

Abstract
Summary: Five of the 10 human Toll‐like receptors (TLRs) (TLR3, TLR4, TLR7, TLR8, and TLR9), and four of the 12 mouse TLRs (TLR3, TLR4, TLR7, TLR9) can trigger interferon (IFN)‐α, IFN‐β, and IFN‐λ, which are critical for antiviral immunity. Moreover, TLR3, TLR7, TLR8, and TLR9 differ from TLR4 in two particularly important ways for antiviral immunity: they can be activated by nucleic acid agonists mimicking compounds produced during the viral cycle, and they are typically present within the cell, along the endocytic pathway, where they sense viral products in the intraluminal space. Investigations in mice have demonstrated that the TLR7/9–IFN and TLR3–IFN pathways are different and critical for protective immunity to various experimental viral infections. Investigations in humans with interleukin‐1 receptor‐associated kinase‐4 (IRAK‐4) deficiency (unresponsive to TLR7, TLR8, and TLR9), UNC‐93B deficiency (unresponsive to TLR3, TLR7, TLR8, and TLR9), and TLR3 deficiency have recently shed light on the role of these two pathways in antiviral immunity in natural conditions. UNC‐93B‐ and TLR3‐deficient patients appear to be specifically prone to herpes simplex virus 1 (HSV‐1) encephalitis, although clinical penetrance is incomplete, whereas IRAK‐4‐deficient patients appear to be normally resistant to most viruses, including HSV‐1. These experiments of nature suggest that the TLR7‐, TLR8‐, and TLR9‐dependent induction of IFN‐α, IFN‐β, and IFN‐λ is largely redundant in human antiviral immunity, whereas the TLR3‐dependent induction of IFN‐α, IFN‐β, and IFN‐λ is critical for primary immunity to HSV‐1 in the central nervous system in children but redundant for immunity to most other viral infections.
Keywords: Toll‐like receptor, interferon, herpes simplex encephalitis, antiviral immunity
Introduction
The first demonstration of Toll involvement in host immunity was provided in 1996 by the observation that Toll‐deficient Drosophila are vulnerable to experimental infection with certain fungi (1). A family of proteins structurally related to Drosophila Toll, known as Toll‐like receptors (TLRs), was subsequently identified in mice and humans (2, 3, 4, 5, 6). Toll and TLR genes have now been described throughout the animal kingdom (7): 10 functional TLRs (from TLR1 to TLR10) have been identified in humans and 11 (from TLR1 to TLR7, TLR9, and from TLR11 to TLR13) in mice (8, 9). The TLR signaling pathway shares multiple components with that of interleukin‐1 receptors (IL‐1Rs), reflecting the intracellular domain common to these receptors, the Toll–IL‐1R (TIR) domain (10). The signaling pathways triggered by TLRs have been dissected in mice and, to a lesser extent, in humans (10, 11, 12) (Fig. 1). TLRs, with the exception of TLR3 (at least in mice), can trigger a ‘classical’ myeloid differentiation factor 88 (MyD88)‐dependent signaling pathway (10, 11, 12), via the TIR‐containing cytosolic adapter MyD88 (13, 14, 15, 16). TLR2 and TLR4 can also trigger this pathway via the TIR domain‐containing adapter protein (TIRAP) (17, 18, 19, 20). Alternatively, TLR3 and TLR4 can trigger a MyD88‐independent ‘alternative’ pathway (10, 11, 12, 21) via the TIR domain‐containing adapter inducing IFN‐β (TRIF) (22, 23, 24); TLR4 may trigger this pathway by association with the TRIF‐related adapter molecule (TRAM), which bridges TLR4 and TRIF (25, 26).
Figure 1.
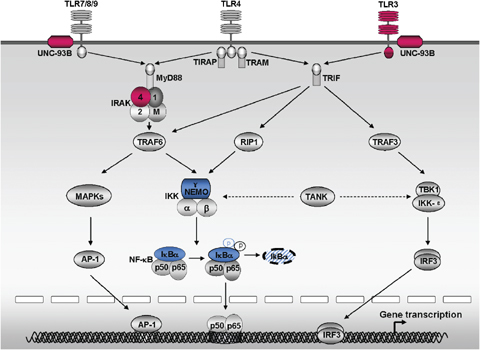
An overview of the signaling pathways triggered by the interferon (IFN)‐inducing Toll‐like receptors (TLRs) (TLR3, TLR4, TLR7, TLR8 and TLR9) and human primary immunodeficiencies involving TLR‐signaling pathways. Five TLRs (TLR3, TLR4, TLR7, TLR8, and TLR9) have been shown to induce the production of IFN‐α, IFN‐β, and IFN‐λ. TLR7, TLR8, and TLR9 trigger the ‘classical’ MyD88‐dependent TLR pathway, via the TIR‐containing cytosolic adapter MyD88. TLR3 triggers the ‘alternative’ MyD88‐independent, TIR domain‐containing adapter inducing IFN‐β (TRIF)‐dependent pathway via the TRIF. TLR4 triggers the MyD88‐dependent TLR pathway, via the TIRAP–MyD88 interaction, and triggers the MyD88‐independent pathway, via the TRIF‐related adapter molecule(TRAM)–TRIF interaction. The MyD88‐dependent pathway results in the activation of both nuclear factor‐κB (NF‐κB) and mitogen‐activated protein kinases (MAPKs), via the interleukin‐1 receptor‐associated kinase (IRAK) complex, which comprises two active kinases (IRAK‐1 and IRAK‐4) and two non‐catalytic subunits (IRAK‐2 and IRAK‐M). The MyD88‐independent pathway results in the activation of IRF3 via two kinases: NF‐κB kinase ɛ (IKK‐ɛ) and TANK‐binding kinase‐1 (TBK‐1). TRAF family member‐associated NF‐κB activator (TANK) interacts with NF‐κB essential modulator (NEMO), TBK1, and IRF3 and may therefore liaise between the MyD88‐dependent and MyD88‐independent pathways. These signaling pathways triggered by TLRs have been dissected in mice and, to a lesser extent, in humans. The three proteins in the TLR signaling pathways (IRAK‐4, UNC‐93B, and TLR3) responsible for the human primary immunodeficiencies discussed in this review are shown in red, the other two proteins in the TLR signaling pathways (NEMO, IκBα) responsible for human primary immunodeficiencies not discussed here are shown in blue. The principal pathways proposed are indicated by solid arrows, and recently described important associations between molecules are indicated with dotted arrows.
The classical MyD88‐dependent pathway (Fig. 1) results in the activation of both nuclear factor‐κB (NF‐κB) and mitogen‐activated protein kinases (MAPKs), via the IL‐1R‐associated kinase (IRAK) complex, which comprises two active kinases (IRAK‐1 and IRAK‐4) and two non‐catalytic subunits (IRAK‐2 and IRAK‐M). This classical pathway leads to the synthesis of inflammatory cytokines and chemokines, such as IL‐1β, IL‐6, IL‐8, IL‐12, and tumor necrosis factor‐α (TNF‐α) (10, 11, 12). The alternative MyD88‐independent pathway (Fig. 1), also referred to as the TRIF‐dependent pathway, results in the activation of IFN regulatory factor 3 (IRF3) via two kinases, the inhibitor of NF‐κB kinase ɛ (IKK‐ɛ) and TANK (TRAF family member‐associated NF‐κB activator)‐binding kinase‐1 (TBK‐1). The MyD88‐independent pathway leads to the transcription of a smaller number of genes, including IFN genes, such as that encoding IFN‐β (10, 11, 12) and possibly also the IFN‐λ gene (27). We now know that the MyD88‐dependent pathway can also activate IRF1 (28), IRF5 (29), and IRF7 (30, 31, 32). The MyD88‐dependent pathway may activate IRF3 via TANK, a molecule that can interact with TNF receptor‐associated factor‐6 (TRAF6), NF‐κB essential modulator (NEMO), TBK‐1, and IKK‐ɛ, linking the MyD88 pathway to IRF‐3 (32, 33, 34, 35, 36). Conversely, the TRIF‐dependent pathway may also activate NF‐κB, via RIP1 (37) or TRAF6 (38, 39) or via interactions between TRAF3, NEMO, and TANK (32, 33, 34, 35, 36). Although the NEMO–TANK interaction seems to bridge the IRF3 and NF‐κB signaling pathways downstream from TLRs (35), the detailed molecular mechanisms underlying these two pathways remain unclear.
