

Abstract
A protease inhibitor, cystatin C (Cst C), is a secreted cysteine protease inhibitor abundantly expressed in body fluids. Clinically, it is mostly used to measure glomerular filtration rate as a marker for kidney function due to its relatively small molecular weight and easy detection. However, recent findings suggest that Cst C is regulated at both transcriptional and post‐translational levels, and Cst C production from haematopoietic cell lineages contributes significantly to the systematic pools of Cst C. Furthermore, Cst C is directly linked to many pathologic processes through various mechanisms. Thus fluctuation of Cst C levels might have serious clinical implications rather than a mere reflection of kidney functions. Here, we summarize the pathophysiological roles of Cst C dependent and independent on its inhibition of proteases, outline its change of expression by various stimuli, and elucidate the regulatory mechanisms to control this disease‐related protease inhibitor. Finally, we discuss the clinical implications of these findings for translational gains.
Cystatin C (Cst C) is a potent extracellular inhibitor of cysteine protease, and has been generally considered a ubiquitously expressed protein as no obvious regulatory elements were found in its gene promoter. 1 Clinically, Cst C is mostly used as a biomarker of kidney function for its relatively lower molecular weight (~13.3 kDa) and easy detection compared to the injection of compounds, radioisotopes or radiocontrast agent 2 to measure glomerular filtration rate (GFR), an index of kidney health, because Cst C is removed from the blood stream by glomerular filtration, whose decline as a result of failed kidney function will lead to increased serum Cst C concentration. Cst C serum levels were claimed to be a more precise index of kidney function than that of creatinine 3 under the assumption that serum input of Cst C is constant, and the main determinant of blood Cst C levels is the rate at which it is filtered at the glomerulus. Furthermore, recent advances have facilitated the use of Cst C as a clinical measure of kidney function (the update on Cst C in recent kidney disease guidelines has been reviewed elsewhere 4 ). Although, the bulk of literature report the use of Cst C for GFR estimation, precaution should be taken when the outcomes of this measurement is interpreted because recent studies indicated that both genetic polymorphisms 5 , 6 and clinical interference 7 , 8 could make Cst C an unreliable index of GFR. Indeed, accumulating reports have documented that subjected to the influence of many factors, serum Cst C levels do vary independent of renal functions, and the synthesis and secretion of Cst C seem to be tightly regulated under different pathophysiological conditions. For example, body composition, thyroid function, glucocorticoid and C‐reactive protein levels or even cigarette smoking and pregnancy status of the candidates examined have been found to affect Cst C blood concentration, 9 , 10 let alone its alteration in patients with cancer, HIV infection, cardiovascular diseases and neurological disorders. 11 , 12 , 13 Moreover, new studies reported direct involvement of Cst C in many pathogenic processes other than renal disorders. 14 , 15 Thus, the oscillation of blood Cst C levels could actually reflect the change of Cst C production, consumption, inactivation or fibrillation (mentioned later) rather than its filtration in the kidney, and the readout of plasma Cst C concentration might have different clinical implications. In this review, we first summarize the major pathophysiological roles of Cst C to illustrate its importance as a functional protein in the body rather than an ubiquitously expressed measuring substance. Second, we outline the impact of different stimuli on its expression and elucidate some of the latest developments on the regulatory mechanisms to explain Cst C variation under certain circumstances. In the end, the clinical implications derived from these studies are also discussed.
Pathophysiological roles of Cst C
Functions dependent of its inhibition of proteases
Cysteine cathepsins play fundamental roles in multiple biological processes such as protein turnover, pro‐protein processing, bone remodeling, antigen presentation and apoptosis. 16 They are also involved in numerous pathological processes such as cardiovascular disease and inflammation. 13 The activities of these enzymes both inside and outside of cells thus need to be tightly controlled by their endogenous inhibitors, of which Cst C is the most abundant and potent member.
Intracellular roles
Apoptosis. Intracellular lysosomes undergo membrane permeabilization in response to extra‐ or intra‐lysosomal stimuli, and the involvement of lysosomal cysteine proteases in apoptosis has been confirmed in several systems. 17 , 18 However, the roles of the endogenous lysosomal cysteine protease inhibitor Cst C in apoptosis remain controversial. Upregulation of Cst C expression was found to correlate with oxidative stress‐induced apoptosis in cultured rat neurons, 19 and Cst C injection into rat hippocampus led to neuronal cell death in the granule cell layer of dentate gyrus in vivo, 20 indicating a possible functional role of Cst C in apoptosis induction. Since, the Cst C‐induced neuronal cell death could be inhibited by simultaneous co‐application of cathepsin B, the inhibitory activity of Cst C on a cysteine protease was proposed to be involved in the process of apoptotic neuronal cell death. 20 Recent studies indicated that the roles of Cst C in neuronal cell apoptosis induction include decreasing B‐cell leukemia‐2 (Bcl‐2) and increasing active caspase‐9 protein levels via the Jun N‐terminal kinase (JNK)‐dependent pathway, 21 and upregulation and accumulation of the insoluble α‐synuclein in oligodendrocytes and neurons. 22 Interestingly, apart from these documented pro‐apoptotic roles, Cst C was also found to have anti‐apoptotic effects on neuronal cells. For example, expression of Cst C in PC12 cells derived from a pheochromocytoma of rat adrenal medulla prevents oxidative stress‐induced death in vitro. 23 In accordance with this finding, the anti‐apoptotic function of Cst C was further demonstrated in vivo in a mouse model of the inherited neurodegenerative disorder, progressive myoclonic epilepsy type 1, where loss of function in cystatin B and enhanced cathepsin B and D activities are the underlying pathologies. Crossbreeding of the mice with either Cst C‐overexpressing transgenic or Cst C‐deficient mice revealed that Cst C levels in vivo can affect neuron degeneration caused by the proteases, indicating that Cst C partially prevents neural cell death in vivo through inhibition of cathepsins activity. 24 (Described in the section of Neuroprotective roles.)
Antigen presentation. Proteolysis of antigen by cysteine cathepsins to generate immunogenic peptides and guide the transit of both major histocompatibility complex class II (MHCII) and MHC‐like molecules through the endocytic compartments are important intracellular activities in antigen‐presenting cells. As a potent inhibitor of cysteine cathepsins, Cst C had initially been implicated in playing a regulatory role in the developmental control of MHCII presentation in dendritic cells (DCs) by inhibiting cathepsin S in invariant chain cleavage. 25 Consistent with the role of Cst C in antigen presentation, a cystatin homolog produced by the filarial nematode parasite was found to inhibit lysosomal cysteine protease activities, which subsequently impeded the generation of human B cells. 26 Further experiments using bone marrow‐derived DCs indicate that interleukin 6 (IL‐6)‐mediated signal transducer and activator of transcription 3 (STAT3) activation decreased Cst C expression and MHCII αβ dimer levels. 27 However, a separate study using mouse primary DCs isolated from Cst C‐deficient mice demonstrated that Cst C is neither necessary nor sufficient to control MHCII expression and antigen presentation in DCs. 28 These discrepant results obtained from different laboratories could be due to the different cell types investigated, and/or to the compensatory roles played by other cysteine protease inhibitors in the absence of Cst C. In support of this view, cell‐specific regulation of cathepsin activity by Cst C was identified in two similar antigen‐presenting cells in the brain: Cst C was found to inhibit cathepsin L activity in astrocytes, but does not regulate cathepsins L and S in microglia. 29
When intracellular localization of the Cst C and its target proteases were examined in human DC differentiated from monocytes in vitro, the different compartmentation of Cst C and cathepsins S, L and H in immature and mature DCs suggests that the regulatory potential of Cst C toward these cathepsins inside DCs is limited, which could explain the inconsistent findings related to the intracellular roles of Cst C. Instead, large secretion of Cst C over cathepsins S, L and H was observed in the culture media, 30 indicating that the extracellular compartment is the primary site for these interactions.
Extracellular roles
The structure of Cst C predisposes this protease inhibitor to extracellular functions as a secreted protein. Accordingly, Cst C is found in all body fluids at significant concentrations, 31 which makes it a major regulator of cysteine protease activity in the extracellular medium.
Atherosclerosis. Atherosclerosis‐based vascular disease is an inflammatory disease characterized by extensive remodeling of the extracellular matrix of the arterial walls. Apart from the well‐known matrix metalloproteinases and serine proteases, lysosomal cysteine proteases were also found to be involved. 32 Present in substantial amounts in the normal vessel walls, the expression of Cst C was found to be severely reduced in both atherosclerostic and aneurysmal lesions, and increased abdominal aortic diameter correlated with lower serum Cst C levels in humans. 13 Furthermore, the pathogenic roles of Cst C were tested in a model of atherosclerosis‐prone mice with apolipoprotein E deficiency (apoE−/−) where the elevated cathepsins were associated with the atherosclerostic process. 33 In two independent studies, Cst C‐ and apoE‐double‐deficient mice were generated, both of them confirmed an anti‐atherosclerostic function of Cst C in the apoE−/− mice, although differences regarding lesion size and composition were found. 34 , 35 These differences are most probably caused by differences in the duration of high‐fat diet of the mice, sex of the mice and anatomic site of analyzed lesions. Interestingly, in line with the anti‐atherosclerostic role of Cst C in this animal model, polymorphisms in the promoter regions of the Cst C gene were found to influence the plasma Cst C concentration in human coronary artery disease, 36 indicating the production of this protease inhibitor could be subject to regulation under the diseased conditions.
