

Abstract
Hepatitis A virus (HAV) and hepatitis C virus (HCV) are two positive‐strand RNA viruses sharing a similar biology, but causing opposing infection outcomes, with HAV always being cleared and HCV establishing persistence in the majority of infections. To gain deeper insight into determinants of replication, persistence, and treatment, we established a homogenous cell‐culture model allowing a thorough comparison of RNA replication of both viruses. By screening different human liver‐derived cell lines with subgenomic reporter replicons of HAV as well as of different HCV genotypes, we found that Huh7‐Lunet cells supported HAV‐ and HCV‐RNA replication with similar efficiency and limited interference between both replicases. HAV and HCV replicons were similarly sensitive to interferon (IFN), but differed in their ability to establish persistent replication in cell culture. In contrast to HCV, HAV replicated independently from microRNA‐122 and phosphatidylinositol 4‐kinase IIIα and β (PI4KIII). Both viruses were efficiently inhibited by cyclosporin A and NIM811, a nonimmunosuppressive analog thereof, suggesting an overlapping dependency on cyclophilins for replication. However, analysis of a broader set of inhibitors revealed that, in contrast to HCV, HAV does not depend on cyclophilin A, but rather on adenosine‐triphosphate–binding cassette transporters and FK506‐binding proteins. Finally, silibinin, but not its modified intravenous formulation, efficiently inhibited HAV genome replication in vitro, suggesting oral silibinin as a potential therapeutic option for HAV infections. Conclusion: We established a cell‐culture model enabling comparative studies on RNA replication of HAV and HCV in a homogenous cellular background with comparable replication efficiency. We thereby identified new host cell targets and potential treatment options for HAV and set the ground for future studies to unravel determinants of clearance and persistence. (Hepatology 2015;62:397–408
Abbreviations
- ABC
adenosine‐triphosphate–binding cassette
- CFA
colony‐formation assay
- CsA
cyclosporin A
- Cyp
cyclophilin
- CypA
cyclophilin A
- DMEM
Dulbecco's modified minimal essential medium
- FKBP
FK506‐binding protein
- gt
genotype
- HAV
hepatitis A virus
- HCV
hepatitis C virus
- IC50
half maximal inhibitory concentration
- IF
immunofluorescence
- IFN
interferon
- ISG
IFN‐stimulated gene
- IV
intravenous
- JFH1
Japanese fulminant hepatitis 1
- MAVS
mitochondrial antiviral‐signaling protein
- miR122
microRNA‐122
- miRNA
microRNA
- NS
nonstructural
- NS5A
nonstructural protein 5A
- NTR
nontranslated region
- PI4K
phosphatidylinositol 4‐kinase
- SFA
sanglifehrin A
- shRNA
short hairpin RNA
- SIL
Legalon‐SIL
- TRIF
TIR‐domain–containing adapter‐inducing interferon beta
- wt
wild‐type
Hepatitis C virus (HCV) and hepatitis A virus (HAV) are two hepatotropic positive‐strand RNA viruses sharing a similar biology, but causing very different infection outcomes. Worldwide, approximately 160 million people are infected with HCV, a member of the Flaviviridae family. HCV establishes persistence in up to 70%‐80% of cases, which often results in severe liver damage. This high rate of persistence is quite unusual for a positive‐strand RNA virus, and the underlying reasons are only poorly understood.1 In contrast, HAV, a picornavirus causing acute self‐limited hepatitis, never leads to chronic infection. Counterintuitive to infection outcomes, HCV usually induces a high, long‐lasting interferon (IFN)‐stimulated gene (ISG) expression, whereas this is very limited in HAV‐infected chimpanzees.2 HAV infections are frequent in developing countries, but also in European countries or in the United States epidemiological HAV outbreaks occur frequently and are mostly derived from contaminated food.3, 4, 5 HAV infection may result in severe or even fulminant hepatitis, a condition that is more likely to develop in patients at higher age or with chronic liver disease,3 including HCV/HAV coinfection.6 However, owing to availability of a vaccine in the early 1990s, research interest in HAV has ceased until recently. This explains the lack of treatment options for HAV infections, though these could shorten and attenuate disease as well as restrict epidemiological outbreaks. In contrast, comprehensive screening programs have brought up a number of highly efficient drugs inhibiting HCV replication by targeting viral and host cell factors, for example, cyclophilin A (CypA) and microRNA‐122 (miR122).7 In addition, picornaviruses related to HAV were reported to depend on the lipid kinase, phosphatidylinositol 4‐kinase (PI4K) IIIβ,8 whereas HCV requires PI4KIIIα for replication.9 These observations argue for potential similarities in host factor usage of the two viruses.
The aim of this study was a comprehensive comparison of HCV and HAV replication in a homogenous cellular background to get deeper insight into HAV replication and identify novel targets for therapy. We used subgenomic reporter replicons of HAV and of HCV genotypes (gt) 1a, 1b, and 2a and found comparable replication efficiencies in Huh7 cells. Replication machineries of HCV and HAV were similarly sensitive to IFNs, but clear differences were found for dependency on PI4K, miR122, and immunophilins. Interestingly, HAV replication was inhibited by silibinin, but not by the intravenous (IV) formulation Legalon‐SIL (SIL), which might suggest oral silibinin as a therapeutic option for treatment of HAV infections.
Materials and Methods
Drug Treatment
Treatment for IFN‐α (PBL InterferonSource, Piscataway, NJ), IFN‐λ (PeproTech GmbH, Hamburg, Germany), IFN‐γ (R&D Systems, Wiesbaden, Germany), SIL (Madaus, Cologne, Germany), silibinin (Sigma‐Aldrich, Steinheim, Germany), PI4KIIIα inhibitor AL‐9 (provided by Raffaele De Francesco, Milan, Italy),10 PI4KIIIβ‐inhibitor PIK93 (Sigma‐Aldrich),11 cyclophilin (Cyp) inhibitors cyclosporin A (CsA; Sigma‐Aldrich),12 NIM811, and sanglifehrin A (SFA; both Novartis, Basel, Switzerland), as well as adenosine‐triphosphate–binding cassette (ABC)B1/ABCC1 inhibitor Reversan,13 ABCB1 and Cyp3A4 inhibitor Piperine (both Sigma‐Aldrich),14 FK506 (InvivoGen, San Diego, CA), and PSC833 (Santa Cruz Biotechnology, Dallas, TX), which inhibits p‐glycoprotein, but not cyclophilins,12 were performed 4 hours after electroporation of in vitro transcribed RNA into Huh7‐Lunet cells by replacing the supernatant with different reagent dilutions until cell lysis at 48 hours. The microRNA (miRNA) inhibitor, mirVana (Invitrogen, Karlsruhe, Germany), and the miRNAs, miR122 and miR122_A4U (Eurofins Genomics, Ebersberg, Germany), were coelectroporated with in vitro transcripts of different reporter replicons. Detailed information about solvents and stock solutions is provided in http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo. Half maximal inhibitory concentrations (IC50s) were calculated using the nonlinear regression (curve fit) tool of GraphPad Prism version 5.03 for Windows (GraphPad Software, San Diego, CA, USA).
