

Summary
Sensitization of the humoral immune response to invading viruses and production of antiviral antibodies forms part of the host antiviral repertoire. Paradoxically, for a number of viral pathogens, under certain conditions, antibodies provide an attractive means of enhanced virus entry and replication in a number of cell types. Known as antibody‐dependent enhancement (ADE) of infection, the phenomenon occurs when virus‐antibody immunocomplexes interact with cells bearing complement or Fc receptors, promoting internalization of the virus and increasing infection. Frequently associated with exacerbation of viral disease, ADE of infection presents a major obstacle to the prevention of viral disease by vaccination and is thought to be partly responsible for the adverse effects of novel antiviral therapeutics such as intravenous immunoglobulins. There is a growing body of work examining the intracellular signaling pathways and epitopes responsible for mediating ADE, with a view to aiding rational design of antiviral strategies. With in vitro studies also confirming ADE as a feature of infection for a growing number of viruses, challenges remain in understanding the multilayered molecular mechanisms of ADE and its effect on viral pathogenesis.
Keywords: Fc receptors, antibody‐dependent enhancement, virus, intravenous immunoglobulins
This article is part of a series of reviews covering Fc Receptors appearing in Volume 268 of Immunological Reviews.
Introduction
Early stages of the viral infection cycle require attachment and entry of viruses into the host cell. Classically, virus attachment is mediated by the specific binding of viral surface proteins to host cell receptor molecules, concentrating virus at the cell plasma membrane. Viruses are often studded with one or multiple surface proteins, each potentially containing numerous subdomains, to promote interaction with cellular receptors and stimulate virus entry into the cell. However, the virus surface is also a highly antigenic structure that can trigger cellular and humoral immune responses that eradicate virus in the host. Antibodies contribute to several levels of antiviral defense effectively to neutralize the virus and reduce infectivity. They neutralize infection by targeting viral glycoproteins (GP) of enveloped viruses or the protein shell of non‐enveloped viruses. In enveloped viruses, they may either block virion binding to cellular receptors, interfere with the fusion machinery or aggregate virus particles; in non‐enveloped viruses, they may prevent uncoating of the viral genome in the endosome or promote cytosolic degradation of incoming virions through a tripartite motif‐containing protein 21 proteasome pathway 1. Two models have been proposed to describe the kinetics of neutralization. The first is a single‐hit model, postulating that binding of a single antibody molecule to a critical site on the virion is sufficient to neutralize the virus, while the second, more widely accepted, multi‐hit model proposes that neutralization is only achieved once an individual virion is bound by a number of antibodies which exceeds the stoichiometric threshold of neutralization 2. As the number of available epitopes for a specific antibody on the virion is known, the stoichiometry of neutralization can be calculated. However, the stoichiometric threshold of neutralization is determined by antibody affinity and epitope accessibility. For example, antibodies specific to poorly accessible epitopes require a higher concentration to exceed the occupancy threshold for neutralization 3.
Paradoxically however, for some time it has been recognized that sub‐neutralizing concentrations of antibodies, under certain conditions, can act to enhance viral infection by aiding viral entry into target cells. This mechanism of improved virus uptake, termed antibody‐dependent enhancement (ADE) of infection, is thought to occur when virus is bound by non‐neutralizing antibodies or sub‐neutralizing concentrations of antibodies that facilitate cell entry in an Fcγ receptor (FcγR)‐dependent manner. Interaction of the protruding antibody Fc of the antibody–virus immunocomplex with the FcγR on myeloid cells such as monocytes, macrophages, dendritic cells (DCs), and certain granulocytes typically leads to phagocytosis, resulting in increased numbers of infected cells – so‐called extrinsic ADE. This form of ADE therefore requires prior sensitization of the humoral immune response, whereby circulating antibodies produced during primary infection recognize and bind to a heterologous serotype of the virus and enhance viral infectivity through internalization of virus–antibody immunocomplexes by cells bearing FcγR, rather than promoting viral neutralization. Once internalized, these immunocomplexes may modulate innate antiviral cells responses to increase virus production substantially in each cell, a process termed intrinsic ADE. Together, extrinsic and intrinsic ADE are thought to prompt the massive release of inflammatory and vasoactive mediators that ultimately contribute to disease severity. Throughout this review we will refer to ADE as the overall process of ADE of infection, and will use the terms ‘intrinsic’ and ‘extrinsic’ when referring specifically to those mechanistic processes.