Tumor metastasis. Metastatic tumor cells invade host tissues through a series of steps that require proteolytic enzymes including cysteine cathepsins to degrade components of the extracellular matrix. 37 As a most abundant endogenous cysteine proteinase inhibitor, Cst C is believed to prevent tumor progression by inhibiting the activities of a family of lysosomal cysteine cathepsins. Results from the first Cst C‐deficient animals indicated that Cst C concentration in vivo might influence tumor metastasis in some tissues. 38 In agreement, the reduced Cst C levels correlate with increased metastasis of different tumors in human tissues or patients. 39 Furthermore, local overexpression of Cst C in the host tissue microenvironment could lead to successful reduction of metastasis via cysteine cathepsin inhibition in an experimental tumor model. 40 This inverse correlation between Cst C and tumor aggressiveness, however, may not always involve inhibition of cysteine proteases. For example, elevated matrix metalloproteinases 2 and crosstalk between Cst C and androgen receptor‐mediated pathways were reportedly implicated in prostate cancer invasion and metastasis, 41 indicating multiple roles of Cst C in tumor metastasis.
Pathogen invasion. Although a secreted protein, Cst C was also reported to be up‐taken by cells of both foreign and endogenous origins in various tissues to regulate both intracellular and extracellular cysteine protease activities. 42 One of the major roles of cystatins is to protect the host against invading microorganisms and parasites that use cysteine proteases to enter the body. 43 Chicken cystatin was first reported to partially block poliovirus replication in infected human cells. 44 Further study demonstrated that a small peptide derivative that mimics part of the proteinase‐binding center of human Cst C could inhibit a cysteine protease specific for the growth of group A streptococci, blocking the growth of these bacteria both in vivo and in vitro. 45 Similarly, recombinant human Cst C was also proven to inhibit the growth of herpes simplex virus 46 and human coronaviruses. 47 Moreover, a family of cathepsin L‐ and B‐like cysteine proteases, found in all species of Leishmania examined, are required for the parasite growth and virulence. 48 Since, these parasite cysteine proteases may not only digest the host extracellular matrix to facilitate their invasion, but also help to ensure a Th2‐like response led to parasite proliferation, 49 cystatin treatment in combination with interferon γ (IFNγ) that leads to reduced parasite numbers, successful Th2 to Th1 conversion, and NO generation, which finally resulted in abrogation of parasite infection in a mouse model of leishmaniasis. 50
Functions independent of its inhibition of proteases
Modulating roles
Most abundantly expressed in tissues, Cst C also serves numerous functions independent of protease inhibition including affecting signaling properties of other molecules. For example, Cst C antagonizes the binding of transforming growth factor‐beta (TGF‐β) to its cell receptors by physically interacting with TGF‐β type II receptor independent of its protease inhibitory activity, as overexpression of Cst C mutant that is impaired in its ability to inhibit cathepsin activity blocked TGF‐β‐dependent invasion of 3T3‐L1 fibroblasts. 51 This novel function of signaling modulation allows Cst C to inhibit the oncogenic activities of TGF‐β through stimulation of mammary epithelial‐mesenchymal transition. 52 Apart from blocking interaction between TGF‐β and its receptor, Cst C was also found to prevent TGF‐β signaling partly by reducing the extent of mothers against decapentaplegic homolog 2 (Smad2), p38 mitogen‐activated protein kinases (p38 MAPKs) and extracellular signal‐regulated kinases 1/2 (Erk1/2) phosphorylation in murine 4T1 breast cancer cells. 53 In addition to affecting TGF‐β signaling, Cst C is involved in the IFNγ signal transduction pathway. It activates nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) p65, induces NO synthase, but downregulates IL‐10 in macrophages. 54
Through interacting with other proteins with effects on their signaling properties, Cst C can also affect biological functions of cells. During brain development, Cst C was identified as a factor to upregulate the glial fibrilllary acidic protein promoter. 55 As a result, addition of human Cst C to the culture medium of primary brain cells increased the number of glial fibrilllary acidic protein‐positive cells and neurospheres formed from the embryonic brain. Again, the promotion of astrocyte development by Cst C appears to be unrelated to its protease inhibitor activity, as another cysteine protease inhibitor did not have this effect. 56 Along the same line, a recent study identified a novel function of Cst C in mediating amyloid β (Aβ) precursor protein‐induced proliferation of neural stem/progenitor cells. 57 Although the mechanism of Aβ precursor protein in stimulating neural stem/progenitor cells to secrete Cst C needs further characterization, this study contributes to a better understanding of multifunctional roles of Cst C in the pathogenesis of Alzheimer's disease.
Amyloidogenic roles
In addition to cysteine protease inhibition, Cst C is also one of the few amyloidogenic proteins that form a fibrillary structure deposited in the vascular walls, affecting the health of blood vessels (angiopathy). The proteins identified in cerebral amyloid angiopathy include beta/A4, transthyretin and Cst C. 58 Cst C amyloidogenesis begin with dimerization by a process known as ‘three‐dimentional domain swapping’, in which two parts of the cystatin structure become separated from each other and next exchanged between two molecules. 59 Interestingly, with their inhibitory region hidden within the dimer interface, Cst C dimers cannot inhibit cysteine proteases. 60 This none‐inhibitory Cst C dimer is required for the formation of the Cst C oligomers, intermediates in fibrillogenesis, because variants of monomeric Cst C, stabilized against domain swapping to block the inhibitory site, fail to produce oligomers, indicating that Cst C fibrils are formed by propagated domain swapping. 61
Domain swapping must be preceded by at least partial unfolding of the molecule, 62 therefore, any factors affecting the stabilization of the molecule could initiate the domain swapping process. For instance, the point mutation with substitution of native leucine in position 68 by glutamine (L68Q) disrupts a network of the hydrophobic interactions, which leads to increased tendency for dimerization and aggregation in vitro. 63 The L68Q replacement is also a naturally occurring point mutation in the Cst C protein sequence with autosomal dominant hereditary. This Cst C variants form fibrils in vivo in the brain vasculature, which cause hemorrhage, dementia and eventually death in people carrying this mutation, a condition known as hereditary cystatin C amyloid angiopathy, 64 also called hereditary cerebral hemorrhage with amyloidosis, icelandic type. 65 The cellular transport of Cst C is impeded by the mutated L68Q variants, resulting in diminished Cst C levels in cerebrospinal fluid 66 , 67 and retained Cst C mutants in blood monocytes from patients. 67 Consistently, in vitro studies demonstrated that clones expressing the gene encoding L68Q Cst C secreted either lower amounts of Cst C, 68 or unstable protein susceptible to a serine protease, 69 contributing to the reduced extracellular Cst C levels.
Cst C amyloid fibrils not only affect brain vasculature but also may lead to toxicity in other tissues. As the most abundant protein, Cst C was found in tissues outside of the brain including the testis, and so was the L68Q variant. 70 A recent study reports that heterozygous transgenic mice that express this pathogenic variant were unable to generate offspring, indicating the L68Q Cst C amyloid affects sperm function. 71 Further analysis of the L68Q mice demonstrated that their epididymal spermatozoa were unable to fertilize oocytes and exhibited poor sperm motility in the presence of Cst C amyloid that were not found in the wild‐type mice. The L68Q epididymal fluid, when depleted of the Cst C amyloids, however, did not impair the motility of wild‐type spermatozoa, suggesting that amyloids in the epididymal fluid can be cytotoxic to the maturing spermatozoa resulting in male infertility. 71 However, two other groups in earlier studies have also generated the transgenic lines expressing this variant form of Cst C, but they were live and fertile, 72 , 73 questioning whether the pathology described is really brought about by the Cst C variants or the site where the transgene was inserted.
Neuroprotective roles
Although mutant Cst C is toxic by amyloidosis, wild‐type Cst C has multiple neuroprotective roles (comprehensively reviewed by Gauthier et al. 74 ). An amyloidogenic protein itself, Cst C has an anti‐toxic role against another amyloid protein, Aβ. Studies in vitro demonstrated that Cst C binds to Aβ and inhibits its oligomerization 75 and amyloid fibril formation. 76 Furthermore, in Aβ precursor protein transgenic mice, Cst C was found to physically associate with the soluble, non‐pathological form of Aβ in vivo, which inhibited the aggregation and deposition of Aβ plaques in the brain. 73 , 77 Moreover, Cst C can also directly protect neuronal cells from amyloid toxicity, as extracellular addition of human Cst C promoted the survival of cultured neuronal cells against the preformed oligomeric or fibrillar Aβ. 78
Autophagy is important for the survival and homeostasis of neurons as they cannot dilute accumulating detrimental substances or damaged organelles by cell division (see latest review 79 for details). Cst C was first reported to induce a fully functional autophagy to protect neuronal cells against various stress via the mammalian target of rapamycin (mTOR) pathway in vitro independent of its inhibitor activity. 80 Consistently, following experimental subarachnoid hemorrhage, exogenous Cst C administration was recently found to activate autophagy pathway ex vivo, which plays a beneficial role in early brain injury in a rat model. 81 , 82 In addition to these apoptotic factors like nutritional deprivation, oxidative stress and hemorrhage, another neurotoxic element that can be counterbalanced by Cst C partly through autophagy is mutant Cu/Zn superoxide dismutase, a frequent cause of inherited amyotrophic lateral sclerosis. 83
Regulation of Cst C by different stimuli
The numerous pathophysiological roles of Cst C foretell that the original application of this small molecular weight (MW) protein as a measurement of the GFR for kidney function might no longer be appropriate, as participation in various pathophysiological processes could lead to consumption and subsequent regulation of this multifunctional protease inhibitor. Indeed, Cst C levels could be altered by many common stimuli under both physiological and diseased conditions.
