

Summary
Zoonotic transmission of Ebola virus (EBOV) to humans causes a severe haemorrhagic fever in afflicted individuals with high case‐fatality rates. Neither vaccines nor therapeutics are at present available to combat EBOV infection, making the virus a potential threat to public health. To devise antiviral strategies, it is important to understand which components of the immune system could be effective against EBOV infection. The interferon (IFN) system constitutes a key innate defence against viral infections and prevents development of lethal disease in mice infected with EBOV strains not adapted to this host. Recent research revealed that expression of the host cell IFN‐inducible transmembrane proteins 1–3 (IFITM1–3) and tetherin is induced by IFN and restricts EBOV infection, at least in cell culture model systems. IFITMs, tetherin and other effector molecules of the IFN system could thus pose a potent barrier against EBOV spread in humans. However, EBOV interferes with signalling events required for human cells to express these proteins. Here, we will review the strategies employed by EBOV to fight the IFN system, and we will discuss how IFITM proteins and tetherin inhibit EBOV infection.
Keywords: Ebola virus, interferon system, tetherin, IFITM, VP24, VP35
Impacts
-
•
Ebola virus infection causes severe haemorrhagic fever in humans. The interferon system can restrict spread and pathogenesis of Ebola virus in mice. Understanding Ebola virus interactions with the human interferon system might allow novel approaches to treatment and prevention.
-
•
In human cells, the Ebola virus proteins VP24 and VP35 inhibit signalling cascades of the interferon system and might thus promote viral evasion of innate immunity.
-
•
IFITM proteins and tetherin are recently discovered antiviral effector molecules of the interferon system, which can inhibit Ebola virus entry and release, respectively, from human cells and might modulate viral spread in patients.
Introduction
The Ebola virus (EBOV) is an enveloped, negative‐stranded RNA virus of the filovirus family. Infection of humans with EBOV causes Ebola haemorrhagic fever. At present, neither antivirals nor vaccines are available to combat this lethal disease, and EBOV is classified as a category A priority pathogen (NIAID, 2011). Four EBOV species have been defined (Kuhn, 2008), Zaire ebolavirus (ZEBOV), Sudan ebolavirus (SEBOV), Côte d’Ivoire ebolavirus (CIEBOV) and Reston ebolavirus (REBOV), and a fifth species has been proposed, Bundibugyo ebolavirus (BEBOV) (Towner et al., 2008). African fruit bats are a natural reservoir of the second filoviral genus, Marburg virus (MARV), and have also been proposed as a natural reservoir of EBOV (Leroy et al., 2005). African fruit bats may transmit the virus to humans either directly or via an intermediate host (Groseth et al., 2007). Outbreaks of ZEBOV, SEBOV, CIEBOV and BEBOV have been recorded in Africa and were associated with case‐fatality rates of up to 90% in larger outbreaks. REBOV has been detected in swine in the Philippines and is believed to be apathogenic for humans with an intact immune system (Barrette et al., 2009; Hartman et al., 2010). The determinants accounting for the differential pathogenicity of the different EBOV species are poorly understood.
The EBOV genome encodes three non‐structural (sGP, ssGP, Δ‐peptide, as discussed later) and seven structural proteins (Fig. 1). The N protein (NP), the L protein, viral protein 30 (VP30) and VP35 are associated with the viral RNA, forming a ribonucleoprotein (RNP) complex. The L protein has a polymerase function and is essential for genome replication and transcription, and these processes are regulated by VP30 and VP35 (Dolnik et al., 2008). The minor matrix protein, VP24, contributes to nucleocapsid formation, while the major matrix protein, VP40, facilitates budding of progeny particles from infected cells (Huang et al., 2002; Hartlieb and Weissenhorn, 2006; Dolnik et al., 2008) (Fig. 1). The structural glycoprotein, GP1,2, mediates binding and infectious entry into host cells (Kawaoka, 2005).
Figure 1.
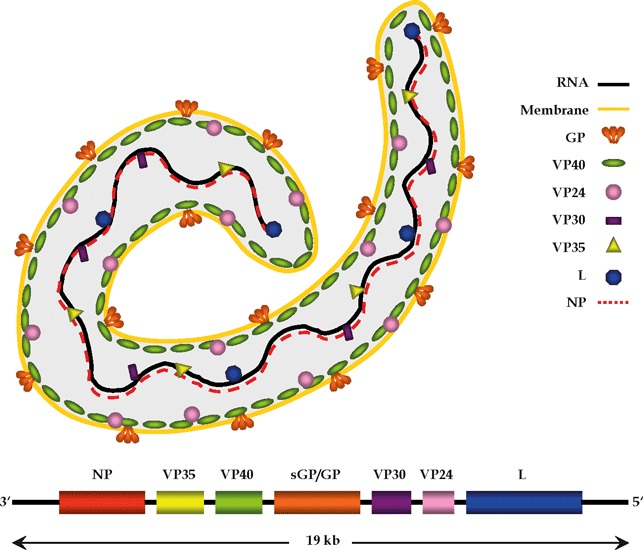
Structural organization of the Ebola virus genome and virion. RNA, ribonucleic acid; GP, glycoprotein; sGP, soluble glycoprotein; VP, viral protein; L, RNA‐dependent RNA polymerase; NP, nucleoprotein.
Innate defences of the host are crucial to successfully fight EBOV and other acute infections. The interferon (IFN) system is an integral part of the innate immunity against viral infections. Sensor molecules of the IFN system recognize viral components and induce signalling that triggers expression of IFNs (Baum and García‐Sastre, 2011). Subsequent binding of IFN to IFN receptors on uninfected cells activates signal transducer and activator of transcription (STAT)‐dependent signalling, which commandeers the cell to express IFN‐stimulated genes (ISGs), which can function as antiviral effectors molecules (Schindler et al., 2007; Sadler and Williams, 2008). Ebola virus strains lethal in humans were found to be unable to produce fatal disease in adult mice. However, when essential components of the IFN system were inactivated in mice, fatal disease was observed (Bray, 2001). Similarly, adaptation of EBOV to efficient replication in adult mice (Bray et al., 1998) resulted in the generation of viruses with mutations allowing efficient interference with components of the murine IFN system (Ebihara et al., 2006). Thus, the IFN system is generally capable of restricting filovirus spread and pathogenesis. However, several EBOV proteins are well adapted to block processes essential for the establishment of a vigorous IFN response in human cells, potentially explaining why the IFN system frequently fails to protect humans from lethal EBOV infection, as discussed below.
The molecular pathways leading to expression of IFN‐induced antiviral effector molecules, and thus to the transition of cells into an antiviral state, are well characterized (Baum and García‐Sastre, 2010). However, the antiviral effector molecules induced by IFN and the molecular mechanisms underlying their antiviral action are incompletely understood. Recent, groundbreaking studies attempted to close this gap (Schoggins et al., 2011) and identified novel ISGs, among them the tetherin and IFN‐induced transmembrane proteins (IFITMs) (Neil et al., 2008; Van Damme et al., 2008; Brass et al., 2009). Tetherin exhibits an unusual topology and restricts release of several enveloped viruses and filovirus‐like particles from infected cells, while IFITM proteins inhibit infection by filoviruses and other enveloped viruses at the stage of viral entry, as discussed below. In the present review, we will summarize current knowledge on EBOV interference with the IFN system. In addition, we will discuss how tetherin and IFITMs block filovirus infection.
The Interferon System
The interferon system constitutes a major innate defence against infections by viruses and other pathogens. The components and signalling pathways of the IFN system have been described in several recent reviews (García‐Sastre and Biron, 2006; Baum and García‐Sastre, 2010; Liu et al., 2011) and are only briefly summarized here. Three classes of IFNs have been defined according to the receptors bound by these cytokines. Type I IFN, including IFNα and IFNβ, are produced by many cell types as a direct result of viral infection. IFNγ, the only type II IFN, is generated by activated T cells and NK cells. Type III IFNs, which include IFNλ1–3, are incompletely characterized, but are believed to regulate the antiviral response. The IFN system integrates two major sensor and signalling networks. In cells exposed to viruses, receptors for pathogen‐associated molecular patterns sense the presence of the invading pathogens. These receptors are located in the cytoplasm, like retinoic acid‐inducible gene I (RIG‐I) and melanoma differentiation‐associated gene 5 (MDA‐5), or in the extracytoplasmic space, like Toll‐like receptor (TLR)‐3 and TLR‐7/8/9, and, upon pathogen recognition, induce IFN regulatory factor (IRF)‐3‐ and IRF‐7‐dependent signalling cascades that lead to the expression of type I IFNs (Fig. 2). Secreted type I IFNs then bind to cells expressing the type I IFN receptor, which consists of two subunits, IFNα receptor 1 (IFNAR1) and IFNAR2. Ligand binding to IFNAR1 and IFNAR2 triggers receptor dimerization and signalling. STAT1 and STAT2 are integral components of the signalling pathway induced by IFNARs (Fig. 2). Homodimers of STAT1 bind to IFNγ‐activated sites (GAS), while STAT1/STAT2 heterodimers recognize IFN‐stimulated response elements, resulting in the transcription of ISGs, many of which have antiviral activity, like the well‐characterized myxovirus resistance guanosine triphosphatases (Mx GTPases) (Haller et al., 2007; Sadler and Williams, 2008). However, the full spectrum of ISGs has only been recently characterized (Schoggins et al., 2011), and novel ISGs that target discrete steps in the viral life cycle, like IFITMs and tetherin, have been identified, as discussed below.
Figure 2.