The first report of ADE was made in 1964 in a study examining arbovirus neutralization by antiviral antibodies 4. A number of viruses from the Flaviviridae and Togaviridae families showed enhanced infectivity when assayed with dilutions of their respective antisera. Later work on fractionated sera confirmed that this enhancement was principally associated with IgG antibodies, which were able to act as a molecular bridge between the virus and FcγR‐expressing cells capable of binding the Fc portion of IgG 5. In a series of reports, Halstead et al. first examined the pathogenic potential of FcγR‐dependent ADE to cause dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) in dengue virus (DENV) infection. Through epidemiological studies and in vivo and in vitro modeling, Halstead et al. established not only the link between severe forms of DENV infection in individuals who have had a previous dengue infection but also the enhanced infectivity of target cells and disease exacerbation caused by DENV antibody opsonins 6, 7, 8. Since then, much of the research surrounding viral ADE has focused on members of the Flaviviridae family, particularly DENV, driven by the lack of a licensed DENV vaccine and effective antiviral agents to treat DENV infection. ADE of infection has been implicated as a major obstacle to vaccine development for a number of viruses that cause a significant human health threat, including DENV.
In 1983, an additional, separate mechanism of extrinsic ADE was identified involving the classical complement pathway. Again identified using a flavivirus, studies found that complement receptor 3 (CR3) was able to mediate IgM‐dependent enhancement of West Nile virus (WNV) infectivity in macrophages 9. Further forms of complement‐mediated ADE were subsequently described in a number of virus‐cell systems including human immunodeficiency virus‐1 (HIV‐1). Enhanced HIV‐1 infection of T‐lymphoblastoid cell lines in the presence of sub‐neutralizing levels of HIV‐specific antibodies was shown to require high levels of CR2 and CD4 expression 10. Although the mechanisms are not thought to be mutually exclusive to enhancing infectivity, complement‐dependent ADE is distinct from FcγR‐dependent ADE. In addition to CR2‐ and CR3‐mediated ADE, a further independent route of complement‐mediated ADE of HIV‐1 infection was shown to require the complement component C1q 11. Interaction of C1q with virus‐antibody immunocomplexes enhanced productive HIV‐1 infection in vitro. A similar mechanism of C1q‐mediated ADE was reported to enhance infection with Ebola virus in non‐monocytic cells by promoting binding virus attachment to the target cell or endocytosis 12. Here, IgG antibodies were reported to bind viral epitopes in close proximity, allowing C1q to bind the Fc portion of the antibodies, with the immunocomplex binding C1q receptors at the cell surface. Enhanced infection of HIV opsonized with complement alone has also been reported.
As with other forms of ADE, complement‐dependent ADE of infection has also been linked to viral pathogenesis. In HIV‐1‐infected patients, levels of enhancing antibody in complement‐restored serum samples correlated with immunosuppression and disease 13. Moreover, a greater variety of cell types express complement receptors than do FcγR, which are generally restricted to cells of the myeloid lineage. It may therefore be possible for complement‐dependent ADE to cause aberrant viral infection of what are typically thought of as non‐target cells. In turn, infection of non‐target cells as a result of complement‐dependent ADE may be a precursor of viral pathogenesis or the atypical and often severe disease symptoms associated with chronic or recurrent viral infection. Indeed, enhanced uptake of human parvovirus B19 into endothelial cells of the myocardium, likely due to ADE mediated by complement factor C1q and its receptor CD93, is associated with acute and chronic inflammatory cardiomyopathies 14. Therefore, despite its strong host tropism for erythroid progenitor cells, complement‐dependent ADE of human parvovirus B19 confers up to a 4000‐fold increase in virus endothelial cell uptake in the presence of human parvovirus B19‐specific antibodies.