Inflammatory cytokines and pathogens
Inflammation is a quite common condition caused by various pathogens that elicit the burst of inflammatory cytokines by the host as a first line of defense. The influence of inflammatory cytokines on the production of Cst C was documented almost 30 years ago. Treatment of resident mouse peritoneal macrophages in vitro with the bacterial compound lipopolysaccharide (LPS) or pro‐inflammatory IFN‐γ downregulates Cst C secretion. 84 Likewise, inflammatory cytokine IL‐6 signaling in vivo was found to decrease Cst C expression in DCs. 27 Moreover, human immunodeficiency virus infection could either inhibit Cst C expression in DCs 12 or reduce the reactivity of Cst C with its target enzyme cathepsin B in macrophages. 85 Along the same line, we found that in an inflammatory mouse model created by intravenous injection of CpG oligodeoxynucleotides, mimics of bacterial and viral DNA responsible for immune stimulation, the synthesis of Cst C in DCs as well as the circulating pools of Cst C in blood were greatly reduced. 86 In non‐hematopoietic cells, however, addition of periodontal pathogens and pro‐inflammatory cytokines to human gingival fibroblasts was found to enhance their Cst C expression, 87 and upregulated levels of Cst C was observed in the ethmoid sinus mucosa of patients with chronic sinusitis, 88 indicating different roles and/or regulatory mechanisms of Cst C might exist in different cell types and tissues. However, the substantial impact of pathogens on immune cells, and our recent finding that serum levels of Cst C could be significantly affected by the replacement of bone marrows 89 highlights the important impact of altered Cst C production from hematopoietic cells on the systematic pools of Cst C during inflammation.
Growth factors and hormones
Growth factors or hormones are naturally occurring substance capable of regulating a variety of cellular processes by stimulating cellular growth, proliferation, healing and differentiation. They are also frequently used in the clinic to adjust the imbalanced cellular process. TGF‐β1 has been reported to upregulate Cst C secretion from vascular smooth muscle cells, murine embryo cells, cultured differentiated podocytes, 3T3‐L1 fibroblasts, and more recently human lung fibroblasts. 13 , 51 , 90 Clinically, a significant influence of circulating Cst C levels by TGF‐β1 were also observed in patients with thyroid dysfunction, and both Cst C mRNA and protein levels were increased by TGF‐β in cultured human hepatoblastoma cells. 91
Dexamethasone is a potent synthetic member of glucocorticoid class commonly used in the clinic to treat many inflammatory and autoimmune conditions. Interestingly, this steroid drug can drive promoter‐mediated upregulation of Cst C gene transcription, which leads to a significant and dose‐dependent increase in the Cst C production by up to 80%. 92 Recently, dexamethasone induced secretions of Cst C from human cancer cells were found to be enhanced by co‐application of cisplatin and 5‐fluorouracil, two agents commonly used in esophageal cancer chemotherapy, 93 adding further factors to the repertoire of Cst C‐altering elements.
Physicochemical damages
Rich in cerebrospinal fluid and brain tissue, Cst C is susceptible to physical and chemical insults that may occur in the central nervous system. For example, enhanced Cst C expression was observed in response to all sorts of neurological injuries, including transient forebrain ischemia 94 and seizure. 95 Consistently, the severity of neuronal damage in the CA1 subfield of the hippocampus correlates with enhanced Cst C immunostaining in microglia, the major Cst C‐expressing cell type in normal brain tissues. 96 Indeed electrical induction of a status epilepticus causes upregulation of Cst C expression in rat neurons and glia. 97 In addition, persistent environmental toxicants, like dieldrin, and neurotoxin MPP (1‐methy1‐4phenyl‐1,2,3,6‐tetrahydropyridine), were also reported to injure dopaminergic neurons and stimulate their secretion of Cst C for microglia activation and neurotoxicity. 98
Another Cst C‐sensitive tissue that is also vulnerable to physical impairment comes from blood vasculature where proteolytic activity of cysteine proteases requires strict regulation by their endogenous inhibitor. Balloon injury was recently reported to increase serum Cst C levels, which correlated with proliferating cell nuclear antigen in smooth muscle cells. 99
Oxidative stress
Interestingly, the two tissues vulnerable to Cst C levels and damages as mentioned above are also sensitive to the disturbances in their normal redox state. Oxidative stress initiates pathological progression through the production of peroxides and free radicals that damage all components of the cells, including proteins, lipids and DNA. Although the exact mechanisms by which Cst C is possibly involved in the disease development remain to be clarified, oxidative stress was repeatedly reported to upregulate Cst C expression in neurological and cardiovascular systems. For example, oxidative stress causes an increase in Cst C expression in cultured rat primary neurons 19 and cerebral microvascular smooth muscle cells. 100 Moreover, 6‐hydroxydopamine induced a temporal and concentration‐dependent increase in Cst C secretion from pheochromocytoma cells. 101 The release of Cst C in response to H2O2 as one of the cytoprotective, anti‐apoptotic factors from the embryonic stem cells were believed to be the mechanism to explain why transplanted embryonic stem cells subsequent to myocardial infarction differentiate into the major cell types in the heart and improve cardiac function. 102 Interestingly, this H2O2 induced release of Cst C was not only observed in embryonic stem cells but was also observed later in cardiomyocytes isolated from rat hearts because coronary artery blocking‐induced myocardial ischemia causes an increase in the levels of Cst C protein in the plasma. 103
Others
Other factors affecting Cst C production include the lung toxicants crystalline silica and arsenic trioxide, which were reported to stimulate Cst C release from rat alveolar macrophages. 104 In cancer patients during malignant progression‐like melanoma and colorectal cancer, high serum concentrations of the cysteine proteases cathepsins B and H induced by the tumors stimulate the production of Cst C for counterbalance. 11 In addition, common factors such as cigarette smoking or C‐reactive protein, an acute‐phase protein in response to inflammation, were also among the lists reported to increase serum Cst C levels independent of renal functions. 9
Mechanisms of Cst C regulation
The dynamic changes of Cst C caused by different stimuli herald the existence of, and prompt people to search for, the regulatory mechanisms of Cst C to keep this multifunctional and disease‐associated protein under check. Like many other proteins, the production of Cst C is subject to both transcriptional and post‐translational regulation (Figure 1).
Figure 1.
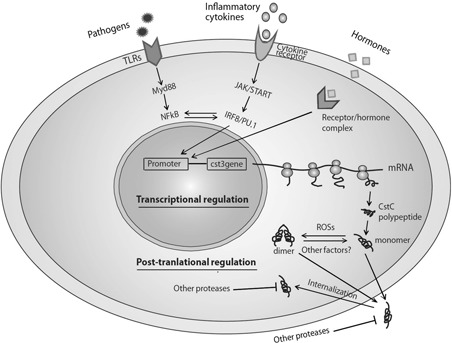
Regulation of cystatin C. Multiple regulatory mechanisms exist to modulate the production and activity of cystatin C inside and outside of cells, at transcriptional and post‐translational levels. JAK, Janus kinase; MyD88, myeloid differentiation primary response gene (88); ROSs, reactive oxygen species; STAT, signal transducer and activator of transcription; TLR, Toll‐like receptor. A full color version of this figure is available at the Immunology and Cell Biology journal online.
Transcriptional regulation
Although several polymorphisms and sequence variations were detected in the promoter region of cst3, the gene coding Cst C, 1 , 105 it generally shares common features with those of housekeeping genes. 1 Interestingly, the promoter region of murine cst3 gene was later found to contain a core sequence of the androgen‐responsive element and two potential binding sites for activator protein 1, 106 a transcription factor that regulates gene expression in response to a variety of stimuli including cytokines, growth factors, stress and bacterial and viral infection. 107 However, the transcriptional regulation of Cst C was not extensively explored probably due to the ubiquitous expression pattern of the protein. With the increasing recognition of the important roles of Cst C in cardiovascular diseases, the association of this elastolytic cysteine protease inhibitor with human coronary artery disease began to be examined at genetic levels. Two common promoter polymorphisms, a G‐to‐C substitution at position −82 and a T‐to‐G substitution at position −78, were found to influence the binding of nuclear factors and affect the basal rate of gene transcription in an allele‐specific manner, which are also associated with the plasma concentration of Cst C in healthy individuals and patients with recent myocardial infarction. 36 In another study, the major haplotype −82G/−5G/+4A of the cst3 gene was found to determine plasma levels of Cst C as the respondents with homozygous genotypes have the highest plasma levels. 108 Collectively, these data suggest that an altered promoter activity of the Cst C gene could be a causal factor for the association between Cst C genotype and plasma Cst C concentration.
