

Abstract
Infectious bronchitis virus (IBV) is a worldwide prevalent RNA virus that causes highly contagious and economically devastating disease in chicken. The virus exists in many different genetic forms which made the disease control very difficult. The present study describes the development and validation of TaqMan probe‐based real‐time reverse transcription‐polymerase chain reaction (real‐time RT‐PCR) targeting the S1 coding region of S gene characteristic for the GII‐1 lineage (formerly the D1466‐like variant) of IBV. These strains are quite different from other European IBV belonging to different lineages of the GI genotype. The developed method was 30‐fold more sensitive than used so far for standard nested RT‐PCR with detection limit of 56 RNA copies per reaction. The specificity of the assay was also evaluated with a panel of different poultry pathogens. Repeatability and reproducibility of the method was very high with coefficients of variation lower than 4%. One hundred and twenty‐seven IBV‐positive samples were tested by this method and GII‐1 strains were detected in four of them (3·15%) which indicate a decrease in the GII‐1 IBV prevalence in Poland. The assay was proven to be a valuable tool for rapid diagnosis of GII‐1 lineage of IBV strains and moreover it enabled the monitoring of viral loads which can be used to assess disease progression.
Significance and Impact of the Study
This study reports a TaqMan probe‐based real‐time reverse transcription‐polymerase chain reaction (real‐time RT‐PCR) for rapid and accurate identification of GII‐1 lineage (formerly D1466‐like variant) of infectious bronchitis virus (IBV). The assay revealed to be more sensitive than standard nested RT‐PCR assay, previously used for this purpose. The developed assay has been tested on numerous field samples and revealed 3·15% prevalence of this lineage of IBV in Polish chicken population. Moreover, this new assay enables the assessment of viral load measurement which might be useful for epidemiology and pathogenesis studies.
Keywords: D1466, GII‐1 lineage, IBV, infectious bronchitis, infectious bronchitis virus, real‐time RT‐PCR
Significance and Impact of the Study: This study reports a TaqMan probe‐based real‐time reverse transcription‐polymerase chain reaction (real‐time RT‐PCR) for rapid and accurate identification of GII‐1 lineage (formerly D1466‐like variant) of infectious bronchitis virus (IBV). The assay revealed to be more sensitive than standard nested RT‐PCR assay, previously used for this purpose. The developed assay has been tested on numerous field samples and revealed 3·15% prevalence of this lineage of IBV in Polish chicken population. Moreover, this new assay enables the assessment of viral load measurement which might be useful for epidemiology and pathogenesis studies.
Introduction
Infectious bronchitis virus (IBV) infects chickens of all ages causing respiratory, urogenital, reproductive and/or digestive systems diseases. Infectious bronchitis virus belongs to the Gammacoronavirus genus (order Nidovirales, family Coronaviridae, subfamily Coronavirinae) (Carstens 2010; Dong et al. 2007) and its genome consists of an approximately 30‐kb‐long single‐stranded, positive‐sense RNA. The genome structure and the biology of the virus predispose for changes in IBV genome (Ovchinnikova et al. 2011; Woo et al. 2009). The most important are changes in the spike protein gene (S), especially in its S1 part which encodes extracellular protein subdomain since they may imply a change in the antigenicity or pathogenicity of the virus. So far, many genotypes, serotypes or protectotypes have been identified, depending on the method used for differentiation. However, even the most commonly used genotyping results were sometimes ambiguous because there were no standard rules that should be followed. That's why new classification based on S1 coding region sequence which distinguished and named 32 lineages arranged in six genotypes has been recently proposed (Valastro et al. 2016).
The GII‐1 lineage (named also D1466 or D212 variant) of IBV was detected for the first time in the Netherlands in the late 1970s as an aetiological factor of problems connected with egg losses (Davelaar et al. 1984; de Wit et al. 2011). Interestingly, the S1 coding region of this variant was different from other European IBV strains which resulted in its classification to the GII genotype together with Dutch V1397 strain (Valastro et al. 2016). For many years the D1466 strains has been only occasionally recognized but results of a molecular survey conducted between 2005 and 2006 indicated increasing problems connected with this variant in most countries of Western Europe (Worthington et al. 2008). In Poland this variant emerged in 2011 and subsequently has been identified in different parts of the country (Domanska‐Blicharz et al. 2012).
