new candidate drugs in the antimalarial pipeline, and it is about to complete phase II clinical trials as a treatment for uncomplicated malaria.13 In addition to its antimalarial activity, FQ also shows an antiviral effect against SARS coronavirus infection.14 This prompted us to test whether FQ exhibits an antiviral activity against HCV. Our data show that FQ inhibits HCV infection at a half‐maximal effective concentration below 1 μM by blocking virus entry at the fusion step.
Figure 1.
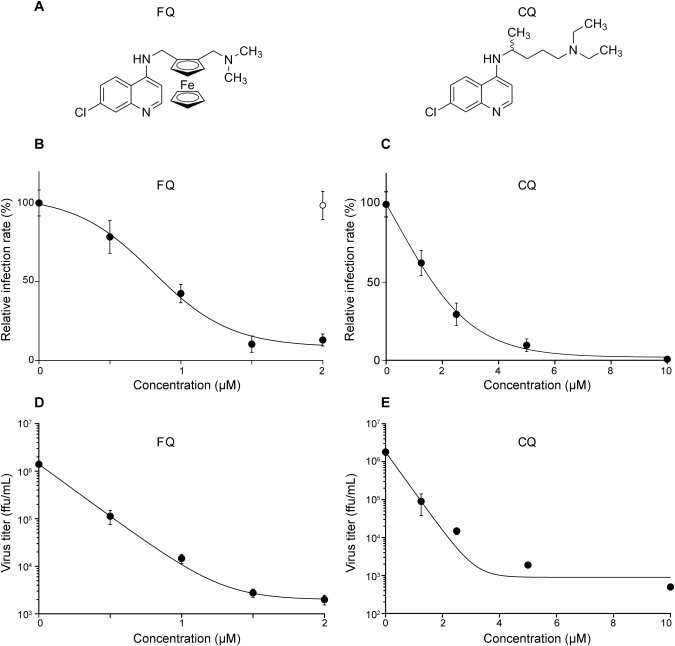
FQ inhibits HCV infection. (A) Chemical structures of FQ and CQ. Huh‐7 cells were pretreated for 1 hour with FQ (B and D) or CQ (C and E) before infection with JFH‐1 (MOI, 1), and infected cells were in contact with drugs until the end of the experiment. At 48 hours postinfection, infected cells were quantified by indirect IF with the anti‐E1 mAb A4 (B and C), and virus released in the supernatant was titrated (D and E). For infected cells, results are expressed as percentage of infection, compared to the control infection, in the presence of the solvent. It is worth noting that the final concentration of solvent was adjusted to be the same for all FQ concentrations, and a control point in the absence of FQ is shown in (B; open circle). Error bars indicate standard errors of the mean values (B and C) or standard deviations (D and E) from at least two independent experiments. Control experiments performed in parallel with characterized HCV inhibitors are presented in Supporting Fig. 3. MOI, multiplicity of infection.
Materials and Methods
Chemicals.
FQ was synthesized as previously described.11, 14 CQ, IFN‐α, heparin, and brefeldin A were purchased from Sigma‐Aldrich (St. Louis). FQ was prepared as 1‐mM stock solutions in dimethyl sulfoxide (DMSO). CQ was prepared as 1‐mM stock solutions in water. Boceprevir was kindly provided by Philippe Halfon (Hôpital Ambroise Paré, Marseille, France). CellTracker Green CMFDA (5‐chloromethylfluorescein diacetate) was from Invitrogen Molecular Probes (Carlsbad, CA).
Cell Culture.
Huh‐7 cells,15 HEK‐293T cells (ATCC CRL‐11268), HeLa cells (ATCC CCL‐2), and RFP‐NLS‐IPS‐Huh7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% of heat‐inactivated fetal calf serum (FCS). Madin‐Darby Bovine Kidney cells (MDBK; ATCC CCL‐22) were cultured in DMEM supplemented with GlutaMAX I and 10% horse serum. Drug toxicity on Huh‐7 cells was evaluated using the MTS assay, following the recommendations of the manufacturer (Promega, Madison, WI).
Antibodies.
Monoclonal antibodies (mAbs) anti‐HCV E1 glycoprotein (A4),16 anti‐HCV E2 glycoprotein (3/11; kindly provided by J. McKeating, University of Birmingham, UK)(17),17 anti–yellow fever virus (YFV) envelope protein (2D12) (ATCC CRL‐1689), anti–bovine viral diarrhea virus (BVDV) NS3 protein (Osc‐23),18 anti‐NS5A antibody (Ab) (AUSTRAL Biologicals, San Ramon, CA), and anti‐CD81 mAb JS‐81 CD81 (BD Pharmingen, San Diego, CA) were used in this study. Cy3‐, Alexa 488‐ and phycoerythrin (PE)‐conjugated secondary Abs were from Jackson ImmunoResearch (West Grove, PA), Invitrogen, and BD Pharmingen, respectively.
Viruses.
To produce cell‐cultured HCV (HCVcc), we used a modified version of the plasmid encoding Japanese fulminant hepatitis type 1 (JFH‐1) genome (genotype 2a; GenBank access no.: AB237837), kindly provided by T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan).19 Briefly, HCVcc was produced in Huh‐7 cells electroporated with in vitro–transcribed RNA of JFH‐1 containing (JFH‐1/Luc) or not, the Renilla luciferase reporter gene, and engineered to express A4 epitope20 and titer‐enhancing mutations.21 JFH‐1 stocks were produced by further amplification in Huh‐7 cells. In the JFH‐1/Luc construct, the Renilla luciferase gene is fused with the viral open reading frame in a monocistronic configuration. With this virus, we verified that our luciferase data were in the linear range of the assay. The JFH‐1/ΔE1E2/Luc plasmid, containing an in‐frame deletion in the E1/E2 region, and the JFH‐1/GND/Luc replication‐defective mutant have been described previously.20, 22 The inhibitory effect of the drugs was determined by quantifying infectivity by indirect immunofluorescence (IF) with the anti‐E1 mAb A416 or by measuring viral titers with the same Ab. For quantitative binding experiments, purified virus was obtained by precipitation of HCVcc‐infected Huh‐7 cells supernatants with 8% polyethylene glycol 6000. Pelleted virus was then loaded onto a continuous 10%‐40% iodixanol gradient. One‐milliter fractions were collected and the most infectious fractions were pooled. The titer of the stock was 5 × 106 focus forming units (ffu)/mL.