The TLR pathways leading to the production of IFN‐α/‐β and IFN‐λ have begun to be unraveled in recent years (11, 12, 40). Five TLRs (TLR3, TLR4, TLR7, TLR8, and TLR9) have been shown to induce the production of IFN‐α, IFN‐β, and IFN‐λ (11, 12, 40). Four of these TLRs (TLR3, TLR7, TLR8, and TLR9) are intracellular in most cell types tested. They are found along the secretory and endocytic pathways and can be stimulated by nucleic acid agonists mimicking those produced during viral infections. TLR3 can be activated by double‐stranded RNA (41); TLR7 and TLR8 (in humans only) can be activated by antiviral derivatives of nucleoside‐like imidazoquinoline (42, 43) and loxoribine (44) and GU‐rich single‐stranded (ss) RNAs (45, 46, 47). TLR9 can be activated by non‐methylated double‐stranded (ds) CpG‐rich DNA (48, 49). Cell surface‐expressed human TLR4 can be stimulated by LPS and, possibly, by some viral proteins, but it is a much less potent inducer of IFN‐α, IFN‐β, and IFN‐λ (50, 51, 52). TLR3 and TLR4 trigger IFN‐β production via TRIF–IRF3 activation, whereas TLR7, TLR8, and TLR9 trigger IFN‐α production via MyD88–IRF7 activation (30, 31) and IFN‐β production via MyD88–IRF1 activation (28) or MyD88–TRAF6‐NEMO–TANK–IRF3 activation (10, 12, 36). The TLR induction of IFN‐α, which are IFN‐β‐inducible (53), has been studied in less detail than the induction of IFN‐β, and little is known about the mechanism of IFN‐λ induction by TLRs.
The innate recognition of viruses by TLRs may lead to the induction of IFNs (12, 54). One key question concerns the importance of the TLR–IFN pathway in protective antiviral immunity. Over the last 10 years, mice lacking individual TLRs and key molecules of the TLR signaling pathways, such as MyD88 and IRAK‐4, have been shown to display diverse viral infectious phenotypes, from susceptibility to resistance, depending on the host gene–pathogen combination (12, 55, 56) (1, 2). For example, TLR7‐deficient mice display impaired immunity to vesicular stomatitis virus (VSV) (47), and TLR9‐deficient mice are susceptible to mouse cytomegalovirus (MCMV) infection (5). However, it remained unclear whether Toll and TLRs played non‐redundant roles, be they beneficial or detrimental, in natural as opposed to experimental infections, particularly in viral infections (57, 58, 59, 60, 61, 62). Five human primary immunodeficiencies resulting in impaired TLR responses have recently been described (Fig. 1): X‐linked recessive NEMO deficiencies in 2001 (63, 64, 65), autosomal dominant IκBα deficiency in 2003 (66), autosomal recessive IRAK‐4 deficiency in 2003 (67), autosomal recessive UNC‐93B deficiency in 2006 (68), and autosomal dominant TLR3 deficiency in 2007 (69). Mutations in NEMO and IκBα impair many pathways other than TLRs, and the morbid alleles are hypomorphic and hypermorphic, respectively, making it difficult to assess antiviral TLR immunity in these patients. We review here the lessons learned from the three other primary immunodeficiencies (IRAK‐4, UNC‐93B and TLR3) that selectively or almost selectively impair TLR responses.
Table 1.
Viral infections of mice deficient for TLR3, TLR7, TLR9
| TLR | Infection | Phenotype | References | ||
|---|---|---|---|---|---|
| Survival rate | Viral load | Others | |||
| TLR3 | Encephalomyocarditis virus (EMCV) | NT | ↑ in the heart | ↓ proinflammatory cytokine and chemokine expression in the heart | (123) |
| Mouse cytomegalovirus (MCMV) | ↓ | ↑ in the spleen | ↓ IFN‐α/β production | (5) | |
| NT | NT | No difference in CD8 T or CD4 T response | (128) | ||
| Respiratory syncytial virus (RSV) | NT | No difference | ↑ Th2‐cytokine production, mucus production and gob5 expression in the airways | (124) | |
| Influenza virus (IAV) | ↑ | ↑ in the lung | ↓ inflammatory mediators (IL‐6, IL‐12) in the bronchalveolar airspace | (125) | |
| Punta Toro virus (PTV) | ↑ | NT | ↓ liver disease ↓ IL‐6, MCP‐1, IFN‐γ and RANTES levels in the liver and serum | (126) | |
| West Nile virus (WNV) | ↑ | ↓ in the brain, ↑ in the periphery | ↓ IL‐6, TNF‐α, IFN‐α, ‐β production in the blood and the brain | (127) | |
| Vesicular stomatitis virus (VSV) | NT | NT | No difference in CD8 T or CD4 T response | (128) | |
| T3 reovirus | NT | NT | No difference in CNS injury | (128) | |
| Lymphocytic choriomenigitis virus (LCMV) | NT | NT | No difference in CD8 T or CD4 T response | (128) | |
| TLR7 | Vesicular stomatitis virus (VSV) | NT | NT | ↓ IFN‐α production | (47) |
| TLR9 | Mouse cytomegalovirus (MCMV) | ↓ | ↑ in the spleen | ↓ IFN‐α/β production and NK cell activation | (5, 79) |
| Herpes simplex virus 1 (HSV‐1) | NT | No difference | (78) | ||
↑ increased; ↓ decreased; NT not tested.
Table 2.
Viral infections of mice deficient for MyD88, IRAK‐4, or UNC‐93B components
| Component | Infection | Phenotype | References | ||
|---|---|---|---|---|---|
| Survival rate | Viral load | Other | |||
| MyD88 | Herpes simplex virus 1 (HSV‐1) | ↓ | ↑ in the brain | (77) | |
| NT | No difference | (78) | |||
| Vesicular stomatitis virus (VSV) | ↓ | ↑ in the brain | ↓ IFN‐α production | (47, 82) | |
| No difference | NT | (82) | |||
| Mouse cytomegalovirus (MCMV) | ↓ | ↑ in the spleen | ↓ IFN‐α/β production and NK cell activation | (5, 79) | |
| Lymphocytic choriomenigitis virus (LCMV) | NT | ↑ | ↓ CD8+ T‐cell response | (80) | |
| Encephalomyocarditis virus (EMCV) | ↓ | NT | ↓ IFN‐α in the serum | (81) | |
| Respiratory virus (RSV) | NT | NT | ↓ eosinophils and mucus production in the pulmonary environment | (83) | |
| Coxsackievirus B3 (CVB3) | ↑ | ↓ in the heart | ↓ inflammatory cytokines, ↑ IFN‐β expression in the heart | (84) | |
| IRAK‐4 | Lymphocytic choriomenigitis virus (LCMV) | NT | NT | ↓ IFN‐γ production | (71) |
| UNC‐93B | Mouse cytomegalovirus (MCMV) | ↓ | ↑ in the spleen | ↓ Production of IFN‐α/β and other cytokines | (92) |
↑ increased; ↓ decreased; NT not tested.
IRAK‐4 deficiency
The four mammalian IRAK family members are orthologues of the serine–threonine kinase Pelle, which is required for Toll signaling in Drosophila (70). IRAK‐4 is the most similar to Pelle and plays an essential role in mediating the signals initiated by IL‐1R and TLR engagement (70, 71, 72). Mouse IRAK‐4 is 87% similar and 84% identical to human IRAK‐4 (70). Like Pelle and the other members of the IRAK family, IRAK‐4 contains an N‐terminal death domain (DD) and a central kinase domain (KD) (71). The crystal structures of the IRAK‐4 DD and KD domains have recently been obtained for mice and humans, revealing structures in these domains of IRAK‐4 very different from those of Pelle and other members of the IRAK family (73, 74). IRAK‐4 acts as a kinase downstream from MyD88 and upstream from IRAK‐1 and TRAF‐6 (72). IRAK‐4‐deficient mouse cells displayed strongly impaired IL‐1R and TLR signaling, and the activation of NF‐κB, c‐Jun N‐terminal kinase (JNK), and p38 MAPK induced by IL‐1 and multiple TLR agonists, including TLR7 and TLR9 agonists and the non‐specific TLR3 agonist polyinosinic‐polycytidylic acid [poly(I:C)], was profoundly inhibited in IRAK‐4‐deficient mouse embryonic fibroblasts (MEFs) and macrophages (71). Knockin mice with an inactivated IRAK‐4 kinase were recently produced. In these mice, TLR7‐ and TLR9‐mediated signaling is abolished in bone marrow (BM)‐derived macrophages and plasmacytoid dendritic cells (pDCs). The TLR7‐ and TLR9‐induced production of IFN‐α and IFN‐β was abolished in pDCs isolated from these mice, and the production of IFN‐λ was not determined. However, TLR3‐mediated signaling was intact in myeloid DCs (mDCs) from IRAK‐4‐deficient and IRAK‐4‐kinase‐inactive knockin mice (75). In mice deficient for IRAK‐1, a molecule immediately downstream from IRAK‐4, TLR7‐, and TLR9‐mediated IFN‐α production was also abolished (76). These in vitro data indicate that the TLR7 and TLR9 pathways governing the production of IFN, or at least of IFN‐α and ‐β, in response to viral infection are IRAK‐4 dependent in mice. In contrast, the TLR3–IFN pathway appeared to be IRAK‐4 independent, at least in mDCs.