ADE is now known to be a feature of infection for many viruses. However, it has proven difficult to rigorously test the occurrence and importance of ADE in vivo and this is an area that requires further work. In addition, little is known about the mechanisms or structural interactions that underlie ADE. FcγR‐dependent ADE is currently thought to be the most commonly used mechanism of ADE among viruses and will be discussed in further detail in this review. Only recently have studies begun to investigate the upstream and downstream molecular signaling events that surround all forms of viral ADE. In light of current data, to be discussed in this review, the effect of these signaling cascades on innate cellular immunity and viral pathogenesis continues to be the focus of research efforts and highlights the recent advances made in the development of permissive model systems with which to examine the role of FcR in mediating viral ADE and FcR‐targeted therapeutic strategies for treatment of viral pathogenesis.
ADE is exploited by a variety of viruses
Arthropod‐borne viruses
The rising rainfall and acceleration of global warming has provided breeding grounds conducive to mosquito vectors, leading to the spread of arthropod‐borne diseases to different parts of the world. Recently, the world has been hit by waves of mosquito‐transmitted diseases caused by alphavirus and flavivirus infections. Arthritogenic alphaviruses such as Chikungunya virus (CHIKV), Ross River virus (RRV), and Sindbis virus (SINV) cause febrile illness and myalgias with polyarthralgias as the clinical hallmark, which can last for months to years after infection 15, 16, 17. RRV and SINV are endemic to Australia and Europe, respectively, while CHIKV causes large outbreaks in many parts of the world including the Americas, Africa, South‐East Asia, India, and Oceania, affecting millions of individuals worldwide 18, 19, 20. Moreover, DENV and WNV are prominent flaviviruses that are major health concerns due to the severity of diseases they cause. DENV has a geographical distribution similar to CHIKV, causing massive worldwide outbreaks, with an estimated 390 million people suffering from DENV infections annually 21, 22. DENV infection is typically self‐limiting; however, in some cases of infection, the disease manifests a more complicated and severe form where patients may suffer from plasma leakage and shock syndrome. Unattended severe cases of DENV infection can lead to a mortality rate of 20% 23. In contrast to DENV, WNV is endemic to the Americas, Europe, Africa, and Middle East 24. Although approximately 80% of WNV infections are subclinical, symptomatic infections known as WNV fever can persist for months post infection. A few – <1% of these patients – develop severe neurological complications that may be fatal 25. Whether this is associated with ADE is not known.
Alphaviruses
Evidence from in vitro study of ADE of alphaviral infections has been reported since the 1980s, where Peiris and Porterfield demonstrated clear antibody‐dependent plaque enhancement in macrophages infected with Semliki Forest virus and SINV in the presence of sub‐neutralizing concentrations of homologous antibody 26. Consistent with this study, Chanas et al. 27 demonstrated that in vitro infection of macrophage‐like cells expressing Fc receptors with SINV, together with high dilutions of anti‐SINV monoclonal antibodies (MAbs), led to increased plaque formation.
Macrophages have been identified as key players in facilitating arthritogenic alphaviral disease progression, largely due to their permissiveness to alphaviral infection 28, 29. The persistence of alphavirus within macrophages during the late phase of infection suggested the likely occurrence of ADE 30, 31. Indeed, several studies on RRV demonstrated ADE in macrophages. Serum recovered from RRV‐infected patients contains anti‐RRV antibodies, which, when diluted to sub‐neutralizing levels, promoted infection of the less‐permissive monocytic cell lines U937 and Mono Mac 6 and enhanced RRV infection in the highly permissive macrophage cell lines J774A.1 and RAW264.7 32. In addition, infection of FcR‐expressing J774A.1 and RAW264.7 cells led to persistent virus production up to 50 days post infection. Further mechanistic studies by Lidbury et al. shed light on the role of the RRV‐induced ADE pathway in modulating host transcription and translation. Immune responses triggered by lipopolysaccharide (LPS) stimulation in macrophage cell cultures prior to RRV infection significantly reduced RRV replication. However, RRV infection in the presence of anti‐RRV antibody abolished the LPS‐induced antiviral effects, allowing RRV‐ADE infection of cells and increased virus replication 33. This phenomenon was due to the inhibition of transcription factors nuclear factor‐κB (NF‐κB) and interferon regulatory factor (IRF)1 and, paradoxically, as expression of these is usually driven through NF‐κB, translational expression of tumor necrosis factor (TNF), and generation of reactive nitrogen intermediates. Downstream studies further identified that active viral replication during RRV‐ADE infection is crucial for the suppression of LPS‐induced activation of transcription factor protein complexes IRF9 and signal transducers and activators of transcription (STAT)1, as well as elevation of interleukin (IL)‐10 expression 34.