HCV pseudotyped retroviral particles (HCVpp) expressing the Firefly luciferase reporter gene were produced in HEK‐293T as previously described.23 The intergenotypic HCV chimera GT3a(452)/JFH‐124 was also used in some experiments. Furthermore, BVDV strain NADL and YFV strain 17D were also used to test the effect of the compounds on other viruses.
Cell‐to‐Cell Inhibition Assay.
HCV cell‐to‐cell transmission was measured by two different approaches, as previously described.25, 26
Indirect IF.
Infected cells grown on glass coverslips were processed for IF detection of viral proteins as previously described.27 Nuclei were stained with 1 μg/mL of 4',6'‐diamidino‐2‐phenylindole. Coverslips were observed with a Zeiss Axiophot microscope (Carl Zeiss, Oberkochen, Germany), and fluorescent signals were collected with a Coolsnap ES camera (Photometrix, Kew, Australia). For quantification of antigen‐positive cells, images of randomly picked areas from each coverslip were recorded.
Quantification of HCV Core Protein.
Huh‐7 cells were inoculated for 2 hours with HCVcc in six‐well plates. At the indicated time, HCV core antigen expressed within cells or secreted into the supernatant was quantified using chemiluminescent microparticle technology (Architect HCV Ag Test; Abbott Diagnostics, Rungis, France), as previously described.28
Quantitative Binding and Virus Internalization Assays.
Virions bound to Huh‐7 cells were determined by quantitative real‐time reverse‐transcription polymerase chain reaction (qRT‐PCR) assay as described previously.29 Internalization was measured as previously described.30
Detection of HCV Receptor Expression by Flow Cytometry and Western Blotting.
Huh‐7 cells were treated with FQ for 48 hours. After incubation with FQ, cells were stained with Abs for flow cytometry and/or western blotting, as previously reported.31
Cell‐Cell Fusion Assay.
Cell‐cell fusion assay was performed as previously reported.32
Selection of a FQ‐Resistant Virus.
Supernatant of HCV‐infected cells were serially passaged under increasing concentrations of FQ. The structural region of HCV genome was amplified by RT‐PCR and sequenced. Amino acid changes that arose during inhibitor selection were identified by analysis of the DNA sequence, compared to the initial and control passages, in the presence of solvent alone. Identified mutations were reintroduced in JFH‐1 plasmid by PCR mutagenesis, and the plasmids were sequenced.
Combination Assays.
Antiviral activity of a range of FQ concentrations alone or combined to IFN‐α or boceprevir was determined by measuring half‐maximal inhibitory concentration (IC50) values. Then, combination index was calculated according to a previously reported method.33 Prichard and Shipman's method34 was also used to analyze interaction effects using a three‐dimensional (3D) approach. This method presents complete drug interactions.
Statistical Analysis.
Prism v5.0c software (GraphPad Software, Inc., La Jolla, CA) was used to prepare graphs, calculate IC50 values, and determine statistical significance of differences between data sets.
Results
FQ Inhibits HCV Infection.
Chemical structures of FQ and CQ are presented in Fig. 1A. To test the effect of FQ on the HCV life cycle, the compound was added to Huh‐7 target cells before, as well as during, infection. FQ exhibited a dose‐dependent inhibition of HCV, indicating that FQ specifically affects the HCV life cycle with an estimated IC50 value of 0.8 μM (± 0.26) and an 90% inhibitory concentration (IC90) of 1.86 μM (± 0.08) (Fig. 1B,D). Similar results were obtained using the HepG2 cell line expressing CD81 (data not shown). The inhibitory effect was not the result of cytotoxicity, because parallel experiments did not show any toxic effect of the drug at the concentrations tested (Supporting Fig. 1). Furthermore, no cytotoxicity was observed in primary human hepatocytes at the concentrations used in our experiments (Supporting Fig. 1). FQ showed a 50% cytotoxic concentration (CC50) of 5.34 μM and a therapeutic index of 6.7. Similar inhibitory effects were observed with FQ‐treated cells infected with a chimeric virus containing the structural proteins of a genotype 3a isolate (Supporting Fig. 2), indicating that the antiviral effect is not specific to JFH‐1 isolate. Parallel control experiments with well‐characterized HCV inhibitors are presented in Supporting Fig. 3. CQ was less effective against HCV (Fig. 1C,E). Indeed, IC50 and IC90 values for CQ were 3.93 (± 1.87) and 4.33 μM (± 0.53), respectively. Furthermore, CQ had a CC50 of 19.58 μM and a therapeutic index of 5. To determine whether HCV is the only member of the Flaviviridae family to be affected by FQ, we tested this compound on two other members of this viral family (BVDV and YFV). BVDV and YFV infections were performed on MDBK and Huh‐7 cells, respectively. FQ showed some antiviral effect on these two viruses, albeit at a much higher concentration. Indeed, IC50 values were 6.74 (± 0.48) and 3.63 μM (± 0.64) for BVDV and YFV, respectively. Altogether, these results indicate that FQ has a potent antiviral activity against HCV.