For IBV detection different molecular assays have been described including conventional reverse transcription‐polymerase chain reaction (RT‐PCR) (Adzhar et al. 1996; Farsang et al. 2002), labelled TaqMan probe‐ or SYBR green‐based real‐time RT‐PCR (real‐time RT‐PCR) assays or reverse transcription loop‐mediated isothermal amplification (RT‐LAMP) (Callison et al. 2006; Chen et al. 2010; Fellahi et al. 2016; Jones et al. 2011; Meir et al. 2010). The next step is identification of detected IBV strain, which relays on amplification, sequencing and blast (Basic Local Alignment Research Tool) or phylogenetic analysis of the sequence of S1 coding region. Recently proposed IBV classification is based on the sequence of the entire S1 part with a length of 1620 nt but this is only possible for highly specialized molecular laboratories (Valastro et al. 2016). The main obstacle is the lack of universal primers and sometimes many primers should be tested before final strain identification which could be a very time and labour consuming. On the other hand, information about IBV circulating in the field should be quickly obtained as it is important for selecting suitable protective vaccine. The real‐time RT‐PCR assays seem to be effective tools for IBV‐type identification and although S1 coding region is prone to mutations, methods aimed in this gene detection has been recently developed and used for GI‐1 (Mass, Connecticut), GI‐9 (Arkansas), GI‐11 (SAI), G1‐16 (ASAII) and GIV‐1 (DE072/GA98) lineages (Acevedo et al. 2013; Marandino et al. 2016; Roh et al. 2014, 2013).
For detection of GII‐1 (D1466‐like) IBV lineage standard nested RT‐PCR assay is commonly applied. However, this method is a time‐consuming and labour intensive as it needs multiple manipulation of genetic material. Moreover, these multiple steps of RNA/DNA handling might result in cross‐contamination and as consequences in false positive results. The real‐time RT‐PCR assay helps to avoid these disadvantages. Here, we describe the development of a TaqMan probe‐based real‐time RT‐PCR for the detection of GII‐1 (D1466‐like) IBV lineage. We also describe the conclusions from the routine use of this method, which was implemented in our laboratory in 2014.
Results and discussion
Optimizing of GII‐1 real‐time RT‐PCR
In the present work we have successfully developed a highly sensitive TaqMan probe‐based real‐time RT‐PCR assay for detection of GII‐1 lineage of IBV. So far, standard nested RT‐PCR assay was commonly used for this purpose (Cavanagh et al. 1999; Worthington et al. 2008). Both methods target similar fragment of S1 coding fragment which is crucial for IBV‐type determination. However, the primers and probe designed for real‐time RT‐PCR met single mismatches in nucleotide sequences of D1466‐like IBV strains previously detected in Poland and they include some degenerative ones (two in the reverse primer and two in the probe) to avoid failure in this genotype detection. The standard curve and linear regression analysis were performed using serial dilutions of known quantity of GII‐1 IBV RNA (Fig. 1). C t values ranged from 17·70 to 37·03 cycles (Table 1) with a coefficient of determination (R 2) of 0·996. The slope of −3·168 reveals a very high RT‐PCR efficiency (106%). The limit of detection of the GII‐1 IBV real‐time RT‐PCRs and universal for all gammacoronavirus were similar at the level of an approximately 56 copies of viral RNA. The sensitivity of the standard nested RT‐PCR was almost 30‐fold lower and detected the presence about 1500 copies of viral RNA (Table 1). Moreover, the assay developed in the present study showed to be highly specific as no fluorescent signals were detected with other tested IBV lineages or chicken RNA viral pathogens. The intra and interassay precision of the new method were assessed using serial dilutions of D1466 IBV RNA ranging from 8·2 × 106 down to 5·6 × 101 copies. The repeatability CVs of D1466 real‐time RT‐PCR ranged from 0·17 to 0·69%, while the reproducibility CVs ranged from 0·99 to 3·55% (Table 2).