Cell Lines
The human hepatoma cell lines, Huh7, Huh6, and Hep3B, were grown in Dulbecco's modified minimal essential medium (DMEM; Life Technologies, Norwalk, CT), supplemented with 2 mM of L‐glutamine, nonessential amino acids, 100 U/mL of penicillin, 100 µg/mL of streptomycin, and 10% fetal calf serum. Huh7‐Lunet are highly permissive for HCV‐RNA replication, whereas Huh7.5 cells support high levels of infection.15 Detailed information of the generation of PI4KIIIβ CRISPR‐Cas9 knockout cells can be found in the Supporting Material and Methods. Generation and cultivation of stable knockdown cell lines expressing a short hairpin RNA (shRNA) targeting cyclophilin A (shCypA) or a nontargeting shRNA (shONT) as well as Huh7‐Lunet shPI4KIIIα cells has been described elsewhere.9, 16
Plasmid Constructs and Cell‐Based Replication Assays
The HAV reporter replicon is based on cell‐culture–adapted strain HM175 18f (GenBank accession no.: M59808) and has been described before.17 HCV reporter replicons of genotype (gt) 1a (H77‐S), 1b (Con1ET), 2a (JFH1), and the gt 2a/1b chimera (Japanese fulminant hepatitis 1 [JFH1] X‐tail Con1) have been described recently18 and will be referred to as H77, Con1, JFH, and R‐LucJFH X‐tail Con1 throughout the article. Methods for in vitro transcription, electroporation of replicon RNA, and reporter assays have been described in a previous work.19
Results
Huh7‐Lunet Cells Support Efficient HAV‐ and HCV‐RNA Replication
Our first aim was to establish a cell‐culture system supporting robust HAV‐ and HCV‐RNA replication, thereby allowing comparative investigation of both viruses. To this end, we used subgenomic HAV and HCV firefly luciferase reporter replicons (Fig. 1A) and measured their replication efficiency by quantifying luciferase reporter activity upon electroporation into various human liver‐derived cell lines. HCV gt2a (JFH) replicated with high efficiency in the human hepatoma cell line, Huh7, and its subclones, Huh7‐Lunet and Huh7.5, whereas HCV gt1 (Con1, H77) RNA replication was highest in Huh7‐Lunet cells and far less efficient in Huh7 and Huh7.5 cells, as reported before15 (Fig. 1B). HAV replicated with similar efficiency in all three cell lines and luciferase activity levels were comparable to gt1 in Huh7‐Lunet, suggesting that this cell clone was well suited for comparative analyses of HCV‐ and HAV‐RNA replication. The human hepatoma cell line, Huh6, only supported RNA replication of HAV and HCV gt2a (JFH), though to a much lesser extent than Huh7‐derived cell lines (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). No transient replication of HAV or HCV replicons could be observed in HepG2, HepaRG, or FrHK4 (data not shown). Furthermore, we performed a colony‐formation assay (CFA) using selectable replicons, which is also well established as a quantitative measure of HCV replication.20 However, albeit replicating with similar efficiency in the transient model, HCV Con1 was 100‐fold more efficient in establishing single‐cell clones (Fig. 1C). This hinted to a higher capability of the HCV replicase to establish persistent replication, as observed in vivo, but precluded the CFA for comparative analysis of HCV and HAV replication efficiency.
Figure 1.
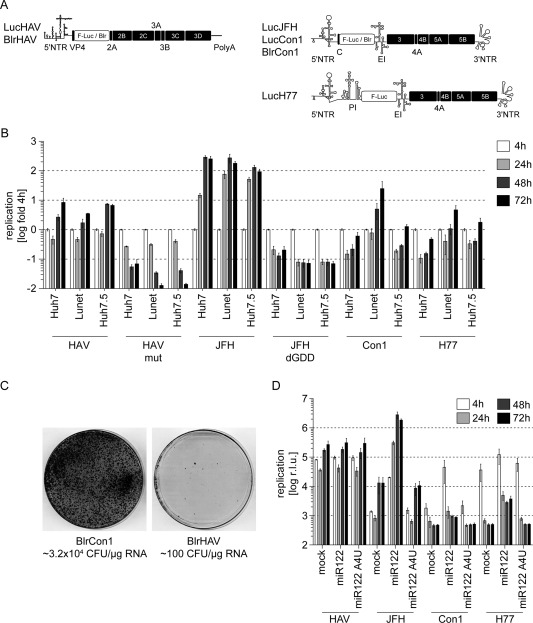
Huh7‐Lunet cells support efficient HAV‐ and HCV‐RNA replication. (A) Schematic representation of subgenomic replicon constructs used for comparative analyses of HAV‐ and HCV‐RNA replication. Boxes represent coding sequences from HAV or HCV (black) or reporter genes / selectable markers (white). Cis‐acting elements are depicted according to their proposed secondary structures. (B) Replication fitness of HAV or HCV subgenomic replicons in Huh7, Huh7‐Lunet, or Huh7.5 hepatoma cells. 4, 24, 48, and 72 hours after electroporation of HAV or HCV reporter replicons, cells were harvested for measurement of luciferase activity. Graph represents mean and standard deviation (SD) of r.l.u. normalized to 4 hours. Data are derived from two independent experiments and depicted in logarithmic scale (n = 2). (C) Huh7‐Lunet cells were transfected with 0.1‐1.0 µg of RNA of selectable subgenomic replicons of HCV (BlrCon1) or HAV (BlrHAV) and selected for blasticidin resistance (2.5 µg/mL). Colonies were stained with Coomassie brilliant blue and colony number per µg RNA was calculated (n = 2). (D) HCV‐, but not HAV‐RNA, replication depends on miR122. Hep3B hepatoma cells, naturally devoid of endogenous miR122, were electroporated with HAV or HCV subgenomic replicons either alone or in combination with miR122 or the inactive mutant, miR122_A4U. Graph shows mean and SD of r.l.u. measured from two independent experiments performed with triplicates (n = 2) at the given time points after electroporation. Data are represented in logarithmic scale and were not normalized owing to the effect of miR122 on HCV‐RNA stability and translation efficiency, which is evident already 4 hours after transfection. Note that owing to presence of the PI upstream of the luciferase coding sequence in the H77 reporter replicon construct, luciferase translation is independent of miR122. Abbreviations: Blr, blasticidin resistance (blasticidin‐S‐deaminase); CFU, colony forming units; EI, encephalomyocarditisvirus IRES; F‐Luc, firefly luciferase; IRES, internal ribosome entry side; PI, poliovirus IRES; r.l.u., relative light units.
