

Summary
Bats have been demonstrated to be natural reservoirs of severe acute respiratory syndrome coronavirus (SARS CoV) and Middle East respiratory syndrome (MERS) CoV. Faecal samples from 248 individuals of 20 bat species were tested for partial RNA‐dependent RNA polymerase gene of CoV and 57 faecal samples from eight bat species were tested positive. The highest detection rate of 44% for Scotophilus kuhlii, followed by 30% for Rhinolophus monoceros. Significantly higher detection rates of coronaviral RNA were found in female bats and Scotophilus kuhlii roosting in palm trees. Phylogenetic analysis classified the positive samples into SARS‐related (SARSr) CoV, Scotophilus bat CoV 512 close to those from China and Philippines, and Miniopterus bat CoV 1A‐related lineages. Coronaviral RNA was also detected in bat guano from Scotophilus kuhlii and Myotis formosus flavus on the ground and had potential risk for human exposure. Diverse bat CoV with zoonotic potential could be introduced by migratory bats and maintained in the endemic bat population in Taiwan.
Keywords: Chiroptera, coronavirus, Taiwan, severe acute respiratory syndrome virus, zoonosis, reverse transcription polymerase chain reaction
Impacts.
Coronavirus (CoV) was detected in 57 individual and 11 ground faecal samples from nine bat species in Taiwan, including severe acute respiratory‐related CoV, Scotophilus bat CoV 512 and Miniopterus bat CoV 1A.
Significantly higher detection rates of coronaviral RNA were found in female bats and Scotophilus kuhlii roosting in palm trees.
High nucleotide identities were shared between bat coronaviruses detected from the same bat species in Taiwan, China, and Philippines indicated the endemic circulation of bat CoV in local bat population by migratory bat species.
Introduction
In May 2015, Middle East respiratory syndrome coronavirus (MERS CoV) caused 185 cases and 36 deaths in the Republic of Korea (Lee and Wong, 2015) when the virus is circulating mainly in Saudi Arabia since September in 2012 (Zaki et al., 2012). One decade ago, severe acute respiratory syndrome (SARS)‐CoV caused total 8096 cases worldwide and 346 cases with 37 deaths in Taiwan (Ksiazek et al., 2003). Phylogenetic analysis showed that MERS‐CoV clustered in the same subgroup with Tylonycteris bat CoV HKU4 and Pipistrellus bat CoV HKU5 (Lau et al., 2013). Later, bat CoV HKU4 was found capable of using the same cell receptor of MERS CoV (Yang et al., 2014b). MERS‐related CoV was also detected in Nycteris bats of Ghana and Pipistrellus bats of Europe, Neoromicia zuluensis bats of South Africa, and Vespertilio superans bats of China (Annan et al., 2013; Ithete et al., 2013; Yang et al., 2014a). Rhinolophus bats have been suggested as natural reservoirs of SARS‐CoV because the SARS related (SARSr) CoV found in Rhinolophus sinicus of Yunnan in China had 99.9% of sequence identity with human SARS‐CoV and were able to use the angiotensin‐converting enzyme 2 (ACE2) of humans, civets and bats as receptors to infect cells from human, civets and bats (Ge et al., 2013). The outbreaks of MERS CoV re‐emphasized the importance of bat‐surveillance programs on CoV since the emergence of SARS CoV.
The organization of CoV genome is in the order of 5‐polymerase‐spike (S)‐envelope (E)‐membrane (M)‐nucleocapsid (N)‐polyA‐3. The polymerase gene encodes 15 to 16 non‐structural proteins including RNA‐dependent RNA polymerase (RdRp) gene, used commonly for phylogenetic analysis (Woo et al., 2012). As the largest enveloped RNA viruses with single plus‐stranded RNA genomes of 26 to 32 kb, CoV are classified into four genera from Alpha‐ to Deltacoronavirus. The genus Betacoronavirus had four subgroups from 2a to 2d, including SARSr CoV from human, civet cats, raccoon dogs and bats belonging to subgroup 2b and MERS CoV belonging to subgroup 2c (Agnihothram et al., 2014). Both Alpha‐ and Betacoronavirus have been found in the countries around Taiwan, including China, Philippines, Thailand and Japan. The most common bat species carrying bat CoV are Rhinolophus, Pipistrellus and Miniopterus bats (Tang et al., 2006; Watanabe et al., 2010; Gouilh et al., 2011; Shirato et al., 2012), which distribute widely in Asia, including Taiwan. They could carry and transmit bat CoV into local bat population of Taiwan via migration even though little is known about the migratory patterns of bats. Closely related CoV can be detected in the same bat species living at locations separated by thousands of miles (Drexler et al., 2010) and different CoV species or genera can be found in different bat species living at the same roosting sites (Chu et al., 2006; Gloza‐Rausch et al., 2008; Tong et al., 2009; Drexler et al., 2010). From a host switch, certain CoV species can be detected in totally different hosts (Gloza‐Rausch et al., 2008). In addition, one bat species can harbour more than one CoV species (Chu et al., 2006; Tong et al., 2009). There are 11 endemic species and six endemic subspecies in 36 bat species in Taiwan (Wu et al., 2012; Csorba et al., 2014; Ruedi et al., 2015). Between two biodiversity hotspots, Southeastern China and Philippines, Taiwan has an impressive biodiversity on unit area, 4.6 and 9 times higher than Thailand and Japan, respectively (Csorba et al., 2014). It is critical for public health to understand the correlation between the prevalence of CoV and the dynamics of endemic and non‐endemic bat species in Taiwan.