Consistent with the potential regulatory mechanisms of human Cst C at genetic levels, we found that Cst C is differentially expressed among mouse cells of the immune system. Specifically, cells of the monocyte/macrophage and DC lineages express it at much higher levels than lymphocytes. 28 , 89 Mouse DC can be divided into two subsets by their surface expression of CD8α (CD8+ and CD8− DC). Further analysis of the DC subsets directly isolated from the spleen demonstrated that the CD8+ DC were the major producer of Cst C. 89 , 109 This was also verified with DCs that had been generated in culture from bone marrow precursors supplemented with Flt3L, in which only the CD8+ DC equivalent, the CD24hi DC subset, contained high levels of Cst C. 110 The differential expression pattern of Cst C among cells of common lineages suggests that its gene could be controlled by cell‐specific transcription factors.
This preferential expression pattern of Cst C in monocyte/macrophage and DCs is of clinical relevance in view of the recent reports showing that these cell types are present in tissues where Cst C plays pathological roles such as atherosclerosis and angiopathy. Vascular DCs (aortic DCs), which increase in number in atherosclerotic lesions as disease progresses, were reported to be related to CD8+DC, 111 and the Flt3L‐signaling‐dependent CD103+/CD11b− (CD8+ equivalent) DCs protect against atherosclerosis. 112 Consistently, our analysis of DC purified from aortas indicates that these cells indeed express Cst C (unpublished observation). Similarly, microglia, also called the monocyte/microphage in the brain, is the major cell type that express Cst C in the brain, 96 where deposition of Cst C fibrils produces angiopathy. 64
To uncover the transcriptional mechanisms underlying the preferential expression of disease‐related Cst C in these cells, a novel cis element for transcription factors, denoted as IRF (interferon regulatory factor)‐Ets composite sequence (IECS), in the promoter region of the cst3 gene was identified in a reporter assay system employing a self‐inactivating retrovirus. 113 This element consists of a core IRF‐binding motif for IRF8, and an Ets‐binding motif for PU.1, an Ets transcription factor and binding partner that facilitates a more stable binding of IRF8 to chromatin. 114 Notably, both IRF8 and PU.1 are essential for the development of DC subsets, 115 , 116 of which IRF8 is required for the development of CD8+ CD103+DC, 115 the major producers of Cst C. This could imply that Cst C expression by these cells might be the direct consequence of the presence of IRF8‐binding motif in the Cst C promoter region in cells that depend on IRF8 for their development. However, this cannot be the only explanation because IRF8 is also required in plasmacytoid DC development, 117 yet this DC type does not express Cst C. 110 Thus, the molecular mechanisms responsible for this phenomenon remain to be revealed.
To investigate the involvement of these two transcription factors in Cst C expression in primary DC in vivo, the physical interaction of IRF8 and PU.1 with the cis element IECS was examined by chromatin immunoprecipitation in three different DC subsets freshly isolated from splenic cells. 86 Consistent with their Cst C expression profile (CD8+DC express Cst C, both CD8−DC and plasmacytoid DC do not express Cst C), the binding of IRF8 to the IECS sequence was detected in CD8+DC, but not in CD8−DC and plasmacytoid DC. Interestingly, in the plasmacytoid DC where PU.1 expression is low but IRF8 expression is the same as CD8+DC, the binding of IRF8 to the IECS sequence is still not detected, indicating a quantitative requirement of PU.1 in IRF8 binding to the chromatin, a result consistent with previous findings with fluorescence recovery after filling to mathematical models. 114 Collectively, the chromatin immunoprecipitation data suggest that IRF8 binding to the IECS of the Cst C promoter in the presence of sufficient PU.1 could drive cst 3 expression. To finally confirm the role of IRF8 in Cst C expression, a CD8+ DC line 1940, where both IRF8 and PU.1 were amply expressed, were transfected with the retrovirus vector LMP encoding shRNAs for irf8 to silence the gene. When the synthesis of IRF8 was reduced in the cells, that of Cst C, but not MHC I, was also compromised. This causal impact of IRF8 on Cst C production, combined with the diminished Cst C expression in bone marrow (BM)‐derived DC from irf8−/− mice compared with wild‐type control, strongly suggest that IRF8 is the key transcription factor regulating Cst C expression. 86
Post‐translational regulation
The translational expression of Cst C in quantity does not necessarily lead to a functional protein product. The activity of Cst C can still be further modulated after its transcription. As discussed before, the reactivity of Cst C with its target protease is lost if it dimerizes. Therefore, the factors that cause this conformational change of Cst C by dimerization can be regarded as a way to regulate the activity of this protease inhibitor. It is intriguing to think that Cst C adopts the unreactive dimer conformation as storage form, which quickly monomerizes in response to the stimulation of its target enzymes. This post‐translational regulation of Cst C by dimerization was first described in transfected Chinese hamster ovary cells, where Cst C is inactivated during the early part of its trafficking through the secretory pathway and then reactivated prior to secretion. 118 Interestingly, we found that steady‐state (immature) CD8+ DCs isolated from primary spleens constitutively contain Cst C homo‐dimers, which can be separated from monomers by size‐exclusion chromatography of cell lysates and their non‐reactivity with the Cst C target enzyme papain, supporting the notion that they are domain‐swapped dimers. Furthermore, when CD8+ DC underwent maturation by incubation in vitro, they no longer produced Cst C dimers. 89 Disappearance of dimer could be the strategy employed by healthy primary cells to process this potentially pathogenic form on their way toward maturation, which might then be disrupted in diseased conditions.
To identify the mechanistic factors leading to Cst C dimerization either as post‐translational regulation of its activity, or as amyloid precursor protein, the intracellular accumulation of reactive oxygen species in the immature and mature states were compared. A strong correlation between reactive oxygen species levels and Cst C dimer was observed not only in same cell type at different developmental stages, but also in different cell types at the same developmental stages. 89 Furthermore, artificial enhancement of the intracellular oxidative status resulted in a time‐dependent Cst C dimer enrichment, which could be prevented by inhibiting mitochondrial activity, indicating the reactive oxygen species released from mitochondria are responsible for the observed constitutive Cst C dimer formation. 89 Although, the exact process by which oxidative stress exerts the conformational changes of Cst C is not fully understood, a recently published report on recombinant human Cst C stabilized by genetically introduced disulfide linkage demonstrated a disappearance of a dithiothreitol‐induced dimer if the concentration of this reducing agent was further increased, 119 suggesting a direct impact of redox environment on the conformational changes of Cst C protein.
Different from type I cystain family members, the type II cystatin Cst C is synthesized with a signal peptide, hence being secreted and consequently found in body fluids. 31 Although its intracellular roles in processing MHCII molecules via lysosomal proteinase in DCs were debatable, the uptake of Cst C in amounts sufficient to affect the activities of intracellular cysteine proteases was described in the eyes, 120 proximal tubule cells 121 and several human cancer cell lines. 122 , 123 Thus, trafficking inside and outside of the cell membrane of Cst C can regulate or fine tune its division of labor between intracellular compartments and the extracellular matrix. However, the structural determinants on the internalized inhibitor required for efficient uptake were not characterized until recently 12 variants of Cst C with substitutions of selected amino acids were generated. 42 Uptake of Cst C in human breast adenocarcinoma cells is dependent on both charged amino acids of the N‐terminal segment and on a hydrophobic amino acid in domains involved in the inhibition of cysteine cathepsins. Furthermore, natural arginine (Arg) residues in positions 24 and 25 are of importance for the uptake process. 42
Another mechanism for post‐translational regulation of Cst C could come from digestion of this cysteine protease inhibitor by proteases of another family. For example, earlier proteomic studies have shown that Cst C is a substrate of matrix metalloproteinase 2 with specific inactivation upon cleavage. 124 In addition, human aspartic endoproteinase cathepsin D was also able to inactivate human Cst C by cutting hydrophobic amino acid residues into several fragments in vitro. 125 Along the same line, the amount of Cst C in the extracellular environment is reduced in the secretome of mouse embryonic fibroblasts stably transfected with human cathepsin D, and the tumor‐derived cathepsin D assists breast cancer progression by inhibiting Cst C activity. 126 Importantly, the relevance of this mechanism was also found in other biological process in which Cst C was shown to be a proteolytic target of cathepsin D, affecting the differentiation of DCs from hematopoietic stem cells. 127
Clinical implications
Given the increasingly discovered roles of Cst C in various pathophysiological processes and identification of its regulatory mechanisms at both transcriptional and post‐transcriptional levels, manipulation of Cst C expression either locally or systematically may have many clinical implications. Characterization of the factors that control Cst C expression at the transcriptional level could provide valuable clues for the treatment of pathologies associated with insufficient control of extracellular proteases. For example, signaling molecules that upregulate Cst C expression could be used to promote Cst C secretion in the case that requires slightly higher Cst C concentration to protect neuronal cells from cell death in Alzheimer's disease, 78 or at sites of inflammation in which excessive protease activity causes tissue damage, as has been suggested to occur in atherosclerosis and aortic aneurysm. 13 However, the factors associated with the Cst C dimerization or cytokines that repress Cst C expression could be developed for the treatment of diseases associated with formation of Cst C amyloid. Moreover, identification of the receptors or signaling pathways that initiate the Cst C uptake on the cell surface could be translated to target this potent inhibitor to intracellular cancer‐promoting proteolysis via the Cst C internalization.