FQ Inhibits HCV Entry.
To determine whether FQ has any effect on HCV entry, the compound was added or removed at different time points before, during, and after inoculation of Huh‐7 cells with JFH‐1 (Fig. 2). The highest decrease in HCVcc infection was observed when FQ was present during viral infection, and only a weak antiviral effect was detected when FQ was added postinfection (Fig. 2A,B). Similar results were obtained with CQ (Fig. 2C,D), and parallel control experiments with well‐characterized HCV inhibitors are presented in Supporting Figs. 4 and 5. These results suggest that FQ inhibits an early step of the HCV life cycle, which is most likely the entry step. To investigate the effect of FQ on HCV entry, we used the HCVpp system. These are retroviral cores carrying HCV glycoproteins in their envelope. In this context, only the early steps of the viral life cycle (i.e., virus interaction with receptors, uptake, and fusion) are HCV specific, whereas all later steps are dependent on retroviral nucleocapsid elements. Using this approach, FQ inhibited HCVpp entry in a dose‐dependent manner (Fig. 2F). Furthermore, this effect was specific of HCV envelope glycoproteins because FQ did not affect the entry of control pseudoparticles containing the envelope glycoprotein of the feline endogenous retrovirus, RD114. Together, these data indicate that FQ is an inhibitor of HCV entry.
Figure 2.
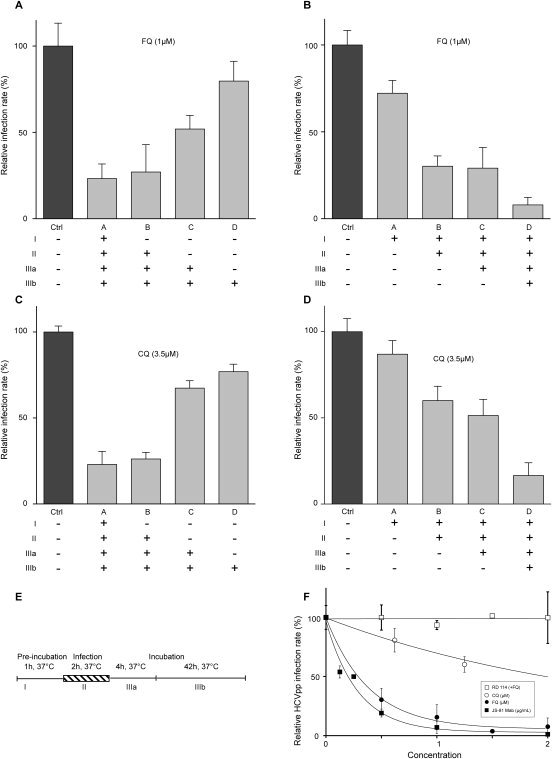
FQ inhibits HCV entry. Huh‐7 cells were infected with JFH‐1 (MOI, approximately 1) and treated for different periods of time with FQ (A and B) or CQ (C and D), as indicated in (E). At 48 hours postinfection, infected cells were quantified. Results are expressed as percentage of infection, relative to the control infection, in the presence of solvent (Ctrl). Error bars indicate standard errors of the mean values from at least three experiments. We controlled in parallel experiments that the drugs had no cellular toxicity in our experimental conditions (data not shown). (F) Antiviral effect of FQ on HCVpp containing the envelope glycoproteins of JFH‐1 isolate. Control experiments with CQ and anti‐CD81 mAb JS‐81 were performed in parallel. Huh‐7 cells were infected with HCVpp or pseudoparticles containing the envelope glycoprotein of the feline endogenous retrovirus, RD114. Cells were treated for 1 hour before infection and for the whole duration of infection. At 48 hours postinfection, cells were lysed to quantify luciferase activity. HCVpp infectivity varied between 105 and 106 relative light units (RLU). Results are expressed as percentage of infection, compared to the control infection, in the presence of solvent. Error bars indicate standard errors of the mean values from at least two independent experiments.
Because FQ specifically affects HCV entry, this prompted us to test its antiviral effect on different HCV genotypes and subtypes in the context of the HCVpp system. FQ inhibited the infectivity of HCVpp from the different genotypes and subtypes tested, indicating that FQ inhibits HCV entry in a genotype‐independent manner (Table 1).
Table 1.
FQ Inhibits HCV Entry in a Genotype‐Independent Manner
| Genotype | IC50 (μM) |
|---|---|
| 1a | 0.85 ± 0.11 |
| 1b | 0.49 ± 0.09 |
| 2a | 0.26 ± 0.06 |
| 2b | 0.28 ± 0.06 |
| 3a | 0.64 ± 0.39* |
| 4 | 0.66 ± 0.09 |
| 5 | 0.69 ± 0.42 |
| 6 | 0.74 ± 0.58 |
| RD114 | No inhibition |
HCVpp expressing E1E2 glycoproteins from different genotypes were tested for their sensitivity to FQ, and IC50 values were determined. Sequences of the following isolates were used to generate HCVpp: H77 (gt1a; GenBank AAB67037); UKN1B‐5.23 (gt1b; AY734976); JFH‐1 (gt2a; AB047639); UKN2B‐1.1 (gt2b; AY734982); UKN3A‐1.28 (gt3a; AY734984); UKN4‐11.1 (gt4; AY734986); and UKN6‐5.340 (gt6; AY736194). Asterisk indicates the IC50 value determined with a chimeric HCVcc virus.
Effect of FQ on HCV Replication and Assembly.