Establishment of HCV replication is highly dependent on liver‐specific miR122. Although comparison of several published genome sequences revealed that the HAV genome does not contain any conserved miR122‐binding sites, we tested whether HAV indirectly depended on miR122. Therefore, we first used the human hepatoma cell line, Hep3B, lacking miR122 and measured HAV and HCV RNA upon coelectroporation of the different replicons with or without miR122 or a defective miR122 mutant (miR122_A4U; Fig. 1D). HAV replication was robust in Hep3B cells, whereas HCV‐RNA replication was strongly dependent on miR122 coelectroporation (Fig. 1D), indicating that HAV‐RNA replication does not require miR122. Because HCV gt1 replication was very low in Hep3B, even in the presence of miR122, we confirmed these results by using a miR122 antagonist (mirVana) in Huh7‐Lunet. Again, all HCV isolates were highly dependent on this miRNA, whereas HAV was not affected by miR122 sequestration (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). In summary, HAV and HCV are able to replicate in an identical set of human hepatoma cell lines (Huh7, Huh6, and Hep3B). However, in contrast to HCV, HAV‐RNA replication was independent of miR122. Because RNA replication of HAV and all HCV isolates proved to be robust and comparable in Huh7‐Lunet cells, we used this cell line for further comparative studies of the two viruses.
HAV and HCV Replicases Possess a Similar IFN Sensitivity
Given that HAV infections are always cleared, whereas HCV infections are associated with viral persistence, we were wondering whether this discrepancy might be, at least partially, explained by differences in their IFN sensitivity. To address this point, we stimulated innate immune responses by addition of type I, II, or III IFNs. HAV and HCV gt1 and gt2a RNA replication were inhibited with similar efficiency by IFN‐α (half maximal inhibitory concentration [IC50]: 3.50‐5.14 IU/mL), IFN‐λ (IC50, 0.07‐0.14 ng/mL), and IFN‐γ (IC50, 0.02‐0.07 ng/mL; Fig. 2A‐C; Table 1). This indicates a comparable accessibility of viral replication complexes to ISGs and a lack of, or an equivalent degree of, countermeasures by the viral nonstructural proteins against this host defense.
Figure 2.

HAV‐ and HCV‐RNA replicases have a similar IFN sensitivity. HAV or HCV (JFH, Con1, and H77) luciferase reporter replicons were electroporated into Huh7‐Lunet cells. First, 4‐hour p.t. medium was exchanged and replaced with DMEM containing the indicated concentrations of (A) IFN‐α (type I), (B) IFN‐λ (type III), or (C) IFN‐γ (type II). Then, 48‐hour p.t. cells were lysed and luciferase activity was determined. Depicted are mean and standard deviation (SD) of r.l.u. normalized to untreated controls. Data are derived from triplicates of two independent experiments and shown in logarithmic scale (n = 2). Abbreviations: p.t., post‐transfection; r.l.u., relative light units.
Table 1.
Compounds Used for Inhibition of HCV‐ or HAV‐RNA Replication and Corresponding IC50 Values
| LucHAV | LucJFH | LucCon1 | LucH77 | |
|---|---|---|---|---|
| Inhibitor | IC50 (95% CI) | IC50 (95% CI) | IC50 (95% CI) | IC50 (95% CI) |
| IFN‐α, IU/mL | 3.50 (2.54‐4.83) | 5.14 (3.95‐6.69) | 2.60 (2.03‐3.32) | 2.58 (2.06‐3.24) |
| IFN‐λ, ng/mL | 0.10 (0.03‐0.37) | 0.14 (0.07‐0.30) | 0.07 (0.03‐0.18) | 0.77 (0.04‐0.16) |
| IFN‐γ, ng/mL | 0.03 (0.03‐0.04) | 0.07 (0.06‐0.08) | 0.02 (0.02‐0.03) | 0.02 (0.02‐0.03) |
| Silibinin, µM | 39.05 (35.65‐42.76) | n.a. | n.a. | n.a. |
| CsA, µM | 5.40 (4.32‐6.75) | 1.36 (1.32‐1.41) | <1.00 | 0.79 (0.71‐0.87) |
| NIM811, µM | 6.77 (4.58‐10.03) | <1.00 | <1.00 | <1.00 |
| SFA, µM | 22.99 (18.98‐27.86) | 6.7 (6.31‐7.19) | <4.00 | 3.16 (2.71‐3.69) |
| Reversan, µM | 6.96 (6.01‐8.08) | n.a. | 2.67 (2.40‐2.972) | 3.11 (2.73‐3.54) |
| Piperine, µM | 28.21 (25.46‐31.26) | n.a. | 22.78 (21.44‐24.20) | n.a. |
| FK506, µM | 6.86 (6.37‐7.40) | n.a. | n.a. | n.a. |
Abbreviations: 95% CI, 95% confidence interval, n.a., not applicable.