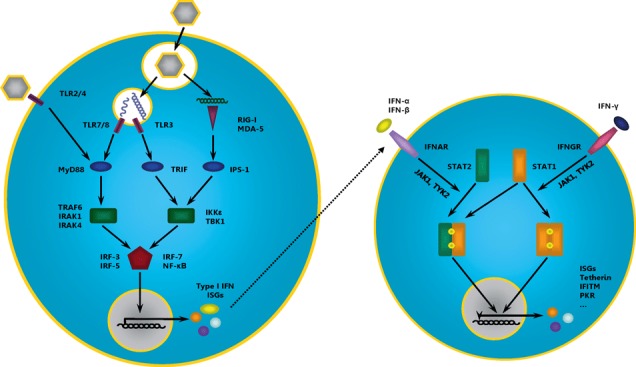
Induction of type I IFNs and ISG expression. Recognition of viral pathogen‐associated molecular patterns (PAMPs) by PRRs (purple) in infected cells initiates a signalling cascade including adaptor molecules (blue) and kinases (green), which activate transcription factors (red) that induce the expression of type I IFNs and ISGs (left panel). Binding of IFN‐α/β and IFN‐γ to their receptors induces phosphorylation (by JAK1/TYK2), dimerization and nuclear translocation of STAT transcription factors, which induce the expression of several ISGs such as tetherin, IFITM, PKR and others (right panel). IFN, interferon; TLR, Toll‐like receptor; RIG‐I, retinoic acid‐inducible gene I; MDA‐5, melanoma differentiation‐associated gene 5; MyD88, myeloid differentiation primary response protein 88; TRIF, TIR domain‐containing adaptor‐inducing IFN‐β; IPS‐1, IFN‐β promoter stimulator 1; TRAF6, tumour necrosis factor receptor‐associated factor 6; IRAK, interleukin‐1 receptor‐associated kinase; IKKε, IκB kinase ε; TBK1, tank‐binding kinase 1; IRF, IFN regulatory factor; NF‐κB, nuclear factor κ light chain enhancer of activated B‐cells; ISG, IFN‐stimulated gene; IFNAR, IFN‐α receptor; IFNGR, IFN‐γ receptor; JAK1, Janus‐activated kinase 1; PRR, pathogen recognition receptor; TYK2, tyrosine kinase 2; STAT, signal transducer and activators of transcription.
How EBOV Antagonizes the Interferon System
VP24
VP24 is the smallest of the seven EBOV‐encoded structural proteins and constitutes the minor matrix protein relative to the major matrix protein VP40 (Han et al., 2003). It is required for the assembly of fully functional nucleocapsids (Huang et al., 2002; Hoenen et al., 2006) and contributes to the budding of virus‐like particles (VLPs) (Han et al., 2003; Licata et al., 2004). Furthermore, VP24 can shut down the host’s IFN‐α/β and IFN‐γ response to viral infection (Reid et al., 2006). For this, VP24 inhibits the nuclear translocation of the transcription factor STAT1 (Reid et al., 2006) (Fig. 3), a key component of the IFN‐induced signalling pathway controlling the expression of ISGs, as discussed below.
Figure 3.
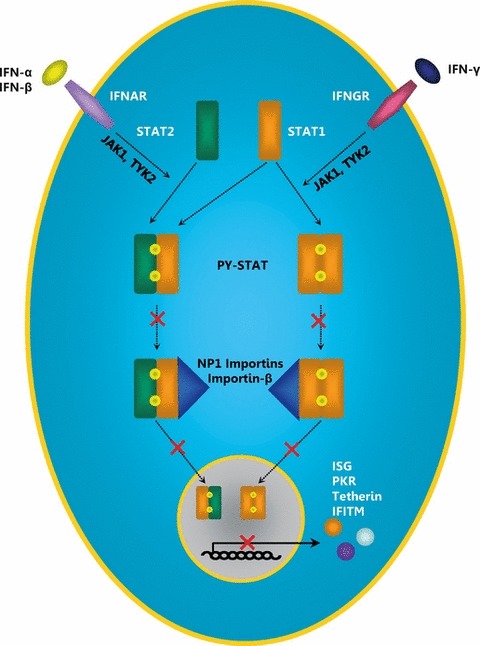
Inhibition of type I and type II IFN signalling by Ebola virus VP24. Upon binding to their receptors, IFNs induce the homo‐ or heterodimerization of STAT1 and STAT2, leading to autophosphorylation of these transcription factors. Exposition of a nuclear localization signal in the phosphorylated STAT proteins (PY‐STAT) allows binding of NP‐1 importins and importin‐β and subsequent nuclear translocation that results in the expression of ISGs. VP24 interferes with the binding of PY‐STAT to NP‐1 importins. PY‐STAT cannot be imported into the nucleus, and the expression of ISGs is suppressed. IFN, interferon; ISG, IFN‐stimulated gene; IFNAR, IFN‐α receptor; IFNGR, IFN‐γ receptor; JAK1, Janus‐activated kinase 1; TYK2, tyrosine kinase 2; STAT, signal transducer and activators of transcription; PY‐STAT, phosphorylated STAT; ISG, IFN‐stimulated gene; PKR, dsRNA‐dependent protein kinase; IFITM, IFN‐induced transmembrane protein.
VP24 inhibits nuclear translocation of STAT1
Upon activation, STAT1 is tyrosine‐phosphorylated (PY‐STAT1) and either heterodimerizes with PY‐STAT2 for type I IFN signalling or homodimerizes with PY‐STAT1 for type II IFN signalling. The dimerization leads to the exposition of a nuclear localization signal (NLS) in STAT1, which is recognized by the nuclear import adaptor protein importin α, specifically importin α5, α6 and α7 (karyopherin α1, α5, α6; NP‐1 subfamily of importins). Importin α is then bound by the nuclear import factor importin β (Sekimoto et al., 1997), which facilitates import of STAT dimers into the nucleus (Fig. 3). However, in the presence of VP24, which like STAT1 binds to importins α5, α6 and α7 (Reid et al., 2006, 2007), the recognition of the PY‐STAT1 NLS by these importins is disrupted, and PY‐STAT1 is not imported into the nucleus. Blockade of nuclear transport of PY‐STAT1 is not because of a global inhibition of nuclear import, as VP24 does not bind to importin α1 (karyopherin α2; RchI subfamily), α3 or α4 (karyopherin α4, α3; Qip1 subfamily) (Reid et al., 2007) and does not affect nuclear import mediated by these factors.
Mutational analyses revealed that VP24 binds to amino acids 458–504 in importin α5, which constitute armadillo repeat (ARM) 10 and comprise part of the binding site for PY‐STAT1 (Reid et al., 2006, 2007). Predictions on VP24 structure offer insights into the mechanisms potentially underlying VP24 inhibition of importin α. (Lee et al., 2009). Thus, it was suggested that VP24, like importin α, importin β and exportins (nuclear export factors important for recycling of importin α to the cytoplasm), belongs to the ARM family of proteins. Furthermore, it was posited that binding of VP24 to importin α mimics that of exportin, which also targets ARM repeat 10 (Lee et al., 2009). As a consequence, VP24 may block release of the autoinhibitory NLS of importin α, and this would inhibit binding of other proteins to the NLS binding site of importin α, including PY‐STAT1. Alternatively, targeting the ARM10 repeat of importin α by VP24 may block binding of PY‐STAT1 because of competitive inhibition (Lee et al., 2009). In VP24, the amino acid residues 42 and 142–146 were found to be crucial for the inhibition of IFN‐β‐induced signalling and PY‐STAT accumulation in the nucleus (Mateo et al., 2010). The lack of inhibition of IFN‐β‐induced signalling by a VP24 mutant with amino acid exchanges at these positions correlated with absence of importin α5 binding, highlighting that binding of VP24 to importin α5 is essential for IFN antagonism (Mateo et al., 2010).
The IFN system can protect immune‐competent mice from lethal EBOV infection (Bray, 2001; Mahanty et al., 2003). Adaptation of ZEBOV to lethal infection of mice was associated with mutations in VP24 and NP (Ebihara et al., 2006). However, both wild‐type VP24 and VP24 of the mouse‐adapted (MA) strain were able to bind to human and mouse NP‐1 importins and to disrupt the interaction with PY‐STAT1 (Reid et al., 2007). Similar findings were documented for VP24 of REBOV, which is believed to be non‐pathogenic for humans, and it was shown that ZEBOV, REBOV and MA VP24 can suppress IFN‐β‐induced gene expression (Reid et al., 2007). Thus, alterations in VP24 interference with the IFN response might not account for the acquisition of virulence of MA ZEBOV in mice and for the lack of virulence of REBOV in humans, respectively.
VP35
The EBOV‐encoded protein VP35 fulfils several functions important for viral amplification. As essential polymerase cofactor, VP35 is involved in the formation of the EBOV RNP complex and therefore crucial for transcription and viral replication (Mühlberger et al., 1999). Furthermore, it is required for nucleocapsid assembly (Huang et al., 2002). Consequently, interference with expression of VP35 attenuates viral growth and virulence (Enterlein et al., 2006; Hartman et al., 2008a; Prins et al., 2010b). Additionally, VP35 blocks multiple steps of the innate antiviral defence (Fig. 4), such as the signalling pathways leading to the expression of type I IFNs and type I IFN‐induced genes, double‐stranded (ds) RNA‐dependent protein kinase (PKR) translation inhibition and RNA silencing (Basler et al., 2003; Cárdenas et al., 2006; Feng et al., 2007; Haasnoot et al., 2007). The key role of VP35 in the defence against innate immunity is exemplified by studies demonstrating that VP35 mutated viruses are unable to block the IFN‐dependent induction of antiviral factors (Cárdenas et al., 2006; Hartman et al., 2008a, 2008b). Moreover, VP35 impairs the maturation of dendritic cells and thus prevents the establishment of an adaptive immune response (Jin et al., 2010), an important feature of EBOV haemorrhagic fever pathogenesis (Hartman et al., 2008a).