Factors correlating with CoV infection include bat species, roosting location, habitat types, age, gender, physical condition and reproductive status (Gloza‐Rausch et al., 2008; Lau et al., 2010; Osborne et al., 2011). Because of the ability of fly and migrate and the tendency to live in the large sizes of social groups, bats are predisposed to maintain and transport viruses to other mammals for the possible outbreaks of epidemics (Bennett, 2006). Habitat types of bats include caves, trees and human‐made constructions. The accumulation of bat faeces in schools or houses posts a potential risk for public health (Dobson, 2005). As a potential zoonotic pathogen, CoV is regarded as particularly significant because its high mutation rate, high frequency of recombination, faecal‐oral transmission, and environment resistance of faeces‐immersed virus particles (Casanova et al., 2010; Graham and Baric, 2010; Geller et al., 2012). The investigation of RNA or infectious particles of CoV in the faeces accumulated on the ground under the roosting locations near or within human community, like Scotophilus bats in schools and houses, can be used to evaluate the exposure risk of CoV to human. Previous studies have shown that young and lactating female bats in maternity roosting sites had significant higher detection rate for bat CoV RNA (Gloza‐Rausch et al., 2008; Lau et al., 2010; Osborne et al., 2011). It is important to analyse the effects of bat physical properties on CoV infection for the risk assessment.
The current study was to determine the prevalence of CoV in faecal samples collected from 21 bat species at 10 sampling locations in Taiwan. Phylogenetical relationships of detected CoV were constructed by RdRp gene fragment. Factors contributing to CoV infection were analysed. The surveillance for CoV on bat reservoirs is important for controlling the potential threat of emerging CoV outbreaks in Taiwan and nearby areas.
Materials and Methods
Sample collection
The protocol for capturing and sampling of bats was approved by the Chung Yuan Christian University Animal Care and Use committee and the agriculture bureau of regional governments. All bat faecal samples were collected from August 2013 to October 2014 at 10 locations marked in Fig. 1 from the woods in Guihou (location 1, 25°11′24″N, 121°40′41″E), Wulai (location 2, 24°51′50″N, 121°33′05″E), Dong'ao (location 3, 24°31′55″N, 121°49′53″E), Nan'ao (location 4, 24°31′9″N, 121°46′2″E), and Miaoli (location 5, 24°25′35″N, 121°00′45″E), from the palm trees in Jhutang (location 6, 23°50′51″N, 120°23′12″E) and Beigang (location 9, 23°34′05″N, 120°17′51″E), from Ficus trees in Shuilin (location 8, 23°37′14″N, 120°15′40″E), and from the irrigation culverts in Dili (location 7, 23°48′24″N, 120°54′51″E) and Dongshan (location 10, 23°19′04″N, 120°25′27″E). A harp trap, butterfly net or mist net was used to catch bats. Species, body weight, gender, reproductive status and maturation status of bats were determined by experienced bat biologists at the Taiwan Endemic Species Research Institute. According to the swelling of nipples, female bats can be classified into lactating, weaning and non‐lactating females. The mating status of male bats include mating, prepared to mate and non‐mating based on the production of semen by the descent testicles into scrotum. The bats with insufficient ephiphyseal–diaphyseal closure in the metacarpal–phalangeal joints were considered younger than 1 year old and others were considered adults. The captured bats were individually placed into cotton bags and allowed to produce faecal pellets. To clarify whether CoV RNA can be maintained in the surrounding environment, the accumulated faeces on clean plastic sheets overnight from Myotis formosus flavus at location 8 and those from Scotophilus kuhlii at location 6 and 9 were collected as ground faecal samples. Faecal samples were collected by 70% ethanol rinsed tweezers into tubes containing RNAlater® RNA stabilization solution (Qiagen, Hilden, Germany) for sample storage and transportation.
Figure 1.
