

Abstract
The current outbreak of a novel severe acute respiratory syndrome‐like coronavirus, 2019_nCoV (now named SARS‐CoV‐2), illustrated difficulties in identifying a novel coronavirus and its natural host, as the coding sequences of various Betacoronavirus species can be highly diverse. By means of whole‐genome sequence comparisons, we demonstrate that the noncoding flanks of the viral genome can be used to correctly separate the recognized four betacoronavirus subspecies. The conservation would be sufficient to define target sequences that could, in theory, classify novel virus species into their subspecies. Only 253 upstream noncoding sequences of Sarbecovirus are sufficient to identify genetic similarities between species of this subgenus. Furthermore, it was investigated which bat species have commercial value in China, and would thus likely be handled for trading purposes. A number of coronavirus genomes have been published that were obtained from such bat species. These bats are used in Traditional Chinese Medicine, and their handling poses a potential risk to cause zoonotic coronavirus epidemics.
Significance and Impact of the Study
The noncoding upstream and downstream flanks of coronavirus genomes allow for rapid classification of novel Betacoronavirus species and correct identification of genetic relationships. Although bats are the likely natural host of 2019_nCoV, the exact bat species that serves as the natural host of the virus remains as yet unknown. Chinese bat species with commercial value were identified as natural reservoirs of coronaviruses and are used in Traditional Chinese Medicine. Since their trading provides a potential risk for spreading zoonoses, a change in these practices is highly recommended.
Keywords: bats, coronavirus, epidemic, Sarbecovirus, Traditional Chinese Medicine, whole‐genome comparison, zoonosis
Introduction
The current outbreak of a novel coronavirus 2019_nCoV (now named SARS‐CoV‐2) that started in December 2019 in the city of Wuhan, Hubei, China, once again demonstrates the risk of emerging zoonotic viral infections in general, and that of coronavirus species in particular. After only 2 months, the epidemic of COVID‐19 is already larger than the severe acute respiratory syndrome (SARS) coronavirus (SARS‐CoV) outbreak, which during 2002/2003 lasted 8 months and infected 8096 people, with a mortality rate of 9·5%. It is a larger threat than the multiple smaller outbreaks of Middle East Respiratory Syndrome (MERS) coronavirus (MERS‐CoV), a zoonotic coronavirus that was discovered in 2012 (Bermingham et al. 2).
Coronaviruses that are closely related to SARS‐CoV are typically found in bats (Li et al. 10). There is evidence that SARS‐CoV originated in bats in China and reached humans most probably after jumping to an intermediate host, the civet (Paguma larvata) (reviewed by Wang and Eaton 18). Similarly, MERS‐CoV is endemic in dromedary camels in the Middle East, from which it can be transmitted to humans, but it may also have originated in bats (Cui et al. 4). The MERS epidemic is ongoing, and as of December 2019, 2468 cases have been reported (Baharoon and Memish 1) and the mortality rate has been as high as 42% (Mohd et al. 13). At the other end of the virulence spectrum are coronavirus species that cause common colds in humans with relatively mild symptoms (reviewed by Su et al. 16).
Some coronaviruses are strictly host‐specific, while others can be found in a range of hosts (Drexler et al. 5). The genome sequences of 2019_nCoV that is responsible for the current outbreak suggest that it too may have originated from bats, though it may have undergone a host jump to another mammal that was sold on a live‐animal market in Wuhan, where the outbreak presumably had started (Zhang et al. 19). While this manuscript was under review, it was published that this animal might have been a pangolin (Manis javanica; Liu et al. 11). Alternatively, though less likely, a highly infectious patient zero may have been present at the market, as we now know that the virus can spread via human‐to‐human contacts (Riou and Althaus 15) and individuals infected with a coronavirus can be ‘super‐spreaders’, as was demonstrated for MERS (Cho et al. 3; Kang et al. 9).