Although the above data indicate that FQ has a strong effect on HCV entry, we cannot exclude additional effects on other steps of the HCV life cycle. To analyze the effect of FQ on HCV genome replication, Huh‐7 cells were electroporated with in vitro–transcribed, assembly‐defective JFH‐1/ΔE1E2/Luc RNA, to bypass the entry step, and avoid any interference with late steps of the HCV life cycle. Furthermore, boceprevir was used in parallel as a control of inhibition of viral replication. FQ had also some effect on HCV replication (Fig. 3A), albeit at higher concentrations (see Figs. 1B and 3A). This observation is in agreement with the additional weak antiviral effect detected when FQ was added postinfection (Fig. 2B).
Figure 3.
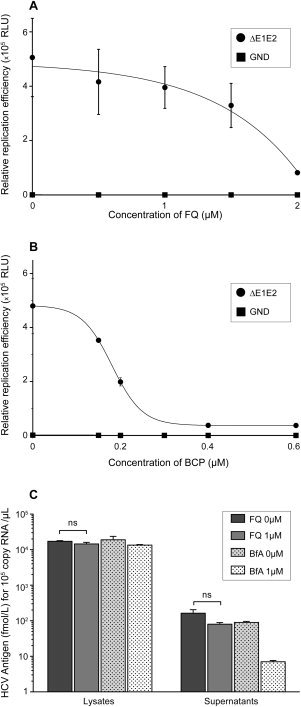
Effect of FQ on HCV replication and assembly. (A and B) JFH‐1/ΔE1E2/Luc RNA was electroporated in Huh‐7 cells and, at 4 hours postelectroporation, cells were treated or not with FQ (A) or boceprevir (BCP) (B). Cells were lysed 48 hours later. Results are expressed as percentage of replication in the absence of drug. Specificity of replication was confirmed with a JFH‐1/GND/Luc (GND) replication‐defective mutant. (C) Huh‐7 cells were inoculated with HCVcc for 2 hours and cultured in the presence or absence of FQ for 48 hours. In parallel, HCV‐infected Huh‐7 cells were treated with brefeldin A for 8 hours before harvest. Supernatants were collected, cells were lysed, and intracellular and extracellular core protein was quantified. Furthermore, intracellular RNA was measured and the data were normalized by HCV replication level to take into account the potential effect of FQ on HCV replication. No significant (ns) difference was observed between FQ‐treated and untreated cells.
To determine whether FQ could have any effect on HCV assembly or secretion, intra‐ and extracellular core protein was quantified in infected cells treated postinfection with 1 μM of FQ. The amount of core in the culture supernatant reflects the quantity of secreted viral particles. The amount of core released in the supernatant of infected cells was not significantly reduced in the presence FQ treatment (Fig. 3C). In contrast, brefeldin A, an inhibitor of HCV release, reduced core secretion by more than 1 log10. These data show that FQ does not inhibit virion assembly and egress.
FQ Inhibits a Late Step of HCV Entry.
Because our data show that FQ has an antiviral activity at an early step of the HCV life cycle, we further investigated its mode of action on HCV entry. To determine whether FQ impairs directly the binding of particles to the cell surface, we analyzed virus binding in the presence of FQ. Cells were inoculated with purified HCVcc at 4°C in the presence of FQ, and the amount of bound virions was determined by quantifying HCV genomic RNA (gRNA). Heparin was used as a control of inhibition of HCV binding. As expected, heparin strongly reduced HCV attachment to the cell surface (Fig. 4A). In the presence of FQ, no effect on virus binding was observed (Fig. 4A), indicating that FQ does not inhibit HCV entry by impairing virus binding to the cell surface.
Figure 4.
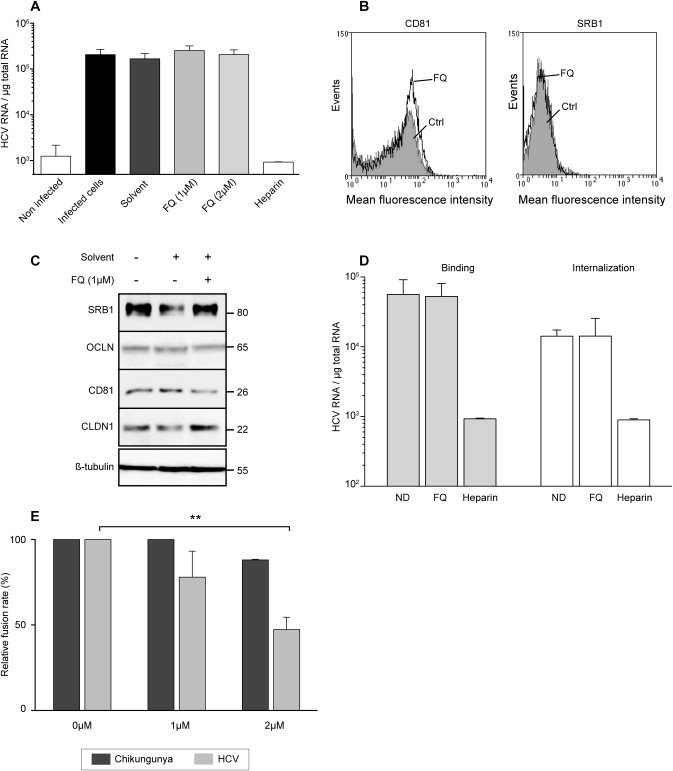
FQ inhibits a late step of HCV entry. (A) To determine the effect of FQ on HCV binding, Huh‐7 cells were inoculated for 1hour at 4°C with purified HCVcc at a MOI of 10 in the presence of solvent (DMSO), FQ, or 500 μg/mL of heparin. Cells were washed thrice with ice‐cold phosphate‐buffered saline, and total RNA was extracted. Bound HCV virions were detected by quantification of HCV gRNA by qRT‐PCR. Mean values ± standard deviation (error bars) of three different experiments are presented. Turkey's multiple comparison test was used for statistical analysis. No significant (ns) difference was observed between treated and untreated cells. Receptors expression was assessed in Huh‐7 cells, exposed (FQ), or not (Ctrl) for 48 hours to 1 μM of FQ, by flow cytometry for CD81 and SRB1 (B), and by western blotting for CD81, SRB1, CLDN1, and OCLN (C). (D) To determine the effect of FQ on HCV internalization, purified HCV particles were bound to Huh‐7 cells, as described in (A), followed by 30‐minute incubation at 37°C. Cells were then chilled on ice, and noninternalized virions were removed by trypsinization for 1 hour at 4°C, and internalized viral particles were determined by quantification of HCV gRNA by qRT‐PCR. To verify efficacy of trypsin treatment, virus was bound to cells at 4°C and then removed by trypsinization (data not shown). (E) Effect of FQ on fusion function of HCV envelope glycoproteins. 293T “donor” cells coexpressing the HCV E1E2 or Chikungunya envelope glycoproteins and a luciferase marker gene under the control of the HIV‐1 promoter were cocultured with Huh‐7‐Tat “indicator” cells expressing the HIV‐1 Tat protein. After 24 hours, cells incubated for 1 hour in the presence or absence of FQ were treated (at pH 5) for 3 minutes, and luciferase activity induced by fusion between donor and indicator cells was measured 72 hours later. Fusion in the absence of drug was taken as 100%. Graphs represent the averages of three independent experiments (**P < 0.01). A control experiment with pseudoparticles containing the envelope glycoproteins of Chikungunya confirmed that this virus is not sensitive to FQ treatment (data not shown). HIV, human immunodeficiency virus.