HAV, but Not HCV‐RNA Replication, Is Independent of PI4KIII α and β
HCV‐RNA replication strictly depends on PI4KIIIα,9 whereas for several members of the Picornaviridae family, as well as for HCV gt1a and 1b, a requirement for PI4KIIIβ has been reported.8, 11 To clarify, whether one of the two PI4KIII isoforms might be involved in HAV‐RNA replication, we utilized the small‐molecule inhibitors, AL‐9 or PIK93, specifically targeting PI4KIIIα or PI4KIIIβ, respectively.10, 11 As expected, AL‐9 strongly inhibited replication of all HCV isolates in absence of cytostatic effects (Fig. 3A and http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). However, HAV remained unaffected by AL‐9, arguing against a role of PI4KIIIα for HAV‐RNA replication (Fig. 3A). This notion was confirmed in Huh7‐Lunet cells stably expressing a shRNA targeting PI4KIIIα expression (shPI4KIIIα; Fig. 3B). To assess whether HAV‐RNA replication depended on PI4KIIIβ, we measured sensitivity of HAV‐ and HCV‐RNA replication to the PI4KIIIβ inhibitor, PIK93. Interestingly, PIK93 treatment resulted in a dose‐dependent and strong inhibition of HCV gt1b (Con1) replication, whereas HCV gt1a and gt2a as well as HAV‐RNA replication remained almost unaffected (Fig. 3C). Only the highest concentration of PIK93 (10 µM) caused a slight reduction in HAV and JFH replication, possibly owing to mild cytostatic effects of the drug at this concentration (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). Additionally, to thoroughly assess a potential involvement of PI4KIIIβ in HAV replication, we applied the CRISPR‐CAS9 technique based on a guideRNA targeting PI4KIIIβ to establish PI4KIIIβ‐knockout cells (Fig. 3D). One cell clone (Clone C1) had a complete knockout of PI4KIIIβ and was therefore chosen for further studies, as well as a cell pool characterized by reduced PI4KIIIβ levels (Pool3) to control for clonal effects (Fig. 3D). HCV gt2a and HAV replication differed only slightly between parental cells, Clone C1 and Pool3 (Fig. 3E), confirming the independence of HAV and JFH replication from PI4KIIIβ. HCV gt1b replication was tremendously impaired in cell lines with a reduced expression of PI4KIIIβ, in contrast to HCV gt2a and in line with data gained from PIK93 treatment (Fig. 3E). HCV gt1a RNA replication was drastically reduced in Clone C1 and in Pool3 as well, hinting at a role of PI4KIIIβ for gt1 in general (Fig. 3E). In conclusion, HCV gt2a RNA replication strongly depended on PI4KIIIα, whereas HCV gt1b and, most likely, gt1a RNA replication relied on both kinases. In contrast to HCV and other picornaviruses, neither PI4KIIIα nor PI4KIIIβ are required for HAV‐RNA replication.
Figure 3.
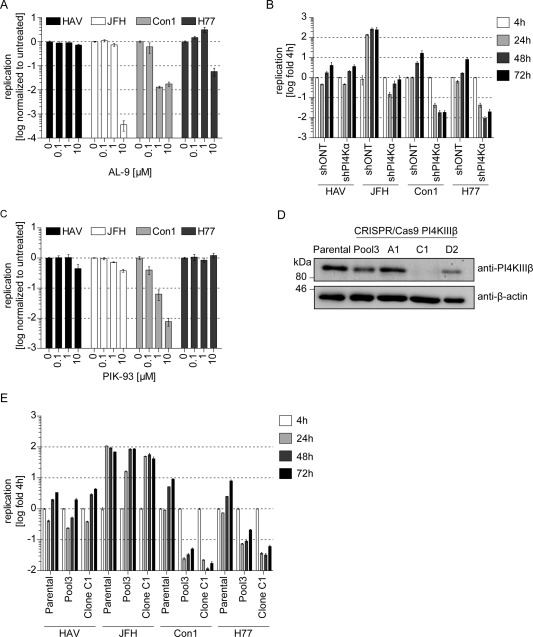
HAV‐RNA replication is independent of PI4KIIIα or β. (A) HAV‐ and HCV‐RNA replication upon treatment with PI4KIIIα inhibitor AL‐9. Huh7‐Lunet cells were electroporated with subgenomic replicon RNA of HAV or HCV (JFH, Con1, and H77) and treated with indicated concentrations of PI4KIIIα inhibitor AL‐9 4 hours p.t. Then, 48 hours upon electroporation, cells were lysed for measurement of luciferase activity. Data are derived from two independent experiments performed with triplicates and normalized to untreated controls. Graph depicts mean and standard deviation (SD) in logarithmic scale (n = 2). (B) HAV‐ and HCV‐RNA replication in PI4KIIIα knockdown cells. HAV or HCV subgenomic replicon RNAs were transfected into Huh7‐Lunet cells stably expressing either a nontargeting shRNA (shONT) or a PI4KIIIα‐specific shRNA (shPI4KIIIα). Then, 4, 24, 48, and 72 hours p.t., cells were lysed to determine luciferase activity. Graph depicts mean and SD of two independent experiments performed with triplicates. Data were normalized to the 4‐hour value and are presented in logarithmic scale (n = 2). (C) HAV‐ and HCV‐RNA replication upon treatment with PI4KIIIβ inhibitor PIK93. Cells were treated with PIK93 and analyzed as described in (A). (D) Western blot analysis of PI4KIIIβ in CRISPR/Cas9‐PI4KIIIβ cells. Parental cells, a pool of CRISPR/Cas9‐PI4KIIIβ cells (Pool3) and three single‐cell clones (A1, C1, and D2), were examined for PI4KIIIβ protein expression by using a mouse monoclonal anti‐PI4KIIIβ antibody. (E) HAV‐ and HCV‐RNA replication in CRISPR/Cas9‐PI4KIIIβ cells. Parental cells, Pool3, or Clone C1 cells were transfected with HAV or HCV subgenomic replicon RNA. Then, 4, 24, 48, and 72 hours upon electroporation, cells were harvested to measure luciferase activity. Graph depicts mean and SD of a representative experiment (n = 2). Data were normalized to the 4‐hour value and are presented in logarithmic scale. Abbreviations: kDa, kilodaltons; p.t., post‐transfection.