Coronaviruses are divided into three genera: Alphacoronavirus, Betacoronavirus and Gammacoronavirus. SARS, MERS and 2019_nCoV are members of Betacoronavirus, which is divided into the subgenera Embecovirus, Hibecovirus, Merbecovirus, Nobecovirus and Sarbecovirus. All coronaviruses have a genome consisting of a positive‐sense single‐strand RNA molecule. A long polycystronic messenger is produced from this that codes for a number of proteins, some of which are posttranslationally cleaved to produce smaller proteins (Cui et al. 4). Approximately two‐thirds of the genome codes for a large open reading frame, often called ‘orf 1ab’ that undergoes a frame shift during translation, and whose product is processed into a number of smaller proteins, while a variable number of smaller open reading frames are located downstream of orf 1ab.
A number of highly similar genome sequences of 2019_nCoV have now been released in the public domain, and these are currently being analysed by multiple groups in an attempt to understand the biology of the virus, although it is still a challenge to predict virulence from viral genome sequences. The fact that Coronaviruses are extremely diverse complicates such analyses.
In order to be better prepared during a future zoonotic coronavirus outbreak, we attempted to identify a DNA target that would be able to detect all known members of Betacoronavirus, and further to identify commonality between members of the subgenera. The analysis identified that a relatively short fragment of the noncoding upstream flank of the genome is sufficient for phylogenetic analysis. Moreover, the types of bats that have commercial value in China were considered as potential sources for zoonotic coronaviruses.
Results and discussion
In a pilot experiment, all coding regions of four virus species representing three subgenera (MERS‐CoV for Merbecovirus, human coronavirus OC43 for Embecovirus and SARS‐CoV and 2019_nCoV for Sarbecovirus) were compared by multiple alignment of nucleotide sequences. This did not identify any region that was sufficiently conserved to potentially design an RNA‐two‐primer amplification detection target for all betacoronavirus genera, which we had set as our initial goal to identify. It is clear that addition of the genomes of more virus species would only decrease conservation further.
This lack of homologous DNA sequences was a bit surprising, as Lu et al. (12) recently published a phylogenetic tree based on 50 genome sequences (including 10 2019_nCoV isolates) with high bootstrap values, which can only be obtained with sufficient degrees of conservation between members of the data set. Their analysis was based on complete genomes (presumably, nucleotide sequences), whereas in our pilot analysis only protein‐coding nucleotide sequences were compared. This initiated an analysis to identify how much of the phylogenetic signal and conservation was actually present in noncoding regions of the viral genomes. In case of coronavirus these are mostly restricted to the 5’‐ and 3‐‘end flanks, as all open reading frames are tightly packed. Therefore, a more extensive comparison was performed, this time with either the 5’‐ or the 3’‐end noncoding flanks, obtained from 31 whole genome sequences covering all subspecies of Betacoronavirus (Table 1). This selection included the most variable genomes identified by Lu and colleagues, complemented with other members of the subgenera, while genomes with high similarity to another genome were discarded to remove redundancy. The phylogenetic trees obtained with these two noncoding regions are shown in Fig. 1. Both fragments could separate the four subspecies, and an unclassified bat isolate from Cameroon could be identified as a member of the Nobecovirus. Another unclassified virus isolated from a Nigerian bat showed high similarity with the only other fully sequenced member of Hibecovirus, at least for its 5’‐ flank (Fig. 1a).
Table 1.