Figure 4.
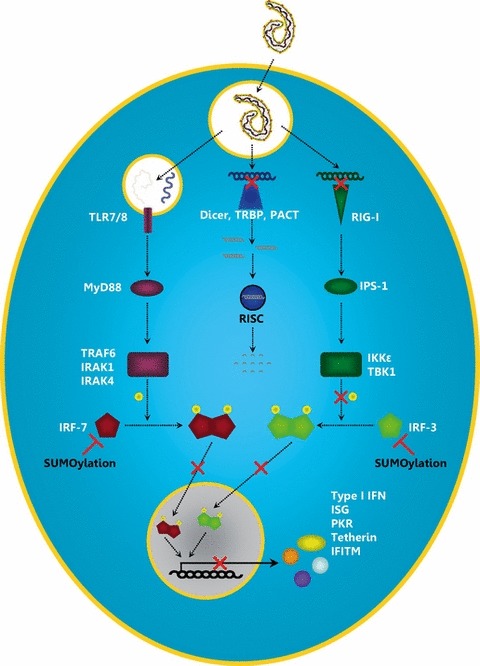
Interference of Ebola virus VP35 with the expression of IFN and ISGs. Transcription of the EBOV genome results in the production of dsRNA intermediates, which are recognized by the cytoplasmic pathogen recognition receptor (PRR) RIG‐I. Single‐stranded RNA is recognized by the endosomal PRRs TLR‐7 and TLR‐8. Activation of these PRRs induces signalling cascades leading to the expression of type I IFNs. In addition, dsRNA is recognized by the RNA interference machinery that facilitates RNA degradation and thereby suppresses the expression of viral genes. VP35 is able to block several of the above‐described processes. Because of its dsRNA‐binding domain, it is able to sequester dsRNA from recognition by RIG‐I and to protect it from degradation through the RNA‐induced silencing complex. Furthermore, it inhibits the phosphorylation of IRF‐3 and therefore IRF‐3 dimerization, translocation into the nucleus and the expression of IFN‐induced genes. In addition, VP35 is able to induce SUMOylation of IRF‐7 and IRF‐3, which interferes with dimerization and nuclear translocation. IFN, interferon; TLR, Toll‐like receptor; TRBP, HIV‐1 trans‐activation response RNA‐binding protein; PACT, protein activator of PKR; RIG‐I, retinoic acid‐inducible gene I; MyD88, myeloid differentiation primary response protein 88; TRIF, TIR domain‐containing adaptor‐inducing IFN‐β; IPS‐1, IFN‐β promoter stimulator 1; TRAF6, tumour necrosis factor receptor‐associated factor 6; IRAK, interleukin‐1 receptor‐associated kinase; IKKε, IκB kinase ε; TBK1, tank‐binding kinase 1; IRF, IFN regulatory factor; ISG, IFN‐stimulated gene; PKR, dsRNA‐dependent protein kinase; IFITM, IFN‐induced transmembrane protein.
VP35 inhibits phosphorylation of IRF‐3
The ability of VP35 to antagonize the type I IFN response was identified by Basler et al. (2000) who showed that VP35 can functionally replace the influenza A virus (FLUAV) non‐structural protein 1 (NS1) protein, a viral type I IFN antagonist. VP35 was found to inhibit the virus‐induced activation of type I IFN promoters by targeting the transcription factor IRF‐3. In contrast, activation of IFN promoters through exogenous IFN was not modulated by VP35 (Basler et al., 2003). Upon activation by viral infection, IRF‐3 is phosphorylated by cellular kinases such as TANK‐binding kinase 1 (TBK‐1) and IκB kinase epsilon (IKKε) (Prins et al., 2009). Subsequently, IFR‐3 dimerizes and translocates into the nucleus where it activates transcription of ISGs (Fig. 4). VP35 blocks IRF‐3 phosphorylation and therefore subsequent nuclear accumulation of IRF‐3 (Basler et al., 2003; Cárdenas et al., 2006). For this, VP35 binds to IKKε and TBK‐1 and inhibits substrate binding and kinase activity (Prins et al., 2009).
Mutational studies revealed that a N‐terminal coiled‐coil domain in VP35 mediates homooligomerization, which was found to be essential for IFN antagonism exhibited by the C‐terminus of the protein (Reid et al., 2005). Several amino acid residues (R305, K309, R312, K319, R322) in the central basic patch of the C‐terminal IFN inhibitory domain (IID) of VP35 were shown to be required for inhibition of virus‐induced activation of IFN‐regulated promoters and production of IFN‐β (Cárdenas et al., 2006; Prins et al., 2010b), with R312 being of particular importance (Hartman et al., 2004; Cárdenas et al., 2006). Mutations in the IID attenuate viral growth in cell lines (Hartman et al., 2006) and the C‐terminal basic patch residues are highly conserved between filoviruses, highlighting their importance for the function of VP35 (Hartman et al., 2004; Prins et al., 2010b). Notably, the disruption of the IRF‐3 inhibitory activity of VP35 does not influence the function of VP35 in viral replication and transcription (Hartman et al., 2006), as amino acid residues essential for polymerase cofactor function of VP35 differ from those required for IFN antagonism (Prins et al., 2010a).
Sequences in the C‐terminus of VP35 resemble the dsRNA‐binding domain of FLUAV NS1, which is important for IFN antagonism (Donelan et al., 2003; Hartman et al., 2004). Indeed, VP35 is able to bind dsRNA, but not single‐stranded (ss) RNA or dsDNA and mutations in the central basic patch of IID, which abrogate IFN antagonism were found to be incompatible with dsRNA binding (Cárdenas et al., 2006; Leung et al., 2010; Prins et al., 2010b). The structure of VP35 IID in complex with dsRNA has been determined at the atomic level (Kimberlin et al., 2010; Leung et al., 2010), and the protein was found to display a unique fold compared with known dsRNA‐binding proteins, including that of FLUAV NS1 (Leung et al., 2009). Interestingly, VP35 displays a bimodal dsRNA‐binding strategy. Upon dsRNA recognition, VP35 builds asymmetric dimers; one monomer binds the dsRNA phosphate backbone, whereas the other binds terminal nucleotides of the dsRNA molecule (Kimberlin et al., 2010; Leung et al., 2010). This dsRNA‐binding mode of VP35 dimers seems to mimic that of RIG‐I, a key cellular sensor of dsRNA, and VP35 and RIG‐I might occupy overlapping binding sites on dsRNA (Kimberlin et al., 2010; Leung et al., 2010). These findings suggest that VP35 may sequester dsRNA from recognition by RIG‐I and potentially other dsRNA‐binding molecules such as MDA‐5 or Dicer (Kimberlin et al., 2010; Leung et al., 2010). In addition, differences in the structural organization and dsRNA binding of ZEBOV and REBOV VP35 were identified, which might contribute to the differential pathogenicity of these viruses in humans (Kimberlin et al., 2010; Leung et al., 2010), although it needs to be noted that both viruses are highly pathogenic in macaques.
VP35 promotes SUMOylation of IFR‐7 via PIAS1
IFR‐7 but not IRF‐3 is critical for the production of type I IFNs (Honda et al., 2005), and a recent study shows that VP35 inhibits IRF‐7 by promoting its SUMOylation (Chang et al., 2009). Thus, VP35 forms a complex with IRF‐7 and PIAS1 (the small ubiquitin‐like modifier (SUMO) E3 ligase protein inhibitor of activated STAT) and promotes IRF‐7 SUMOylation via PIAS1 (Chang et al., 2009) (Fig. 4). This study also revealed that VP35 displays an IFN inhibitory activity independent of its ability to recognize dsRNA, and this activity was mapped to the N‐terminus, which is essential for interactions with IRF‐7 and PIAS1.
VP35 inhibits the dsRNA‐dependent protein kinase (PKR)
The interferon‐inducible dsRNA‐dependent protein kinase (PKR) recognizes dsRNA produced in the context of infection by RNA viruses and DNA viruses encoding opposite open reading frames from which overlapping mRNA sequences are generated (George et al., 2009). Subsequently, it phosphorylates the eukaryotic translation initiation factor 2 α (eIF2α), which results in the arrest of protein synthesis from cellular and viral mRNAs (García et al., 2007). In the context of EBOV infection, it was noted that the expression of VP35 and eIF‐2α phosphorylation inversely correlate and that autophosphorylation of PKR is disrupted in the presence of VP35 (Feng et al., 2007). Thus, VP35 inhibits PKR activation (Fig. 5). In fact, ZEBOV was shown to not only block, but to reverse activation of PKR (Schumann et al., 2009). Feng et al. (2007) suggested that VP35 inhibition of PKR might not involve interactions of these proteins and might not depend on dsRNA binding by VP35. Instead, a role of the N‐terminal sequences in VP35 was suggested. Subsequent work by Schumann et al. (2009) indicates that mutations in the IID can be sufficient to relief the block to PKR activation imposed by VP35 and that PRK antagonism by VP35 is therefore functionally separate from dsRNA binding and IRF‐3 inhibition.
Figure 5.