HCV‐ and HAV‐RNA Replication Is Sensitive to CsA, but Involves a Differential Set of Target Proteins
CypA, a member of the immunophilin family of peptidyl‐prolyl cis trans isomerases, represents another cellular factor involved in HCV replication. Hence, we probed whether HAV also depended on members of the immunophilin family and therefore analyzed inhibition of viral RNA replication by various immunophilin inhibitors. As shown before, CsA, a cyclic undecapeptide inhibiting a wide range of Cyps,12, 21 blocked RNA replication of all HCV isolates in a dose‐dependent manner (Fig. 4A). In addition CsA inhibited HAV replication, albeit with lower efficiency, indicating that different immunophilins might be involved in HCV and HAV replication, respectively (Fig. 4A). To analyze the dependency of HAV on CypA and other Cyps, we used NIM811, a nonimmunosuppressive CsA analog with similar structure,12, 22 and SFA, a structurally unrelated Cyp inhibitor,12 which is unlikely to have further common targets with CsA and NIM811 apart from Cyps. Furthermore, we applied specific knockdown of CypA, which is the Cyp most important for HCV. As expected, NIM811 inhibited HCV replication with even higher efficiency than CsA, whereas its effect on HAV replication was comparable to CsA‐induced inhibition (Fig. 4B; Table 1 and http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). In contrast, SFA treatment inhibited all HCV isolates to a similar extent as CsA and NIM811, but resulted only in a marginal decrease of HAV replication. This suggested either that HAV utilized immunophilins, which were not efficiently inhibited by SFA, or that HAV replication might be independent from cyclophilins (Fig. 4C; Table 1 and http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). Indeed, HCV replication was dramatically reduced for all HCV isolates in shCypA cells,16 whereas HAV replicated with equal efficiency in shCypA and shONT cells, arguing against a role of CypA for HAV‐RNA replication (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). Given that CsA not only inhibits Cyps, but also ABC transporters,23 we implemented further inhibitors to narrow down potential targets involved in the CsA‐mediated inhibition of HAV replication. PSC833 is a CsA analog not blocking Cyp activity, but still inhibiting ABC transporters such as p‐glycoprotein (ABCB1).12, 23 Reversan and Piperine inhibit p‐glycoprotein and other ABC transporters without being structurally related to CsA.13, 14
Figure 4.
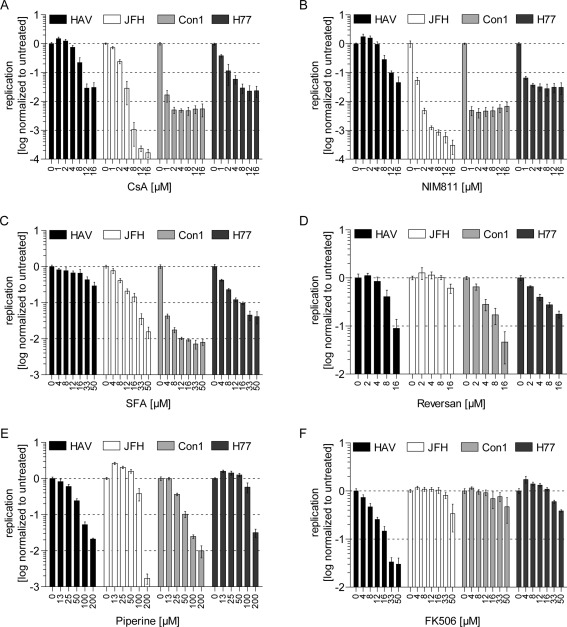
HAV‐ and HCV‐RNA replication are sensitive toward an overlapping set of inhibitors. (A‐F) Huh7‐Lunet cells were electroporated with subgenomic HAV or HCV (JFH, Con1, and H77) luciferase reporter replicons. First, 4 hours p.t., different compounds were added at indicated concentrations. Then, 48 hours p.t., cells were lysed and luciferase activity was measured. Data are derived from two independent experiments performed with triplicates and depicted in logarithmic scale. Graphs present mean and standard deviation of r.l.u. normalized to untreated controls (n = 2). Abbreviations: p.t., post‐transfection; r.l.u., relative light units.
Indeed, PSC833 inhibited HAV and not HCV replication (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo), but strong cytostatic effects on Huh7‐Lunet questioned the specificity of the inhibition (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). However, the involvement of certain ABC transporters in HAV‐RNA replication was further supported by the dose‐dependent inhibition upon treatment with Reversan (Fig. 4D) and Piperine (Fig. 4E; Table 1 and http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo), which exerted less‐pronounced cytostatic effects (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). Interestingly, Piperine and Reversan additionally inhibited HCV isolate Con1 and the latter, also to a lesser extent, H77, indicating that ABC transporters might play a role for HCV gt1 as well (Fig. 4D,E; Table 1 and http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo). Owing to the large number of human ABC transporters (49 encoded in the human genome), most of which are poorly characterized,24 and considering the lack of specific inhibitors, we did not undertake further efforts to identify distinct candidates. Instead, we focused on FK506‐binding proteins (FKBPs), another class of immunophilins with peptidyl‐prolyl cis trans isomerase activity, which can be inhibited by the immunosuppressant, FK506 (Tacrolimus).21 We observed a dose‐dependent and strong inhibition of HAV‐RNA replication by FK506 at noncytostatic concentrations, whereas HCV replication was not affected by this drug, suggesting that HAV‐RNA replication depends on FKBPs rather than Cyps (Fig. 4F).
In summary, we confirmed the strong dependence of HCV‐RNA replication on CypA and found evidence for a role of ABC transporters in HCV gt1(b) RNA replication. In contrast, HAV replication appears to depend on ABC transporters and FKBPs, but not on Cyps.
Limited Competition Between HAV and HCV Replicases
Thus far, our data revealed no individual common host factor of HCV and HAV replication, but rather pointed to a related set of functions exploited by both viruses. To get a general overview on cellular resources occupied by HCV and HAV, we examined viral competition by cotransfecting firefly luciferase (LucHAV, LucJFH) and renilla luciferase (R‐LucJFH) reporter replicons into Huh7‐Lunet cells (Fig. 5A). In case of largely overlapping sets of host factors, we expected a strong interference between both replicons, whereas a lack of interference would indicate that both viruses replicate in different subcellular niches. Given that JFH1 replication was much more efficient and faster than HAV, we included a JFH1 replicon containing a chimeric 3’ nontranslated region (NTR) that attenuates replication efficiency.18 Indeed, the presence of wild‐type (wt) JFH strongly suppressed HAV‐RNA replication, which remained unaffected, when combined with the replication‐deficient control, JFHdGDD (Fig. 5B). In contrast, R‐LucJFH RNA replication was not affected by coelectroporation of HAV RNA (Fig. 5C). However, the JFH chimera (R‐LucJFH X‐tail Con1/JFH X‐tail Con1), with reduced replication fitness, only marginally impaired HAV‐RNA replication (Fig. 5B), arguing against largely overlapping sets of limiting host factors between the two viruses. This result rather suggested limitations of global cellular resources in the presence of JFH wt, which reaches maximal replication levels already at 24 hours after transfection (Fig. 5C). In support of this view, JFH wt had a stronger relative impact on replication of R‐LucJFH X‐tail Con1 than on HAV, probably owing to the additional competition for limiting host factors and replication within the same subcellular compartment (Fig. 5B,D). To confirm that cells indeed were transfected with both replicons, we performed immunofluorescence (IF) analysis (Fig. 5E). Almost all cells were positive for HCV nonstructural (NS) protein 5A (NS5A), whereas only a few cells stained positive for HAV, most likely owing to lower replication levels of HAV and the limited sensitivity of the anti‐3C antibody. However, several cells were clearly double positive for HAV and HCV, confirming that both replicons were able to replicate within the same cell and corroborating the notion of only limited competition between viral replicases (Fig. 5E). In support of this assumption, HCV NS5A and HAV 3C also did not colocalize substantially in double‐positive cells, which was further confirmed in an expression model using HCV NS3‐5B and HAV 2A‐3D (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo,C), also showing that HAV 3C primarily colocalized with mitochondria (http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo,C).