Since Cst C dimerization loses its activity as a protease inhibitor, which could also be an initial step for the amyloid formation in hereditary cystatin C amyloid angiopathy patients, the development of Cst C dimer/oligomer specific antibodies could be used to make diagnostic screening of hereditary cystatin C amyloid angiopathy family members for early prevention, or selectively remove Cst C dimers from biological fluids containing both dimers and monomers in patients with hereditary cystatin C amyloid angiopathy. In addition, the mechanistic factors affecting the inhibitive activity of Cst C can also be harnessed for therapeutic gains. Drugs to improve the intracellular redox environment by removal of reactive oxygen species and compounds may be beneficial in not only reducing the Cst C amyloidogenesis, but also regulating the elastic proteolysis in the diseased locus as elevated protease activity in the local body fluids is partly responsible for the tissue destruction in the disease associated with Cst C amyloid. 64
Last but not least, identification of transplantable cellular sources for major Cst C production will also be of great clinical value. Since Cst C is involved in inflammatory diseases, in which immune cells accumulate and play important roles, bone marrow‐derived cells are an important cellular source for Cst C manipulation. Along this line, we found that hematopoietic cells contribute significantly to the systematic pools of Cst C. 89 Therefore, bone marrow transplantation would be an applicable approach in clinic to treat patients with Cst C amyloidogenesis or reduction.
Conclusion
With the recent identification of regulatory elements in the promoter region of Cst C and increasing reports of factors affecting its production and/or activities, precaution should be taken when Cst C is used as an index of GFR because its blood concentration is subject to changes caused by many factors independent of kidney function. These factors include cigarette smoking, body composition, viral infection, tumor malignancy or gene mutations. Ultimately, the altered Cst C levels or activity could lead to pathological processes in cardiovascular diseases, neurological disorders or even mortality (Figure 2). Such an important disease‐associated protein thus should be effectively regulated for therapeutic gains. Further characterization of the signaling pathways leading to Cst C expression will help to develop antagonists and/or synagonists for medical intervention or regulation of the abnormal production and/or activity of this disease‐associated enzyme inhibitor. Likewise, identification of the post‐translational modifications in the microheterogeneity of the pathogenic species will also assist in understanding the mechanisms or direct factors triggering Cst C dimerization. The knowledge should provide the information for a better understanding of the mysterious mechanisms underlying the diseases associated with Cst C abnormalities.
Figure 2.
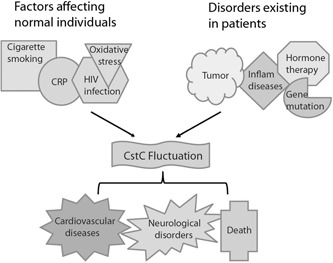
Multiple factors are involved in the progression of cystatin C‐related diseases. A scheme summarizing factors leading to the fluctuation of cystatin C levels, which triggles or participates in the pathogenic processes of several diseases. CRP, C‐reactive protein. A full color version of this figure is available at the Immunology and Cell Biology journal online.
ACKNOWLEDGEMENTS
This work was funded by a Project Grant from National Health and Medical Research Council (NHMRC) of Australia (no. 1006428), and a start‐up fund from Anhui Normal University of China to YX (no. 004061439).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1. Abrahamson M, Olafsson I, Palsdottir A, Ulvsback M, Lundwall A, Jensson O et al Structure and expression of the human cystatin C gene. Biochem J 1990; 268: 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roos JF, Doust J, Tett SE, Kirkpatrick CM. Diagnostic accuracy of cystatin C compared to serum creatinine for the estimation of renal dysfunction in adults and children—a meta‐analysis. Clin Biochem 2007; 40: 383–391. [DOI] [PubMed] [Google Scholar]
- 3. Dharnidharka VR, Kwon C, Stevens G. Serum cystatin C is superior to serum creatinine as a marker of kidney function: a meta‐analysis. Am J Kidney Dis 2002; 40: 221–226. [DOI] [PubMed] [Google Scholar]
- 4. Shlipak MG, Mattes MD, Peralta CA. Update on cystatin C: incorporation into clinical practice. Am J Kidney Dis 2013; 62: 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akerblom A, Eriksson N, Wallentin L, Siegbahn A, Barratt BJ, Becker RC et al Polymorphism of the cystatin C gene in patients with acute coronary syndromes: results from the PLATelet inhibition and patient Outcomes study. Am Heart J 2014; 168: 96–102 e2. [DOI] [PubMed] [Google Scholar]
- 6. O'Seaghdha CM, Tin A, Yang Q, Katz R, Liu Y, Harris T et al Association of a cystatin C gene variant with cystatin C levels, CKD, and risk of incident cardiovascular disease and mortality. Am J Kidney Dis 2014; 63: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oc MA, Demir H, Cekmen MB, Isgoren S, Gorur GD, Bilgili U. Correlation of cystatin‐C and radionuclidic measurement method of glomerular filtration rate in patients with lung cancer receiving cisplatin treatment. Ren Fail 2014; 36: 1043–1050. [DOI] [PubMed] [Google Scholar]
- 8. Laskin BL, Nehus E, Goebel J, Furth S, Davies SM, Jodele S. Estimated versus measured glomerular filtration rate in children before hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014; 20: 2056–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knight EL, Verhave JC, Spiegelman D, Hillege HL, de Zeeuw D, Curhan GC et al Factors influencing serum cystatin C levels other than renal function and the impact on renal function measurement. Kidney Int 2004; 65: 1416–1421. [DOI] [PubMed] [Google Scholar]
- 10. Risch L, Herklotz R, Blumberg A, Huber AR. Effects of glucocorticoid immunosuppression on serum cystatin C concentrations in renal transplant patients. Clin Chem 2001; 47: 2055–2059. [PubMed] [Google Scholar]
- 11. Kos J, Stabuc B, Cimerman N, Brunner N. Serum cystatin C, a new marker of glomerular filtration rate, is increased during malignant progression. Clin Chem 1998; 44: 2556–2557. [PubMed] [Google Scholar]
- 12. Harman AN, Kraus M, Bye CR, Byth K, Turville SG, Tang O et al HIV‐1‐infected dendritic cells show 2 phases of gene expression changes, with lysosomal enzyme activity decreased during the second phase. Blood 2009; 114: 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi GP, Sukhova GK, Grubb A, Ducharme A, Rhode LH, Lee RT et al Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J Clin Invest 1999; 104: 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimura T, Jiang H, Konno T, Seto M, Iwanaga K, Tsujihata M et al Bunina bodies in motor and non‐motor neurons revisited: a pathological study of an ALS patient after long‐term survival on a respirator. Neuropathology 2014; 34: 392–397. [DOI] [PubMed] [Google Scholar]
- 15. Xu N, Zhang YY, Lin Y, Bao B, Zheng L, Shi GP et al Increased levels of lysosomal cysteinyl cathepsins in human varicose veins: a histology study. Thromb Haemost 2014; 111: 333–344. [DOI] [PubMed] [Google Scholar]
- 16. Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B et al Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 2012; 1824: 68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D et al Lysosomal membrane permeabilization induces cell death in a mitochondrion‐dependent fashion. J Exp Med 2003; 197: 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C et al Cathepsin B contributes to TNF‐alpha‐mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest 2000; 106: 1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishio C, Yoshida K, Nishiyama K, Hatanaka H, Yamada M. Involvement of cystatin C in oxidative stress‐induced apoptosis of cultured rat CNS neurons. Brain Res 2000; 873: 252–262. [DOI] [PubMed] [Google Scholar]
- 20. Nagai A, Terashima M, Sheikh AM, Notsu Y, Shimode K, Yamaguchi S et al Involvement of cystatin C in pathophysiology of CNS diseases. Front Biosci 2008; 13: 3470–3479. [DOI] [PubMed] [Google Scholar]
- 21. Liang X, Nagai A, Terashima M, Sheikh AM, Shiota Y, Mitaki S et al Cystatin C induces apoptosis and tyrosine hydroxylase gene expression through JNK‐dependent pathway in neuronal cells. Neurosci Lett 2011; 496: 100–105. [DOI] [PubMed] [Google Scholar]