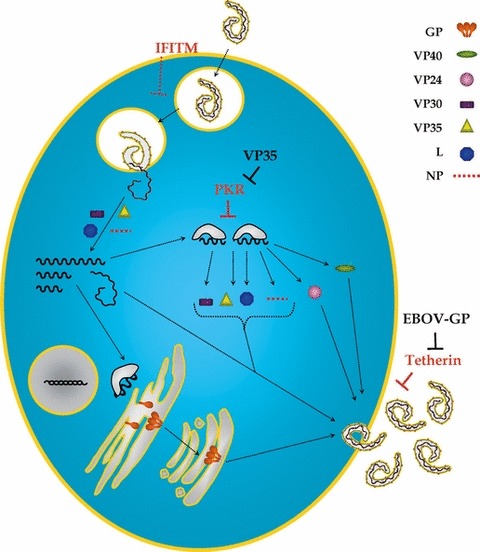
Countermeasures of Ebola virus against IFN‐induced antiviral proteins. Ebola virus infection commences with viral uptake and transport of virions into endosomal compartments, where the EBOV‐GP is activated by cathepsins. Subsequently, GP mediates fusion of the viral envelope with the endosomal membrane, allowing release of the viral genome into the cytosol. Subsequently, the viral genome is transcribed into mRNAs and replicated. Viral proteins are translated and transported to the cellular membrane for assembly of new particles, which eventually bud from the host cell membrane. Virus entry is inhibited by the IFN‐induced IFITM proteins. The exact step that is blocked by IFITMs remains to be defined. The protein kinase R, which senses dsRNA intermediates of viral replication, phosphorylates the translation initiation factor eIF2α, which results in the arrest of translation of viral and cellular mRNAs. The tetherin protein tethers budding EBOV‐like particles to the cell surface and is counteracted by EBOV‐GP. GP, glycoprotein; VP, viral protein; L, RNA‐dependent RNA polymerase; NP, nucleoprotein; IFITM, IFN‐induced transmembrane protein; PKR, dsRNA‐dependent protein kinase; EBOV‐GP, Ebola virus glycoprotein.
VP35 inhibits RNAi by targeting the components of the RNA‐induced silencing complex
RNA interference allows cells to specifically recognize and destroy viral RNA and thus to combat viral infection. The observation that several viruses encode RNAi silencing suppressors (RSS), like the NS1 protein of FLUAV or the Tat protein of the human immunodeficiency virus type 1 (HIV‐1), underlines the potency of this cellular defence mechanism (Bivalkar‐Mehla et al., 2011). Haasnoot et al. (2007) demonstrated that EBOV VP35 is an RSS, which inhibits shRNA‐mediated suppression of reporter gene expression and rescues production of tat‐defective HIV‐1 from suppression by RNAi. The RSS activity of VP35 was dependent on its dsRNA‐binding capability (Haasnoot et al., 2007), suggesting that VP35 may sequester and protect dsRNA from recognition by dicer, a central molecule of the RNAi pathway (Fig. 4). However, a subsequent study demonstrated that VP35 interacts with components of the RNA‐induced silencing complex (RISC) independent of the presence of siRNA (Fabozzi et al., 2011). Specifically, VP35 was found to interact with the two dsRNA‐binding proteins HIV‐1 trans‐activation response RNA‐binding protein (TRBP) and protein activator of PKR (PACT), which are part of the RISC complex, but not directly with dicer (Fabozzi et al., 2011). In addition, evidence was presented that also VP30 and VP40 act as RSS, and VP30 like VP35 was shown to bind to components of the RISC complex, while no such interactions were seen for VP40, which might employ a different strategy to inhibit RNAi (Fabozzi et al., 2011).
Novel Interferon‐Induced Antiviral Proteins that Target EBOV
Tetherin
The tetherin protein (HM1.24, BST‐2, CD317) was identified as a type I interferon‐inducible host cellular factor, which restricts release of progeny HIV‐1 particles from infected cells (Neil et al., 2008; Van Damme et al., 2008). Tetherin is counteracted by the HIV‐1 accessory protein Vpu (viral protein u), which allows efficient HIV‐1 release form tetherin‐expressing cells (Neil et al., 2008; Van Damme et al., 2008). The antiviral action of tetherin is not limited to HIV‐1; several recent studies found that tetherin restricts release of VLPs and progeny particles of several enveloped viruses, including members of the Retroviridae, Arenaviridae, Paramyxoviridae, Filoviridae, Rhabdoviridae and Herpesviridae families (Jouvenet et al., 2009; Mansouri et al., 2009; Sakuma et al., 2009; Pardieu et al., 2010; Radoshitzky et al., 2010; Weidner et al., 2010; Watanabe et al., 2011; Yondola et al., 2011) (Fig. 5). The broad spectrum antiviral action of tetherin is intimately linked to its unusual structural organization. Tetherin encodes a short cytoplasmic domain at its N‐terminus, followed by a single transmembrane (TM) domain, an extracellular coiled‐coil region and a C‐terminal glycosylphosphatidylinositol (GPI) anchor (Kupzig et al., 2003). Thus, the protein spans the membrane twice and is able to insert one end into the cellular membrane and the other end into the viral envelope. By this mechanism, tetherin retains budding virions at the surface of the infected cell and inhibits their transmission to new target cells (Perez‐Caballero et al., 2009; Hammonds et al., 2010). An artificially constructed tetherin, which displays the same topology as the original molecule, but is assembled from completely unrelated sequences, is able to inhibit virus spread as well, confirming that it is tetherin’s unusual architecture that is responsible for tethering virions to cells (Perez‐Caballero et al., 2009).
Antagonism of tetherin is not limited to Vpu. HIV‐2 uses its envelope (Env) protein to counteract tetherin (Le Tortorec and Neil, 2009), while the simian immunodeficiency virus (SIV) uses either the accessory protein Nef or Vpu or Env, depending on the origin of the virus (Gupta et al., 2009; Sauter et al., 2009). The Kaposi’s sarcoma‐associated herpes virus encodes the E3 ubiquitin ligase K5 as a tetherin antagonist (Mansouri et al., 2009). For the EBOV, the GP1,2 serves as tetherin antagonist (Kaletsky et al., 2009). This ability of GP1,2 is conserved between the GPs of the different EBOV species (Zaire, Sudan, Côte d’Ivoire and Reston), the proposed species BEBOV and the second filoviral genus MARV (Kaletsky et al., 2009; Lopez et al., 2010; Radoshitzky et al., 2010; Kühl et al., 2011a, 2011b).
The EBOV glycoprotein and HIV‐1 Vpu employ different strategies to counteract tetherin
The EBOV glycoprotein (EBOV‐GP1,2) is the only viral surface protein and mediates viral entry into the host cell that requires binding of GP1,2 to the endosomal membrane protein Niemann‐Pick C1 (NPC1) (Carette et al., 2011; Côté et al., 2011). EBOV‐GP1,2 is synthesized as a precursor protein, which is post‐translationally cleaved by subtilisin‐like proteases into its two subunits GP1 and GP2 (Volchkov et al., 1998). The large and heavily glycosylated, extracellular domain GP1 mediates attachment to the host cell; the smaller TM unit GP2 facilitates fusion of the viral envelope with the membrane of host cell endosomes (Takada et al., 1997; Wool‐Lewis and Bates, 1998). A recent study demonstrated that GP1,2 also inactivates one of the cell`s innate defences against infection, the tetherin protein (Kaletsky et al., 2009). Vpu allows HIV‐1 to evade tetherin by mediating cell surface downregulation and relocalization of tetherin into intracellular compartments (Van Damme et al., 2008). In addition, Vpu facilitates degradation of tetherin in lysosomal or proteasomal compartments (Douglas et al., 2009; Goffinet et al., 2009, 2010; Mangeat et al., 2009; Mitchell et al., 2009; Dubé et al., 2010). In contrast, no evidence for downregulation, relocalization away from the cell surface or degradation of tetherin was observed in the presence of EBOV‐GP1,2 or MARV‐GP1,2 (Kaletsky et al., 2009; Lopez et al., 2010; Radoshitzky et al., 2010; Kühl et al., 2011a). Furthermore, EBOV‐GP1,2 is active against tetherin homologues from different monkeys, in accordance with the ability of EBOV to infect several non‐human primates (Kühl et al., 2011a). In contrast, tetherin antagonism by Vpu is limited to tetherin of human, chimpanzee and gorilla origin, and these species are infected by HIV‐1 and Vpu encoding SIV, respectively (Goffinet et al., 2009; Jia et al., 2009; McNatt et al., 2009). Thus, Vpu and EBOV‐GP1,2 have a different specificity for tetherin and employ different strategies to counteract tetherin.
Tetherin interacts with the GP2 subunit of the EBOV‐GP1,2
Four different proteins are expressed from the EBOV‐GP gene: (i) The primary transcript from the GP gene is sGP, a soluble, secreted form of the GP (Volchkov et al., 1995; Sanchez et al., 1996), which has an anti‐inflammatory property (Wahl‐Jensen et al., 2005). (ii) A soluble Δ‐peptide is released upon proteolytic processing of sGP by furin (Volchkova et al., 1999). (iii) The full‐length, membrane‐bound form GP1,2, which is incorporated into the virions, is produced by RNA editing of the primary transcript (Volchkov et al., 1995; Sanchez et al., 1996). (iv) Furthermore, a small soluble GP (ssGP) has recently been identified, but the function of this protein is at present unknown (Mehedi et al., 2011). In addition, the TNF‐α converting enzyme induces shedding of GP1,2Δ from the surface of infected cells by cleaving GP1,2 near the TM domain (Dolnik et al., 2004). All different forms of the GP might play a role in combating the host’s immune system as, for example the shedded form of GP might be able to act as a decoy for neutralizing antibodies (Dolnik et al., 2008). However, neither sGP nor GP1,2Δ are able to counteract tetherin (Kaletsky et al., 2009). In addition, a GP1,2 mutant that is retained in the ER failed to counteract tetherin, suggesting that the correct localization of the EBOV‐GP1,2 at sites of viral budding is crucial for tetherin antagonism (Kaletsky et al., 2009). Indeed, because of the expression of the GPI‐anchor, tetherin is localized to lipid rafts, which are used by HIV‐1 and EBOV as platforms for budding (Ono and Freed, 2001; Bavari et al., 2002).