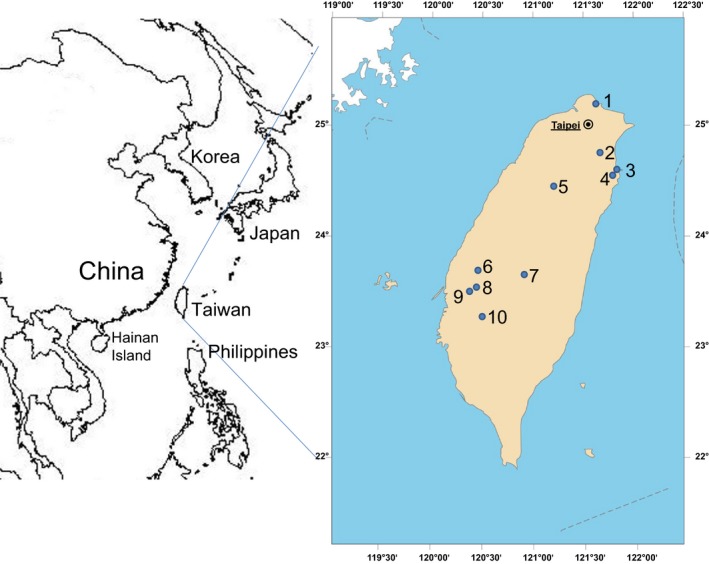
Map of East Asia (left) with enlarged view of Taiwan Island (right). The dots labeled with Arabic number indicated sampling locations 1 to 10. Name/habitat type/geographic coordinate: location 1/Guihou/woods/25°11′24″N/121°40′41″E, location 2/Wulai/woods/24°51′50″N/121°33′05″E, location 3/Dong'ao/woods/24°31′55″N/121°49′53″E, location 4/Nan'ao/woods/24°31′9″N/121°46′2″E, location 5/Miaoli/woods/24°25′35″N/121°00′45″E, location 6/Jhutang/palm trees in an elementary school/23°50′51″N/120°23′12″E, location 7/Dili/irrigation culvert/23°48′24″N/120°54′51″E, location 8/Shuilin/Ficus trees in an elementary school/23°37′14″N/120°15′40″E, location 9/Beigang/palm trees in a sugar factory/23°34′05″N/120°17′51″E, and location 10/Dongshan/irrigation culvert/23°19′04″N/120°25′27″E.
Coronaviral sequence detection and analysis
Viral RNA was extracted from the 140‐μL supernatant of each faecal sample by using the QIAamp Viral RNA Mini Kit (Qiagen) and reverse‐transcribed into cDNA by using iScript Select DNA Synthesis kit (Bio‐Rad, Hercules, CA, USA) with the mixture of random primers and oligo dT primers. Nested reverse transcription polymerase chain reaction (RT‐PCR) with primers targeting a 440‐bp RdRp fragment were performed on all samples according to previous study (Poon et al., 2005). Sequences of the nested PCR products were determined by Sanger sequencing method (Genomics Company, New Taipei City, Taiwan). The consensus RdRp sequence (375‐bp) of 24 bat CoV samples were aligned using the Clustal W method, and the phylogenetic trees based on nucleotide sequences were constructed using the neighbour‐joining model with a 1000 bootstrap test and pairwise deletion option in the maximum likelihood method through the mega6 program (http://www.megasoftware.net). The pairwise distance of nucleotide sequences was analysed using mega6 with the parameters of a 1000 bootstrap test, a p‐distance model, and pairwise deletion. Representative CoV sequences selected from NCBI for phylogenetic analysis were listed in Table S1.
Statistical analysis
Comparisons of coronaviral RdRp gene detection results between groups were performed using Fisher's exact test for categorical variables (bat species, gender, maturation status, reproductive status, sampling location and habitat type) and anova test for continuous variables (body weight) in the R project (R Core Team, 2013). P values of <0.05 were regarded as statistically significance.
Results
Detection of bat CoV RNA in faecal samples
A total of 248 individual and 64 ground faecal samples from 21 bat species were collected. Information regarding bat samples is listed in Table 1. Overall prevalence of CoV in all individual bats was 23% with detection rates of 10% in Kerivoula titania (K) to 43.8% in Scotophilus kuhlii (S). Coronaviral RNA can be detected in 17% (11/64) of ground faecal samples: 25% (2/8) from Scotophilus kuhlii at location 6 in September 2013, 23% (6/26) from Scotophilus kuhlii at location 9 in September 2014, and 10% (3/30) in Myotis formosus flavus at location 8 in June 2014. Of nine bat species tested positive for CoV RNA, five were endemic species and one was endemic subspecies in Taiwan (Table 1). The bats roosting in palm trees had significantly higher detection rates than those in irrigation culvert (P = 0.0243), followed by those in woods (P = 0.0127). Of 10 bat species collected from more than two sampling locations, Myotis fimbriatus taiwanesis had significantly higher detection rate (57%) at location 2 than those (7%) at location 10 (P = 0.0207) and Miniopterus fuliginosus showed significantly higher detection rate (50%) at location 10 than those in other locations (P = 0.0005), including 20% at location 1 and 0% at both location 5 and 7.
Table 1.