Figure 1.
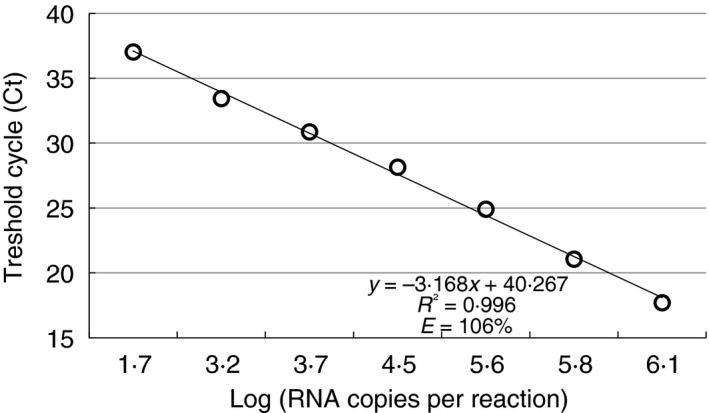
Standard curve of real‐time RT‐PCR assay for the detection of GII‐1 lineage of Infectious bronchitis virus (IBV). Each point represents dilution of vaccinal D1466 variant of IBV containing from 8·2 × 104 to 5·6 × 101 RNA copies/reaction.
Table 1.
Sensitivity of standard nested RT‐PCR, universal and D1466 real‐time RT‐PCR assays
| Viral copies | Standard nested RT‐PCR | Universal real‐time RT‐PCR* | D1466 real‐time RT‐PCRa |
|---|---|---|---|
| 8·2 × 106 | pos | 18·29 | 17·70 |
| 6·1 × 105 | pos | 21·59 | 21·06 |
| 3·7 × 105 | pos | 25·05 | 24·91 |
| 3·5 × 104 | pos | 28·36 | 28·15 |
| 5·1 × 103 | pos | 31·27 | 30·87 |
| 1·5 × 103 | pos | 33·24 | 33·44 |
| 5·6 × 101 | neg | 36·92 | 37·03 |
nd, not done; pos, positive result; neg, negative result.
Mean C t value obtained in the assays.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 2.
Intra‐ and interassay variability in the D1466 real‐time RT‐PCR
| Viral copies | C t mean | Intra‐assay (repeatability) | C t mean | Interassay (reproducibility) | ||
|---|---|---|---|---|---|---|
| CV | SD | CV | SD | |||
| 8·2 × 106 | 17·70 | 0·69 | 0·12 | 17·76 | 2·39 | 0·42 |
| 6·1 × 105 | 21·06 | 0·10 | 0·02 | 21·32 | 3·55 | 0·75 |
| 3·7 × 105 | 24·91 | 0·04 | 0·01 | 24·87 | 1·27 | 0·31 |
| 3·5 × 104 | 28·15 | 0·36 | 0·10 | 28·03 | 1·12 | 0·31 |
| 5·1 × 103 | 30·87 | 0·16 | 0·05 | 30·85 | 1·24 | 0·38 |
| 1·5 × 103 | 33·44 | 0·53 | 0·18 | 32·60 | 2·93 | 0·95 |
| 5·6 × 101 | 37·03 | 0·17 | 0·06 | 37·59 | 0·99 | 0·37 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
GII‐1 IBV detection in field samples
The assay developed in this study has been tested on numerous field tissues and swabs samples. The total of 190 field samples from chicken (broilers, commercial layers and broiler breeders) were tested by gammacoronavirus universal real‐time RT‐PCR assay in the period between 2014 and 2016. One hundred and twenty‐seven (66·8%) were positive for IBV. When these samples were tested by the GII‐1 real‐time RT‐PCR, 4 of 127 (3·15%) were positive. Interestingly, no D1466‐positive sample was detected in 2014 but three samples in 2015 and one in 2016 were identified. Molecular studies carried out between 2005 and 2006 in Western European countries showed growing problems associated with D1466 genotype (Worthington et al. 2008). In 2005, the average frequency of the D1466‐like virus in Belgium, the Netherlands and Germany was about 3–5%, and in 2006 the presence of this variant increased to 7, 10 and 16% respectively. In Poland, in the period from November 2011, when the first case of D1466 IBV was detected, to December 2013, the standard nested RT‐PCR was used and this assay enabled detection of 26 positive samples indicating the presence of this variant at 11·7% of studied chicken flocks. Our study clearly shows a gradual decrease in the GII‐1 IBV prevalence in Poland.