To further analyze the mechanism by which FQ inhibits HCV entry, we assessed the expression of known essential HCV entry factors CD81, SRB1, CLDN1, and OCLN. Huh‐7 cells were treated with FQ at 1 μM for 48 hours. Then, CD81, SRB1, CLDN1, and OCLN expression was assessed by western blotting and/or flow cytometry. Expression levels of all four entry factors were unaltered, indicating that FQ does not act through their down‐regulation (Fig. 4B,C).
Because FQ does not inhibit the binding of HCV particles to the cell surface and because it has no effect on the expression of HCV receptors, we also analyzed the effect of this molecule on the internalization of the viral particle. HCV internalization was not affected by FQ treatment, indicating that this molecule blocks a postinternalization step (Fig. 4D). It is also worth noting that FQ has no effect on IFN induction (Supporting Fig. 6).
To determine the effect of FQ on the fusion process, we used a cell‐cell fusion assay that has been previously described.32 FQ induced a dose‐dependent decrease of fusion activity of HCV envelope glycoproteins, whereas no effect was observed on control Chikungunya virus envelope glycoproteins (Fig. 4E). Together, these results indicate that FQ inhibits the fusion step during the HCV entry process.
FQ Resistance Maps to E1 Glycoprotein.
To further investigate the mechanism of action of FQ, we selected a partially resistant mutant by propagation for several passages in the presence of increasing concentrations of drug. After 16 passages, we did not observe any amino acid change in E2, whereas two mutations were identified in E1 glycoprotein (Y297H and S327A). Interestingly, reverse genetics experiments indicate that the S327A mutation is able, by itself, to confer some resistance to FQ (Fig. 5). It is worth noting that serine 327 is well conserved in genotypes 1‐6.
Figure 5.
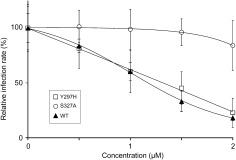
Mutation S327A in E1 confers resistance to FQ treatment. Huh‐7 cells were pretreated for 1 hour with FQ before infection with JFH‐1 or mutant viruses Y297H or S327A (MOI, approximately 1), and infected cells were in contact with drug until the end of the experiment. At 48 hours postinfection, infected cells were quantified. Results are expressed as percentage of infection in the presence of solvent. Error bars indicate standard errors of the mean values from at least three independent experiments.
FQ Inhibits Cell‐To‐Cell Transmission.
Subsequent to infection of Huh‐7 cells with HCVcc, progeny viruses are transmitted to adjacent cells, resulting in focal areas of spreading infection (foci). This mode of transmission is refractory to neutralization by anti‐E2 Abs.9 To determine whether FQ can block cell‐to‐cell spread, HCV‐infected RFP‐NLS‐IPS‐Huh‐7 cells were cocultured with naïve Huh‐7 cells in the presence or absence of FQ, as previously described26 (Fig. 6A). In a second approach, HCV‐infected Huh‐7 cells were labeled with CMFDA and cocultured with naïve target cells in the presence or absence of FQ, as previously described25 (Fig. 6B). A strong decrease in cell‐to‐cell transmission was clearly observed in both approaches (Fig. 6).
Figure 6.
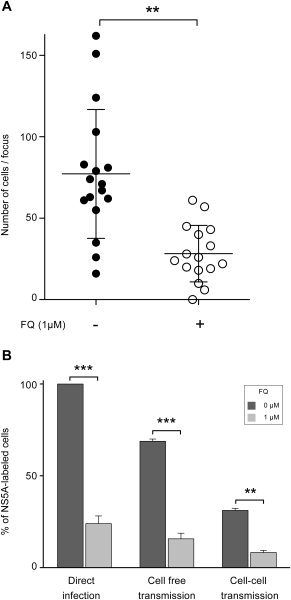
FQ inhibits cell‐to‐cell transmission. (A) Cocultures of naïve Huh‐7 target cells with JFH‐1‐infected RFP‐NLS‐IPS‐Huh‐7 cells were treated with neutralizing mAb 3/11 (50 μg/mL) in the presence or absence of FQ. Numbers of infected target cells were measured by IF in at least 16 foci for each condition. Bars indicate mean values. (B) Huh‐7 donor cells were infected with JFH‐1 and stained with CMFDA. Acceptor cells are naïve Huh‐7 cells. Coculture of donor and acceptor cells with or without neutralizing mAb 3/11 allowed for monitoring cell‐to‐cell or total transmission of HCV. Percentage of cell‐free transmission was obtained by subtracting the percentage of cells infected in the presence of mAb 3/11 from the percentage of cells infected in the absence of Ab. After 48 hours, cells were labeled with anti‐NS5A mAb, followed by PE‐conjugated secondary Ab and analyzed by flow cytometry. In these conditions, newly infected cells are negative for CMFDA staining and positive for PE staining. Results are reported as the percentage of HCV‐infected cells from two independent experiments. **P < 0.01; ***P < 0.001.