Overview of bat faecal samples collected in Taiwan for detection of coronavirus
| Bat Species (abbreviation) | Total No. (positive) | Females (positive) | Locationa | Time (month/year) |
|---|---|---|---|---|
| Family Hipposideridae | ||||
| Coelops frithii formosanus (CF)c | 1 (0) | 1 (0) | 5 | 9/14 |
| Hipposideros armiger terasensis (H)b | 7 (1) | 5 (1) | 5, 10 | 11/2013; 7, 10/2014 |
| Family Rhinolophidae | ||||
| Rhinolophus monoceros (R)b | 50 (15) | 34 (13) | 5, 7, 10 | 11/2013; 7, 10/2014 |
| Family Miniopteridae | ||||
| Miniopterus fuliginosus (M) | 54 (11) | 24 (7) | 1, 5, 7, 10 | 8, 9, 11/2013; 1, 7, 10/2014 |
| Family Vespertilionidae | ||||
| Barbastella darjelingesis (BL) | 2 (0) | 0 | 5 | 7, 8/2014 |
| Harpiola isodon (HI) | 1 (0) | 1 (0) | 5 | 8/14 |
| Kerivoula titania (K) | 10 (1) | 6 (1) | 3, 4, 5 | 9/2013; 7, 9, 10/2014 |
| Murina gracilis (MG)b | 2 (0) | 0 | 5 | 9, 10/2014 |
| Murina puta (MP)b | 17 (0) | 2 (0) | 3, 4, 5 | 4, 7, 8, 9, 10/2014 |
| Murina recondita (MR)b | 4 (1) | 3 (1) | 4, 5 | 4, 6, 10/2014 |
| Myotis fimbriatus taiwanensis (MT)c | 22 (5) | 4 (1) | 2, 10 | 3, 10/2014 |
| Myotis laniger (MS) | 2 (0) | 1 (0) | 5 | 8/14 |
| Myotis rufoniger (MW) | 2 (0) | 1 (0) | 3 | 8/14 |
| Myotis secundus (MY)b | 9 (0) | 6 (0) | 4, 5 | 4, 6, 7/2014 |
| Pipistrellus abramus (P) | 1 (0) | 1 (0) | 6 | 9/13 |
| Pipistrellus montanus (PM) | 3 (0) | 0 | 5 | 7/14 |
| Pipistrellus taiwanesis (PS) | 3 (0) | 2 (0) | 5 | 8, 9/2014 |
| Plecotus taivanus (PT)b | 7 (1) | 2 (0) | 5 | 7, 9/2014 |
| Scotophilus kuhlii (S) | 48 (21) | 28 (11) | 6, 9 | 9/2013; 9/2014 |
| Submyotodon latirostris (ML)b | 3 (1) | 0 | 4, 5 | 6, 9, 10/2014 |
| Total individual faecal samples | 248 (57) | 121 (35) | ||
| Scotophilus kuhlii (S) | 34 (8) | N.D. | 6, 9 | 9/2013; 9/2014 |
| Myotis formosus flavus (MF)c | 30 (3) | N.D. | 8 | 6/14 |
| Total ground faecal samples | 61 (11) | |||
Name/habitat type/geographic coordinate: location 1/Guihou/woods/25°11′24″N/121°40′41″E, location 2/Wulai/woods/24°51′50″N/121°33′05″E, location 3/Dong'ao/woods/24°31′55″N/121°49′53″E, location 4/Nan'ao/woods/24°31′9″N/121°46′2″E, location 5/Miaoli/woods/24°25′35″N/121°00′45″E, location 6/Jhutang/palm trees in an elementary school/23°50′51″N/120°23′12″E, location 7/Dili/irrigation culvert/23°48′24″N/120°54′51″E, location 8/Shuilin/Ficus trees in an elementary school/23°37′14″N/120°15′40″E, location 9/Beigang/palm trees in a sugar factory/23°34′05″N/120°17′51″E, and location 10/Dongshan/irrigation culvert/23°19′04″N/120°25′27″E.
Taiwan endemic species.
Taiwan endemic subspecies.
N.D. means ‘not determine’.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Factors contributing for CoV detection were analysed and shown in Table 2. There was no sampling bias on genders because approximately half of the captured bats were females (49%) but the detection rate of CoV in females was 29%, significantly higher than that (17%) in males (P = 0.0348). Exceptionally, male Scotophilus kuhlii had a higher detection rate (50%) than females did (39%). By examining the nipples of female bats, 20 of 71 female bats evaluated have been lactating and four of them tested positive (20%) for CoV RNA. According to the descent of testicles into scrotum, 37 of 67 male bats evaluated have been mating and only six of them were positive for CoV RNA (16%).
Table 2.