Recently, animal studies conducted using CHIKV also demonstrated evidence of ADE of CHIKV infection. A single immunization of mice with a CHIKV vaccine candidate led to low titers of anti‐CHIKV IgG production by the host, giving rise to early disease onset upon challenge represented by acute foot swelling and inflammation 35, 36. To date, no therapeutic strategies or vaccines are currently available for the treatment of arthrtitogenic alphaviral diseases. In light of the occurrence of ADE of infection with these alphaviruses, caution should be exercised during vaccine clinical trials to avoid adverse health impacts.
Flaviviruses
DENV is made up of four different serotypes – DENV‐1, DENV‐2, DENV‐3, and DENV‐4. Halstead and O'Rourke first identified ADE of DENV infection, observing that non‐DENV susceptible human‐ or rhesus monkey‐derived primary peripheral blood mononuclear cells (PBMCs) could be successfully infected with DENV in the presence of a high dilution of anti‐DENV antibody 37. Cross‐reactivity of anti‐DENV antibodies generated from exposure to different DENV serotypes was later shown to mediate ADE 38. The occurrence of ADE has long been associated with the severity of DENV disease pathogenesis, giving rise to severe forms of disease – DHF and DSS – typically during secondary infections 23, most commonly in the order of DENV‐1 followed by secondary infection with DENV‐2 or DENV‐3 39, 40, 41. As discussed in more detail later, primary DENV infection of infants who have non‐neutralizing titers of anti‐DENV antibodies acquired through maternal transmission from a DENV‐immune mother, has reportedly led to severe DHF 42, 43. Other evidence of ADE exacerbating DENV infection was reported by Moi et al., where serum specimens were collected from DENV patients during primary or secondary infections. Virus titers measured in serum specimens from secondary DENV‐infected patients were approximately 10‐fold higher than those in the serum of patients with primary infection, detected using FcγR‐expressing BHK cells 44.
To date, DENV is one of the most extensively studied viruses in relation to ADE infection, due in part to the global prevalence of dengue infections. Since the 1980s, other flaviviruses such as WNV have also been associated with ADE infection. As with DENV, the key event in facilitating WNV‐mediated ADE infection is the binding of virions to antibodies at sub‐neutralizing concentration 45. Cardosa et al. 46 previously characterized the dual role of anti‐WNV antibodies in neutralization and enhancement of WNV infection depending on the antibody titers, class and isotype, correlating with an earlier study conducted by Peiris et al. 47.
Respiratory viruses
Besides the most devastating pandemic of the 20th century, caused in 1918 by an influenza virus, and the two subsequent episodes in 1957 and 1968 that caused several millions of deaths worldwide, respiratory viruses have continued to pose serious threats to public health in recent years. This has been illustrated by the global impact of the severe acute respiratory syndrome coronavirus (SARS‐CoV) outbreak in 2003 48, 49. Equally concerning are confirmed human cases of avian influenza in many countries, particularly across Asia, with the H7N7 strain of avian influenza that has jumped into humans in China 50, 51, 52, the emergence of the 2009 H1N1 influenza pandemic 53, as well as a novel coronavirus that is the causative agent of the Middle East respiratory syndrome (MERS‐CoV) 54. These events have triggered scores of investigations to gain insight into the pathogenic mechanisms underlying the severity of symptoms and high fatality rates of SARS, MERS, and avian influenza. One of the concepts that has driven research in this area has been the recognition that the host immune system, which is essential to clear infections, may sometimes mediate excessive and dysregulated responses contributing to more severe clinical symptoms and prognosis in affected individuals. These immunopathological responses are exemplified by the so‐called cytokine storm that is triggered by the highly pathogenic avian influenza and is considered to be one of the primary causes of the severity of the Spanish flu pandemic 55, 56, 57. The pathology in SARS patients also appears to be at least partially immune‐mediated and, therefore, several studies have investigated whether the host immune response may be involved in the development of severe respiratory diseases 58, 59.