Vpu and tetherin interact via their TM domains, and the interaction is critical for tetherin antagonism (McNatt et al., 2009). In contrast, the sequences of tetherin’s cytoplasmic tail (CT) and TM domain do not determine counteraction by EBOV‐GP1,2. Thus, tetherin chimeras in which the TM region and the N‐terminus of tetherin were exchanged against similar domains of the transferrin receptor type 1 (TfR) displayed antiviral activity and were counteracted by EBOV‐GP1,2 (Lopez et al., 2010). Nevertheless, ZEBOV‐GP1,2 is able to efficiently coimmunoprecipitate human tetherin (Kaletsky et al., 2009), and the GP2 subunit of EBOV‐GP1,2 was shown to be critical for the interaction (Kühl et al., 2011a). Which domain of tetherin interacts with ZEBOV‐GP2 remains to be determined, and, because of topological constraints, the TM unit and the CT are interesting candidates. However, the TM and CT have been shown to be irrelevant for tetherin counteraction by ZEBOV‐GP1,2 (Lopez et al., 2010), and the importance of tetherin binding for tetherin inhibition by EBOV‐GP1,2 remains to be determined.
The EBOV‐GP1,2 might relocalize tetherin within the plasma membrane or interfere with the structural integrity of tetherin
Tetherin and VP40, which is essential for EBOV budding, colocalize at the plasma membrane and release of VP40‐based VLPs is inhibited by tetherin (Jouvenet et al., 2009). It is thus possible that the EBOV‐GP1,2 rescues the block to particle release by interfering with the integrity of tetherin at filoviral budding sites (Kühl et al., 2011a). Alternatively, the extracellular, heavily glycosylated EBOV‐GP1 subunit might interfere with the formation of the ‘tetherin‐clamp’ between the cellular and the viral membrane, because of steric hindrance. Finally, EBOV‐GP1,2 might relocalize tetherin within the plasma membrane, thereby excluding it from membrane domains used by EBOV for budding. Indeed, it was observed that tetherin is excluded from plasma membrane sites positive for GP1,2 in EBOV‐infected cells (Radoshitzky et al., 2010), lending support to the idea that EBOV‐GP1,2 blocks tetherin’s antiviral action by inducing its mislocalization within the plasma membrane.
Regardless of the mechanism underlying tetherin counteraction by EBOV‐GP1,2, it remains to be determined whether endogenous tetherin can reduce EBOV release from infected cells, as modest effects were observed in one study (Kühl et al., 2011a) but not in another applying a substantially higher multiplicity of infection (MOI) (Radoshitzky et al., 2010), and might thus restrict viral spread in the infected host. In addition, it will be interesting to determine whether African fruit bats, the potential natural reservoir of EBOV, encode a tetherin‐like protein and whether this tetherin homologue restricts EBOV spread.
IFITMs
The IFITM 1, 2 and 3 were recently discovered as inhibitors of host cell entry of several enveloped viruses, including EBOV (Fig. 5). IFITMs 1, 2 and 3 are ubiquitously expressed in human cells and tissues upon exposure to type I (α) and type II (γ) IFN, and homologous proteins are present in many vertebrates (Siegrist et al., 2011). IFITM proteins were shown to play a role in early development, cell adhesion and control of cell growth (Siegrist et al., 2011). Their antiviral activity was discovered in a siRNA screen designed to identify host cell factors modulating FLUAV infection (Brass et al., 2009). IFITM3 was identified as a potent inhibitor of host cell entry of FLUAV and members of the family Flaviviridae, West Nile virus and Dengue virus serotype 2, but not hepatitis C Virus (Brass et al., 2009; Jiang et al., 2010), and the induction of IFITM3 expression was shown to be largely responsible for the blockade to FLUAV entry imposed by treatment of target cells with IFNs (Brass et al., 2009). IFITM1 and 2 were also shown to restrict viral infection although to a lower extent, and the antiviral activity of IFITMs was conserved between human proteins and murine orthologues (Brass et al., 2009). Thus, IFITMs are novel IFN‐induced antiviral effector proteins, which could modulate viral spread in humans and animals.
IFITMs restrict viral entry into host cells
While antiviral activity of IFITMs was initially reported for influenza and flaviviruses (Brass et al., 2009; Jiang et al., 2010), subsequent studies showed that IFITMs inhibit entry of additional enveloped viruses such as vesicular stomatitis Indiana virus (VSIV), severe acute respiratory syndrome coronavirus (SARS‐CoV) and filoviruses (Weidner et al., 2010; Huang et al., 2011), which, very much like FLUAV and flaviviruses, depend on endo‐/lysosomal acidic pH for host cell entry. Inhibition of filoviruses and SARS‐CoV was demonstrated employing lentiviral vectors pseudotyped with the respective viral GPs and with replication competent virus (Huang et al., 2011), but the inhibitory efficiency was modest. In contrast to FLUAV, IFITM1 showed the most prominent antiviral effects against replication competent EBOV and MARV, while inhibition by IFITM3 was less efficient (Huang et al., 2011). Entry of MARV and EBOV was reduced by the expression of several IFITM orthologues of mouse and chicken, although with different efficacies compared to the human proteins (Huang et al., 2011). Depletion of IFITM3 by shRNA was sufficient to rescue entry of FLUAV pseudotypes, whereas a knockdown of both, IFITM1 and IFITM3, was required to increase entry of EBOV and MARV pseudotypes (Huang et al., 2011). Finally, a different study also detected inhibition of HIV‐1 infection (Lu et al., 2011), which does not depend on low pH, and it is at present unclear which structure or process shared by the viruses listed above for host cell entry is targeted by IFITMs.
Determinants of the antiviral activity of IFITM proteins
Domains and modifications important for antiviral activity of the IFITMs have been identified, although most studies were not conducted in the context of filovirus infection. Abrogation of S‐palmitoylation of IFITM3 by mutation of crucial cysteine residues inhibited IFITM3 clustering in membrane compartments as well as the antiviral activity of IFITM3 against FLUAV (Yount et al., 2010). The sites of S‐palmitoylation are highly conserved in members of the IFITM protein family, suggesting a general role in the antiviral activity. In addition, the sequence of the N‐terminus is distinct for the different IFITMs as IFITM1 is lacking an N‐terminal amino acid stretch present in IFITM2 and 3. Part of the N‐terminus might be crucial for their different antiviral activity (Siegrist et al., 2011). Indeed, the characterization of IFITM1/IFITM3 chimeras revealed that sequences within the N‐ and C‐terminus of IFITM3 are important for antiviral activity against VSIV (Weidner et al., 2010). Furthermore, Lu et al. (2011) could show that only IFITM‐2 and IFITM‐3 inhibit the HIV‐1 life cycle at the step of viral entry, and that deletion of the N‐terminal region abrogated this ability. IFITM1, in contrast, needs an intact intracellular domain to inhibit HIV‐1 replication, whereas the N‐ and C‐terminus are dispensable (Lu et al., 2011).
IFITM block cellular entry after transport of viruses into endosomal compartments
What is known about the mechanism underlying IFITM dependent inhibition of host cell entry of EBOV and other viruses? One possibility is that IFITMs interfere with receptor expression. However, IFITM proteins do not interfere with the level of surface expression of sialic acids and ACE2, the receptors for FLUAV and SARS‐CoV, respectively (Brass et al., 2009; Huang et al., 2011). IFITM expression is compatible with FLUAV access to low pH compartments, and inhibition of SARS‐CoV by IFITMs could be rescued by forcing the virus to fuse with the plasma membrane instead of an internal membrane (Huang et al., 2011). Thus, viruses might reach internal compartments in IFITM expressing cells in which fusion of viral and compartment membrane could normally occur, but membrane fusion might be blocked by IFITMs. One possibility could be that IFITMs interfere with the activity of cathepsins, pH‐dependent endo‐/lysosomal proteases essential for proteolytic activation of SARS‐CoV and filoviruses in vitro (Chandran et al., 2005; Simmons et al., 2005). No appreciable decrease of cathepsin activity was observed in IFITM expressing cells (Huang et al., 2011). The endosomal membrane protein NPC1 has recently been reported as an essential host cell factor for EBOV entry (Carette et al., 2011; Côté et al., 2011). According to the model of Côté et al., NPC1 expression or activity is required for endosomal fusion after cathepsin‐mediated processing of GP1,2. It is conceivable that IFITMs interfere with NPC1 expression or activity. Furthermore, it is possible that a step in the filovirus life cycle different from membrane fusion could be affected by IFITMs, as it was reported that IFITM2 and 3 inhibit HIV‐1 entry, whereas IFITM1 acts later in the viral life cycle by suppressing Gag translation (Lu et al., 2011). Thus, the mechanisms underlying inhibition of filoviruses and other viruses remain to be defined, including the possibility that IFITMs require cellular cofactors to exert their antiviral effects.
In sum, IFITMs inhibit filovirus entry into host cells, and the level of constitutive IFITM expression might shape the choice of early target cells in filovirus infection.