The assay here developed enables the estimation of the amount of viral RNA copies, which could be a useful indicator of the infection phase and correlate with viraemia or clinical disease (Mackay et al. 2002). The real‐time RT‐PCR is able to detect as few as 56 RNA molecules of this IBV lineage. When testing field samples, the C t values obtained were relatively high (mean C t values of 32·4 ± 3·8) which address the mean amount of virus RNA about 1500 copies per reaction. The low number of GII‐1 IBV RNA copies in most cases did not parallel the C t values obtained in universal real‐time RT‐PCR which were much lower. This was due to the presence of other IBV lineages in these samples. Subsequent applied assays aimed at the identification of other IBV lineages revealed the presence GI‐1 (Mass‐like), GI‐13 (793B‐like), GI‐19 (QX‐like) or GI‐23 (Var2‐like) in almost all of them. There was only one case where the C t values in both assays were equal indicating a low number of RNA copies (C t about 32). It seems that this variant replicates poorly in the tissues of chickens and its excretion with the faeces is also low. The poorly replicating viruses are unlikely to cause disease symptoms. In fact some of the D1466‐positive chicken flocks were described as healthy by the maintenance staff. Therefore, pathogenicity and virulence of the GII‐1 strains seem to be insignificant.
We also revealed the usefulness the RT‐ddPCR for RNA concentration determination. Usually, standard curves are synthetically generated with plasmids or transcript molecules with the same sequence as the gene of interest. However, its concentration depends not only on the dilution but also on the quantification methods which themselves are prone to uncertainties. The RT‐ddPCR is a method that greatly facilitates RNA measurement and overcomes these limitations. The primers and probe constructed in this study for real‐time RT‐PCR were successfully used in RT‐ddPCR. However, due to the fact that only few laboratories use this technology routinely, our efforts have focused on real‐time RT‐PCR.
In conclusion, the TaqMan probe‐based real‐time RT‐PCR assay described here is a time‐saving, specific, sensitive and reliable method of detection of GII‐1 lineage (D1466‐like) of IBV which could successfully replace standard nested RT‐PCR. Moreover, this new assay enables the assessment of viral load measurement which might be useful for epidemiology and pathogenesis studies.
Materials and methods
Primers and probe design
The S1 coding fragment sequences of D1466‐like IBV strains were retrieved from the GenBank database and aligned using mega6 (http://www.megasoftware.net, Molecular Evolutionary Genetics Analysis ver. 6.0) software package. Primers and probe were designed manually based on a conserved region within the aligned S1 part of S gene sequences of 12 strains of GII‐1 IBV lineage. Forward primer D5‐f (5′‐TTACAGCCTGGCAATGTCTT‐3′) located at nucleotide position 458–477 according to the sequence of D1466 strain (GenBank accession no. M21971); reverse primer D6‐r (5′‐CAACATCCTCmrTAAAGTTAGAAC‐3′) located at nucleotide position 521–544, and TaqMan D‐probe (FAM‐CyAGTGTGTTTCTAAAyGGCAACCTT‐BHQ1) located at nucleotide position 488–514 amplified and detected 87‐bp fragment. In silico specificity of the primers and probe was assessed by blastn search function (http://www.ncbi.nlm.nih.gov/BLAST). The primers and probe were synthesized by Genomed Sp. z o.o. (Warsaw, Poland).