Genome sequences used in this study
| GenBank accession | Name (‘coronavirus’ is abbreviated as CoV in all) | Subspecies | Isolated from |
|---|---|---|---|
| KT368891 | Camel/Riyadh/Ry123/2015 | Embecovirus * | Camel, Saudi Arabia |
| FJ938067 | Human enteric CoV strain 4408 | Embecovirus | Human |
| AY391777 | HCoV‐OC43 | Embecovirus | Human, UK |
| JN874559 | Rabbit CoV HKU14 | Embecovirus † | Rabbit, China |
| KY370046 | RtMruf‐CoV‐2/JL2014 | Embecovirus † | Vole: Myodes rufocanus, China |
| LC061274 | Equine CoV strain Obihiro12‐2 | Embecovirus | Horse, Japan |
| KM349744 | HKU24 strain HKU24‐R05010I | Embecovirus † | Rat: Rattus norvegicus, China |
| MK167038 | Human CoV HKU1 str. SC2521 | Embecovirus † | Human, USA |
| FJ647223 | Murine CoV MHV‐1 | Embecovirus | Mouse, USA |
| MG693170 | Bat CoV isolate CMR66 | Unclassified | Bat: Eidolon helvum, Cameroon |
| KU762338 | CoV isolate GCCDC1 356 | Nobecovirus | Bat: Rousettus leschenaulti, China |
| EF065513 | Bat CoV HKU9‐1 | Nobecovirus | Bat, China |
| MK492263 | Bat CoV strain BtCoV92 | Nobecovirus | Bat: Cynopterus brachyotis, Singapore |
| KC545386 | ErinaceusCoV/2012‐216/GER/2012 | Merbecovirus | Hedgehog: Erinaceus europaeus, Germany |
| MK679660 | Hedgehog CoV 1 | Merbecovirus | Hedgehog: Erinaceus europaeus, UK |
| KJ473820 | BtPa‐BetaCoV/GD2013 | Merbecovirus † | Bat: Pipistrellus abramus, China |
| JX869059 | MERS, HCoV‐EMC/2012 | Merbecovirus | Human, Saudi Arabia |
| NC038294 | Betacoronavirus England 1 | Merbecovirus | Human, UK |
| MN611519 | CoV HKU4‐related isolate GZ131656 | Merbecovirus | Bat: Tylonycteris pachypus, China |
| KU182965 | CoV isolate JPDB144 | Merbecovirus † | Bat: Myotis daubentonii, China |
| KX442565 | CoV HKU25 isolate NL140462 | Merbecovirus † | Bat: Hypsugo pulveratus, China |
| KJ473821 | BtVs‐BetaCoV/SC2013 | n.s. | Bat: Vespertilio superans, China |
| KF636752 | Bat Hp‐betacoronavirus/Zhejiang2013 | Hibecovirus | Bat: Hipposideros pratti, China |
| HQ166910 | Zaria bat CoV strain ZBCoV | Unclassified | Bat: Hipposideros commersoni, Nigeria |
| MG772933 | bat‐SL‐CoVZC45 | Sarbecovirus | Bat: Rhinolophus sinicus, China |
| NC045512 | 2019_nCoV, isolate Wuhan‐Hu‐1 | Sarbecovirus | Human, China |
| GU190215 | Bat CoV BM48‐31/BGR/2008 | Sarbecovirus † | Bat: Rhinolophus blasii, Bulgaria |
| KY352407 | SARS‐like CoV strai nBtKY72 | Sarbecovirus † | Bat: Rhinolophus sp., Kenya |
| FJ882963 | SARS CoV P2 | Sarbecovirus | Human, USA |
| MK211376 | BtRs‐BetaCoV/YN2018B | Sarbecovirus † | Bat: Rhinolophus affinis, China |
| MN611520 | Bat CoV HKU5‐related isolate BY140568 | Merbecovirus | Bat: Pipistrellus abramus, China |
| KU182966‡ | Bat CoV P1ab | Unclassified | Bat, Murina leucogaster, China |
| DQ412042‡ | Bat SARS CoV Rf1 | Sarbecovirus | Bat: Rhinolophus ferrumequinum, China |
| KP886808 | Bat SARS‐like CoV YNLF_31C | Sarbecovirus | Bat: Rhinolophus ferrumequinum, China |
| KJ473813 | BtRf‐BetaCoV/SX2013 | n.s. | Bat: Rhinolophus ferrumequinum, China |
| Y417150 | SARS‐like CoV isolate Rs4874 | n.s. | Bat: Rhinolophus sinicus, China |
| KF367457 | Bat SARS‐like CoV WIV1 | Sarbecovirus | Bat: Rhinolophus sinicus, China |
| KF569996 | Rhinolophus affinis CoV isolate LYRa11 | Sarbecovirus | Bat: Rhinolophus affinis, China |
| KY417146 | Bat SARS‐like CoV isolate Rs4231 | n.s. | Bat: Rhinolophus sinicus, China |
| MK211379 | BtRt‐BetaCoV/GX2018 | Unclassified | Bat: Rhinolophus affinis, China |
| GQ153539 | Bat SARS CoV HKU3‐4 | Sarbecovirus | Bat, China |
| KY770858 | Bat CoV isolate Anlong‐103 | Unclassified | Bat: Rhinolophus sinicus, China |
| KF294457 | SARS‐related bat CoV isolate Longquan‐140 | Sarbecovirus | Bat: Rhinolophus monoceros, China |
n.s., not specified.