IRAK4‐deficient mice have been challenged only with lymphocytic choriomenigitis virus (LCMV). Following infection with LCMV, IFN‐γ production was found to be severely impaired in IRAK‐4‐deficient mice, but it remains unclear whether IRAK‐4‐deficient mice are more susceptible to LCMV infection than the wildtype (WT) mice (Table 2) (71). Mice lacking MyD88, a molecule acting immediately upstream from IRAK‐4, are prone to herpes simplex virus 1 (HSV‐1) encephalitis (HSE) following intranasal infection (77). In another model of footpad or corneal infection, no differences were found between MyD88‐deficient and WT mice (78). MyD88‐deficient mice have been shown to be susceptible to MCMV (5, 79), LCMV (80), encephalomyocarditis virus (EMCV) (81), and VSV in a route‐dependent (intranasal but not intravenous) manner (47, 82). They are also susceptible, although to a lesser extent, to respiratory syncytial virus (RSV) (83). However, MyD88‐deficient mice have been shown to be more resistant to coxsackievirus B3 infection than WT mice (84) (Table 2). Mice lacking IRAK‐4 or MyD88 display impaired TLR7‐ and TLR9‐mediated induction of IFNs, and they are more susceptible than WT mice to experimental viral infections (5, 47, 77, 78, 79, 80, 81, 82). The only known exceptions concern MyD88‐deficient mice infected with HSV‐1 (78), coxsakievirus B3 (84), and VSV (intravenous route) (82) in certain experimental conditions (Table 2). However, the broader impairment of immunity in these mice, affecting multiple TLR and IL‐1R responses, makes it difficult to draw firm conclusions as to whether the susceptibility of these mice to viruses results from the impairment of TLR7‐ and TLR9‐mediated induction of IFNs.
Human IRAK‐4 deficiency was first reported in 2003, in three children with pyogenic bacterial diseases (67). Up to 28 such patients have since been reported (85). IRAK‐4‐deficient cells from these patients, including whole blood cells, peripheral blood mononuclear cells (PBMCs), individual myeloid and lymphoid leukocyte subsets, and skin‐derived fibroblasts, displayed impaired response to IL‐1 and the TLR agonists tested, with the exception of TLR3 responses and TLR4‐mediated IFN‐α, ‐β, and ‐λ responses (85, 86) (Fig. 2). In particular, the induction of IFN‐α, ‐β, and ‐λ in response to the stimulation of TLR7, TLR8, or TLR9 was found to be completely abolished in IRAK4‐deficient PBMCs (Fig. 2). Unlike MyD88‐deficient mice, IRAK‐4‐deficient patients appear to be resistant to most viruses. None of the known 28 IRAK‐4‐deficient patients suffered from severe viral disease, although at least 20 common viruses have a high prevalence among children worldwide. These viruses include HSV‐1, cytomegalovirus, Epstein–Barr virus, varicella–zoster virus, human herpes virus‐6, parvovirus B19, RSV, rotavirus, adenovirus, influenza virus, papilloma virus, enterovirus, and coronavirus (86) (Table 3). IRAK‐4‐deficient patients are not susceptible to HSE (85, 86). These data indicate that the IRAK‐4‐dependent, TLR7‐, TLR8‐, and TLR9‐mediated IFN responses are redundant for protective immunity to most viruses in humans.
Figure 2.
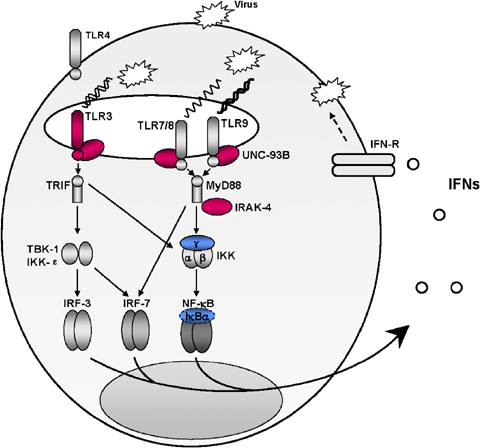
Toll‐like receptors (TLR)3‐, TLR7‐, TLR8‐, and TLR9‐dependent induction of interferon (IFN)‐α, ‐β, and ‐λ is largely redundant in human antiviral immunity. Four of the five IFN‐inducing TLRs (TLR3, TLR7, TLR8, and TLR9) are intracellular in most cell types tested. Viral nucleic acids, including double stranded (ds) RNA, single stranded (ss) RNA, and DNA, produced during virus replication are thought to be detected by TLR3, TLR7/8, and TLR9, respectively, and to induce IFN production. The production of IFN‐α, ‐β, and ‐λ in response to TLR7, TLR8, and TLR9 stimulation is impaired in interleukin‐1 receptor‐associated kinase (IRAK)‐4‐deficient patients, who appear to be resistant to most viruses. The induction of IFN‐α, ‐β, and ‐λ by TLR3, TLR7, TLR8, and TLR9 is impaired in UNC‐93B‐deficient patients, who are prone to herpes simplex virus 1 encephalitis (HSE) but appear to be resistant to most viruses. The UNC‐93B–TLR3–IFN‐α, ‐β, and ‐λ pathway is presumably essential for primary immunity to HSV‐1 in the central nervous system (CNS) in children but is redundant for immunity to most other viral infections. The three proteins in the TLR signaling pathways (IRAK‐4, UNC‐93B, and TLR3) responsible for the human primary immunodeficiencies discussed in this review are shown in red; the other two proteins in the TLR signaling pathways (NEMO, IκBα) responsible for human primary immunodeficiencies not discussed here are shown in blue.
Table 3.
Viral serological data for patients deficient for IRAK‐4, UNC‐93B, or TLR3
| Genetic defect | Infection | Positive serological tests (with vaccines) | References | |||
|---|---|---|---|---|---|---|
| dsDNA | ssDNA | ss−RNA | ss+RNA | |||
| IRAK‐4 (nine patients) | Gram‐positive bacteria | HSV‐1, EBV, CMV VZV, HHV‐6 Metapneumovirus | Parvovirus B19 | Mumps, measles RSV | CoxB1, rubella | (67, 85, 86) |
| UNC‐93B (two patients) | HSE | HSV‐1, EBV, CMV VZV, HHV‐6 | Parvovirus B19 | Mumps, measles RSV Influenza A and B parainflenzae 1 | CoxB1, rubella poliovirus | (68) |
| TLR3 (two patients) | HSE | HSV‐1, EBV, CMV VZV, HHV‐6 HBV | Mumps, measles | CoxB1, rubella | (69) | |
Note: Positive serological results in at least one patient are indicated.
HSE, herpes simplex encephalitis; HSV‐1, herpes simplex virus‐1; VZV, varicella zoster virus; RSV, respiratory syncytial virus; EBV, Epstein–Barr virus; HHV‐6, human herpes virus‐6; CoxB1, coxsackievirus B1; CMV, cytomegalovirus; HBV, hepatitis B virus.