Sampling and sample preparation
Tissue samples (kidney, oviduct, respiratory and intestinal tracts) and oropharyngeal/cloacal swabs from various broiler chickens, commercial layers and broiler breeder farms were submitted for laboratory diagnosis. In the period between 2014 and 2016 a total of 190 flocks from different regions of Poland were obtained. For molecular assays optimization the live attenuated IB vaccine (Nobilis IB D1466; MSD Animal Health, Boxmeer, the Netherlands) was used. Tissue samples were homogenized in 10–20% (w/v) suspension and vaccine and swabs hydrated in phosphate‐buffered saline (PBS) with addition of antibiotics (100 μ penicillin and 100 μg streptomycin per ml), centrifuged at 3000g for 15 min. Total RNA was extracted from 250 μl of tissues homogenates, vaccine and swabs (five pooled swabs) supernatants into 50 μl RNase‐free water using commercial kits (RNeasy Mini Kit; Qiagen, Hilden, Germany) according to the manufacturer's instruction.
Standard nested RT‐PCR and real‐time RT‐PCR assays
Standard nested RT‐PCR amplification was performed using the primers described by Cavanagh et al. (1999). The first reaction was carried out with OneStep RT‐PCR Kit (Qiagen) in a 25‐μl final reaction mixture volume containing 5 μl 5× OneStep RT‐PCR buffer, 5 μl 5xQ solution, primers to a final concentration of 600 nmol l−1, 400 nmol l−1 of each dNTP, 2·5 μl RNA template and nuclease‐free water. The nested reaction was carried out with Taq DNA Polymerase Kit (Eurx, Gdansk, Poland) in a 25‐μl volume containing 12·5 μl 2× Taq PCR Master Mix, nested primers to a final concentration of 600 nmol l−1, 2·5 μl cDNA from the first reaction and nuclease‐free water. Thermocycler running conditions were: 94°C for 30 s, 49°C for 30 s and 72°C for 30 s, repeated 35 times and thereafter a final extension step at 72°C for 5 min. Additionally the first reaction was preceded by a reverse transcription at 50°C for 30 min and by polymerase activation at 95°C for 15 min. PCR products were visualized and documented by electrophoresis in a 1·8% agarose gel stained with ethidium bromide under UV light using a MiniBIS Pro System (DNR Bio‐Imaging Systems Ltd., Neve Yamin, Israel).
The real‐time RT‐PCR assays were performed using 7500 Fast real‐time PCR system (Applied Biosystems) in a 96‐well optical plate format. Amplification of D1466‐like variant S1 cDNA fragment was carried out in a 25‐μl mixture volume containing 12·5 μl 2× QuantiTect Probe RT‐PCR Master Mix (QuantiTect Probe RT‐PCR Kit, Qiagen), 0·25 μl QuantiTect RT Mix, primers to a final concentration of 400 nmol l−1, probe to a final concentration of 200 nmol l−1, 2·5 μl RNA template and nuclease free water. Thermal cycling conditions included one cycle at 50°C for 30 min for reverse transcription, one cycle at 95°C for 15 min for Taq polymerase activation and 40 cycles of 95°C for 10 s, and 60°C for 60 s for cDNA amplification. Fluorescence was acquired during each extension step. Negative controls contained PCR‐grade water.
Universal real‐time RT‐PCR assay for all gammacoronaviruses detection was run with the same chemistry and condition, but with the primers and probe according to Callison et al. (2006).
Standard curve generation for analytical testing and specificity assay
The reverse transcription droplet digital PCR (RT‐ddPCR) with the same primers and probe as in D1466 real=time RT‐PCR was used to quantify the RNA concentration. The RNA isolated from 10‐fold dilutions of D1466 IB vaccine were analysed using a Droplet Digital PCR QX100 System (Bio‐Rad Laboratories, Inc., Hercules, CA, USA) courtesy of the Department of Microbiology, National Veterinary Institute (Uppsala, Sweden). The 25 μl RT‐ddPCR reaction mixture contained 1 × Supermix, 2 U μl−1 reverse transcriptase, 300 nmol l−1 DTT (Bio‐Rad Laboratories, Inc.), D1466 primers (400 nmol l−1)/probes (200 nmol l−1) and 8·2 μl nucleic acid. Reverse transcription was carried out at 50°C for 30 min followed by denaturation at 95°C for 10 min, and DNA was amplified with 40 PCR cycles at 95°C (30 s) and 60°C (1 min). Read‐out of positive vs negative droplets was performed with the droplet reader and the absolute quantification of PCR target was executed using quantasoft v1.6 software (BioRad Laboratories, Inc.). Each dilution was tested in duplicate and the amount of RNA were medium value from the measurements obtained. Then, the same dilutions of viral RNA containing a known number of molecules were tested in real‐time RT‐PCR.