* In the GenBank file described as unclassified Alphacoronavirus.
† As per Lu et al. (12).
‡ Upstream or downstream noncoding sequences were not available.
Figure 1.
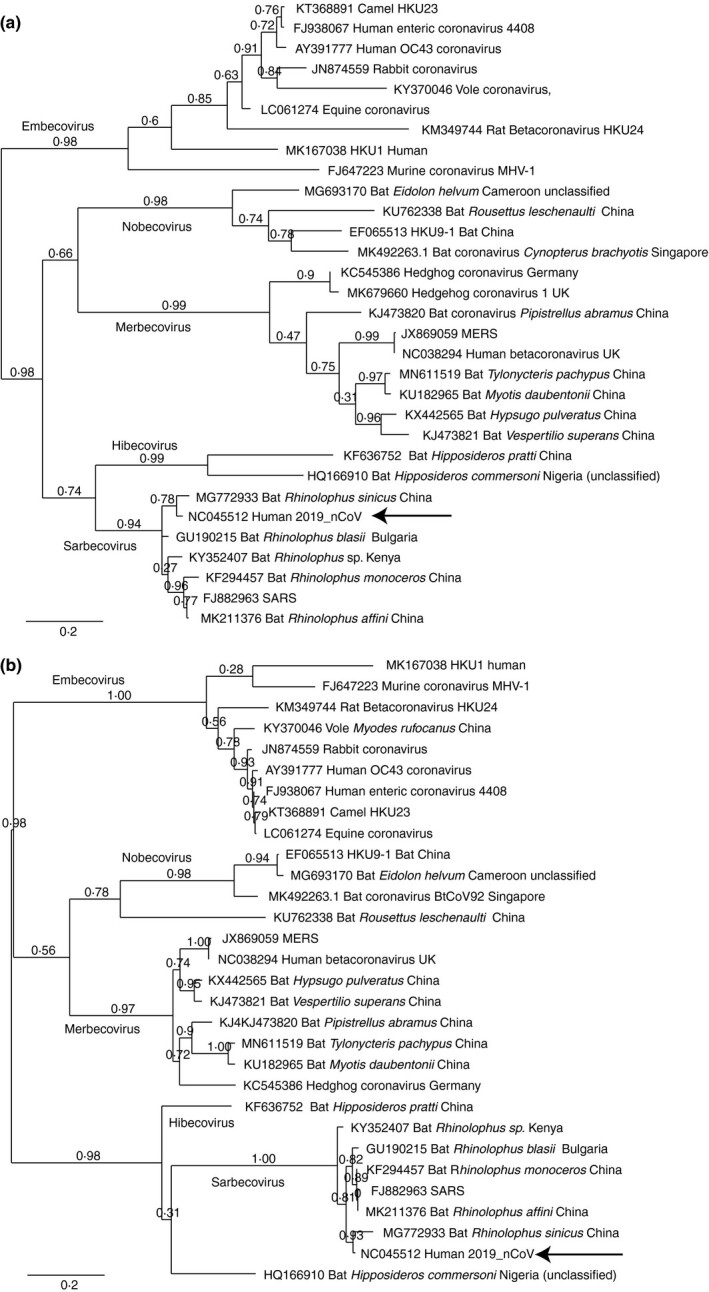
Phylogenetic trees of the 5’‐end (a) and 3’‐end (b) noncoding regions of Betacoronusvirus genomes. The arrow points to the position of 2019_nCoV. Numbers show bootstrap values.