Consistent with IRAK‐4‐deficient patients being no more susceptible than control subjects to most viruses, IFN production by IRAK‐4‐deficient PBMCs and fibroblasts has been shown to be normal or only slightly subnormal following stimulation with most of the viruses tested, with the possible exception of mumps virus and coxsackie virus (86). The functional TLR3 pathway in IRAK‐4‐deficient patients may contribute to the control of viruses in these patients. Upon activation by poly(I:C), IRAK‐4‐deficient fibroblasts display normal activation of IRF3, NF‐κB, and MAPK [p38, extracellular signal‐regulated kinases 1 and 2 (ERK1/2), JNK1/2](86). Moreover, IFN‐α, ‐β, and ‐λ are produced normally following poly(I:C) stimulation in IRAK‐4‐deficient PBMCs and fibroblasts. Induction of the IFN‐inducible monocyte chemotactic protein‐1 (MCP‐1) and IP‐10 in PBMCs and of CD40, CD80, and CD86 in monocyte‐derived DCs are also normal upon activation by poly(I:C) (85). However, poly(I:C) is a non‐specific TLR3 agonist. The question of the TLR3 dependence of poly(I:C) responses could not be addressed in individual human cell types until description of the TLR3‐deficient human fibrosarcoma cell line P2.1 (87) and of human patients bearing a germline dominant‐negative TLR3 mutation (69). In any event, whether by means of TLR3‐ and IRAK‐4‐independent responses to dsRNA, TLR3‐dependent but IRAK‐4‐independent responses to dsRNA, or responses to other viral intermediates using IRAK‐4‐independent pathways, IRAK‐4‐deficient patients appear to be normally resistant to most viruses.
UNC‐93B deficiency
Unc‐93 was first cloned in Caenorhabditis elegans in 1980 (88). It encodes a regulatory subunit of a SUP‐9 two‐pore potassium channel and coordinates muscle contraction (89, 90). Unc‐93 has been shown to be conserved in several distantly related species, including C. elegans, Arabidopsis thaliana, Drosophila melanogaster, mice, and humans (90). The human UNC93B1 gene has a sequence similar to that of its orthologues in A. thaliana, C. elegans, D. melanogaster, and mice (91). The human and mouse UNC‐93B proteins each have 12 transmembrane domains and are found mostly in the endoplasmic reticulum (92, 93). The crystal structure of UNC‐93B has not yet been described, and the function of this protein remains unclear. In humans, UNC‐93B protein levels are high in the heart and brain (91). The role of UNC‐93B in immunity was revealed by identification of the ‘3d’ gene in mice with a triple defect (3d) of impaired responses to TLR3, TLR7, and TLR9 (92, 93) (Table 1). The human orthologue of this gene was subsequently found to control TLR3, TLR7, TLR8, and TLR9 responses (68). UNC‐93B interacts directly with these TLRs, as it has been shown to bind the transmembrane domains of TLR3 and TLR9 but not that of TLR4 (93). However, the way in which UNC‐93B participates in TLR signaling remains to be determined (93).
The H412R mutation in Unc93b1 was found in 3d mice and was shown to be responsible for the 3d phenotype (92). UNC‐93B‐deficient mouse macrophages display impaired responses to the intracellular, nucleic acid‐sensing TLRs (TLR3, TLR7, and TLR9) for the production of several cytokines, including IFN‐α and ‐β. The 3d mouse also has a defect in exogenous antigen processing, as conventional major histocompatibility class II (MHC‐II) presentation and cross‐presentation by class I MHC (MHC‐I) are impaired. The impairment of cross‐presentation, to some extent, may reflect impaired IFN‐α/β production (94). 3d mice are highly susceptible to MCMV, which was used to identify 3d mutant mice generated by random mutagenesis (56). These mice are also susceptible to the Gram‐positive extracellular bacterium Staphylococcus aureus and the Gram‐positive intracellular bacterium Listeria monocytogenes. The particular susceptibility of 3d mice to MCMV may be accounted for by the impaired activation of TLR3 and TLR9, as mutations affecting signaling via the TLR3 and TLR9 pathways have also been shown to increase susceptibility to MCMV in C57BL/6 mice, which normally show robust resistance to MCMV (5) (Table 2). 3d mice have yet to be challenged with other viruses.
In humans, homozygous germline mutations in UNC93B1 were reported in 2006, in two unrelated patients who suffered from HSE, from different consanguineous kindreds (68). The identification of human UNC‐93B deficiency provided the first genetic etiology of HSE, the most common sporadic viral encephalitis in Western countries, caused by the almost ubiquitous and typically innocuous virus HSV‐1 (95). The identification of human UNC‐93B deficiency also provided the first evidence that HSE can result from a monogenic trait. The 1034del4 and 781G>A mutations in the coding region of UNC93B1 led to a complete loss of the expression and function of UNC‐93B. PBMCs from UNC‐93B‐deficient patients produce no IFN‐α, ‐β, or ‐λ in response to TLR7, TLR8, or TLR9 stimulation (Fig. 2). The production of IFN‐α, ‐β, and ‐λ by the patients' PBMCs in response to HSV‐1 and several other viruses is impaired but not abolished. Fibroblasts from the patients display impaired IFN‐β and ‐λ production following stimulation with HSV‐1, VSV, and the TLR‐3 agonist poly(I:C), and the activation of IRF3, NF‐κB, and MAPK (p38) in response to poly(I:C) stimulation is also impaired. Together with the lack of poly(I:C) response in the TLR3‐deficient fibrosarcoma cell line P2.1 (87), the normal poly(I:C) response in UNC‐93B‐deficient PBMCs and impaired poly(I:C) response in fibroblasts suggested a cell type‐specific poly(I:C) response: TLR3 dependent in fibroblasts and TLR independent in PBMCs (68). UNC‐93B‐deficient mice and patients have a similar overall biological phenotype, with abolished TLR3, TLR7, TLR8 (in human), and TLR9 responses, including those involving IFN‐α, ‐β, and ‐λ production (Fig. 2).
IRAK‐4‐deficient individuals are normally resistant to HSV‐1, do not develop HSE, and respond normally to TLR3 but not to TLR7, TLR8, and TLR9 agonists (85, 86). Signal transducer and activator of transcription 1 (Stat‐1)‐deficient patients with impaired cellular responses to IFN‐α, ‐β, and ‐λ are prone to HSE (96). These observations, together with the impaired TLR3, TLR7, TLR8, and TLR9 responses of UNC‐93B‐deficient patients prone to HSE, strongly suggest that impaired TLR3‐triggered, UNC‐93B‐dependent, IFN‐α, ‐β, and ‐λ induction may be involved in HSE. Unlike Stat‐1 (96, 97, 98) and Tyk‐2 (99) deficiencies, which affect most cells and confer predisposition to multiple viral diseases, human UNC‐93B deficiency seems to confer a relatively narrow predisposition to viruses, so far limited to HSE. HSE is not accompanied by the dissemination of HSV‐1 disease via the bloodstream or epithelium (95). Moreover, HSE patients are typically resistant to other infections, both before and after HSE, as serological tests have shown these patients to have been exposed to many other viruses, with no severe clinical manifestations (68, 69) (Table 3). This finding suggests that UNC‐93B‐dependent, virus‐induced IFN‐α, ‐β, and ‐λ production may be involved principally in primary immunity to HSV‐1 in the central nervous system (CNS) in children but redundant for immunity to most other viral infections. It is currently difficult to compare the infectious phenotypes of UNC‐93B‐deficient mice and humans, as such mice have been inoculated with few pathogens and only two patients have been described. However, the infectious phenotype seems to be much narrower in humans than in mice.
TLR3 deficiency
TLR3 mRNA production has been demonstrated in all vertebrates studied, including mice and humans (100, 101, 102). TLR3, like other TLRs, consists of an extracellular leucine‐rich repeat (LRR) motif, a transmembrane (TM) domain, and an intracellular TIR domain (103). The crystal structure of the TLR3 ectodomain (ECD) has been determined. This domain has been shown to take the form of a large, horseshoe‐shaped solenoid, potentially providing a large surface area for ligand interaction and recognition (104, 105). The ECD of TLR3 is essential for ligand binding and multimerization (104, 105, 106, 107, 108, 109). TRIF recruitment is mediated by interaction with the TLR3 TIR domain, leading to IRF3 and NF‐κB activation (10, 11, 12). TLR3 expression has been shown to be cell type specific, with this receptor preferentially expressed in DCs (110), fibroblasts (111), epithelial cells (112, 113, 114, 115), and CNS‐resident cells (116, 117, 118, 119, 120). Many in vitro studies have shown that a functional TLR3 is expressed in these cell types, which produce IFN‐α, ‐β, ‐λ and other cytokines and chemokines in response to poly(I:C) stimulation, presumably via TLR3 (68, 112, 113, 115, 118, 119, 121). TLR3 is intracellular in most cell types (122) but has been detected on the surface of a few cell types, including fibroblasts (111). The cellular compartment in which dsRNA is efficiently recognized by TLR3 has not yet been identified. It is also unclear whether the natural dsRNA produced during viral infection can actually stimulate TLR3. However, as TLR3 recognizes dsRNA, an almost universal viral intermediate potentially generated during most viral infections, it is thought that TLR3 may play a broad role in antiviral immunity.