Additionally, the amplification efficiency and coefficient of determination were calculated for the GII‐1 real‐time RT‐PCR. To assess repeatability and reproducibility, each dilution was tested on different dates and by two different laboratory technicians (in triplicate in each combination). Obtained fluorescence threshold crossing point values (C t values) from each run was then used for calculation of the mean, standard deviation and coefficient of variation.
To evaluate the specificity of the real‐time RT‐PCR, the RNA preparations from other common poultry viral pathogens (GI‐1, GI‐12, GI‐13 and GI‐19 lineages of IBV, infectious bursal disease virus, Newcastle disease virus, avian influenza virus, avian reovirus, astrovirus and rotavirus) were used.
Funding Information
This work was partially funded by KNOW (Leading National Research Centre) Scientific Consortium “Healthy Animal ‐ Safe Food”, decision of Ministry of Science and Higher Education No. 05‐1/KNOW2/2015.
Conflict of Interest
No conflict of interest declared.
Acknowledgements
We thank the staff of the Department of Microbiology of the National Veterinary Institute (Uppsala, Sweden) for allowing analyses on Droplet Digital PCR QX100 System (Bio‐Rad Laboratories, Inc.) for RNA concentration determination.
References
- Acevedo, A.M. , Perera, C.L. , Vega, A. , Rios, L. , Coronado, L. , Relova, D. , Frias, M.T. , Ganges, L. et al (2013) A duplex SYBR green I‐based real‐time RT‐PCR assay for the simultaneous detection and differentiation of Massachusetts and non‐Massachusetts serotypes of infectious bronchitis virus. Mol Cell Probes 27, 184–192. [DOI] [PubMed] [Google Scholar]
- Adzhar, A. , Shaw, K. , Britton, P. and Cavanagh, P. (1996) Universal oligonucleotides for detection of infectious bronchitis virus by polymerase chain reaction. Avian Pathol 25, 817–836. [DOI] [PubMed] [Google Scholar]
- Callison, S.A. , Hilt, D.A. , Boynton, T.O. , Sample, B.F. , Robison, R. , Swayne, D.E. and Jackwood, M.W. (2006) Development and evaluation of a real‐time Taqman RT‐PCR assay for the detection of infectious bronchitis virus from infected chickens. J Virol Meth 138, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens, E.B. (2010) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2009). Arch Virol 155, 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh, D. , Mawditt, K. , Britton, P. and Naylor, C.J. (1999) Longitudinal field studies of infectious bronchitis virus and avian pneumovirus in broilers using type‐specific polymerase chain reactions. Avian Pathol 28, 593–605. [DOI] [PubMed] [Google Scholar]
- Chen, H.T. , Zhang, J. , Ma, Y.P. , Ma, L.N. , Ding, Y.Z. , Liu, X.T. , Cai, X.P. , Ma, L.Q. et al (2010) Reverse transcription loop‐mediated isothermal amplification for the rapid detection of infectious bronchitis virus in infected chicken tissues. Mol Cell Probes 24, 104–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davelaar, F.G. , Kouwenhoven, B. and Burger, A.G. (1984) Occurrence and significance of infectious bronchitis virus variant strains in egg and broiler production in the Netherlands. Vet Quart 6, 114–120. [DOI] [PubMed] [Google Scholar]
- Domanska‐Blicharz, K. , Lisowska, A. , Jatczak, J. , Mamczur, J. and Minta, Z. (2012) D1466‐like genotype of infectious bronchitis virus responsible for a new epidemic in chickens in Poland. Vet Rec 171, 351. [DOI] [PubMed] [Google Scholar]