Assigning a betacoronavirus to a subgenus does not implicate information about its virulence, host specificity or geographical origin. However, the two analysed fragments were relatively strongly conserved within a subgenus, as illustrated by multiple alignments shown in Supporting Information. Since only two, highly similar, genome sequences of Hibecovirus members were available for analysis, their comparison was not included. The identified conserved regions could provide potential targets for PCR or rt‐PCR, allowing for rapid classification of novel coronaviruses, in particular for the Sarbecovirus group.
Although any section of the viral genome that is conserved within but not between subspecies would result in correct classification, the conservation of such regions within coding sequences of Sarbecovirus members was generally weaker than we observed for the noncoding flanks. Using coding sequences of this same small selection of seven Sarbecovirus members, the maximum conservation at nucleotide level for gene 1a/b was 85% within a sliding window of 60 bases, compared to 93% for the downstream and 90% for the upstream flank. The longest stretch of conserved nucleotides was 22 in the upstream flank, 33 in the downstream flank and only 20 nucleotides in orf 1ab (results not shown). Only the gene coding for envelope protein E was as strongly conserved as the noncoding flanks (maximum conservation 95% over a window of 60 bases, with a longest conserved stretch of 32 nucleotides) for the seven genomes analysed here. As conservation of coding sequences decreases with third‐base variation, it is likely that this degree of conservation in gene E would decrease as more genomes would be included in the comparison. Thus, restricting analyses to only comparing coding regions may reduce the chance a proposed detection target might be able to detect a novel Sarbecovirus species. Instead, we recommend inclusion of the noncoding genome regions in such attempts. Although it was not our aim to develop improved detection methods (for which we would have to experimentally test any proposed target region), our findings can be a starting point for such studies.
It has been proposed, though not yet proven, that 2019_nCoV naturally propagates in bats, but the bat species in which the population mainly resides was unknown at the time of writing. The food market of Wuhan may or may not have been selling bats for food; however, bats and their excrements are often used in Traditional Chinese Medicine (TCM), which may be a reason for their legal or illegal trading. It is also possible that the virus had infected another mammal that was traded at the market and served as the source of the infection. Such a case has been proposed for Malayan pangolins (Liu et al. 11), a meat delicacy that was most likely sold in the exotic meat market. Such cross‐infection could also have occurred during handling, storage or selling prior to animals reaching the food market. The case for pangolins being the actual source of 2019_nCoV has not yet been proven, but was referred from sequence similarities (Liu et al. 11). Even in this scenario there must have been a reason why an infected bat came in the vicinity of a pangolin that ended up in the market, for which only bat species with local commercial value come into question.
Of an extensive list of bat species that historically have been used in TCM (Riccucci 14), five were found to carry coronaviruses, as reported in a surveillance study (Tang et al. 17): Pipistrellus abramus, Murina leucogaster, Rhinolophus ferrumequinum, Scotophilus kuhlii and Scotomanes ornatus. Of these bat species, the first three can be found in the province of Hubei (source: www.iucnredlist.org); the habitat of S. ornatus reaches to the south of the province but does not cover the Wuhan area, while S. kuhlii is not endemic in this region. A number of whole genome Sarbecovirus sequences isolated from these bats and other bat species were selected and their 5’‐flank was compared to that of 2019_nCoV (Fig. 2). This produced the highest identity to the same isolate that Lu and coworkers had identified (Lu et al. 12), demonstrating that an analysis of 253 nucleotides directly upstream of the start codon of the polycystronic messenger suffices to identify genetic relationships between Sarbecovirus species.
Figure 2.