Antiviral Effect of FQ in Combination With IFN‐α or Boceprevir.
We tested whether FQ could be combined with other anti‐HCV compounds currently used in hepatitis C treatment. We used boceprevir, a drug targeting directly HCV protease NS3/4A, and IFN‐α, which has been used for several decades in anti‐HCV treatment. These drug combinations were analyzed using the combination index (CI) as well as Prichard and Shipman's method.34 Each drug was combined with FQ at different fractions of their IC50 value. Combination of FQ with boceprevir and IFN‐α resulted in an additive effect, as reflected by a CI of 0.97 (± 0.03) and 1.08 (± 0.18), respectively. Furthermore, synergy was also observed for some higher concentrations, as measured by Prichard and Shipman's method34 (Fig. 7).
Figure 7.
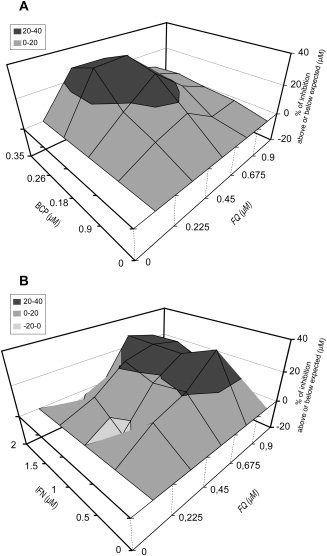
3D analysis of in vitro interaction between FQ and IFN‐α or boceprevir (BCP). Combinations on JFH‐1 infection were evaluated using Prichard and Shipman's method.34 Combination studies for each pair of compounds were performed in triplicate. The theoretical additive effect is calculated from the dose‐response curves of individual compounds by the equation, Z = X + Y(1‐X), where X and Y represent the inhibition produced by the individual compounds and Z represents the effect produced by the combination of compounds. The theoretical additive surface is subtracted from the actual experimental surface, resulting in a horizontal surface that equals the zero plane when the combination is additive. A surface raising more than 20% above the zero plane indicates a synergistic effect of the combination, and a surface dropping lower than 20% below the zero plane indicates antagonism.
Discussion
HCV entry represents an attractive target for antiviral intervention, with opportunities to prevent multiple virus‐receptor interactions and interfere with virus‐cell membrane fusion.35 In this study, we showed that FQ exhibits a genotype‐independent antiviral activity against HCV by inhibiting a postbinding and postinternalization step of HCV entry into target cells and by blocking cell‐to‐cell spread between neighboring cells.
Although FQ is an analog of CQ, its mechanism of action is potentially different.12 The mechanism of inhibition by CQ involves impaired endosomal‐mediated virus entry, most likely through the prevention of endocytosis and/or endosomal acidification. In contrast, FQ has weaker base properties, compared to CQ.36 This difference could potentially explain the lack of antiviral activity of FQ against viruses such as vesicular stomatitis virus, influenza virus, or Sindbis virus,14 whereas CQ blocks cell entry of these viruses as well as other pH‐dependent viruses.37, 38, 39 Other interesting features of FQ are its higher lipophilicity (at pH 7.4) and the peculiar conformation provided by an intramolecular hydrogen bond present in nonpolar conditions, which result in a better potency for FQ to cross membranes.40 In addition, FQ has also been shown to specifically generate reactive oxygen species and induce lipid peroxidation.40, 41 Recently, it has been shown that HCV envelope proteins form large molecular complexes stabilized by intermolecular disulfide bonds.42 This observation strongly suggests that the entry process necessitates a rearrangement of these disulfide bonds for the fusion process to take place. Therefore, it is possible that the oxidative properties of FQ in acidic conditions could inhibit the fusion process by affecting reorganization of the disulfide bonds within endosomes.
FQ inhibits a postbinding and postinternalization step of HCV entry into target cells. Indeed, FQ does not affect binding of HCVcc to Huh‐7 cells or the expression of specific cellular entry factors. Furthermore, the effect of FQ on HCVpp strongly suggests that FQ affects the entry function of HCV envelope proteins and not the lipoprotein moiety associated with the virion. Our data also show that FQ blocks HCV entry by inhibiting the fusion process. Finally, the S327A‐resistant mutation identified in this work suggests that E1 could be the target of FQ.
In addition to its effect on HCV entry, FQ can also affect HCV RNA replication, albeit at higher concentrations. This inhibitory effect is similar to the inhibition of an HCV subgenomic replicon by CQ.43 HCV is known to exploit autophagy for its replication,44 and inhibition of replication by CQ targets virus‐associated autophagy.43 However, it remains to be determined whether inhibition of HCV RNA replication by FQ depends on the same mechanism.