Further research is needed to define the basal expression of IFITMs in filovirus target cells and to elucidate the mechanism by which the different IFITM proteins restrict filovirus infection.
Conclusions
The IFN system can potently restrict EBOV spread and pathogenesis in infected mice, but is tuned down by viral proteins in infected humans. VP24 and VP35 play key roles in suppressing the IFN response by preventing nuclear translocation of STAT1 and by targeting IRF‐3 and IRF‐7, respectively. Tetherin and IFITM proteins are novel ISGs, which could suppress EBOV infectious entry into target cells (IFITMs) and release of EBOV particles from infected cells (tetherin). Efficient suppression of EBOV release has so far only been demonstrated with VLPs and remains to be shown with infectious EBOV. For this, the sequences in GP1,2, which facilitate tetherin antagonism, need to be identified and altered, which should render EBOV susceptible to inhibition by tetherin. The GP2 subunit is an excellent candidate for these endeavours (Kühl et al., 2011a). The mechanism underlying EBOV inhibition by IFITMs is at present unknown, and its elucidation might require the identification of potential interaction partners of GP1,2 and IFITMs in host cell endosomes. In addition, it will be interesting to determine to which extent basal (in the absence of IFN) expression of IFITMs and tetherin impact EBOV spread. A recent study demonstrated that basal tetherin expression is broader than initially appreciated and raised doubts concerning the sole regulation of tetherin expression by IFNs (Erikson et al., 2011). Moreover, the role of the IFN system in the EBOV infection of the potential reservoir host, fruit bats (Leroy et al., 2005), is of high interest. Thus, EBOV infection of these animals does not seem to induce disease (Swanepoel et al., 1996; Hayman et al., 2010), and it is tempting to speculate that the bat IFN system efficiently controls viral replication. Finally, the interesting insights into EBOV interference with the IFN system open new avenues to the development of filovirus inhibitors. Proof of concept comes from studies with HIV‐1, which demonstrated that stabilization of antiviral effectors molecules of the IFN system or inhibition of viral IFN antagonists by small molecules can suppress viral spread (Cen et al., 2010; Jiang et al., 2010).
Conflicts of interest
The authors have no potential conflicts to declare.
Acknowledgements
We thank T.F. Schulz for support.
References
- Barrette, R. W. , Metwally S. A., Rowland J. M., Xu L., Zaki S. R., Nichol S. T., Rollin P. E., Towner J. S., Shieh W. J., Batten B., Sealy T. K., Carrillo C., Moran K. E., Bracht A. J., Mayr G. A., Sirios‐Cruz M., Catbagan D. P., Lautner E. A., Ksiazek T. G., White W. R., and McIntosh M. T., 2009: Discovery of swine as a host for the Reston ebolavirus. Science 325, 204–206. [DOI] [PubMed] [Google Scholar]
- Basler, C. F. , Wang X., Mühlberger E., Volchkov V., Paragas J., Klenk H. D., García‐Sastre A., and Palese P., 2000: The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. U.S.A. 97, 12289–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler, C. F. , Mikulasova A., Martínez‐Sobrido L., Paragas J., Mühlberger E., Bray M., Klenk H. D., Palese P., and García‐Sastre A., 2003: The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 77, 7945–7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, A. , and García‐Sastre A., 2010: Induction of type I interferon by RNA viruses: cellular receptors and their substrates. Amino Acids 38, 1283–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, A. , and García‐Sastre A., 2011: Differential recognition of viral RNA by RIG‐I. Virulence 2, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavari, S. , Bosio C. M., Wiegand E., Ruthel G., Will A. B., Geisbert T. W., Hevey M., Schmaljohn C., Schmaljohn A., and Aman M. J., 2002: Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 195, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivalkar‐Mehla, S. , Vakharia J., Mehla R., Abreha M., Kanwar J. R., Tikoo A., and Chauhan A., 2011: Viral RNA silencing suppressors (RSS): novel strategy of viruses to ablate the host RNA interference (RNAi) defense system. Virus Res. 155, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass, A. L. , Huang I. C., Benita Y., John S. P., Krishnan M. N., Feeley E. M., Ryan B. J., Weyer J. L., van der W. L., Fikrig E., Adams D. J., Xavier R. J., Farzan M., and Elledge S. J., 2009: The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, M. , 2001: The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 82, 1365–1373. [DOI] [PubMed] [Google Scholar]
- Bray, M. , Davis K., Geisbert T., Schmaljohn C., and Huggins J., 1998: A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 178, 651–661. [DOI] [PubMed] [Google Scholar]
- Cárdenas, W. B. , Loo Y. M., Gale M. Jr, Hartman A. L., Kimberlin C. R., Martínez‐Sobrido L., Saphire E. O., and Basler C. F., 2006: Ebola virus VP35 protein binds double‐stranded RNA and inhibits alpha/beta interferon production induced by RIG‐I signaling. J. Virol. 80, 5168–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette, J. E. , Raaben M., Wong A. C., Herbert A. S., Obernosterer G., Mulherkar N., Kuehne A. I., Kranzusch P. J., Griffin A. M., Ruthel G., Dal Cin P., Dye J. M., Whelan S. P., Chandran K., and Brummelkamp T. R., 2011: Ebola virus entry requires the cholesterol transporter Niemann‐Pick C1. Nature, 477, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen, S. , Peng Z. G., Li X. Y., Li Z. R., Ma J., Wang Y. M., Fan B., You X. F., Wang Y. P., Liu F., Shao R. G., Zhao L. X., Yu L., and Jiang J. D., 2010: Small molecular compounds inhibit HIV‐1 replication through specifically stabilizing APOBEC3G. J. Biol. Chem. 285, 16546–16552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran, K. , Sullivan N. J., Felbor U., Whelan S. P., and Cunningham J. M., 2005: Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308, 1643–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, T. H. , Kubota T., Matsuoka M., Jones S., Bradfute S. B., Bray M., and Ozato K., 2009: Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 5, e1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté, M. , Misasi J., Ren T., Bruchez A., Lee K., Filone C. M., Hensley L., Li Q., Ory D., Chandran K., and Cunningham J., 2011: Small molecule inhibitors reveal Niemann‐Pick C1 is essential for Ebola virus infection. Nature 477, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolnik, O. , Volchkova V., Garten W., Carbonnelle C., Becker S., Kahnt J., Ströher U., Klenk H. D., and Volchkov V., 2004: Ectodomain shedding of the glycoprotein GP of Ebola virus. EMBO J. 23, 2175–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolnik, O. , Kolesnikova L., and Becker S., 2008: Filoviruses: interactions with the host cell. Cell. Mol. Life Sci. 65, 756–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donelan, N. R. , Basler C. F., and García‐Sastre A., 2003: A recombinant influenza A virus expressing an RNA‐binding‐defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 77, 13257–13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas, J. L. , Viswanathan K., McCarroll M. N., Gustin J. K., Früh K., and Moses A. V., 2009: Vpu directs the degradation of the human immunodeficiency virus restriction factor BST‐2/Tetherin via a {beta}TrCP‐dependent mechanism. J. Virol. 83, 7931–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé, M. , Roy B. B., Guiot‐Guillain P., Binette J., Mercier J., Chiasson A., and Cohen E. A., 2010: Antagonism of tetherin restriction of HIV‐1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 6, e1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara, H. , Takada A., Kobasa D., Jones S., Neumann G., Theriault S., Bray M., Feldmann H., and Kawaoka Y., 2006: Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enterlein, S. , Warfield K. L., Swenson D. L., Stein D. A., Smith J. L., Gamble C. S., Kroeker A. D., Iversen P. L., Bavari S., and Mühlberger E., 2006: VP35 knockdown inhibits Ebola virus amplification and protects against lethal infection in mice. Antimicrob. Agents Chemother. 50, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erikson, E. , Adam T., Schmidt S., Lehmann‐Koch J., Over B., Goffinet C., Harter C., Bekeredjian‐Ding I., Sertel S., Lasitschka F., and Keppler O. T., 2011: In vivo expression profile of the antiviral restriction factor and tumor‐targeting antigen CD317/BST‐2/HM1.24/tetherin in humans. Proc. Natl. Acad. Sci. U.S.A. 108, 13688–13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabozzi, G. , Nabel C. S., Dolan M. A., and Sullivan N. J., 2011: Ebolavirus proteins suppress the effects of small interfering RNA by direct interaction with the mammalian RNA interference pathway. J. Virol. 85, 2512–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Z. , Cerveny M., Yan Z., and He B., 2007: The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double‐stranded RNA‐dependent protein kinase PKR. J. Virol. 81, 182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García, M. A. , Meurs E. F., and Esteban M., 2007: The dsRNA protein kinase PKR: virus and cell control. Biochimie 89, 799–811. [DOI] [PubMed] [Google Scholar]
- García‐Sastre, A. , and Biron C. A., 2006: Type 1 interferons and the virus‐host relationship: a lesson in detente. Science 312, 879–882. [DOI] [PubMed] [Google Scholar]
- George, C. X. , Li Z., Okonski K. M., Toth A. M., Wang Y., and Samuel C. E., 2009: Tipping the balance: antagonism of PKR kinase and ADAR1 deaminase functions by virus gene products. J. Interferon Cytokine Res. 29, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffinet, C. , Allespach I., Homann S., Tervo H. M., Habermann A., Rupp D., Oberbremer L., Kern C., Tibroni N., Welsch S., Krijnse‐Locker J., Banting G., Kräusslich H. G., Fackler O. T., and Keppler O. T., 2009: HIV‐1 antagonism of CD317 is species specific and involves Vpu‐mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5, 285–297. [DOI] [PubMed] [Google Scholar]
- Goffinet, C. , Homann S., Ambiel I., Tibroni N., Rupp D., Keppler O. T., and Fackler O. T., 2010: Antagonism of CD317 restriction of human immunodeficiency virus type 1 (HIV‐1) particle release and depletion of CD317 are separable activities of HIV‐1 Vpu. J. Virol. 84, 4089–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groseth, A. , Feldmann H., and Strong J. E., 2007: The ecology of Ebola virus. Trends Microbiol. 15, 408–416. [DOI] [PubMed] [Google Scholar]