Figure 5.
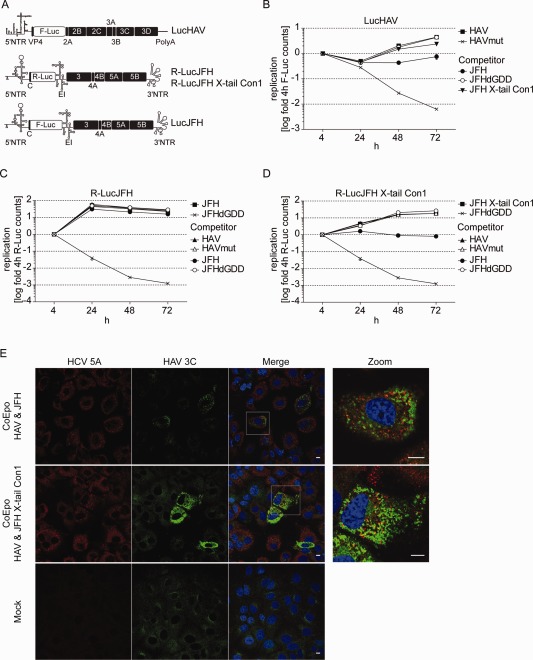
Low level of viral competition argues against a strong overlap of host factors between HAV and HCV. (A) Schematic representation of subgenomic reporter replicon constructs used for coelectroporation. The R‐LucJFH X‐tail Con1 chimera is characterized by reduced replication efficiency. (B‐D) Coelectroporation of HAV and HCV points to a low level of viral replicase competition. Huh7‐Lunet cells were either electroporated with single‐reporter replicons or coelectroporated with different combinations of HAV and HCV subgenomic reporter replicons (CoEpo). To distinguish between different coelectroporated constructs, firefly and renilla luciferase reporter replicons were combined. Then, 4, 24, 48, and 72 hours upon electroporation, firefly and renilla luciferase activity were determined, normalized to the 4‐hour value, and depicted in logarithmic scale (B: firefly luciferase activity; C and D: renilla luciferase activity). Graph depicts mean and standard deviation of a representative experiment (n = 2). (B) LucHAV, (C) R‐LucJFH, (D) R‐LucJFH X‐tail Con1, (E) IF analysis of Huh7‐Lunet cells 72 hours upon coelectroporation with HAV and HCV replicon constructs (upper two panels) or mock electroporation (lower panel) using mouse monoclonal anti‐HCV‐NS5A antibodies and a rabbit polyclonal anti‐HAV‐3C serum. Right panel represents a magnification of the boxed areas. Scale bar, 10 µm. Abbreviations: F‐Luc/Luc, firefly luciferase; R‐Luc, renilla luciferase.
HCV and HAV Are Sensitive to Different Formulations of Silibinin
Silibinin, a flavonolignan isolated from the milk thistle, Sylibum marianum, has been reported to exert hepatoprotective effects,25 and, in contrast to the oral formulation, the IV formulation, Legalon‐SIL, has been shown to inhibit HCV gt1 replication in vitro and in vivo.18 We wanted to address whether HAV replication could also be inhibited by this compound. However, HAV‐RNA replication was fully resistant to SIL, whereas HCV gt1a and gt1b replication, in contrast to gt2a, were efficiently suppressed (Fig. 6A), as published earlier.18 Surprisingly, the oral formulation, silibinin, blocked HAV‐, but not HCV‐RNA, replication with an IC50 value of 18.84 µg/mL, corresponding to 39.05 µM (Fig. 6B; Table 1), suggesting that oral silibinin might represent a potential treatment option for HAV‐infected patients.
Figure 6.
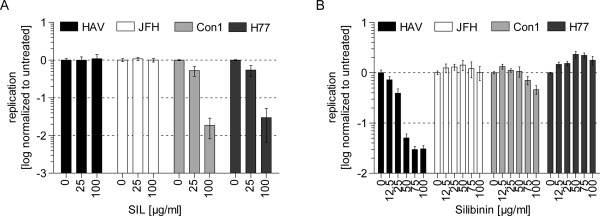
HAV‐ and HCV‐RNA replication can be inhibited by flavonolignans. Huh7‐Lunet cells were electroporated with HAV or HCV (JFH, Con1, and H77) reporter replicons. First, 4 hours p.t., medium was replaced by DMEM containing SIL (A) or silibinin (B) at indicated concentrations. Then, 48 hours p.t., cells were harvested for measurement of luciferase activity. Graph shows mean and standard deviation of triplicate values of two independent experiments (n = 2). Values are normalized to untreated controls and depicted in logarithmic scale. Abbreviations: p.t., post‐transfection.