TLR3‐deficient mice are susceptible to some experimental viral infections (Table 1). In particular, they are susceptible to EMCV, displaying impaired induction of proinflammatory cytokines and chemokines and a significantly higher viral load in the heart than that of WT mice following infection with this virus (123). TLR3‐deficient mice are also susceptible to MCMV in some experimental conditions, as shown by the impaired production of cytokines, including IFN‐α and ‐βin vivo, higher viral load in the spleen, and lower survival rate of TLR3‐deficient mice than that of WT mice (5). TLR3‐deficient mice are also susceptible to RSV infection, but to a lesser extent (124). Remarkably, TLR3‐deficient mice appear to be more resistant to other infections than WT mice (Table 1). Following infection with influenza A virus (IAV) (125), Punta Toro virus (PTV) (126), or West Nile virus (WNV) (127), TLR3‐deficient mice produce smaller amounts of inflammatory mediators, such as IFN‐α and ‐β, than WT mice, but had a higher survival rate. TLR3‐deficient mice did not differ from controls in their ability to generate both CD8+ T and CD4+ T IFN‐γ responses to LCMV, VSV, and MCMV; they were also similar to WT mice in terms of T3 reovirus‐induced injury in the CNS (128). Most studies have reported low levels of induction of cytokines or IFN in TLR3‐deficient mice, suggesting that TLR3 is involved in the balance of inflammatory mediators. Nevertheless, infectious phenotypes varied greatly, from enhanced susceptibility to enhanced resistance. It is therefore important to determine whether TLR3 plays a significant role in antiviral immunity during natural infections.
A germline TLR3 mutation was recently identified in two children with HSE (69). The impairment of TLR3‐triggered, UNC‐93B‐dependent IFN‐α, ‐β, and ‐λ induction therefore seems to be involved in HSE (68). In these two children, from non‐consanguineous families, a heterozygous 1660C>T mutation (P554S) was found in TLR3 (69). There was no founder gene effect, as the two 1660C>T mutations were in different TLR3 haplotypes. The 1660C>T mutant allele is dominant negative for the response to poly(I:C) in fibroblasts, in terms of IFN‐β and ‐λ induction. Fibroblasts, monocyte‐differentiated DCs, NK cells, and CD8+ T cells from the patients displayed an impaired response to poly(I:C). A defect in the production of IFN‐β and ‐λ by fibroblasts stimulated with HSV‐1, VSV, and poly(I:C) has been observed in TLR3 heterozygous fibroblasts, like in UNC93B‐deficient fibroblasts. Higher virus titers and enhanced cell mortality upon HSV‐1 and VSV infection were observed in both TLR3 heterozygous fibroblasts and UNC93B‐deficient fibroblasts complemented with IFN‐α and ‐β and, less efficiently, IFN‐λ. Impaired TLR3 signaling thus results in abnormally weak IFN‐α, ‐β, and ‐λ production, enhanced viral replication, and enhanced cell death in fibroblasts. By inference, this is a plausible pathogenesis of HSE in the CNS. Indeed, TLR3 is the most abundantly and widely expressed TLR in CNS‐resident cells, including neurons (118), microglia (116, 119), astrocytes (116, 119), and oligodendrocytes (116).
The development of HSE in otherwise healthy patients with TLR3 and UNC‐93B deficiency indicates that the UNC‐93B–TLR3–IFN‐α, ‐β, and ‐λ pathway is essential for primary immunity to HSV‐1 in the CNS in children (Fig. 2). Nonetheless, clinical penetrance is incomplete, as five of the seven TLR3‐deficient individuals and one of the three UNC‐93B‐deficient individuals did not develop HSE despite HSV‐1 infection (69). Interestingly, UNC‐93B and TLR3 deficiencies seem to confer predisposition to HSE alone, as the patients do not suffer from other viral diseases (Table 3) or from HSV‐1 infections outside the CNS (69). TLR3‐independent IFN responses to dsRNA, even in some cells and tissues that express TLR3, may confer protection against a wide range of viruses in TLR3‐ and UNC‐93B‐deficient patients (54). We have shown that mDCs and keratinocytes (which normally express TLR3) and pDCs (which do not normally express TLR3) produce IFN‐α, ‐β, and ‐λ in response to poly(I:C), in a TLR3‐independent manner. The IFN response to viruses other than VSV and HSV‐1 also is strong in TLR3‐heterozygous and UNC‐93B‐deficient fibroblasts. Moreover, the production of IFN‐α, ‐β, and ‐λ has been shown to be normal in PBMCs from TLR3‐deficient patients, following stimulation with the viruses tested (69). These observations account for the TLR3‐IFN pathway being apparently largely redundant for protective immunity to most viruses, although less so than the TLR7/8/9‐IFNs pathway, which is even redundant for protective immunity to HSV‐1.
Concluding remarks and perspectives
Studies on human IRAK‐4 deficiency, UNC‐93B deficiency, and TLR3 deficiency have shed light on the function of the human TLR‐IFN signaling pathways in protective immunity, particularly that to viruses. They have indicated that the IRAK‐4‐ and UNC‐93B‐dependent, TLR7‐, TLR8‐, and TLR9‐mediated induction of IFN‐α, ‐β, and ‐λ is redundant for protective immunity to most viruses (Fig. 2). This finding by no means suggests that these pathways are not involved in antiviral immunity; they may be, although in a redundant manner. Similarly, the IRAK‐4‐independent, UNC‐93B‐dependent, TLR3‐mediated induction of IFN‐α, ‐β, and ‐λ is redundant for protective immunity to most viruses. Nonetheless, this pathway seems to be selectively critical for protective immunity to HSV‐1 in the CNS upon primary HSV‐1 infection, in at least some children (Fig. 2). It is remarkable that HSV‐1 does not spread via the epithelia or bloodstream in patients with no functional TLR3 or UNC‐93B. It would be interesting to challenge TLR3‐ and UNC‐93B‐deficient mice with HSV‐1. The considerable redundancy observed in humans contrasts with the findings of many studies in mice showing that both the TLR7/8/9–UNC‐93B–MyD88–IRAK‐4–IFN pathway and the TLR3–UNC‐93B–TRIF–IFN pathway protect against experimental viral infections. Data for humans are more consistent with these pathways having a redundant effect on immunity to most viruses, and it may even well be that UNC‐93B‐ and TLR3‐deficient patients are more resistant to some viruses than the general population. In our view, the differences between data for mice and humans probably result mostly from the differences between experimental and natural infections (57, 61), although intrinsic differences between the species may also have some effect.
The small number of IRAK‐4‐, UNC‐93B‐, and TLR3‐deficient patients studied and the ascertainment bias in the recruitment of patients make it impossible to rule out the possibility that other patients with any of these three disorders may present with other infectious diseases, including viral diseases. The precise role of the two TLR–IFN pathways in protective immunity to viruses has therefore not been definitively determined. Receptors other than TLRs involved in sensing viral infections have been described recently, including cytosolic RNA helicases, retinoic acid‐inducible gene‐I (RIG‐I), which can be triggered by dsRNA and 5′‐phosphated ssRNA (129, 130, 131), and melanoma differentiation‐associated gene 5 (MDA‐5), which can be triggered by dsRNA (132). IFN‐inducing cytoplasmic recognition receptors may be critical for the control of viral infections, as suggested by in vitro and in vivo investigations in mice (54, 130, 133, 134). Other viral sensors, such as the IFN‐inducing cytoplasmic dsDNA sensor (135, 136, 137), have also been identified and may also be involved in antiviral immunity. We aim to evaluate the involvement of these molecules in natural antiviral immunity, by searching for germline mutations in the genes encoding these molecules, in patients with unexplained viral diseases. As viruses have developed multiple strategies for counteracting host antiviral IFN‐mediated immunity (138, 139), these investigations may improve our understanding of host–virus interaction and evolution.