- Gupta, R. K. , Mlcochova P., Pelchen‐Matthews A., Petit S. J., Mattiuzzo G., Pillay D., Takeuchi Y., Marsh M., and Towers G. J., 2009: Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST‐2/CD317 by intracellular sequestration. Proc. Natl. Acad. Sci. U.S.A. 106, 20889–20894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haasnoot, J. , de Vries W., Geutjes E. J., Prins M., de Haan P., and Berkhout B., 2007: The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 3, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller, O. , Staeheli P., and Kochs G., 2007: Interferon‐induced Mx proteins in antiviral host defense. Biochimie 89, 812–818. [DOI] [PubMed] [Google Scholar]
- Hammonds, J. , Wang J. J., Yi H., and Spearman P., 2010: Immunoelectron microscopic evidence for Tetherin/BST2 as the physical bridge between HIV‐1 virions and the plasma membrane. PLoS Pathog. 6, e1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, Z. , Boshra H., Sunyer J. O., Zwiers S. H., Paragas J., and Harty R. N., 2003: Biochemical and functional characterization of the Ebola virus VP24 protein: implications for a role in virus assembly and budding. J. Virol. 77, 1793–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlieb, B. , and Weissenhorn W., 2006: Filovirus assembly and budding. Virology 344, 64–70. [DOI] [PubMed] [Google Scholar]
- Hartman, A. L. , Towner J. S., and Nichol S. T., 2004: A C‐terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA‐binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology 328, 177–184. [DOI] [PubMed] [Google Scholar]
- Hartman, A. L. , Dover J. E., Towner J. S., and Nichol S. T., 2006: Reverse genetic generation of recombinant Zaire Ebola viruses containing disrupted IRF‐3 inhibitory domains results in attenuated virus growth in vitro and higher levels of IRF‐3 activation without inhibiting viral transcription or replication. J. Virol. 80, 6430–6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman, A. L. , Bird B. H., Towner J. S., Antoniadou Z. A., Zaki S. R., and Nichol S. T., 2008a: Inhibition of IRF‐3 activation by VP35 is critical for the high level of virulence of Ebola virus. J. Virol. 82, 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman, A. L. , Ling L., Nichol S. T., and Hibberd M. L., 2008b: Whole‐genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol. 82, 5348–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman, A. L. , Towner J. S., and Nichol S. T., 2010: Ebola and marburg hemorrhagic fever. Clin. Lab. Med. 30, 161–177. [DOI] [PubMed] [Google Scholar]
- Hayman, D. T. , Emmerich P., Yu M., Wang L. F., Suu‐Ire R., Fooks A. R., Cunningham A. A., and Wood J. L., 2010: Long‐term survival of an urban fruit bat seropositive for Ebola and Lagos bat viruses. PLoS ONE 5, e11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenen, T. , Groseth A., Kolesnikova L., Theriault S., Ebihara H., Hartlieb B., Bamberg S., Feldmann H., Ströher U., and Becker S., 2006: Infection of naive target cells with virus‐like particles: implications for the function of Ebola virus VP24. J. Virol. 80, 7260–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda, K. , Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., and Taniguchi T., 2005: IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 434, 772–777. [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Xu L., Sun Y., and Nabel G. J., 2002: The assembly of Ebola virus nucleocapsid requires virion‐associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol. Cell 10, 307–316. [DOI] [PubMed] [Google Scholar]
- Huang, I. C. , Bailey C. C., Weyer J. L., Radoshitzky S. R., Becker M. M., Chiang J. J., Brass A. L., Ahmed A. A., Chi X., Dong L., Longobardi L. E., Boltz D., Kuhn J. H., Elledge S. J., Bavari S., Denison M. R., Choe H., and Farzan M., 2011: Distinct patterns of IFITM‐mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 7, e1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, B. , Serra‐Moreno R., Neidermyer W., Rahmberg A., Mackey J., Fofana I. B., Johnson W. E., Westmoreland S., and Evans D. T., 2009: Species‐specific activity of SIV Nef and HIV‐1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5, e1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, D. , Weidner J. M., Qing M., Pan X. B., Guo H., Xu C., Zhang X., Birk A., Chang J., Shi P. Y., Block T. M., and Guo J. T., 2010: Identification of five interferon‐induced cellular proteins that inhibit west nile virus and dengue virus infections. J. Virol. 84, 8332–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, H. , Yan Z., Prabhakar B. S., Feng Z., Ma Y., Verpooten D., Ganesh B., and He B., 2010: The VP35 protein of Ebola virus impairs dendritic cell maturation induced by virus and lipopolysaccharide. J. Gen. Virol. 91, 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet, N. , Neil S. J., Zhadina M., Zang T., Kratovac Z., Lee Y., McNatt M., Hatziioannou T., and Bieniasz P. D., 2009: Broad‐spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 83, 1837–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaletsky, R. L. , Francica J. R., Agrawal‐Gamse C., and Bates P., 2009: Tetherin‐mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 106, 2886–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaoka, Y. , 2005: How Ebola virus infects cells. N. Engl. J. Med. 352, 2645–2646. [DOI] [PubMed] [Google Scholar]
- Kimberlin, C. R. , Bornholdt Z. A., Li S., Woods V. L. Jr, MacRae I. J., and Saphire E. O., 2010: Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc. Natl. Acad. Sci. U.S.A. 107, 314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühl, A. , Banning C., Marzi A., Votteler J., Steffen I., Bertram S., Glowacka I., Konrad A., Stürzl M., Guo J. T., Schubert U., Feldmann H., Behrens G., Schindler M., and Pöhlmann S., 2011a: The Ebola virus glycoprotein and HIV‐1 Vpu employ different strategies to counteract the antiviral factor tetherin. J. Infect. Dis. 204 (Suppl. 3), S850–S860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühl, A. , Hoffmann M., Müller M. A., Munster V. J., Kiene M., Tsegaye T. S., Behrens G., Herrler G., Feldmann H., Drosten C., and Pöhlmann S., 2011b: Comparative analysis of Ebola virus glycoprotein interactions with human and bat cells. J. Infect. Dis. 204 (Suppl. 3), S840–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, J. H. , 2008: Filoviruses. A compendium of 40 years of epidemiological, clinical, and laboratory studies. Arch. Virol. Suppl. 20, 13–360. [PubMed] [Google Scholar]
- Kupzig, S. , Korolchuk V., Rollason R., Sugden A., Wilde A., and Banting G., 2003: Bst‐2/HM1.24 is a raft‐associated apical membrane protein with an unusual topology. Traffic 4, 694–709. [DOI] [PubMed] [Google Scholar]
- Le Tortorec, A. , and Neil S. J., 2009: Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 83, 11966–11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. S. , Lebeda F. J., and Olson M. A., 2009: Fold prediction of VP24 protein of Ebola and Marburg viruses using de novo fragment assembly. J. Struct. Biol. 167, 136–144. [DOI] [PubMed] [Google Scholar]
- Leroy, E. M. , Kumulungui B., Pourrut X., Rouquet P., Hassanin A., Yaba P., Délicat A., Paweska J. T., Gonzalez J. P., and Swanepoel R., 2005: Fruit bats as reservoirs of Ebola virus. Nature 438, 575–576. [DOI] [PubMed] [Google Scholar]
- Leung, D. W. , Ginder N. D., Fulton D. B., Nix J., Basler C. F., Honzatko R. B., and Amarasinghe G. K., 2009: Structure of the Ebola VP35 interferon inhibitory domain. Proc. Natl. Acad. Sci. U.S.A. 106, 411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, D. W. , Prins K. C., Borek D. M., Farahbakhsh M., Tufariello J. M., Ramanan P., Nix J. C., Helgeson L. A., Otwinowski Z., Honzatko R. B., Basler C. F., and Amarasinghe G. K., 2010: Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat. Struct. Mol. Biol. 17, 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata, J. M. , Johnson R. F., Han Z., and Harty R. N., 2004: Contribution of Ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus‐like particles. J. Virol. 78, 7344–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. Y. , Sanchez D. J., and Cheng G., 2011: New developments in the induction and antiviral effectors of type I interferon. Curr. Opin. Immunol. 23, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez, L. A. , Yang S. J., Hauser H., Exline C. M., Haworth K. G., Oldenburg J., and Cannon P. M., 2010: Ebola virus glycoprotein counteracts BST‐2/Tetherin restriction in a sequence‐independent manner that does not require tetherin surface removal. J. Virol. 84, 7243–7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Pan Q., Rong L., He W., Liu S. L., and Liang C., 2011: The IFITM proteins inhibit HIV‐1 infection. J. Virol. 85, 2126–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty, S. , Gupta M., Paragas J., Bray M., Ahmed R., and Rollin P. E., 2003: Protection from lethal infection is determined by innate immune responses in a mouse model of Ebola virus infection. Virology 312, 415–424. [DOI] [PubMed] [Google Scholar]
- Mangeat, B. , Gers‐Huber G., Lehmann M., Zufferey M., Luban J., and Piguet V., 2009: HIV‐1 Vpu neutralizes the antiviral factor Tetherin/BST‐2 by binding it and directing its beta‐TrCP2‐dependent degradation. PLoS Pathog. 5, e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri, M. , Viswanathan K., Douglas J. L., Hines J., Gustin J., Moses A. V., and Früh K., 2009: Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma‐associated herpesvirus. J. Virol. 83, 9672–9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo, M. , Reid S. P., Leung L. W., Basler C. F., and Volchkov V. E., 2010: Ebolavirus VP24 binding to karyopherins is required for inhibition of interferon signaling. J. Virol. 84, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNatt, M. W. , Zang T., Hatziioannou T., Bartlett M., Fofana I. B., Johnson W. E., Neil S. J., and Bieniasz P. D., 2009: Species‐specific activity of HIV‐1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 5, e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehedi, M. , Falzarano D., Seebach J., Hu X., Carpenter M. S., Schnittler H. J., and Feldmann H., 2011: A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J. Virol. 85, 5406–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, R. S. , Katsura C., Skasko M. A., Fitzpatrick K., Lau D., Ruiz A., Stephens E. B., Margottin‐Goguet F., Benarous R., and Guatelli J. C., 2009: Vpu antagonizes BST‐2‐mediated restriction of HIV‐1 release via beta‐TrCP and endo‐lysosomal trafficking. PLoS Pathog. 5, e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlberger, E. , Weik M., Volchkov V. E., Klenk H. D., and Becker S., 1999: Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Virol. 73, 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil, S. J. , Zang T., and Bieniasz P. D., 2008: Tetherin inhibits retrovirus release and is antagonized by HIV‐1 Vpu. Nature, 451, 425–430. [DOI] [PubMed] [Google Scholar]
- NIAID , 2011: NIAID category A, B, and C priority pathogens. Available at: http://www.niaid.nih.gov/topics/biodefenserelated/biodefense/research/pages/cata.aspx (accessed on 10 August 2011).