Discussion
In search for a suitable cell‐culture model for comparative analysis of HAV and HCV, we identified Huh7‐Lunet cells, originally selected for optimal HCV‐RNA replication, as a cell line, supporting HAV replication to similar levels as replication of HCV gt1 reporter replicons. However, in a CFA, reflecting the ability to establish persistent replication, HCV replicons were substantially more efficient. Although we cannot rule out an impact of the differing replicon design, this result indicates that the replicases of both viruses might have a different capability to maintain long‐term replication in the same cells or that persistent HAV replication exerts cytopathic effects. It will be the subject of future studies to see how these phenotypes in cell culture contribute to the differing natural history of infection in vivo. Furthermore, availability of a homogenous cell‐culture system for both viruses will enable studies of activation and blockade of innate immune responses by mitochondrial antiviral‐signaling protein (MAVS) and TIR‐domain–containing adapter‐inducing IFN‐β (TRIF) cleavage, which is achieved by proteases of HAV and HCV.26 Despite being capable of blocking innate immune responses, HCV induces high ISG levels in acute and chronic infection, preferentially in infected cells,27, 28 whereas almost no ISGs are induced in livers of HAV‐infected chimpanzees,2 suggesting that HAV either has a lower capability of stimulating innate immunity or is more efficient in cleaving the critical adaptor proteins. However, induction of ISGs seems to be a factor promoting persistence, given that we found very similar sensitivity of both replicases to type I, II, and III IFNs, in line with studies on HCV and HAV infection in vitro.29, 30 This seems somehow counterintuitive, but reducing antigen loads by a well‐dosed induction of ISGs in the case of HCV might dampen efficiency of recognition by the adaptive immune system, thereby preventing eradication of infected cells.
PI4KIIIα is an essential host factor for HCV replication,31 whereas PI4KIIIβ plays an important role for the life cycle representatives of the Picornaviridae family (genera enterovirus and kobuvirus) and HCV gt1.31 Our results now indicate that HAV‐RNA replication, in contrast to HCV and other picornaviruses, is independent of PI4KIIIα and PI4KIIIβ, corroborating the notion of its unique position among Picornaviridae members32 and in line with a previous report.30
Immunophilins comprise two large groups of peptidyl‐prolyl cis trans isomerases, namely, Cyps and FKBPs, which play a role in the life cycle of many different viruses. There are at least 16 different human Cyps and 12 human FKBPs, which are involved in various cellular processes.21 Furthermore, Cyps are involved in replication of a wide range of viruses, including human immunodeficiency virus, influenza virus, hepatitis B virus, vesicular stomatitis virus, and several coronaviruses,21 whereas also FKBPs play a role for the latter.33 Furthermore, CypA represents an essential host factor for HCV.16 We assessed the dependency of HAV on different immunophilin members by usage of a diverse set of inhibitors. From the inhibition profiles, we deduced that most Cyps are not involved in HAV‐RNA replication. Still, we cannot exclude Cyps being utilized, which are not blocked or inhibited, to a lesser extent, by the inhibitors. However, it seems more likely that the block of HAV replication by CsA and structurally related compounds is a result of inhibition of ABC transporters such as p‐glycoprotein,34 which are additional targets of CsA, but most likely not of SFA.35 Furthermore, the inhibitory effect of Reversan and Piperine, two more specific inhibitors of ABC transporters, supports the concept of a potential role of ABC transporters for HAV‐RNA replication. Given that HAV replication is inhibited by nonimmunosuppressive CsA analogs, which are in clinical development for HCV therapy,36 such compounds might represent a future treatment option for HAV as well.
The herbal compound, silibinin, which is the main constituent of the milk thistle extract, silymarin, is widely consumed because of its hepatoprotective properties. Recently, it became apparent that the IV formulation, Legalon‐SIL, exerts antiviral activity against HCV,37 likely by targeting formation of viral replication sites.18 We observed no such activity against HAV replication. However, the oral formulation, silibinin, efficiently inhibited HAV‐, but not HCV‐RNA, replication, suggesting different modes of action for oral silibinin and Legalon‐SIL. Importantly, silibinin, which does not induce significant side effects, even in high doses (up to 2.1 g/day for 24 weeks),38 might therefore represent a safe, effective, and cheap treatment option for HAV infections. Indeed, protection from HAV infection might have contributed to the proverbial hepatoprotective effects of silibinin. However, in vivo potency of silibinin has to be confirmed in a cell‐based virus assay and, in particular, in clinical studies. In addition, it would be interesting to clarify its mechanism of action and analyze occurrence of viral resistance.
In conclusion, we found evidence for possible new host factors for HAV‐RNA replication (FKBP and ABC transporters) and identified oral silibinin as a potential new antiviral for treatment of HAV infection. Furthermore, we have established and characterized a transient replication model for HCV and HAV, allowing a side‐by‐side comparison of both viruses with similar efficiency. Future studies employing full‐length viruses and focusing on the differential ISG induction by HAV and HCV will shed light on the high capability of HCV to establish chronic infections and help to understand general features of viral persistence.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo.
Supporting Information
Acknowledgment
The authors especially thank Rahel Klein and Ulrike Herian for excellent technical assistance and Ji Young Lee for help with analysis of confocal images. The authors are grateful to F. Peri, R. de Francesco, and P. Neddermann for providing AL‐9. The authors thank Novartis for supply of NIM811 and sanglifehrin A. Huh7.5 cells were a generous gift from Charlie Rice; HCV isolates H77 and JFH1 were provided by Jens Bukh and T. Wakita, respectively.
Potential conflict of interest: Nothing to report.
This project was funded, in part, by grants from the Deutsche Forschungsgemeinschaft (LO 1556/1‐2 and FOR1202, TP3).