Acknowledgements
We thank all members of the laboratory of Human Genetics of Infectious Diseases for helpful discussions. We also warmly thank the patients and their families worldwide, as well as our collaborators. These studies were supported by grants from the Schlumberger Foundation, the BNP‐Paribas Foundation, the GIS Maladies Rares, the Action Concertée Incitative de Microbiologie, the March of Dimes, and the ANR. Shen‐Ying Zhang is supported by the Howard Hughes Medical Institute. Jean‐Laurent Casanova is an International Scholar of the Howard Hughes Medical Institute.
References
- 1. Lemaitre B, et al The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996;86:973–983. [DOI] [PubMed] [Google Scholar]
- 2. Medzhitov R, Preston‐Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997;388:394–397. [DOI] [PubMed] [Google Scholar]
- 3. Rock FL, et al A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci USA 1998;95:588–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Poltorak A, et al Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998;282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 5. Tabeta K, et al Toll‐like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA 2004; 101:3516–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang D, et al A toll‐like receptor that prevents infection by uropathogenic bacteria. Science 2004;303:1522–1526. [DOI] [PubMed] [Google Scholar]
- 7. Roach JC, et al The evolution of vertebrate Toll‐like receptors. Proc Natl Acad Sci USA 2005;102:9577–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takeda K, Kaisho T, Akira S. Toll‐like receptors. Annu Rev Immunol 2003;21:335–376. [DOI] [PubMed] [Google Scholar]
- 9. Kawai T, Akira S. TLR signaling. Cell Death Differ 2006;13:816–825. [DOI] [PubMed] [Google Scholar]
- 10. O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007;7:353–364. [DOI] [PubMed] [Google Scholar]
- 11. Akira S, Takeda K. Toll‐like receptor signalling. Nat Rev Immunol 2004; 4:499–511. [DOI] [PubMed] [Google Scholar]
- 12. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783–801. [DOI] [PubMed] [Google Scholar]
- 13. Wesche H, et al MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity 1997;7:837–847. [DOI] [PubMed] [Google Scholar]
- 14. Muzio M, et al IRAK (Pelle) family member IRAK‐2 and MyD88 as proximal mediators of IL‐1 signaling. Science 1997;278: 1612–1615. [DOI] [PubMed] [Google Scholar]
- 15. Medzhitov R, et al MyD88 is an adaptor protein in the hToll/IL‐1 receptor family signaling pathways. Mol Cell 1998;2: 253–258. [DOI] [PubMed] [Google Scholar]
- 16. Muzio M, et al The human toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6). J Exp Med 1998;187: 2097–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol 2001;2:835–841. [DOI] [PubMed] [Google Scholar]
- 18. Fitzgerald KA, et al Mal (MyD88‐adapter‐like) is required for Toll‐like receptor‐4 signal transduction. Nature 2001;413: 78–83. [DOI] [PubMed] [Google Scholar]
- 19. Yamamoto M, et al Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 2002;420: 324–329. [DOI] [PubMed] [Google Scholar]
- 20. Horng T, et al The adaptor molecule TIRAP provides signalling specificity for Toll‐like receptors. Nature 2002;420:329–333. [DOI] [PubMed] [Google Scholar]
- 21. Mata‐Haro V, et al The vaccine adjuvant monophosphoryl lipid A as a TRIF‐biased agonist of TLR4. Science 2007;316: 1628–1632. [DOI] [PubMed] [Google Scholar]
- 22. Yamamoto M, et al Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol 2002;169: 6668–6672. [DOI] [PubMed] [Google Scholar]
- 23. Oshiumi H, et al TICAM‐1, an adaptor molecule that participates in Toll‐like receptor 3‐mediated interferon‐beta induction. Nat Immunol 2003;4:161–167. [DOI] [PubMed] [Google Scholar]
- 24. Yamamoto M, et al Role of adaptor TRIF in the MyD88‐independent Toll‐like receptor signaling pathway. Science 2003;301: 640–643. [DOI] [PubMed] [Google Scholar]
- 25. Fitzgerald KA, et al LPS‐TLR4 signaling to IRF‐3/7 and NF‐kappaB involves the toll adapters TRAM and TRIF. J Exp Med 2003;198:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamamoto M, et al TRAM is specifically involved in the Toll‐like receptor 4‐mediated MyD88‐independent signaling pathway. Nat Immunol 2003;4:1144–1150. [DOI] [PubMed] [Google Scholar]
- 27. Onoguchi K, et al Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem 2007;282:7576–7581. [DOI] [PubMed] [Google Scholar]
- 28. Negishi H, et al Evidence for licensing of IFN‐gamma‐induced IFN regulatory factor 1 transcription factor by MyD88 in Toll‐like receptor‐dependent gene induction program. Proc Natl Acad Sci USA 2006;103: 15136–15141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takaoka A, et al Integral role of IRF‐5 in the gene induction programme activated by Toll‐like receptors. Nature 2005;434: 243–249. [DOI] [PubMed] [Google Scholar]
- 30. Kawai T, et al Interferon‐alpha induction through Toll‐like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 2004;5:1061–1068. [DOI] [PubMed] [Google Scholar]
- 31. Honda K, et al Role of a transductional‐transcriptional processor complex involving MyD88 and IRF‐7 in Toll‐like receptor signaling. Proc Natl Acad Sci USA 2004;101:15416–15421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oganesyan G, et al Critical role of TRAF3 in the Toll‐like receptor‐dependent and ‐independent antiviral response. Nature 2006;439:208–211. [DOI] [PubMed] [Google Scholar]
- 33. Chariot A, et al Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. J Biol Chem 2002;277:37029–37036. [DOI] [PubMed] [Google Scholar]
- 34. Guo B, Cheng G. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem 2007;282:11817–11826. [DOI] [PubMed] [Google Scholar]
- 35. Zhao T, et al The NEMO adaptor bridges the nuclear factor‐kappaB and interferon regulatory factor signaling pathways. Nat Immunol 2007;8:592–600. [DOI] [PubMed] [Google Scholar]
- 36. Barton GM, Medzhitov R. Linking Toll‐like receptors to IFN‐alpha/beta expression. Nat Immunol 2003;4:432–433. [DOI] [PubMed] [Google Scholar]
- 37. Meylan E, et al RIP1 is an essential mediator of Toll‐like receptor 3‐induced NF‐kappa B activation. Nat Immunol 2004;5:503–507. [DOI] [PubMed] [Google Scholar]
- 38. Sato S, et al Toll/IL‐1 receptor domain‐containing adaptor inducing IFN‐beta (TRIF) associates with TNF receptor‐associated factor 6 and TANK‐binding kinase 1, and activates two distinct transcription factors, NF‐kappa B and IFN‐regulatory factor‐3, in the Toll‐like receptor signaling. J Immunol 2003;171: 4304–4310. [DOI] [PubMed] [Google Scholar]
- 39. Jiang Z, et al Toll‐like receptor 3‐mediated activation of NF‐kappaB and IRF3 diverges at Toll‐IL‐1 receptor domain‐containing adapter inducing IFN‐beta. Proc Natl Acad Sci USA 2004;101:3533–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol 2006;7:131–137. [DOI] [PubMed] [Google Scholar]
- 41. Alexopoulou L, et al Recognition of double‐stranded RNA and activation of NF‐kappaB by Toll‐like receptor 3. Nature 2001;413:732–738. [DOI] [PubMed] [Google Scholar]
- 42. Hemmi H, et al Small anti‐viral compounds activate immune cells via the TLR7 MyD88‐dependent signaling pathway. Nat Immunol 2002;3:196–200. [DOI] [PubMed] [Google Scholar]
- 43. Jurk M, et al Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R‐848. Nat Immunol 2002;3:499. [DOI] [PubMed] [Google Scholar]
- 44. Heil F, et al The Toll‐like receptor 7 (TLR7)‐specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol 2003;33: 2987–2997. [DOI] [PubMed] [Google Scholar]
- 45. Diebold SS, et al Innate antiviral responses by means of TLR7‐mediated recognition of single‐stranded RNA. Science 2004;303: 1529–1531. [DOI] [PubMed] [Google Scholar]
- 46. Heil F, et al Species‐specific recognition of single‐stranded RNA via toll‐like receptor 7 and 8. Science 2004;303: 1526–1529. [DOI] [PubMed] [Google Scholar]