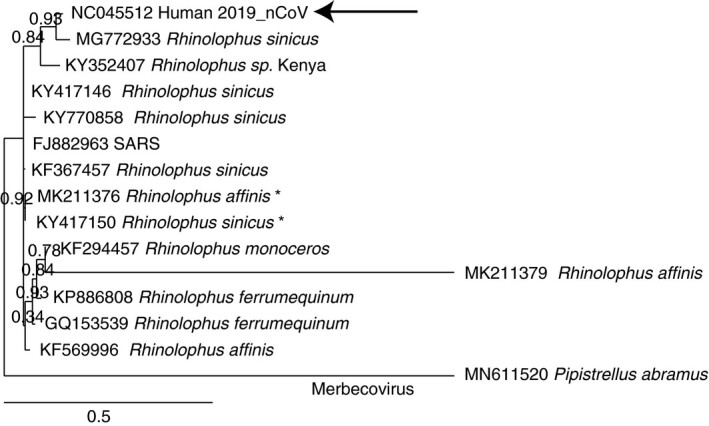
Phylogenetic tree of the 253 5’‐end noncoding sequences of Sarbecovirus species from bats. A Merbecovirus was included as an outlier. The asterisks indicate highly similar CoV sequences obtained from different bat species.
The bat CoV to which 2019_nCoV has the highest similarity was isolated from Rhinolophus sinicus (MG772933, described in Hu et al. 7), which might indicate that this species may also have been the original source of 2019_nCoV. However, the database is highly biased towards particular bat species that were specifically sampled in search of the source of SARS. Highly similar coronaviruses can be detected in different bat species (as identified by asterisks in Fig. 2), so that even a high similarity match to a bat isolate may not always identify the correct bat species that was the cause of a given zoonotic outbreak. This caution can be extended to other possible animal hosts that have been or will be claimed as sources of the 2019_nCoV without a proven epidemiological link.
The use of bats in TCM is of great concern, and the use of the Greater horseshoe bat, Rhinolophus ferrumequinum, is of particular interest. The faeces of this bat (Yè ming shǎ in Chinese, marketed as Vespertilionis, see for instance https://www.bestplant.shop/products/ye-ming-sha-bat-feces-bat-dung-bat-guano) is used to cure eye conditions, while body parts are dried and added to wine or ground into a powder for oral intake as a means to ‘detoxify’ the body. Both practices could be highly risky in case an animal was infected with a coronavirus, particularly the first use, as the virus can be present in faeces and can enter a host via the eye. We speculate that a live or recently deceased infected bat species was handled by traders because of its value in TCM, and that such an infected individual, or the still infective bat or bat products, may have been the route by which the virus entered the exotic meat market in Wuhan. Alternatively, during the trade chain, a host jump occurred between an infected bat (handled for TCM purposes) and another mammal, that then was the source of infection. Less likely, because the material is usually dried significantly, the outbreak may have started by medicinal use of bat‐derived, contaminated TCM material. In this respect it is interesting to note that the first known onset of symptoms (on 1 December 2019) were observed in a patient with no known epidemiological links to the Wuhan food market (Huang et al. 8).
The possibility that bat‐derived materials sold for TCM practices may be involved in this outbreak, has severe implications. We point out there is a serious risk of producing bat‐derived materials sold for TCM practices as it involves handling of and trading with wild bats. Even when the selling of live wild animals at food markets would be completely prohibited in China, the trading and handling of bats for traditional medicinal practices would remain a serious risk for future zoonotic coronavirus epidemics.
Materials and methods
All genome sequences used in this study were extracted from GenBank (Table 1). In all cases, the reading frame of gene 1a/b was corrected for slippage. Open reading frames missing from the annotation in some of the genomes were corrected.
Multiple alignments were performed with Muscle at www.ebi.ac.uk. Phylogenetic Maximum Likelihood trees were produced with PhyMLy ver. 3.0 (Guindon et al. 6) at www.phylogeny.fr with default settings except for skipping Gblock.
Acknowledgements
The authors thank the anonymous reviewers for their quick feedback, and Dr Gergory Buzard for his stimulating discussions and critical reading of the manuscript.
Conflict of Interest
The authors have no conflict of interest to declare.
Supplementary Material
Data S1. Multiple alignment of non‐coding flanks of Betacoronavirus genomes identify strongly conserved regions.