Coronaviruses
SARS‐CoV is a member of the Coronaviridae family of single‐stranded, positive‐sense RNA viruses that cause a wide variety of illnesses ranging from respiratory, gastrointestinal to systemic diseases in a broad range of hosts, including birds, felines, pigs, cows, turkeys, and dogs 60. It is usually agreed that SARS‐CoV, in addition to infection of the respiratory tract, may also directly infect immune cells, which do not express the angiotensin I‐converting enzyme 2 (ACE2), the receptor that recognizes the SARS‐CoV spike protein to mediate virus entry 61, 62. This raises the possibility that antibody‐mediated entry may provide SARS‐CoV with an opportunity to broaden its target options. This possibility was first suggested in a study describing that a SARS‐CoV vaccine candidate based on recombinant, full‐length SARS‐CoV spike protein – the major antigenic protein of the four virus structural proteins inducing neutralizing and protective antibodies 63, 64 – triggered infection of human B cell lines despite eliciting a neutralizing and protective immune response in rodents 65. By monitoring the susceptibility of human PBMCs to infection with SARS‐CoV spike‐pseudotyped lentiviral particles (SARS‐CoVpp) as well as replication‐competent SARS‐CoV, the occurrence of ADE was established in different cell lines 66, 67 and circulating immune cell types, among which the monocytic lineage (CD68+ cells) was the primary target 68. In addition to monocytes, human macrophages were also infected by SARS‐CoV in the presence of anti‐spike antibodies. ADE‐mediated infection of macrophages, however, did not support productive replication of the virus, nor did it significantly affect the expression profile of cytokines/chemokines that are involved in the initial inflammatory response 68.
Mechanistic studies to decipher the molecular correlate of ADE of viral infections have focused on either identifying the (immune) receptor(s) and/or serum component(s) allowing penetration of the pathogen into the target cell (i.e. extrinsic ADE), or the outcome response(s) of the target cell downstream to extrinsic ADE infection (i.e. intrinsic ADE) 69, 70. In vitro infection by SARS‐CoV opsonized with anti‐spike serum was enhanced via FcγRIIa and, to a lesser extent FcγRIIb1 68. Similarly, infection of primary feline macrophages was enhanced when feline infectious peritonitis virus was opsonized with anti‐spike antibody and abrogated by blocking antibodies against Fc receptor 71, 72. Construction of truncated and chimeric FcR molecules further revealed that binding of SARS‐CoVpp to cells via the extracellular domain of FcγRs is not sufficient to trigger ADE of infection as entry requires an intact intracellular domain 68. Although the relationship between internalization of immune complexes and ADE infection by SARS‐CoV via FcγRIIs appears to be a complex process, the observation of a change in tropism of SARS‐CoV in the presence of antiviral immune serum distinguishes the ADE of SARS‐CoV infection from many other examples of antibody‐mediated viral infection, where the presence of antiviral antibodies increases the viral load due to infection of a higher number of already susceptible cells 69. Of note, neutralization of the endosomal acidic pH led to exacerbation of the antibody‐mediated infection 66, in contrast to that previously described for SARS‐CoV infection mediated by its receptor, ACE2 73. It is possible that viral particles infecting cells through the ADE pathway remain trapped in an acidic compartment and the inhibition of endosomal acidification prevents their degradation or, alternatively, that the ability of Spike to mediate cell membrane fusion at neutral pH 74 could allow more antibody‐opsonized SARS‐CoVpp to fuse with the endosomal membranes. Regardless of the mechanism, the biochemical requirements of FcγR‐mediated infection are markedly distinct from those underlying virus receptor‐dependent entry and illustrate a novel mechanism in addition to ACE2 by which SARS‐CoV can enter target cells.
To date, whether ADE occurs in SARS‐CoV infection in infected individuals is still controversial. Several clinical studies have reported that SARS‐CoV‐specific antibodies found in SARS patients are not harmful 75, 76, whereas others have observed poor disease outcomes in early seroconverted SARS patients 77, 78. To address this issue directly, it would be necessary to screen SARS patient sera, keeping in mind the possibility that ADE may only occur within a narrow window during the course of an infection and only in a subset of infected patients. One should remain aware, however, of the possible consequences of this alternative infection pathway, as other coronaviruses besides SARS‐CoV appear to be able to jump species and vaccination remains one of the few options available to prevent the spread of infections caused by this family of viruses.