Possible influence factors for coronavirus detection in Taiwan
| Factors | Category | % (CoV positive/total No.) | P value |
|---|---|---|---|
| Gender | Female | 29% (35/121) | 0.0348 |
| Male | 17% (22/127) | ||
| Age | <1 year olda | 8% (2/25) | 0.0778 |
| Adult | 25% (55/223) | ||
| Lactation (Females) | Lactating | 0% (0/6) | 0.1417 |
| Weaning | 29% (4/14) | ||
| Non‐lactating | 39% (20/51) | ||
| Mating (Males) | Mating | 19% (5/27) | 0.9137 |
| Prepare to mate | 10% (1/10) | ||
| Non‐mating | 20% (6/30) |
Age of <1 year old was determined by epiphyseal–diaphyseal closure in the metacarpal–phalangeal joints of bats.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Phylogenetic analysis of bat CoV sequences
A 375‐bp RdRp gene fragment from 24 bat CoV samples was sequenced and assigned GenBank accession number from KT381902 to KT381925 for bat CoV sample S1, S3, S5, S6, S8, S9, S11, S13, S34, S37, S38, R6, R9, R11, R14, M4, M8, M16, M22, M23, M24, M26, K9 and ML1 by the National Center for Biotechnology Information. The phylogenetic tree based on nucleotide sequences was constructed. Three distinct lineages were defined and noted with the identities of nucleotide sequences to the closest CoV strains in Fig. 2. Bat CoV sample CYCU‐R9/TW/2013 from Rhinolophus monoceros of Dongshan (location 10) belonged to SARSr CoV‐related lineage in the genus Betacoronavirus because of the 88% identity to those of human SARS Co Tor2 strain, bat CoV HKU3, and SARSr CoV Rp3 found in Rhinolophus bat of China. Scotophilus bat CoV 512‐related lineage contained 20 samples from four bat species and Miniopterus bat CoV 1A‐related lineage included three samples from two bat species. Both lineages belonged to the genus Alphacoronavirus. Bat CoV RNA isolated in all five Miniopterus bat samples from location 10 in southern Taiwan belonged to Scotophilus bat CoV 512‐related lineage and those in two Miniopterus bat samples from location 2 in northern Taiwan belonged to Miniopterus bat CoV 1A‐related lineage. The same colony of Rhinolophus monoceros in the same irrigation culvert at location 10 was found bat CoV RNA belonging to SARSr CoV and Scotophilus bat CoV 512‐related lineages.
Figure 2.
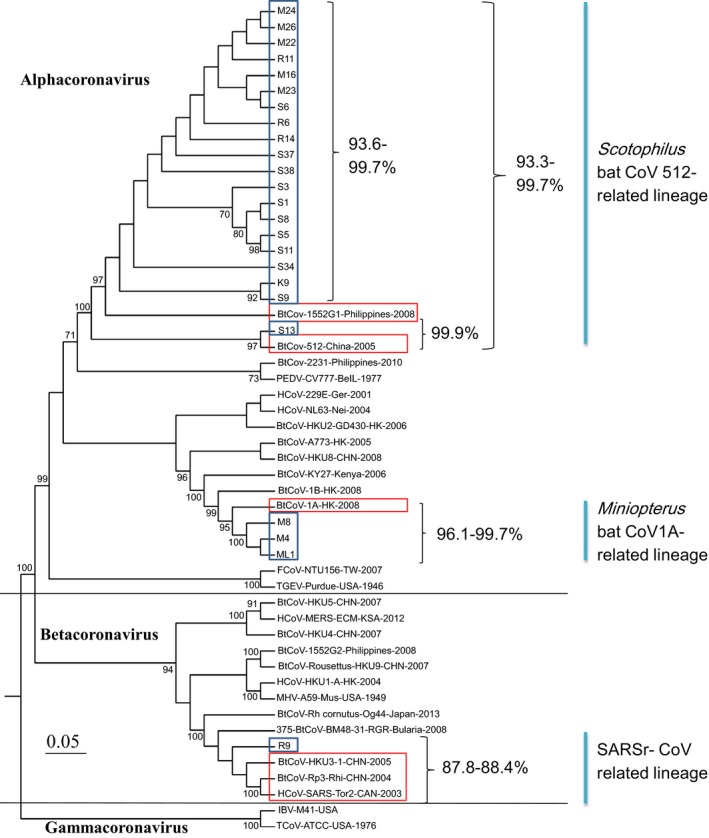
Phylogenetic tree of three bat coronavirus lineages detected in Taiwan. The consensus 375 nucleotide sequences of partial RNA‐dependent RNA polymerase gene from 24 bat CoV samples in Taiwan and selected sequences from Genbank were aligned using the Clustal W method and the tree was constructed using the neighbour‐joining model with a 1000 bootstrap test and pairwise deletion option in the maximum likelihood method by mega6 program. Nucleotide identities were indicated next to the bracket.
Discussion
The current study has revealed a diverse bat CoV population circulating in Taiwan, following our previous report on Scotophilus bat CoV in central Taiwan (Su et al., 2016). Compared to the detection range of 5–73% for CoV RNA in bats worldwide, the CoV prevalence of 23% in Taiwan confirmed the widely distribution of CoV (Tang et al., 2006; Tong et al., 2009; Rihtaric et al., 2010; Balboni et al., 2011; Shirato et al., 2012; Woo et al., 2012; Góes et al., 2013; Su et al., 2016). Female bats had significantly higher detection rate for bat CoV in this study, especially Rhinolophus monoceros (38% in females and 8% in males). In breeding season, female bats and their offspring tend to roost together as maternity colony and viruses are easier to circulate in large number (Gloza‐Rausch et al., 2008). However, male and female Scotophilus bats roost together so no significance was found in the detection rate of CoV RNA for different genders, reproductive and maturation status of Scotophilus bats in this study. The presence of bat CoV RNA in 17% of ground faecal samples could post a risk for potential interspecies transmission through gene recombination, such as the emergence of human CoV (HCoV) NL63, HCoV HKU1, SARS CoV and MERS CoV (Bolles et al., 2011), if the faeces containing bat CoV are ingested by animals or humans. Further investigation on the survival of infectious bat CoV in the environment is needed.