In clinical studies with healthy human volunteers, it has been shown that FQ is safe and very well tolerated with oral doses of 400‐1,600 mg FQ, and the mean estimate for blood apparent terminal half‐life of FQ was 16 days.45 In addition, a maximum blood concentration of 487 ng/mL (or 1.1 μM) was observed for the highest dose of FQ, which is slightly above the IC50 value calculated for HCV in cell culture. Although such a concentration would probably not be high enough to eliminate HCV completely, it would likely be sufficient in combination therapies with other drugs showing a synergistic or additive effect. Further studies will be necessary to determine the in vivo potency of FQ against HCV.
FQ may provide a new approach to prevent HCV infection, especially in the setting of liver transplantation (LT) of chronically infected HCV patients. Indeed, a major problem for LT resulting from HCV is the reinfection of the graft, which is always observed with an accelerated progression of liver disease.46 Thus, the ability of FQ at inhibiting HCV cell‐to‐cell transmission is a major asset for an entry inhibitor. Furthermore, FQ exhibits an antiviral activity against all HCV genotypes, tested in the HCVpp system, increasing its potential interest as a general anti‐HCV agent. The combination of entry, replication, and polyprotein‐processing inhibitors in a context of a multidrug therapy might be the way to reduce the risk of emergence of resistant viruses. FQ might thus be a valuable option to be tested in low‐cost anti‐HCV combinations. Finally, these findings highlight the potential interest of FQ use in countries where malaria coinfection with HCV can occur.
Supporting information
Additional Supporting Information may be found in the online version of this article.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Supporting Information
Acknowledgements
The authors thank Julie Potel, Yves Rouillé, and Karin Séron for their scientific input.
Potential conflict of interest: Nothing to report.
This work was supported by the French “Agence Nationale de la Recherche sur le Sida et les hépatites virales” (ANRS). T.V. was supported by a fellowship from the French Ministry of Research (MESR). N.C. was successively supported by a fellowship from the MESR and the ANRS. A.W. was supported by a postdoctoral fellowship of the ANRS. S.B. was supported by a Marie Curie International Reintegration Grant (PIRG‐GA‐2009‐256300). The authors are grateful to J.K. Ball, R. Bartenschlager, F.L. Cosset, P. Halfon, J. McKeating, and T. Wakita for providing essential reagents. The immunofluorescence analyses were performed with the help of the imaging core facility of the BioImaging Center Lille Nord‐de‐France.
References
- 1. Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect 2011; 17: 107‐115. [DOI] [PubMed] [Google Scholar]
- 2. Asselah T, Marcellin P. New direct‐acting antivirals' combination for the treatment of chronic hepatitis C. Liver Int 2011; 31( Suppl 1): 68‐77. [DOI] [PubMed] [Google Scholar]
- 3. Gane EJ, Roberts SK, Stedman CA, Angus PW, Ritchie B, Elston R, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM‐1): a randomised, double‐blind, placebo‐controlled, dose‐escalation trial. Lancet 2010; 376: 1467‐1475. [DOI] [PubMed] [Google Scholar]
- 4. Lok AS, Gardiner DF, Lawitz E, Martorell C, Everson GT, Ghalib R, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366: 216‐224. [DOI] [PubMed] [Google Scholar]
- 5. Lindenbach BD, Thiel HJ, Rice CM: Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, eds. Fields Virology, 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007: 1101‐1152. [Google Scholar]
- 6. Andre P, Komurian‐Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, et al. Characterization of low‐ and very‐low‐density hepatitis C virus RNA‐containing particles. J Virol 2002; 76: 6919‐6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belouzard S, Cocquerel L, Dubuisson J. Hepatitis C virus entry into the hepatocyte. Cent Eur J Biol 2011; 6: 933‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. Hepatitis C virus entry depends on clathrin‐mediated endocytosis. J Virol 2006; 80: 6964‐6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, et al. Hepatitis C virus cell‐cell transmission in hepatoma cells in the presence of neutralizing antibodies. HEPATOLOGY 2008; 47: 17‐24. [DOI] [PubMed] [Google Scholar]
- 10. Witteveldt J, Evans MJ, Bitzegeio J, Koutsoudakis G, Owsianka AM, Angus AG, et al. CD81 is dispensable for hepatitis C virus cell‐to‐cell transmission in hepatoma cells. J Gen Virol 2009; 90: 48‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biot C, Glorian G, Maciejewski LA, Brocard JS. Synthesis and antimalarial activity in vitro and in vivo of a new ferrocene‐chloroquine analogue. J Med Chem 1997; 40: 3715‐3718. [DOI] [PubMed] [Google Scholar]
- 12. Dubar F, Bohic S, Slomianny C, Morin JC, Thomas P, Kalamou H, et al. In situ nanochemical imaging of label‐free drugs: a case study of antimalarials in Plasmodium falciparum‐infected erythrocytes. Chem Commun (Camb) 2012; 48: 910‐912. [DOI] [PubMed] [Google Scholar]
- 13. Biot C, Nosten F, Fraisse L, Ter‐Minassian D, Khalife J, Dive D. The antimalarial ferroquine: from bench to clinic. Parasite 2011; 18: 207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Biot C, Daher W, Chavain N, Fandeur T, Khalife J, Dive D, De Clercq E. Design and synthesis of hydroxyferroquine derivatives with antimalarial and antiviral activities. J Med Chem 2006; 49: 2845‐2849. [DOI] [PubMed] [Google Scholar]
- 15. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 1982; 42: 3858‐3863. [PubMed] [Google Scholar]