- Ono, A. , and Freed E. O., 2001: Plasma membrane rafts play a critical role in HIV‐1 assembly and release. Proc. Natl. Acad. Sci. U.S.A. 98, 13925–13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardieu, C. , Vigan R., Wilson S. J., Calvi A., Zang T., Bieniasz P., Kellam P., Towers G. J., and Neil S. J., 2010: The RING‐CH ligase K5 antagonizes restriction of KSHV and HIV‐1 particle release by mediating ubiquitin‐dependent endosomal degradation of tetherin. PLoS Pathog. 6, e1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Caballero, D. , Zang T., Ebrahimi A., McNatt M. W., Gregory D. A., Johnson M. C., and Bieniasz P. D., 2009: Tetherin inhibits HIV‐1 release by directly tethering virions to cells. Cell 139, 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins, K. C. , Cárdenas W. B., and Basler C. F., 2009: Ebola virus protein VP35 impairs the function of interferon regulatory factor‐activating kinases IKKepsilon and TBK‐1. J. Virol. 83, 3069–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins, K. C. , Binning J. M., Shabman R. S., Leung D. W., Amarasinghe G. K., and Basler C. F., 2010a: Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J. Virol. 84, 10581–10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins, K. C. , Delpeut S., Leung D. W., Reynard O., Volchkova V. A., Reid S. P., Ramanan P., Cárdenas W. B., Amarasinghe G. K., Volchkov V. E., and Basler C. F., 2010b: Mutations abrogating VP35 interaction with double‐stranded RNA render Ebola virus avirulent in guinea pigs. J. Virol. 84, 3004–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoshitzky, S. R. , Dong L., Chi X., Clester J. C., Retterer C., Spurgers K., Kuhn J. H., Sandwick S., Ruthel G., Kota K., Boltz D., Warren T., Kranzusch P. J., Whelan S. P., and Bavari S., 2010: Infectious Lassa virus, but not filoviruses, is restricted by BST‐2/tetherin. J. Virol. 84, 10569–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, S. P. , Cárdenas W. B., and Basler C. F., 2005: Homo‐oligomerization facilitates the interferon‐antagonist activity of the ebolavirus VP35 protein. Virology, 341, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, S. P. , Leung L. W., Hartman A. L., Martinez O., Shaw M. L., Carbonnelle C., Volchkov V. E., Nichol S. T., and Basler C. F., 2006: Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 80, 5156–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, S. P. , Valmas C., Martinez O., Sanchez F. M., and Basler C. F., 2007: Ebola virus VP24 proteins inhibit the interaction of NPI‐1 subfamily karyopherin alpha proteins with activated STAT1. J. Virol. 81, 13469–13477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler, A. J. , and Williams B. R., 2008: Interferon‐inducible antiviral effectors. Nat. Rev. Immunol. 8, 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma, T. , Noda T., Urata S., Kawaoka Y., and Yasuda J., 2009: Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 83, 2382–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, A. , Trappier S. G., Mahy B. W., Peters C. J., and Nichol S. T., 1996: The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U.S.A. 93, 3602–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter, D. , Schindler M., Specht A., Landford W. N., Münch J., Kim K. A., Votteler J., Schubert U., Bibollet‐Ruche F., Keele B. F., Takehisa J., Ogando Y., Ochsenbauer C., Kappes J. C., Ayouba A., Peeters M., Learn G. H., Shaw G., Sharp P. M., Bieniasz P., Hahn B. H., Hatziioannou T., and Kirchhoff F., 2009: Tetherin‐driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV‐1 strains. Cell Host Microbe 6, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler, C. , Levy D. E., and Decker T., 2007: JAK‐STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063. [DOI] [PubMed] [Google Scholar]
- Schoggins, J. W. , Wilson S. J., Panis M., Murphy M. Y., Jones C. T., Bieniasz P., and Rice C. M., 2011: A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann, M. , Gantke T., and Muhlberger E., 2009: Ebola virus VP35 antagonizes PKR activity through its C‐terminal interferon inhibitory domain. J. Virol. 83, 8993–8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekimoto, T. , Imamoto N., Nakajima K., Hirano T., and Yoneda Y., 1997: Extracellular signal‐dependent nuclear import of Stat1 is mediated by nuclear pore‐targeting complex formation with NPI‐1, but not Rch1. EMBO J. 16, 7067–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist, F. , Ebeling M., and Certa U., 2011: The small interferon‐induced transmembrane genes and proteins. J. Interferon Cytokine Res. 31, 183–197. [DOI] [PubMed] [Google Scholar]
- Simmons, G. , Gosalia D. N., Rennekamp A. J., Reeves J. D., Diamond S. L., and Bates P., 2005: Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U.S.A. 102, 11876–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanepoel, R. , Leman P. A., Burt F. J., Zachariades N. A., Braack L. E., Ksiazek T. G., Rollin P. E., Zaki S. R., and Peters C. J., 1996: Experimental inoculation of plants and animals with Ebola virus. Emerg. Infect. Dis. 2, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada, A. , Robison C., Goto H., Sanchez A., Murti K. G., Whitt M. A., and Kawaoka Y., 1997: A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 94, 14764–14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towner, J. S. , Sealy T. K., Khristova M. L., Albariño C. G., Conlan S., Reeder S. A., Quan P. L., Lipkin W. I., Downing R., Tappero J. W., Okware S., Lutwama J., Bakamutumaho B., Kayiwa J., Comer J. A., Rollin P. E., Ksiazek T. G., and Nichol S. T., 2008: Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 4, e1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme, N. , Goff D., Katsura C., Jorgenson R. L., Mitchell R., Johnson M. C., Stephens E. B., and Guatelli J., 2008: The interferon‐induced protein BST‐2 restricts HIV‐1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchkov, V. E. , Becker S., Volchkova V. A., Ternovoj V. A., Kotov A. N., Netesov S. V., and Klenk H. D., 1995: GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 214, 421–430. [DOI] [PubMed] [Google Scholar]
- Volchkov, V. E. , Feldmann H., Volchkova V. A., and Klenk H. D., 1998: Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. U.S.A. 95, 5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchkova, V. A. , Klenk H. D., and Volchkov V. E., 1999: Delta‐peptide is the carboxy‐terminal cleavage fragment of the nonstructural small glycoprotein sGP of Ebola virus. Virology 265, 164–171. [DOI] [PubMed] [Google Scholar]
- Wahl‐Jensen, V. M. , Afanasieva T. A., Seebach J., Ströher U., Feldmann H., and Schnittler H. J., 2005: Effects of Ebola virus glycoproteins on endothelial cell activation and barrier function. J. Virol. 79, 10442–10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, R. , Leser G. P., and Lamb R. A., 2011: Influenza virus is not restricted by tetherin whereas influenza VLP production is restricted by tetherin. Virology 417(1), 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner, J. M. , Jiang D., Pan X. B., Chang J., Block T. M., and Guo J. T., 2010: Interferon‐induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 84, 12646–12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool‐Lewis, R. J. , and Bates P., 1998: Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor‐deficient cell lines. J. Virol. 72, 3155–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yondola, M. A. , Fernandes F., Belicha‐Villanueva A., Uccelini M., Gao Q., Carter C., and Palese P., 2011: Budding capability of the influenza virus neuraminidase can be modulated by tetherin. J. Virol. 85, 2480–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount, J. S. , Moltedo B., Yang Y. Y., Charron G., Moran T. M., Lopéz C. B., and Hang H. C., 2010: Palmitoylome profiling reveals S‐palmitoylation‐dependent antiviral activity of IFITM3. Nat. Chem. Biol. 6, 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]