Both SARSr CoV and Scotophilus bat CoV 512 were found in the same colony of Rhinolophus monoceros. Although the bat detected positive for SARSr CoV appeared healthy, it had lower body weight of 4 g when the body weights of other collected Rhinolophus bats ranged from 4 to 6.5 g. Similar observation was noted in the Rhinolophus pearsoni carrying SARSr CoV Rp3 in Guangxi of China, which caused acute and self‐limiting infection with viral clearance occurring between 2 weeks and 4 month (Lau et al., 2010). Recombination between SARSr CoV Rp3 from Guangxi and Rf1 from Hubei may generate SARSr CoV SZ3 isolated from civets. A 4‐year investigation on the migration pattern and genome epidemiology showed that Rhinolophus bats can migrate from 1.86 to 17 km and carry bat CoV HKU2 or SARSr CoV Rp3 for further co‐infection and frequent recombination (Lau et al., 2010). Scotophilus bat CoV 512 was only detected in one bat species, Scotophilus kuhlii, in Hainan Island of China and the Philippines previously (Tang et al., 2006; Watanabe et al., 2010). In this study, endemic Rhinolophus monoceros, endemic Kerivoula titania and migratory Miniopterus fuliginosus were detected positive for bat CoV RNA belonging to Scotophilus bat CoV 512 related lineage beside Scotophilus kuhlii. Transmission of Scotophilus bat CoV 512 between migratory and endemic bat species was implied because some of them share the same roosting site.
Bat CoVs sharing 96% of nucleotide identity to those of Miniopterus bat CoV 1A were not only detected in migratory Miniopterus fuliginosus but also endemic Submyotodon latirostris. It is also the first report that Miniopterus bat CoV 1 A can be detected in a non‐Miniopterus bat species. These two bat species did not share roosting location although all bats tested positive came from northern Taiwan. Another unique finding is that two different bat CoV lineages were detected in two different populations of Miniopterus fuliginosus whose living areas did not overlap. The distribution patterns of bat CoV in Miniopterus bats are very complicated. In Hong Kong, phylogenetically related Miniopterus bat CoV 1A and 1B have distinct host specificities to Miniopterus magnate and Miniopterus pusillus, respectively, even though these two bat species co‐inhabit the same cave. On the other hand, co‐infection of bat CoV 1B and HKU8 was detected in Miniopterus pusillus. No evidence of recombination was detected between bat CoV 1A, 1B, HKU8, 512, HKU2 and SARSr CoV isolated from Miniopterus bats (Chu et al., 2006). In Kenya, bat CoV 1A was isolated in Miniopterus africanus, Miniopterus minor and Miniopterus natalensis, and both bat CoV 1A and HKU8 can be isolated in Miniopterus inflatus (Tong et al., 2009). In Bulgaria, bat CoV 1A, HKU‐7, and HKU‐8 were detected in Miniopterus schreibersii but not Rhinolophus and Hipposideros bats although all three bat species shared the same cave (Woo et al., 2012). Since Miniopterus bats are migratory and the distance of migration is reported to be several hundreds of kilometers (Ramos Pareira et al., 2009), it is implied that Miniopterus bat CoV might be transmitted between Hong Kong, Japan, even Europe as a result of bat migration. Further investigations on the distribution of bat CoV and interactions between migratory and endemic bat species are required for the close monitoring on SARSr‐CoV and other bat CoV in Taiwan.
This study partially filled a knowledge gap regarding the prevalence and distribution of CoV in different bat species of Taiwan. Three bat CoV lineages identified as SARSr CoV, Scotophilus bat CoV 512 and Miniopterus bat CoV 1A, are very common in countries around Taiwan. Host switch may play a very important role in the distribution of different bat CoV from migratory bat species to Taiwan endemic bat species. Ongoing surveillance on bat CoV in different bat populations is necessary to understand the zoonotic potential of bat CoV.
Supporting information
Table S1. The 28 coronavirus sequences from NCBI for phylogenetic analysis.
Acknowledgements
This work was funded by the Ministry of Science and Technology of Taiwan (NSC 102‐2313‐B‐033‐001). We thank Chia‐Hong Chen at Shei‐Pa National Park Headquarters at Miaoli for the foraging bat samples and Heng‐Chia Chang for the ground faeces from Shuilin.