- Dong, B.Q. , Liu, W. , Fan, X.H. , Vijaykrishna, D. , Tang, X.C. , Gao, F. , Li, L.F. , Li, G.J. et al (2007) Detection of a novel and highly divergent coronavirus from Asian leopard cats and Chinese ferret badgers in Southern China. J Virol 81, 6920–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsang, A. , Ros, C. , Renstrom, L.H. , Baule, C. , Soos, T. and Belak, S. (2002) Molecular epizootiology of infectious bronchitis virus in Sweden indicating the involvement of a vaccine strain. Avian Pathol 31, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellahi, S. , El Harrak, M. , Kuhn, J. , Sebbar, G. , Bouaiti, E. , Khataby, K. , Fassi Fihri, O. , El Houadfi, M. et al (2016) Comparison of SYBR green I real‐time RT‐PCR with conventional agarose gel‐based RT‐PCR for the diagnosis of infectious bronchitis virus infection in chickens in Morocco. BMC Res Notes 9, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, R.M. , Ellis, R.J. , Cox, W.J. , Errington, J. , Fuller, C. , Irvine, R.M. and Wakeley, P.R. (2011) Development and validation of RT‐PCR tests for the detection and S1 genotyping of infectious bronchitis virus and other closely related gammacoronaviruses within clinical samples. Transbound Emerg Dis 58, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, I.M. , Arden, K.E. and Nitsche, A. (2002) Real‐time PCR in virology. Nucleic Acids Res 30, 1292–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marandino, A. , Tomas, G. , Hernandez, M. , Panzera, Y. , Craig, M.I. , Vagnozzi, A. , Vera, F. , Techera, C. et al (2016) Development of RT‐qPCR assays for the specific identification of two major genotypes of avian infectious bronchitis virus. J Virol Methods 235, 21–25. [DOI] [PubMed] [Google Scholar]
- Meir, R. , Maharat, O. , Farnushi, Y. and Simanov, L. (2010) Development of a real‐time TaqMan RT‐PCR assay for the detection of infectious bronchitis virus in chickens, and comparison of RT‐PCR and virus isolation. J Virol Methods 163, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikova, E.V. , Bochkov, Y.A. , Shcherbakova, L.O. , Nikonova, Z.B. , Zinyakov, N.G. , Elatkin, N.P. , Mudrak, N.S. , Borisov, A.V. et al (2011) Molecular characterization of infectious bronchitis virus isolates from Russia and neighbouring countries: identification of intertypic recombination in the S1 gene. Avian Pathol 40, 507–514. [DOI] [PubMed] [Google Scholar]
- Roh, H.J. , Hilt, D.A. , Williams, S.M. and Jackwood, M.W. (2013) Evaluation of infectious bronchitis virus Arkansas‐type vaccine failure in commercial broilers. Avian Dis 57, 248–259. [DOI] [PubMed] [Google Scholar]
- Roh, H.J. , Hilt, D.A. and Jackwood, M.W. (2014) Detection of infectious bronchitis virus with the use of real‐time quantitative reverse transcriptase‐PCR and correlation with virus detection in embryonated eggs. Avian Dis 58, 398–403. [DOI] [PubMed] [Google Scholar]
- Valastro, V. , Holmes, E.C. , Britton, P. , Fusaro, A. , Jackwood, M.W. , Cattoli, G. and Monne, I. (2016) S1 gene‐based phylogeny of infectious bronchitis virus: an attempt to harmonize virus classification. Infect Gen Evol 39, 349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit, J.J. , Cook, J.K.A. and van der Heijden, H.M.J.F. (2011) Infectious bronchitis virus variants: a review of the history, current situation and control measures. Avian Pathol 40, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P.C.Y. , Lau, S.K.P. , Huang, Y. and Yuen, K.Y. (2009) Coronavirus diversity, phylogeny and interspecies jumping. Exp Biol Med 234, 1117–1127. [DOI] [PubMed] [Google Scholar]
- Worthington, K.J. , Currie, R.J. and Jones, R.C. (2008) A reverse transcriptase‐polymerase chain reaction survey of infectious bronchitis virus genotypes in Western Europe from 2002 to 2006. Avian Pathol 37, 247–257. [DOI] [PubMed] [Google Scholar]