- 47. Lund JM, et al Recognition of single‐stranded RNA viruses by Toll‐like receptor 7. Proc Natl Acad Sci USA 2004;101: 5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bauer S, et al Human TLR9 confers responsiveness to bacterial DNA via species‐specific CpG motif recognition. Proc Natl Acad Sci USA 2001;98: 9237–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hemmi H, et al A Toll‐like receptor recognizes bacterial DNA. Nature 2000;408:740–745. [DOI] [PubMed] [Google Scholar]
- 50. Coccia EM, et al Viral infection and Toll‐like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte‐derived dendritic cells. Eur J Immunol 2004;34:796–805. [DOI] [PubMed] [Google Scholar]
- 51. Kato A, et al Lipopolysaccharide‐binding protein critically regulates lipopolysaccharide‐induced IFN‐beta signaling pathway in human monocytes. J Immunol 2004;172:6185–6194. [DOI] [PubMed] [Google Scholar]
- 52. Toshchakov V, et al TLR4, but not TLR2, mediates IFN‐beta‐induced STAT1alpha/beta‐dependent gene expression in macrophages. Nat Immunol 2002;3: 392–398. [DOI] [PubMed] [Google Scholar]
- 53. Smith PL, Lombardi G, Foster GR. Type I interferons and the innate immune response – more than just antiviral cytokines. Mol Immunol 2005;42:869–877. [DOI] [PubMed] [Google Scholar]
- 54. Meylan E, Tschopp J. Toll‐like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol Cell 2006;22: 561–569. [DOI] [PubMed] [Google Scholar]
- 55. Qureshi ST, Medzhitov R. Toll‐like receptors and their role in experimental models of microbial infection. Genes Immun 2003;4:87–94. [DOI] [PubMed] [Google Scholar]
- 56. Beutler B, et al Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol 2006;24:353–389. [DOI] [PubMed] [Google Scholar]
- 57. Casanova JL, Abel L. The human model: a genetic dissection of immunity to infection in natural conditions. Nat Rev Immunol 2004;4:55–66. [DOI] [PubMed] [Google Scholar]
- 58. Casanova JL, Abel L. Inborn errors of immunity to infection: the rule rather than the exception. J Exp Med 2005;202: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abel L, Casanova JL. Human genetics of infectious diseases: fundamental insights from clinical studies. Semin Immunol 2006;18:327–329. [DOI] [PubMed] [Google Scholar]
- 60. Casanova JL, Abel L. Human genetics of infectious diseases: a unified theory. EMBO J 2007;26:915–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quintana‐Murci L, Alcaïs A, Abel L, Casanova JL. Immunology in natural: clinical, epidemiological, and evolutionary genetics of infectious diseases. Nat Immunol 2007, in press. [DOI] [PubMed] [Google Scholar]
- 62. Casanova JL, Abel L. Primary immunodeficiencies: a field in infancy. Science 2007; 317:617–619. [DOI] [PubMed] [Google Scholar]
- 63. Doffinger R, et al X‐linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF‐kappaB signaling. Nat Genet 2001; 27:277–285. [DOI] [PubMed] [Google Scholar]
- 64. Jain A, et al Specific missense mutations in NEMO result in hyper‐IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol 2001;2:223–228. [DOI] [PubMed] [Google Scholar]
- 65. Mansour S, et al Incontinentia pigmenti in a surviving male is accompanied by hypohidrotic ectodermal dysplasia and recurrent infection. Am J Med Genet 2001;99:172–177. [DOI] [PubMed] [Google Scholar]
- 66. Courtois G, et al A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest 2003;112:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Picard C, et al Pyogenic bacterial infections in humans with IRAK‐4 deficiency. Science 2003;299:2076–2079. [DOI] [PubMed] [Google Scholar]
- 68. Casrouge A, et al Herpes simplex virus encephalitis in human UNC‐93B deficiency. Science 2006;314:308–312. [DOI] [PubMed] [Google Scholar]
- 69. Zhang SY, et al TLR3 deficiency in otherwise healthy patients with herpes simplex encephalitis. Science 2007; 317:1522–1527. [DOI] [PubMed] [Google Scholar]
- 70. Li S, et al IRAK‐4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci USA 2002; 99:5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Suzuki N, et al Severe impairment of interleukin‐1 and Toll‐like receptor signalling in mice lacking IRAK‐4. Nature 2002;416:750–756. [DOI] [PubMed] [Google Scholar]
- 72. Suzuki N, Saito T. IRAK‐4 – a shared NF‐kappaB activator in innate and acquired immunity. Trends Immunol 2006;27: 566–572. [DOI] [PubMed] [Google Scholar]
- 73. Lasker MV, Gajjar MM, Nair SK. Cutting edge: molecular structure of the IL-1R-associated kinase-4 death domain and its implications for TLR signaling. J Immunol 2005;175:4175–4179. [DOI] [PubMed] [Google Scholar]
- 74. Kuglstatter A, et al Cutting edge: IL-1 receptor-associated kinase 4 structures reveal novel features and multiple conformations. J Immunol 2007; 178:2641–2645. [DOI] [PubMed] [Google Scholar]
- 75. Kim TW, et al A critical role for IRAK4 kinase activity in Toll‐like receptor‐mediated innate immunity. J Exp Med 2007;204: 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Uematsu S, et al Interleukin‐1 receptor‐associated kinase‐1 plays an essential role for Toll‐like receptor (TLR)7‐ and TLR9‐mediated interferon‐{alpha} induction. J Exp Med 2005;201:915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mansur DS, et al Lethal encephalitis in myeloid differentiation factor 88‐deficient mice infected with herpes simplex virus 1. Am J Pathol 2005;166:1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Krug A, et al Herpes simplex virus type 1 activates murine natural interferon‐producing cells through toll‐like receptor 9. Blood 2004;103:1433–1437. [DOI] [PubMed] [Google Scholar]
- 79. Delale T, et al MyD88‐dependent and ‐independent murine cytomegalovirus sensing for IFN‐alpha release and initiation of immune responses in vivo. J Immunol 2005;175:6723–6732. [DOI] [PubMed] [Google Scholar]
- 80. Zhou S, et al MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol 2005;35:822–830. [DOI] [PubMed] [Google Scholar]
- 81. Honda K, et al IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 2005;434:772–777. [DOI] [PubMed] [Google Scholar]
- 82. Zhou S, et al Role of MyD88 in route‐dependent susceptibility to vesicular stomatitis virus infection. J Immunol 2007;178:5173–5181. [DOI] [PubMed] [Google Scholar]
- 83. Rudd BD, et al MyD88‐mediated instructive signals in dendritic cells regulate pulmonary immune responses during respiratory virus infection. J Immunol 2007;178: 5820–5827. [DOI] [PubMed] [Google Scholar]
- 84. Fuse K, et al Myeloid differentiation factor‐88 plays a crucial role in the pathogenesis of Coxsackievirus B3‐induced myocarditis and influences type I interferon production. Circulation 2005;112:2276–2285. [DOI] [PubMed] [Google Scholar]
- 85. Ku CL, et al Human IRAK‐4 deficiency: a selective predisposition to life-threatening pyogenic bacterial infections during childhood reveals an otherwise redundant role for TLRs in protective immunity. J Exp Med 2007, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yang K, et al Human TLR‐7‐, ‐8‐, and ‐9‐mediated induction of IFN‐alpha/beta and ‐lambda Is IRAK‐4 dependent and redundant for protective immunity to viruses. Immunity 2005;23:465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sun Y, Leaman CW. Ectopic expression of toll‐like receptor‐3 (TLR‐3) overcomes the double‐stranded RNA (dsRNA) signaling defects of P2.1 cells. J Interferon Cytokine Res 2004;24:350–361. [DOI] [PubMed] [Google Scholar]
- 88. Greenwald IS, Horvitz HR. unc‐93(e1500): a behavioral mutant of Caenorhabditis elegans that defines a gene with a wild-type null phenotype. Genetics 1980;96:147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Levin JZ, Horvitz HR. The Caenorhabditis elegans unc‐93 gene encodes a putative transmembrane protein that regulates muscle contraction. J Cell Biol 1992; 117:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. De La Cruz IP, et al sup‐9, sup‐10, and unc‐93 may encode components of a two‐pore K+ channel that coordinates muscle contraction in Caenorhabditis elegans . J Neurosci 2003;23:9133–9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kashuba VI, et al hUNC93B1: a novel human gene representing a new gene family and encoding an unc-93-like protein. Gene 2002;283:209–217. [DOI] [PubMed] [Google Scholar]