References
- Agnihothram, S. , Gopal R., Yount B. L., Donaldson E. F., Menachery V. D., Graham R. L., Scobey T. D., Gralinski L. E., Denison M. R., Zambon M., and Baric R. S., 2014: Evaluation of serologic and antigenic relationships between middle eastern respiratory syndrome coronavirus and other coronaviruses to develop vaccine platforms for the rapid response to emerging coronaviruses. J. Infect. Dis. 209, 995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annan, A. , Baldwin H. J., Corman V. M., Klose S. M., Owusu M., Nkrumah E. E., Badu E. K., Anti P., Agbenyega O., Meyer B., Oppong S., Sarkodie Y. A., Ealko E. K., Lina P. H., Godlevska E. V., Reusken C., Seebens A., Gloza‐Rausch F., Vallo P., Tschapka M., Drosten C., and Drexler J. F., 2013: Human Betacoronavirus 2c EMC/2012‐related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 19, 456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balboni, A. , Palladini A., Bogliani G., and Battilani M., 2011: Detection of a virus related to Betacoronaviruses in Italian greater horseshoe bats. Epidemiol. Infect. 139, 216–219. [DOI] [PubMed] [Google Scholar]
- Bennett, M. , 2006: Bats and human emerging diseases. Epidemiol. Infect. 134, 905–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolles, M. , Donaldson E., and Baric R., 2011: SARS‐CoV and emergent coronaviruses: viral determinants of interspecies transmission. Curr. Opin. Virol. 1, 624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova, L. M. , Jeon S., Rutala W. A., Weber D. J., and Sobsey M. D., 2010: Effects of air temperature and relative humidity on coronavirus survival on surfaces. Appl. Environ. Microbiol. 76, 2712–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, D. K. , Poon L. L., Chan K. H., Chen H., Guan Y., Yuen K. Y., and Peiris J. S., 2006: Coronaviruses in bent‐winged bats (Miniopterus spp.). J. Gen. Virol. 87, 2461–2466. [DOI] [PubMed] [Google Scholar]
- Csorba, G. , Chou C. H., Ruedi M., Görföl T., Motokawa M., Wiantoro S., Thong V. D., Son N. T., Lin L. K., and Furey N., 2014: The reds and the yellows: a review of Asian Chrysopteron Jentink, 1910 (Chiroptera: Vespertilionidae: Myotis). J. Mammal. 95, 663–678. [Google Scholar]
- Dobson, A. P. , 2005: Virology. What links bats to emerging infectious diseases? Science 310, 628–629. [DOI] [PubMed] [Google Scholar]
- Drexler, J. F. , Gloza‐Rausch F., Glende J., Corman V. M., Muth D., Goettsche M., Seebens A., Niedrig M., Pfefferle S., Yordanov S., Zhelyazkov L., Hermanns U., Vallo P., Lukashev A., Muller M. A., Deng H., Herrler G., and Drosten C., 2010: Genomic characterization of severe acute respiratory syndrome‐related coronavirus in European bats and classification of coronaviruses based on partial RNA‐dependent RNA polymerase gene sequences. J. Virol. 84, 11336–11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, X. Y. , Li J. L., Yang X. L., Chmura A. A., Zhu G., Epstein J. H., Mazet J. K., Hu B., Zhang W., Peng C., Zhang Y. J., Luo C. M., Tan B., Wang N., Zhu Y., Crameri G., Zhang S. Y., Wang L. F., Daszak P., and Shi Z. L., 2013: Isolation and characterization of a bat SARS‐like coronavirus that uses the ACE2 receptor. Nature 503, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller, C. , Varbanov M., and Duval R. E., 2012: Human coronaviruses: insights into environmental resistance and its influence on the development of new antiseptic strategies. Viruses 4, 3044–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloza‐Rausch, F. A. , Ipsen A., Seebens M., Gottsche M., Panning J. F., Drexler N., Petersen A., Annan K., Grywna M., Muller S. Pfefferle., and Drosten C., 2008: Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 14, 626–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Góes, L. G. , Ruvalcaba S. G., Campos A. A., Queiroz L. H., de Carvalho C., Jerez J. A., Durigon E. L., Dávalos L. I., and Dominguez S. R., 2013: Novel bat coronaviruses, Brazil and Mexico. Emerg. Infect. Dis. 19, 1711–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouilh, M. A. , Puechmaille S. J., Gonzalez J. P., Teeling E., Kittayapong P., and Manuguerra J. C., 2011: SARS‐Coronavirus ancestor's foot‐prints in South‐East Asian bat colonies and the refuge theory. Infect. Genet. Evol. 11, 1690–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, R. L. , and Baric R. S., 2010: Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross‐species transmission. J. Virol. 84, 3134–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ithete, N. L. , Stoffberg S., Corman V. M., Cottontail V. M., Richards L. R., Schoeman M. C., Drosten C., Drexler J. F., and Preiser W., 2013: Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 19, 1697–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek, T. G. , Erdman D., Goldsmith C. S., Zaki S. R., Peret T., and Emery S., 2003: A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966. [DOI] [PubMed] [Google Scholar]
- Lau, S. K. , Li K. S., Huang Y., Shek C. T., Tse H., Wang M., Choi G. K., Xu H., Lam C. S., Guo R., Chan K. H., Zheng B. J., Woo P. C., and Yuen K. Y., 2010: Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome‐related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self‐limiting infection that allows recombination events. J. Virol. 84, 2808–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, S. K. , Li K. S., Tsang A. K., Lam C. S., Ahmed S., Chen H., Chan K. H., Woo P. C., and Yuen K. Y., 2013: Genetic characterization of Betacoronavirus lineage c viruses in bats reveals marked sequence divergence in the spike protein of Pipistrellus bat coronavirus HKU5 in Japanese Pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J. Virol. 87, 8638–8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. S. , and Wong N. S., 2015: Probable transmission chains of Middle East respiratory syndrome coronavirus and the multiple generations of secondary infection in South Korea. Int. J. Infect. Dis. 38, 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne, C. , Cryan P. M., O'Shea T. J., Oko L. M., Ndaluka C., Calisher C. H., Berglund A. D., Klavetter M. L., Bowen R. A., Holmes K. V., and Dominguez S. R., 2011: Alphacoronaviruses in new world bats: prevalence, persistence, phylogeny, and potential for interaction with humans. PLoS One 6, e19156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, L. L. , Chu D. K., Chan K. H., Wong O. K., Ellis T. M., Leung Y. H., Lau S. K., Woo P. C., Suen K. Y., Yuen K. Y., Guan Y., and Peiris J. S., 2005: Identification of a novel coronavirus in bats. J. Virol. 79, 2001–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R_project.org/.