- 22. Suzuki Y, Jin C, Yazawa I. Cystatin C triggers neuronal degeneration in a model of multiple system atrophy. Am J Pathol 2014; 184: 790–799. [DOI] [PubMed] [Google Scholar]
- 23. Nishiyama K, Konishi A, Nishio C, Araki‐Yoshida K, Hatanaka H, Kojima M et al Expression of cystatin C prevents oxidative stress‐induced death in PC12 cells. Brain Res Bull 2005; 67: 94–99. [DOI] [PubMed] [Google Scholar]
- 24. Kaur G, Mohan P, Pawlik M, DeRosa S, Fajiculay J, Che S et al Cystatin C rescues degenerating neurons in a cystatin B‐knockout mouse model of progressive myoclonus epilepsy. Am J Pathol 2010; 177: 2256–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pierre P, Mellman I. Developmental regulation of invariant chain proteolysis controls MHC class II trafficking in mouse dendritic cells. Cell 1998; 93: 1135–1145. [DOI] [PubMed] [Google Scholar]
- 26. Manoury B, Gregory WF, Maizels RM, Watts C. Bm‐CPI‐2, a cystatin homolog secreted by the filarial parasite Brugia malayi, inhibits class II MHC‐restricted antigen processing. Curr Biol 2001; 11: 447–451. [DOI] [PubMed] [Google Scholar]
- 27. Kitamura H, Kamon H, Sawa S, Park SJ, Katunuma N, Ishihara K et al IL‐6‐STAT3 controls intracellular MHC class II alphabeta dimer level through cathepsin S activity in dendritic cells. Immunity 2005; 23: 491–502. [DOI] [PubMed] [Google Scholar]
- 28. El‐Sukkari D, Wilson NS, Hakansson K, Steptoe RJ, Grubb A, Shortman K et al The protease inhibitor cystatin C is differentially expressed among dendritic cell populations, but does not control antigen presentation. J Immunol 2003; 171: 5003–5011. [DOI] [PubMed] [Google Scholar]
- 29. Gresser O, Weber E, Hellwig A, Riese S, Regnier‐Vigouroux A. Immunocompetent astrocytes and microglia display major differences in the processing of the invariant chain and in the expression of active cathepsin L and cathepsin S. Eur J Immunol 2001; 31: 1813–1824. [DOI] [PubMed] [Google Scholar]
- 30. Zavasnik‐Bergant T, Repnik U, Schweiger A, Romih R, Jeras M, Turk V et al Differentiation‐ and maturation‐dependent content, localization, and secretion of cystatin C in human dendritic cells. J Leukoc Biol 2005; 78: 122–134. [DOI] [PubMed] [Google Scholar]
- 31. Abrahamson M, Grubb A, Olafsson I, Lundwall A. Molecular cloning and sequence analysis of cDNA coding for the precursor of the human cysteine proteinase inhibitor cystatin C. FEBS Lett 1987; 216: 229–233. [DOI] [PubMed] [Google Scholar]
- 32. Liu J, Sukhova GK, Sun JS, Xu WH, Libby P, Shi GP. Lysosomal cysteine proteases in atherosclerosis. Arterioscler Thromb Vasc Biol 2004; 24: 1359–1366. [DOI] [PubMed] [Google Scholar]
- 33. Jormsjo S, Wuttge DM, Sirsjo A, Whatling C, Hamsten A, Stemme S et al Differential expression of cysteine and aspartic proteases during progression of atherosclerosis in apolipoprotein E‐deficient mice. Am J Pathol 2002; 161: 939–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sukhova GK, Wang B, Libby P, Pan JH, Zhang Y, Grubb A et al Cystatin C deficiency increases elastic lamina degradation and aortic dilatation in apolipoprotein E‐null mice. Circ Res 2005; 96: 368–375. [DOI] [PubMed] [Google Scholar]
- 35. Bengtsson E, To F, Hakansson K, Grubb A, Branen L, Nilsson J et al Lack of the cysteine protease inhibitor cystatin C promotes atherosclerosis in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol 2005; 25: 2151–2156. [DOI] [PubMed] [Google Scholar]
- 36. Eriksson P, Deguchi H, Samnegard A, Lundman P, Boquist S, Tornvall P et al Human evidence that the cystatin C gene is implicated in focal progression of coronary artery disease. Arterioscler Thromb Vasc Biol 2004; 24: 551–557. [DOI] [PubMed] [Google Scholar]
- 37. Mignatti P, Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev 1993; 73: 161–195. [DOI] [PubMed] [Google Scholar]
- 38. Huh CG, Hakansson K, Nathanson CM, Thorgeirsson UP, Jonsson N, Grubb A et al Decreased metastatic spread in mice homozygous for a null allele of the cystatin C protease inhibitor gene. Mol Pathol 1999; 52: 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kolwijck E, Kos J, Obermajer N, Span PN, Thomas CM, Massuger LF et al The balance between extracellular cathepsins and cystatin C is of importance for ovarian cancer. Eur J Clin Invest 2010; 40: 591–599. [DOI] [PubMed] [Google Scholar]
- 40. Kopitz C, Anton M, Gansbacher B, Kruger A. Reduction of experimental human fibrosarcoma lung metastasis in mice by adenovirus‐mediated cystatin C overexpression in the host. Cancer Res 2005; 65: 8608–8612. [DOI] [PubMed] [Google Scholar]
- 41. Wegiel B, Jiborn T, Abrahamson M, Helczynski L, Otterbein L, Persson JL et al Cystatin C is downregulated in prostate cancer and modulates invasion of prostate cancer cells via MAPK/Erk and androgen receptor pathways. PLoS ONE 2009; 4: e7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wallin H, Abrahamson M, Ekstrom U. Cystatin C properties crucial for uptake and inhibition of intracellular target enzymes. J Biol Chem 2013; 288: 17019–17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Turk B, Turk D, Salvesen GS. Regulating cysteine protease activity: essential role of protease inhibitors as guardians and regulators. Curr Pharm Des 2002; 8: 1623–1637. [DOI] [PubMed] [Google Scholar]
- 44. Korant BD, Brzin J, Turk V. Cystatin, a protein inhibitor of cysteine proteases alters viral protein cleavages in infected human cells. Biochem Biophys Res Commun 1985; 127: 1072–1076. [DOI] [PubMed] [Google Scholar]
- 45. Bjorck L, Akesson P, Bohus M, Trojnar J, Abrahamson M, Olafsson I et al Bacterial growth blocked by a synthetic peptide based on the structure of a human proteinase inhibitor. Nature 1989; 337: 385–386. [DOI] [PubMed] [Google Scholar]
- 46. Bjorck L, Grubb A, Kjellen L. Cystatin C, a human proteinase inhibitor, blocks replication of herpes simplex virus. J Virol 1990; 64: 941–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Collins AR, Grubb A. Inhibitory effects of recombinant human cystatin C on human coronaviruses. Antimicrob Agents Chemother 1991; 35: 2444–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mottram JC, Brooks DR, Coombs GH. Roles of cysteine proteinases of trypanosomes and Leishmania in host‐parasite interactions. Curr Opin Microbiol 1998; 1: 455–460. [DOI] [PubMed] [Google Scholar]
- 49. Descoteaux A. Leishmania cysteine proteinases: virulence factors in quest of a function. Parasitol Today 1998; 14: 220–221. [DOI] [PubMed] [Google Scholar]
- 50. Das L, Datta N, Bandyopadhyay S, Das PK. Successful therapy of lethal murine visceral leishmaniasis with cystatin involves up‐regulation of nitric oxide and a favorable T cell response. J Immunol 2001; 166: 4020–4028. [DOI] [PubMed] [Google Scholar]
- 51. Sokol JP, Schiemann WP. Cystatin C antagonizes transforming growth factor beta signaling in normal and cancer cells. Mol Cancer Res 2004; 2: 183–195. [PubMed] [Google Scholar]
- 52. Sokol JP, Neil JR, Schiemann BJ, Schiemann WP. The use of cystatin C to inhibit epithelial‐mesenchymal transition and morphological transformation stimulated by transforming growth factor‐beta. Breast Cancer Res 2005; 7: R844–R853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tian M, Schiemann WP. Preclinical efficacy of cystatin C to target the oncogenic activity of transforming growth factor beta in breast cancer. Transl Oncol 2009; 2: 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Frendeus KH, Wallin H, Janciauskiene S, Abrahamson M. Macrophage responses to interferon‐gamma are dependent on cystatin C levels. Int J Biochem Cell Biol 2009; 41: 2262–2269. [DOI] [PubMed] [Google Scholar]
- 55. Kumada T, Hasegawa A, Iwasaki Y, Baba H, Ikenaka K. Isolation of cystatin C via functional cloning of astrocyte differentiation factors. Dev Neurosci 2004; 26: 68–76. [DOI] [PubMed] [Google Scholar]
- 56. Hasegawa A, Naruse M, Hitoshi S, Iwasaki Y, Takebayashi H, Ikenaka K. Regulation of glial development by cystatin C. J Neurochem 2007; 100: 12–22. [DOI] [PubMed] [Google Scholar]
- 57. Hu Y, Hung AC, Cui H, Dawkins E, Bolos M, Foa L et al Role of cystatin C in amyloid precursor protein‐induced proliferation of neural stem/progenitor cells. J Biol Chem 2013; 288: 18853–18862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Haan J, Maat‐Schieman ML, Roos RA. Clinical aspects of cerebral amyloid angiopathy. Dementia 1994; 5: 210–213. [DOI] [PubMed] [Google Scholar]