- 92. Tabeta K, et al The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll‐like receptors 3, 7 and 9. Nat Immunol 2006;7:156–164. [DOI] [PubMed] [Google Scholar]
- 93. Brinkmann MM, et al The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol 2007;177:265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kuchtey J, et al Enhancement of dendritic cell antigen cross‐presentation by CpG DNA involves type I IFN and stabilization of class I MHC mRNA. J Immunol 2005;175: 2244–2251. [DOI] [PubMed] [Google Scholar]
- 95. Whitley RJ. Herpes simplex encephalitis: adolescents and adults. Antiviral Res 2006;71:141–148. [DOI] [PubMed] [Google Scholar]
- 96. Dupuis S, et al Impaired response to interferon‐alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 2003;33:388–391. [DOI] [PubMed] [Google Scholar]
- 97. Chapgier A, et al Human complete Stat‐1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol 2006;176:5078–5083. [DOI] [PubMed] [Google Scholar]
- 98. Chapgier A, et al Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet 2006;2:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Minegishi Y, et al Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 2006;25:745–755. [DOI] [PubMed] [Google Scholar]
- 100. Heinz S, et al Species‐specific regulation of Toll‐like receptor 3 genes in men and mice. J Biol Chem 2003;278:21502–21509. [DOI] [PubMed] [Google Scholar]
- 101. Baoprasertkul P, et al Toll‐like receptor 3 and TICAM genes in catfish: species-specific expression profiles following infection with Edwardsiella ictaluri. Immunogenetics 2006;58:817–830. [DOI] [PubMed] [Google Scholar]
- 102. Dhara A, et al Molecular characterization of coding sequences and analysis of Toll‐like receptor 3 mRNA expression in water buffalo (Bubalus bubalis) and nilgai (Boselaphus tragocamelus). Immunogenetics 2007;59:69–76. [DOI] [PubMed] [Google Scholar]
- 103. Bell JK, et al Leucine‐rich repeats and pathogen recognition in Toll‐like receptors. Trends Immunol 2003;24:528–533. [DOI] [PubMed] [Google Scholar]
- 104. Choe J, Kelker MS, Wilson IA. Crystal structure of human Toll‐like receptor 3 (TLR3) ectodomain. Science 2005; 309:581–585. [DOI] [PubMed] [Google Scholar]
- 105. Bell JK, et al The molecular structure of the Toll‐like receptor 3 ligand‐binding domain. Proc Natl Acad Sci USA 2005;102: 10976–10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bell JK, et al The dsRNA binding site of human Toll‐like receptor 3. Proc Natl Acad Sci USA 2006;103:8792–8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. De Bouteiller O, et al Recognition of double‐stranded RNA by human toll‐like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem 2005;280: 38133–38145. [DOI] [PubMed] [Google Scholar]
- 108. Ranjith‐Kumar CT, et al Biochemical and functional analyses of the human Toll‐like receptor 3 ectodomain. J Biol Chem 2007;282:7668–7678. [DOI] [PubMed] [Google Scholar]
- 109. Takada E, et al C‐terminal LRRs of human Toll‐like receptor 3 control receptor dimerization and signal transmission. Mol Immunol 2007;44:3633–3640. [DOI] [PubMed] [Google Scholar]
- 110. Kadowaki N, et al Subsets of human dendritic cell precursors express different toll‐like receptors and respond to different microbial antigens. J Exp Med 2001;194: 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Matsumoto M, et al Establishment of a monoclonal antibody against human Toll‐like receptor 3 that blocks double‐stranded RNA‐mediated signaling. Biochem Biophys Res Commun 2002;293: 1364–1369. [DOI] [PubMed] [Google Scholar]
- 112. Kumar MV, et al Innate immunity in the retina: Toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. J Neuroimmunol 2004;153:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kumar A, Zhang J, Yu FS. Toll‐like receptor 3 agonist poly(I:C)‐induced antiviral response in human corneal epithelial cells. Immunology 2006;117:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tohyama M, et al dsRNA‐mediated innate immunity of epidermal keratinocytes. Biochem Biophys Res Commun 2005;335: 505–511. [DOI] [PubMed] [Google Scholar]
- 115. Lebre MC, et al Human keratinocytes express functional Toll‐like receptor 3, 4, 5, and 9. J Invest Dermatol 2007;127: 331–341. [DOI] [PubMed] [Google Scholar]
- 116. Bsibsi M, et al Broad expression of Toll‐like receptors in the human central nervous system. J Neuropathol Exp Neurol 2002;61:1013–1021. [DOI] [PubMed] [Google Scholar]
- 117. Jackson AC, Rossiter JP, Lafon M. Expression of Toll‐like receptor 3 in the human cerebellar cortex in rabies, herpes simplex encephalitis, and other neurological diseases. J Neurovirol 2006;12:229–234. [DOI] [PubMed] [Google Scholar]
- 118. Prehaud C, et al Virus infection switches TLR‐3‐positive human neurons to become strong producers of beta interferon. J Virol 2005;79:12893–12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Jack CS, et al TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol 2005;175: 4320–4330. [DOI] [PubMed] [Google Scholar]
- 120. McKimmie CS, et al Viruses selectively upregulate Toll‐like receptors in the central nervous system. Biochem Biophys Res Commun 2005;336:925–933. [DOI] [PubMed] [Google Scholar]
- 121. Bsibsi M, et al Toll‐like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 2006;53: 688–695. [DOI] [PubMed] [Google Scholar]
- 122. Iwasaki A, Medzhitov R. Toll‐like receptor control of the adaptive immune responses. Nat Immunol 2004;5:987–995. [DOI] [PubMed] [Google Scholar]
- 123. Hardarson HS, et al Toll‐like receptor 3 is an essential component of the innate stress response in virus‐induced cardiac injury. Am J Physiol Heart Circ Physiol 2007;292:H251–H258. [DOI] [PubMed] [Google Scholar]
- 124. Rudd BD, et al Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J Immunol 2006;176:1937–1942. [DOI] [PubMed] [Google Scholar]
- 125. Le Goffic R, et al Detrimental contribution of the Toll‐like receptor (TLR)3 to influenza A virus‐induced acute pneumonia. PLoS Pathogen 2006;2:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gowen B, et al TLR3 deletion limits mortality and disease severity due to Phlebovirus infection. J Immunol 2006;177:6301–6307. [DOI] [PubMed] [Google Scholar]
- 127. Wang T, et al Toll‐like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 2004;10: 1366–1373. [DOI] [PubMed] [Google Scholar]
- 128. Edelmann KH, et al Does Toll‐like receptor 3 play a biological role in virus infections? Virology 2004;322:231–238. [DOI] [PubMed] [Google Scholar]
- 129. Yoneyama M, et al The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 2004;5:730–737. [DOI] [PubMed] [Google Scholar]
- 130. Kato H, et al Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature 2006;441:101–105. [DOI] [PubMed] [Google Scholar]
- 131. Pichlmair A, et al RIG‐I‐mediated antiviral responses to single‐stranded RNA bearing 5′‐phosphates. Science 2006;314: 997–1001. [DOI] [PubMed] [Google Scholar]
- 132. Gitlin L, et al Essential role of MDA‐5 in type I IFN responses to polyriboinosinic: polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA 2006;103:8459–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kato H, et al Cell type‐specific involvement of RIG‐I in antiviral response. Immunity 2005;23:19–28. [DOI] [PubMed] [Google Scholar]
- 134. Siren J, et al Retinoic acid inducible gene‐I and MDA‐5 are involved in influenza A virus‐induced expression of antiviral cytokines. Microbes Infect 2006;8: 2013–2020. [DOI] [PubMed] [Google Scholar]
- 135. Ishii KJ, et al A Toll‐like receptor‐independent antiviral response induced by double‐stranded B‐form DNA. Nat Immunol 2006;7:40–48. [DOI] [PubMed] [Google Scholar]
- 136. Okabe Y, et al Toll‐like receptor‐independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med 2005;202:1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3‐dependent innate immune response. Immunity 2006;24:93–103. [DOI] [PubMed] [Google Scholar]
- 138. Katze MG, He Y, Gale M Jr. Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2002;2: 675–687. [DOI] [PubMed] [Google Scholar]