- Ramos Pareira, M. J. , Salgueiro P., Rodrigues L., Coelho M. M., and Palmeirim J. M., 2009: Population structure of a cave‐dwelling bat, Miniopterus schreibersii: does it reflect history and social organization? J. Hered. 100, 533–544. [DOI] [PubMed] [Google Scholar]
- Rihtaric, D. , Hostnik P., Steyer A., Grom J., and Toplak I., 2010: Identification of SARS‐like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol. 155, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruedi, M. , Csorba G., Lin L. K., and Chou C. H., 2015: Molecular phylogeny and morphological revision of Myotis bats (Chiroptera: Vespertilionidae) from Taiwan and adjacent China. Zootaxa 3920, 301–342. [DOI] [PubMed] [Google Scholar]
- Shirato, K. , Maeda K., Tsuda S., Suzuki K., Watanabe S., Shimoda H., Ueda N., Iha K., Taniguchi S., Kyuwa S., Endoh D., Matsuyama S., Kurane I., Saijo M., Morikawa S., Yoshikawa Y., Akashi H., and Mizutani T., 2012: Detection of bat coronaviruses from Miniopterus fuliginosus in Japan. Virus Genes 44, 40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, B. G. , Chen H. C., Cheng H. C., and Chen Y. N., 2016: Detection of bat coronavirus and specific antibodies in chestnut bat (Scotophilus kuhlii) population in central Taiwan. Taiwan Vet. J. 42, 1–8. [Google Scholar]
- Tang, X. C. , Zhang J. X., Zhang S. Y., Wang P., Fan X. H., Li L. F., Li G., Dong B. Q., Liu W., Cheung C. L., Xu K. M., Song W. J., Vijaykrishna D., Poon L. L., Peiris J. S., Smith G. J., Chen H., and Guan Y., 2006: Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 80, 7481–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, S. , Conrardy C., Ruone S., Kuzmin I. V., Guo X., Tao Y., Niezgoda M., Haynes L., Agwanda B., Breiman R. F., Anderson L. J., and Rupprecht C. E., 2009: Detection of novel SARS‐like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15, 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, S. , Masangkay J. S., Nagata N., Morikawa S., Mizutani T., Fukushi S., Alviola P., Omatsu T., Ueda N., Iha K., Taniguchi S., Fujii H., Tsuda S., Endoh M., Kato K., Tohya Y., Kyuwa S., Yoshikawa Y., and Akashi H., 2010: Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 16, 1217–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau S. K., Lam C. S., Lau C. C., Tsang A. K., Lau J. H., Bai R., Teng J. L., Tsang C. C., Wang M., Zheng B. J., Chan K. H., and Yuen K. Y., 2012: Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus . J. Virol. 86, 3995–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Li Y., Lin L. K., Harada M., Chen Z., and Motokawa M., 2012: New records of Kerivoula titania (Chiroptera: Vespertilionidae) from Hainan Island and Taiwan. Mammal Study 37, 69–72. [Google Scholar]
- Yang, L. , Wu Z., Ren X., Yang F., Zhang J., He G., Dong J., Sun L., Zhu Y., Zhang S., and Jin Q., 2014a: MERS‐related Betacoronavirus in Vespertilio superans bats, China. Emerg. Infect. Dis. 20, 1260–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Du L., Liu C., Wang L., Ma C., Tang J., Baric R. S., Jiang S., and Li F., 2014b: Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat‐to‐human transmission of MERS coronavirus. Proc. Natl Acad. Sci. USA 111, 12516–12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki, A. M. , van Boheemen S., Bestebroer T. M., Osterhaus A. D., and Fouchier R. A., 2012: Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The 28 coronavirus sequences from NCBI for phylogenetic analysis.