- 59. Janowski R, Kozak M, Jankowska E, Grzonka Z, Grubb A, Abrahamson M et al Human cystatin C, an amyloidogenic protein, dimerizes through three‐dimensional domain swapping. Nat Struct Biol 2001; 8: 316–320. [DOI] [PubMed] [Google Scholar]
- 60. Ekiel I, Abrahamson M. Folding‐related dimerization of human cystatin C. J Biol Chem 1996; 271: 1314–1321. [DOI] [PubMed] [Google Scholar]
- 61. Wahlbom M, Wang X, Lindstrom V, Carlemalm E, Jaskolski M, Grubb A. Fibrillogenic oligomers of human cystatin C are formed by propagated domain swapping. J Biol Chem 2007; 282: 18318–18326. [DOI] [PubMed] [Google Scholar]
- 62. Liu HL, Lin YM, Zhao JH, Hsieh MC, Lin HY, Huang CH et al Molecular dynamics simulations of human cystatin C and its L68Q varient to investigate the domain swapping mechanism. J Biomol Struct Dyn 2007; 25: 135–144. [DOI] [PubMed] [Google Scholar]
- 63. Abrahamson M, Grubb A. Increased body temperature accelerates aggregation of the Leu‐68—>Gln mutant cystatin C, the amyloid‐forming protein in hereditary cystatin C amyloid angiopathy. Proc Natl Acad Sci USA 1994; 91: 1416–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Abrahamson M, Jonsdottir S, Olafsson I, Jensson O, Grubb A. Hereditary cystatin C amyloid angiopathy: identification of the disease‐causing mutation and specific diagnosis by polymerase chain reaction based analysis. Human genetics 1992; 89: 377–380. [DOI] [PubMed] [Google Scholar]
- 65. Gudmundsson G, Hallgrimsson J, Jonasson TA, Bjarnason O. Hereditary cerebral haemorrhage with amyloidosis. Brain 1972; 95: 387–404. [DOI] [PubMed] [Google Scholar]
- 66. Olafsson I, Gudmundsson G, Abrahamson M, Jensson O, Grubb A. The amino terminal portion of cerebrospinal fluid cystatin C in hereditary cystatin C amyloid angiopathy is not truncated: direct sequence analysis from agarose gel electropherograms. Scand J Clin Lab Invest 1990; 50: 85–93. [DOI] [PubMed] [Google Scholar]
- 67. Asgeirsson B, Haebel S, Thorsteinsson L, Helgason E, Gudmundsson KO, Gudmundsson G et al Hereditary cystatin C amyloid angiopathy: monitoring the presence of the Leu‐68—>Gln cystatin C variant in cerebrospinal fluids and monocyte cultures by MS. Biochem J 1998; 329: 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bjarnadottir M, Wulff BS, Sameni M, Sloane BF, Keppler D, Grubb A et al Intracellular accumulation of the amyloidogenic L68Q variant of human cystatin C in NIH/3T3 cells. Mol Pathol 1998; 51: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wei L, Berman Y, Castano EM, Cadene M, Beavis RC, Devi L et al Instability of the amyloidogenic cystatin C variant of hereditary cerebral hemorrhage with amyloidosis, Icelandic type. J Biol Chem 1998; 273: 11806–11814. [DOI] [PubMed] [Google Scholar]
- 70. Palsdottir A, Snorradottir AO, Thorsteinsson L. Hereditary cystatin C amyloid angiopathy: genetic, clinical, and pathological aspects. Brain Pathol 2006; 16: 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Whelly S, Serobian G, Borchardt C, Powell J, Johnson S, Hakansson K et al Fertility defects in mice expressing the L68Q variant of human cystatin C: a role for amyloid in male infertility. J Biol Chem 2014; 289: 7718–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pawlik M, Sastre M, Calero M, Mathews PM, Schmidt SD, Nixon RA et al Overexpression of human cystatin C in transgenic mice does not affect levels of endogenous brain amyloid beta peptide. J Mol Neurosci 2004; 22: 13–18. [DOI] [PubMed] [Google Scholar]
- 73. Mi W, Pawlik M, Sastre M, Jung SS, Radvinsky DS, Klein AM et al Cystatin C inhibits amyloid‐beta deposition in Alzheimer's disease mouse models. Nat Genet 2007; 39: 1440–1442. [DOI] [PubMed] [Google Scholar]
- 74. Gauthier S, Kaur G, Mi W, Tizon B, Levy E. Protective mechanisms by cystatin C in neurodegenerative diseases. Front Biosci 2011; 3: 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Selenica ML, Wang X, Ostergaard‐Pedersen L, Westlind‐Danielsson A, Grubb A. Cystatin C reduces the in vitro formation of soluble Abeta1‐42 oligomers and protofibrils. Scand J Clin Lab Invest 2007; 67: 179–190. [DOI] [PubMed] [Google Scholar]
- 76. Sastre M, Calero M, Pawlik M, Mathews PM, Kumar A, Danilov V et al Binding of cystatin C to Alzheimer's amyloid beta inhibits in vitro amyloid fibril formation. Neurobiol Aging 2004; 25: 1033–1043. [DOI] [PubMed] [Google Scholar]
- 77. Kaeser SA, Herzig MC, Coomaraswamy J, Kilger E, Selenica ML, Winkler DT et al Cystatin C modulates cerebral beta‐amyloidosis. Nat Genet 2007; 39: 1437–1439. [DOI] [PubMed] [Google Scholar]
- 78. Tizon B, Ribe EM, Mi W, Troy CM, Levy E. Cystatin C protects neuronal cells from amyloid‐beta‐induced toxicity. J Alzheimers Dis 2010; 19: 885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Damme M, Suntio T, Saftig P, Eskelinen EL. Autophagy in neuronal cells: general principles and physiological and pathological functions. Acta Neuropathol, (e‐pub ahead of print 4 November 2014; doi: 10.1007/s00401-014-1361-4. [DOI] [PubMed] [Google Scholar]
- 80. Tizon B, Sahoo S, Yu H, Gauthier S, Kumar AR, Mohan P et al Induction of autophagy by cystatin C: a mechanism that protects murine primary cortical neurons and neuronal cell lines. PLoS ONE 2010; 5: e9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu Y, Cai H, Wang Z, Li J, Wang K, Yu Z et al Induction of autophagy by cystatin C: a potential mechanism for prevention of cerebral vasospasm after experimental subarachnoid hemorrhage. Eur J Med Res 2013; 18: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu Y, Li J, Wang Z, Yu Z, Chen G. Attenuation of early brain injury and learning deficits following experimental subarachnoid hemorrhage secondary to cystatin C: possible involvement of the autophagy pathway. Mol Neurobiol 2014; 49: 1043–1054. [DOI] [PubMed] [Google Scholar]
- 83. Watanabe S, Hayakawa T, Wakasugi K, Yamanaka K. Cystatin C protects neuronal cells against mutant copper‐zinc superoxide dismutase‐mediated toxicity. Cell Death Dis 2014; 5: e1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Warfel AH, Zucker‐Franklin D, Frangione B, Ghiso J. Constitutive secretion of cystatin C (gamma‐trace) by monocytes and macrophages and its downregulation after stimulation. J Exp Med 1987; 166: 1912–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rodriguez‐Franco EJ, Cantres‐Rosario YM, Plaud‐Valentin M, Romeu R, Rodriguez Y, Skolasky R et al Dysregulation of macrophage‐secreted cathepsin B contributes to HIV‐1‐linked neuronal apoptosis. PLoS ONE 2012; 7: e36571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xu Y, Schnorrer P, Proietto A, Kowalski G, Febbraio MA, Acha‐Orbea H et al IL‐10 controls cystatin C synthesis and blood concentration in response to inflammation through regulation of IFN regulatory factor 8 expression. J Immunol 2011; 186: 3666–3673. [DOI] [PubMed] [Google Scholar]
- 87. Tsai CH, Yang SF, Huang FM, Chang YC. The upregulation of cystatin C in human gingival fibroblasts stimulated with cyclosporine A. J Periodontal Res 2009; 44: 459–464. [DOI] [PubMed] [Google Scholar]
- 88. Lee CW, Kim TH, Lee HM, Lee SH, Lee SH, Yoo JH et al Upregulation of elafin and cystatin C in the ethmoid sinus mucosa of patients with chronic sinusitis. Arch Otolaryngol Head Neck Surg 2009; 135: 771–775. [DOI] [PubMed] [Google Scholar]
- 89. Xu Y, Lindemann P, Vega‐Ramos J, Zhang JG, Villadangos JA. Developmental regulation of synthesis and dimerization of the amyloidogenic protease inhibitor cystatin C in the hematopoietic system. J Biol Chem 2014; 289: 9730–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kasabova M, Joulin‐Giet A, Lecaille F, Gilmore BF, Marchand‐Adam S, Saidi A et al Regulation of TGF‐beta1‐driven differentiation of human lung fibroblasts: emerging roles of cathepsin B and cystatin C. J Biol Chem 2014; 289: 16239–16251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kotajima N, Yanagawa Y, Aoki T, Tsunekawa K, Morimura T, Ogiwara T et al Influence of thyroid hormones and transforming growth factor‐beta1 on cystatin C concentrations. J Int Med Res 2010; 38: 1365–1373. [DOI] [PubMed] [Google Scholar]
- 92. Bjarnadottir M, Grubb A, Olafsson I. Promoter‐mediated, dexamethasone‐induced increase in cystatin C production by HeLa cells. Scand J Clin Lab Invest 1995; 55: 617–623. [DOI] [PubMed] [Google Scholar]