- 16. Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 1994; 68: 6147‐6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flint M, Maidens C, Loomis‐Price LD, Shotton C, Dubuisson J, Monk P, et al. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J Virol 1999; 73: 6235‐6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boulanger D, Waxweiler S, Karelle L, Loncar M, Mignon B, Dubuisson J, et al. Characterization of monoclonal antibodies to bovine viral diarrhoea virus: evidence of a neutralizing activity against the gp 48 in the presence of a goat anti‐mouse immunoglobulin serum. J Gen Virol 1991; 72: 1195‐1198. [DOI] [PubMed] [Google Scholar]
- 19. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 2005; 11: 791‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, et al. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 2010; 84: 773‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouillé Y, Dubuisson J, et al. Robust production of infectious viral particles in Huh‐7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol 2007; 88: 2495‐2503. [DOI] [PubMed] [Google Scholar]
- 22. Rocha‐Perugini V, Montpellier C, Delgrange D, Wychowski C, Helle F, Pillez A, et al. The CD81 partner EWI‐2wint inhibits hepatitis C virus entry. PLoS One 2008; 3: e1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Op De Beeck A, Voisset C, Bartosch B, Ciczora Y, Cocquerel L, Keck Z, et al. Characterization of functional hepatitis C virus envelope glycoproteins. J Virol 2004; 78: 2994‐3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 2006; 103: 7408‐7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brimacombe CL, Grove J, Meredith LW, Hu K, Syder AJ, Flores MV, et al. Neutralizing antibody‐resistant hepatitis C virus cell‐to‐cell transmission. J Virol 2011; 85: 596‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meuleman P, Albecka A, Belouzard S, Vercauteren K, Verhoye L, Wychowski C, et al. Griffithsin has antiviral activity against hepatitis C virus. Antimicrob Agents Chemother 2011; 55: 5159‐5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rouille Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E, et al. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol 2006; 80: 2832‐2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alsaleh K, Delavalle PY, Pillez A, Duverlie G, Descamps V, Rouille Y, et al. Identification of basic amino acids at the N‐terminal end of the core protein that are crucial for hepatitis C virus infectivity. J Virol 2010; 84: 12515‐12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castelain S, Descamps V, Thibault V, Francois C, Bonte D, Morel V, et al. TaqMan amplification system with an internal positive control for HCV RNA quantitation. J Clin Virol 2004; 31: 227‐234. [DOI] [PubMed] [Google Scholar]
- 30. Albecka A, Belouzard S, de Beeck AO, Descamps V, Goueslain L, Bertrand‐Michel J, et al. Role of low‐density lipoprotein receptor in the hepatitis C virus life cycle. HEPATOLOGY 2012; 55: 998‐1007. [DOI] [PubMed] [Google Scholar]
- 31. Ciesek S, Steinmann E, Iken M, Ott M, Helfritz FA, Wappler I, et al. Glucocorticosteroids increase cell entry by hepatitis C virus. Gastroenterology 2010; 138: 1875‐1884. [DOI] [PubMed] [Google Scholar]
- 32. Lavillette D, Pecheur EI, Donot P, Fresquet J, Molle J, Corbau R, et al. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol 2007; 81: 8752‐8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res 2004; 10: 7994‐8004. [DOI] [PubMed] [Google Scholar]
- 34. Prichard MN, Shipman C, Jr . A three‐dimensional model to analyze drug‐drug interactions. Antiviral Res 1990; 14: 181‐205. [DOI] [PubMed] [Google Scholar]
- 35. Ploss A, Dubuisson J. New advances in the molecular biology of hepatitis C virus infection: towards the identification of new treatment targets. Gut 2012; 61( Suppl 1): i25‐i35. [DOI] [PubMed] [Google Scholar]
- 36. Biot C, Taramelli D, Forfar‐Bares I, Maciejewski LA, Boyce M, Nowogrocki G, et al. Insights into the mechanism of action of ferroquine. Relationship between physicochemical properties and antiplasmodial activity. Mol Pharm 2005; 2: 185‐193. [DOI] [PubMed] [Google Scholar]
- 37. Picard‐Maureau M, Jarmy G, Berg A, Rethwilm A, Lindemann D. Foamy virus envelope glycoprotein‐mediated entry involves a pH‐dependent fusion process. J Virol 2003; 77: 4722‐4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cassell S, Edwards J, Brown DT. Effects of lysosomotropic weak bases on infection of BHK‐21 cells by Sindbis virus. J Virol 1984; 52: 857‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yoshimura A, Kuroda K, Kawasaki K, Yamashina S, Maeda T, Ohnishi S. Infectious cell entry mechanism of influenza virus. J Virol 1982; 43: 284‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dubar F, Egan TJ, Pradines B, Kuter D, Ncokazi KK, Forge D, et al. The antimalarial ferroquine: role of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem Biol 2011; 6: 275‐287. [DOI] [PubMed] [Google Scholar]
- 41. Chavain N, Vezin H, Dive D, Touati N, Paul JF, Buisine E, Biot C. Investigation of the redox behavior of ferroquine, a new antimalarial. Mol Pharm 2008; 5: 710‐716. [DOI] [PubMed] [Google Scholar]
- 42. Vieyres G, Thomas X, Descamps V, Duverlie G, Patel AH, Dubuisson J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J Virol 2010; 84: 10159‐10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mizui T, Yamashina S, Tanida I, Takei Y, Ueno T, Sakamoto N, et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus‐associated autophagy. J Gastroenterol 2010; 45: 195‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dreux M, Chisari FV. Impact of the autophagy machinery on hepatitis C virus infection. Viruses 2011; 3: 1342‐1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Supan C, Mombo‐Ngoma G, Dal‐Bianco MP, Ospina Salazar CL, Issifou S, Mazuir F, et al. Pharmacokinetics of ferroquine, a novel 4‐aminoquinoline, in asymptomatic carriers of Plasmodium falciparum infections. Antimicrob Agents Chemother 2012; 56: 3165‐3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brown RS. Hepatitis C and liver transplantation. Nature 2005; 436: 973‐978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
Supporting Information Figure 5.
Supporting Information Figure 6.
Supporting Information