Contributor Information
T.M. Wassenaar, Molecular Microbiology and Genomics Consultants Zotzenheim Germany .
Y. Zou, SciPaperEdit Chuangkexing Wuhan China
References
- Baharoon,S. and Memish,Z.A. (2019) MERS‐CoV as an emerging respiratory illness: a review of prevention methods. Travel Med Infect Dis 32. 10.1016/j.tmaid.2019.101520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham,A., Chand,M.A., Brown,C.S., Aarons,E., Tong,C., Langrish,C., Hoschler,K., Brown,K. et al. (2012) Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East, September 2012. Euro Surveill 17, 20290. [PubMed] [Google Scholar]
- Cho,S.Y., Kang,J.M., Ha,Y.E., Park,G.E., Lee,J.Y., Ko,J.H., Lee,J.Y., Kim,J.M. et al. (2016) MERS‐CoV outbreak following a single patient exposure in an emergency room in South Korea: an epidemiological outbreak study. Lancet 388, 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui,J., Li,F. and Shi,Z.L. (2019) Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler,J.F., Corman,V.M. and Drosten,C. (2014) Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res 101, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon,S., Dufayard,J.F., Lefort,V., Anisimova,M., Hordijk,W. and Gascuel,O. (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Hu,D., Zhu,C., Ai,L., He,T., Wang,Y., Ye,F., Yang,L., Ding,C. et al. (2018) Genomic characterization and infectivity of a novel SARS‐like coronavirus in Chinese bats. Emerg Microbes Infect 7, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang,C., Wang,Y., Li,X., Ren,L., Zhao,J., Hu,Y., Zhan,L., Fan,G. et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang,C.K., Song,K.H., Choe,P.G., Park,W.B., Bang,J.H., Kim,E.S., Park,S.W., Kim,H.B. et al. (2017) Clinical and epidemiologic characteristics of spreaders of middle east respiratory syndrome coronavirus during the 2015 outbreak in Korea. J Korean Med Sci 32, 744–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li,W., Shi,Z., Yu,M., Ren,W., Smith,C., Epstein,J.H., Wang,H., Crameri,G. et al. (2005) Bats are natural reservoirs of SARS‐like coronaviruses. Science 310, 676–679. [DOI] [PubMed] [Google Scholar]
- Liu,P., Chen,W. and Chen,J.P. (2019) Viral metagenomics revealed sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses 11. 10.3390/v11110979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu,R., Zhao,X., Li,J., Niu,P., Yang,B., Wu,H., Wang,W., Song,H. et al. (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohd,H.A., Al‐Tawfiq,J.A. and Memish,Z.A. (2016) Middle east respiratory syndrome coronavirus (MERS‐CoV) origin and animal reservoir. Virol J 13, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccucci,M. (2012) Bats as materia medica: an ethnomedical review and implications for conservation. Vespertilio 16, 249–270. [Google Scholar]
- Riou,J. and Althaus,C.L. (2020) Pattern of early human‐to‐human transmission of Wuhan 2019 novel coronavirus (2019‐nCoV), December 2019 to January 2020. Eurosurveillance 25. 10.2807/1560-7917.ES.2020.25.4.2000058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su,S., Wong,G., Shi,W., Liu,J., Lai,A.C.K., Zhou,J., Liu,W., Bi,Y. et al. (2016) Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 24, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang,X.C., Zhang,J.X., Zhang,S.Y., Wang,P., Fan,X.H., Li,L.F., Li,G., Dong,B.Q. et al. (2006) Prevalence and genetic diversity of coronaviruses in bats from China. J Virol 80, 7481–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang,L.F. and Eaton,B.T. (2007) Bats, civets and the emergence of SARS. Curr Top Microbiol Immunol 315, 325–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang,N., Wang,L., Deng,X., Liang,R., Su,M., He,C., Hu,L. et al. (2020) Recent advances in the detection of respiratory virus infection in humans. J Med Virol. 10.1002/jmv.25674 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Multiple alignment of non‐coding flanks of Betacoronavirus genomes identify strongly conserved regions.