- 93. Yamawaki C, Takahashi M, Takara K, Kume M, Hirai M, Yasui H et al Effect of dexamethasone on extracellular secretion of cystatin C in cancer cell lines. Biomed Rep 2013; 1: 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Palm DE, Knuckey NW, Primiano MJ, Spangenberger AG, Johanson CE. Cystatin C, a protease inhibitor, in degenerating rat hippocampal neurons following transient forebrain ischemia. Brain Res 1995; 691: 1–8. [DOI] [PubMed] [Google Scholar]
- 95. Hendriksen H, Datson NA, Ghijsen WE, van Vliet EA, da Silva FH, Gorter JA et al Altered hippocampal gene expression prior to the onset of spontaneous seizures in the rat post‐status epilepticus model. Eur J Neurosci 2001; 14: 1475–1484. [DOI] [PubMed] [Google Scholar]
- 96. Lukasiuk K, Pirttila TJ, Pitkanen A. Upregulation of cystatin C expression in the rat hippocampus during epileptogenesis in the amygdala stimulation model of temporal lobe epilepsy. Epilepsia 2002; 43: 137–145. [DOI] [PubMed] [Google Scholar]
- 97. Aronica E, van Vliet EA, Hendriksen E, Troost D, Lopes da Silva FH, Gorter JA. Cystatin C, a cysteine protease inhibitor, is persistently up‐regulated in neurons and glia in a rat model for mesial temporal lobe epilepsy. Eur J Neurosci 2001; 14: 1485–1491. [DOI] [PubMed] [Google Scholar]
- 98. Dutta G, Barber DS, Zhang P, Doperalski NJ, Liu B. Involvement of dopaminergic neuronal cystatin C in neuronal injury‐induced microglial activation and neurotoxicity. J Neurochem 2012; 122: 752–763. [DOI] [PubMed] [Google Scholar]
- 99. Wu XJ, Dong ZQ, Lu QH. The role of cystatin C in vascular remodeling of balloon‐injured abdominal aorta of rabbits. Mol Biol Rep 2014; 41: 6225–6231. [DOI] [PubMed] [Google Scholar]
- 100. Wang Z, Wu D, Vinters HV. Hypoxia and reoxygenation of brain microvascular smooth muscle cells in vitro: cellular responses and expression of cerebral amyloid angiopathy‐associated proteins. APMIS 2002; 110: 423–434. [DOI] [PubMed] [Google Scholar]
- 101. Lee DC, Close FT, Goodman CB, Jackson IM, Wight‐Mason C, Wells LM et al Enhanced cystatin C and lysosomal protease expression following 6‐hydroxydopamine exposure. Neurotoxicology 2006; 27: 260–276. [DOI] [PubMed] [Google Scholar]
- 102. Singla DK, McDonald DE. Factors released from embryonic stem cells inhibit apoptosis of H9c2 cells. Am J Physiol Heart Circ Physiol 2007; 293: H1590–H1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Xie L, Terrand J, Xu B, Tsaprailis G, Boyer J, Chen QM. Cystatin C increases in cardiac injury: a role in extracellular matrix protein modulation. Cardiovasc Res 2010; 87: 628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Huaux F, Lasfargues G, Lauwerys R, Lison D. Lung toxicity of hard metal particles and production of interleukin‐1, tumor necrosis factor‐alpha, fibronectin, and cystatin‐c by lung phagocytes. Toxicol Appl Pharmacol 1995; 132: 53–62. [DOI] [PubMed] [Google Scholar]
- 105. Balbin M, Abrahamson M. PCR assay for a polymorphic DdeI site in the promoter region of the human cystatin C gene. Hum Genet 1992; 88: 710. [DOI] [PubMed] [Google Scholar]
- 106. Huh C, Nagle JW, Kozak CA, Abrahamson M, Karlsson S. Structural organization, expression and chromosomal mapping of the mouse cystatin‐C‐encoding gene (Cst3). Gene 1995; 152: 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hess J, Angel P, Schorpp‐Kistner M. AP‐1 subunits: quarrel and harmony among siblings. J Cell Sci 2004; 117: 5965–5973. [DOI] [PubMed] [Google Scholar]
- 108. Loew M, Hoffmann MM, Koenig W, Brenner H, Rothenbacher D. Genotype and plasma concentration of cystatin C in patients with coronary heart disease and risk for secondary cardiovascular events. Arterioscler Thromb Vasc Biol 2005; 25: 1470–1474. [DOI] [PubMed] [Google Scholar]
- 109. Zhan Y, Vega‐Ramos J, Carrington EM, Villadangos JA, Lew AM, Xu Y. The inflammatory cytokine, GM‐CSF, alters the developmental outcome of murine dendritic cells. Eur J Immunol 2012; 42: 2889–2900. [DOI] [PubMed] [Google Scholar]
- 110. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M et al Cutting edge: generation of splenic CD8+ and CD8‐ dendritic cell equivalents in Fms‐like tyrosine kinase 3 ligand bone marrow cultures. J Immunol 2005; 174: 6592–6597. [DOI] [PubMed] [Google Scholar]
- 111. Choi JH, Do Y, Cheong C, Koh H, Boscardin SB, Oh YS et al Identification of antigen‐presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med 2009; 206: 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Choi JH, Cheong C, Dandamudi DB, Park CG, Rodriguez A, Mehandru S et al Flt3 signaling‐dependent dendritic cells protect against atherosclerosis. Immunity 2011; 35: 819–831. [DOI] [PubMed] [Google Scholar]
- 113. Tamura T, Thotakura P, Tanaka TS, Ko MS, Ozato K. Identification of target genes and a unique cis element regulated by IRF‐8 in developing macrophages. Blood 2005; 106: 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Laricchia‐Robbio L, Tamura T, Karpova T, Sprague BL, McNally JG, Ozato K. Partner‐regulated interaction of IFN regulatory factor 8 with chromatin visualized in live macrophages. Proc Natl Acad Sci USA 2005; 102: 14368–14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tsujimura H, Tamura T, Gongora C, Aliberti J, Reis e Sousa C, Sher A et al ICSBP/IRF‐8 retrovirus transduction rescues dendritic cell development in vitro . Blood 2003; 101: 961–969. [DOI] [PubMed] [Google Scholar]
- 116. Anderson KL, Perkin H, Surh CD, Venturini S, Maki RA, Torbett BE. Transcription factor PU.1 is necessary for development of thymic and myeloid progenitor‐derived dendritic cells. J Immunol 2000; 164: 1855–1861. [DOI] [PubMed] [Google Scholar]
- 117. Tsujimura H, Tamura T, Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN‐producing plasmacytoid dendritic cells. J Immunol 2003; 170: 1131–1135. [DOI] [PubMed] [Google Scholar]
- 118. Merz GS, Benedikz E, Schwenk V, Johansen TE, Vogel LK, Rushbrook JI et al Human cystatin C forms an inactive dimer during intracellular trafficking in transfected CHO cells. J Cell Physiol 1997; 173: 423–432. [DOI] [PubMed] [Google Scholar]
- 119. Ostner G, Lindstrom V, Hjort Christensen P, Kozak M, Abrahamson M, Grubb A. Stabilization, characterization, and selective removal of cystatin C amyloid oligomers. J Biol Chem 2013; 288: 16438–16450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wasselius J, Johansson K, Hakansson K, Abrahamson M, Ehinger B. Cystatin C uptake in the eye. Graefes Arch Clin Exp Ophthalmol 2005; 243: 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kaseda R, Iino N, Hosojima M, Takeda T, Hosaka K, Kobayashi A et al Megalin‐mediated endocytosis of cystatin C in proximal tubule cells. Biochem Biophys Res Commun 2007; 357: 1130–1134. [DOI] [PubMed] [Google Scholar]
- 122. Ekstrom U, Wallin H, Lorenzo J, Holmqvist B, Abrahamson M, Aviles FX. Internalization of cystatin C in human cell lines. FEBS J 2008; 275: 4571–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wallin H, Bjarnadottir M, Vogel LK, Wasselius J, Ekstrom U, Abrahamson M. Cystatins—extra‐ and intracellular cysteine protease inhibitors: high‐level secretion and uptake of cystatin C in human neuroblastoma cells. Biochimie 2010; 92: 1625–1634. [DOI] [PubMed] [Google Scholar]
- 124. Dean RA, Butler GS, Hamma‐Kourbali Y, Delbe J, Brigstock DR, Courty J et al Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP‐2) in cell‐based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotrophin) and VEGF/connective tissue growth factor angiogenic inhibitory complexes by MMP‐2 proteolysis. Mol Cell Biol 2007; 27: 8454–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lenarcic B, Krasovec M, Ritonja A, Olafsson I, Turk V. Inactivation of human cystatin C and kininogen by human cathepsin D. FEBS Lett 1991; 280: 211–215. [DOI] [PubMed] [Google Scholar]
- 126. Laurent‐Matha V, Huesgen PF, Masson O, Derocq D, Prebois C, Gary‐Bobo M et al Proteolysis of cystatin C by cathepsin D in the breast cancer microenvironment. FASEB J 2012; 26: 5172–5181. [DOI] [PubMed] [Google Scholar]
- 127. Martino S, Tiribuzi R, Ciraci E, Makrypidi G, D'Angelo F, di Girolamo I et al Coordinated involvement of cathepsins S, D and cystatin C in the commitment of hematopoietic stem cells to dendritic cells. Int J Biochem Cell Biol 2011; 43: 775–783. [DOI] [PubMed] [Google Scholar]