References
- 1. Thimme R, Binder M, Bartenschlager R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol Rev 2012;36:663‐683. [DOI] [PubMed] [Google Scholar]
- 2. Lanford RE, Feng Z, Chavez D, Guerra B, Brasky KM, Zhou Y, et al. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc Natl Acad Sci U S A 2011;108:11223‐11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. FitzSimons D, Hendrickx G, Vorsters A, Van Damm P. Hepatitis A and E: update on prevention and epidemiology. Vaccine 2010;28:583‐588. [DOI] [PubMed] [Google Scholar]
- 4. Fitzgerald M, Thornton L, O'Gorman J, O'Connor L, Garvey P, Boland M, et al. Outbreak of hepatitis A infection associated with the consumption of frozen berries, Ireland, 2013—linked to an international outbreak. Euro Surveill 2014;19(43). pii: 20942. [DOI] [PubMed] [Google Scholar]
- 5. Collier MG, Khudyakov YE, Selvage D, Adams‐Cameron M, Epson E, Cronquist A, et al. Outbreak of hepatitis A in the USA associated with frozen pomegranate arils imported from Turkey: an epidemiological case study. Lancet Infect Dis 2014;14:976‐981. [DOI] [PubMed] [Google Scholar]
- 6. Vento S. Fulminant hepatitis associated with hepatitis A virus superinfection in patients with chronic hepatitis C. J Viral Hepat 2000;7(Suppl 1):7‐8. [DOI] [PubMed] [Google Scholar]
- 7. Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol 2013;11:482‐496. [DOI] [PubMed] [Google Scholar]
- 8. Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010;141:799‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reiss S, Rebhan I, Backes P, Romero‐Brey I, Erfle H, Matula P, et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011;9:32‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bianco A, Reghellin V, Donnici L, Fenu S, Alvarez R, Baruffa C, et al. Metabolism of phosphatidylinositol 4‐kinase IIIalpha‐dependent PI4P Is subverted by HCV and is targeted by a 4‐anilino quinazoline with antiviral activity. PLoS Pathog 2012;8:e1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Borawski J, Troke P, Puyang X, Gibaja V, Zhao S, Mickanin C, et al. Class III phosphatidylinositol 4‐kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J Virol 2009;83:10058‐10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaither LA, Borawski J, Anderson LJ, Balabanis KA, Devay P, Joberty G, et al. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology 2010;397:43‐55. [DOI] [PubMed] [Google Scholar]
- 13. Burkhart CA, Watt F, Murray J, Pajic M, Prokvolit A, Xue C, et al. Small‐molecule multidrug resistance‐associated protein 1 inhibitor reversan increases the therapeutic index of chemotherapy in mouse models of neuroblastoma. Cancer Res 2009;69:6573‐6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhardwaj RK, Glaeser H, Becquemont L, Klotz U, Gupta SK, Fromm MF. Piperine, a major constituent of black pepper, inhibits human P‐glycoprotein and CYP3A4. J Pharmacol Exp Ther 2002;302:645‐650. [DOI] [PubMed] [Google Scholar]
- 15. Binder M, Quinkert D, Bochkarova O, Klein R, Kezmic N, Bartenschlager R, et al. Identification of determinants involved in initiation of hepatitis C virus RNA synthesis by using intergenotypic replicase chimeras. J Virol 2007;81:5270‐5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, et al. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog 2009;5:e1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gauss‐Muller V, Kusov YY. Replication of a hepatitis A virus replicon detected by genetic recombination in vivo. J Gen Virol 2002;83(Pt 9):2183‐2192. [DOI] [PubMed] [Google Scholar]
- 18. Esser‐Nobis K, Romero‐Brey I, Ganten TM, Gouttenoire J, Harak C, Klein R, et al. Analysis of hepatitis C virus resistance to silibinin in vitro and in vivo points to a novel mechanism involving nonstructural protein 4B. Hepatology 2013;57:953‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmitt M, Scrima N, Radujkovic D, Caillet‐Saguy C, Simister PC, Friebe P, et al. A comprehensive structure‐function comparison of hepatitis C virus strain JFH1 and J6 polymerases reveals a key residue stimulating replication in cell culture across genotypes. J Virol 2011;85:2565‐2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lohmann V. Hepatitis C virus RNA replication. Curr Top Microbiol Immunol 2013;369:167‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frausto SD, Lee E, Tang H. Cyclophilins as modulators of viral replication. Viruses 2013;5:1684‐1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gallay PA. Cyclophilin inhibitors. Clin Liver Dis 2009;13:403‐417. [DOI] [PubMed] [Google Scholar]
- 23. Watanabe T, Kokubu N, Charnick SB, Naito M, Tsuruo T, Cohen D. Interaction of cyclosporin derivatives with the ATPase activity of human P‐glycoprotein. Br J Pharmacol 1997;122:241‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2008;9:105‐127. [DOI] [PubMed] [Google Scholar]
- 25. Loguercio C, Festi D. Silybin and the liver: from basic research to clinical practice. World J Gastroenterol 2011;17:2288‐2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martin A, Lemon SM. Hepatitis A virus: from discovery to vaccines. Hepatology 2006;43(2 Suppl 1):S164‐S172. [DOI] [PubMed] [Google Scholar]
- 27. Wieland S, Makowska Z, Campana B, Calabrese D, Dill MT, Chung J, et al. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 2014;59:2121‐2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, Ploss A, et al. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe 2014;15:190‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, et al. Complete replication of hepatitis C virus in cell culture. Science 2005;309:623‐626. [DOI] [PubMed] [Google Scholar]
- 30. Debing Y, Kaplan GG, Neyts J, Jochmans D. Rapid and convenient assays to assess potential inhibitory activity on in vitro hepatitis A replication. Antiviral Res 2013;98:325‐331. [DOI] [PubMed] [Google Scholar]
- 31. Delang L, Paeshuyse J, Neyts J. The role of phosphatidylinositol 4‐kinases and phosphatidylinositol 4‐phosphate during viral replication. Biochem Pharmacol 2012;84:1400‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang X, Ren J, Gao Q, Hu Z, Sun Y, Li X, et al. Hepatitis A virus and the origins of picornaviruses. Nature 2015;517:85‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carbajo‐Lozoya J, Muller MA, Kallies S, Thiel V, Drosten C, von Brunn A. Replication of human coronaviruses SARS‐CoV, HCoV‐NL63 and HCoV‐229E is inhibited by the drug FK506. Virus Res 2012;165:112‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fu J, Tjandra M, Becker C, Bednarczyk D, Capparelli M, Elling R, et al. Potent nonimmunosuppressive cyclophilin inhibitors with improved pharmaceutical properties and decreased transporter inhibition. J Med Chem 2014;57:8503‐8516. [DOI] [PubMed] [Google Scholar]
- 35. Sweeney ZK, Fu J, Wiedmann B. From chemical tools to clinical medicines: nonimmunosuppressive cyclophilin inhibitors derived from the cyclosporin and sanglifehrin scaffolds. J Med Chem 2014;57:7145‐7159. [DOI] [PubMed] [Google Scholar]
- 36. Gallay PA, Lin K. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des Devel Ther 2013;7:105‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Polyak SJ, Oberlies NH, Pecheur EI, Dahari H, Ferenci P, Pawlotsky JM. Silymarin for HCV infection. Antivir Ther 2013;18:141‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fried MW, Navarro VJ, Afdhal N, Belle SH, Wahed AS, Hawke RL, et al. Effect of silymarin (milk thistle) on liver disease in patients with chronic hepatitis C unsuccessfully treated with interferon therapy: a randomized controlled trial. JAMA 2012;308:274‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.27847/suppinfo.
Supporting Information