Influenza virus
Influenza viruses types A, B, and C are enveloped viruses with a negative, single‐stranded RNA segmented genome. They belong to the Orthomyxoviridae family and are responsible for a wide range of illnesses. Because of their endemicity, demonstrated pandemic potential, and the annual vaccination of millions of people worldwide, the possible occurrence of ADE has important implications. More than 30 years have passed since the publication of the first study reporting ADE of influenza in an experimental rat model with heterologous challenge following immunization 79. This initial observation was soon followed by similar reports in mouse macrophage cell lines and it was noticed that antibodies against hemagglutinin (HA) or neuraminidase (NA), the two main structural components of the viral envelope, were able to induce ADE 80, 81. Influenza viruses have 18 HA and 11 NA – including the distinct lineage of influenza viruses identified in New World bats 82 – and, therefore, several subtypes were tested, revealing that the enhancing activity was strain‐dependent and FcR‐mediated 83, 84, 85, 86. Although almost all studies were carried out using mouse cell lines, several instances of increased severity of clinical disease were observed in immunized animals, especially pigs 87 and, more recently, in ferrets 88, 89, the animal model regarded as most accurately reproducing human infection when challenged with a heterologous strain 90. Although it was not directly tested, ADE was presented as the most likely explanation.
The occurrence of infection‐enhancing antibodies in humans was suggested in one study comparing the impact of sera from vaccinated or naturally infected individuals on the uptake of influenza virus 91. These experiments showed that both natural infection and attenuated vaccine‐induced antibodies enhanced uptake of homologous virus, as well as H1N1 virus isolated several years later, and that viral titers were significantly higher when challenging cells in the presence of sera from the vaccinated group, pointing to the possible induction of ADE 91. Despite the lack of evidence of vaccine‐induced ADE in humans, during the H1N1 pandemic of 2009, several observational studies raised the possibility that prior anti‐influenza vaccination was associated with enhanced risk of medically attended, laboratory‐confirmed disease 92, 93. These findings were further extended in a randomized, blinded, placebo‐controlled ferret study whose conclusions corroborated the hypothesis of direct, adverse effect of prior administration of the 2008–2009 trivalent inactivated influenza vaccine on subsequent infection with the 2009 pandemic virus A(H1N1)pdm 89. Although the authors did not assess whether ADE was involved and cautioned that results in ferrets may not be extrapolated to explain human cases, those observations warranted further investigations in different experimental models.
This issue was revisited by comparing infection in both mouse macrophage‐like cell lines and different subsets of human macrophages with a panel of sera from patients who had received different influenza vaccines. The data indicate that heterologous antibodies facilitate entry of lentiviruses pseudotyped with highly pathogenic H5 HA and of A(H1N1)pdm viruses in the P388D1 murine cell line 94, 95. Infection with different influenza A virus strains showed that both classically and alternatively activated primary human macrophages (M1 and M2, respectively) were permissive to viral infection in the absence of antibody, more so to avian H5N1 and H9N2/G1 viruses than to A(H1N1)pdm. However, there was no evidence of ADE of influenza with either subset, indicating that this event may not pose a threat to vaccinated individuals 95. In addition, other studies reported significant protection induced by the 2009–2010 trivalent inactivated vaccine 96. Clearly, the safety of universal influenza vaccines remains an issue, as studies in pigs have identified cross‐reacting antibodies induced by heterologous inactivated influenza A vaccination that target an immunodominant epitope close to the fusion peptide, leading to enhanced A(H1N1)pdm virus fusion 97. The authors dubbed this phenomenon vaccination‐associated enhanced respiratory disease and refrained from referring to ADE as an underlying mechanism, although they presented evidence of cross‐reactive antibodies leading to enhanced infection due to virus fusion after pH‐induced conformational changes of HA in the endosomal compartment. Despite some limitations, as acknowledged by the authors, the identification of such antibodies was correlated with vaccine‐induced enhancement of viral infection and, therefore, their presence should be monitored in evaluating the immune response during development of universal influenza vaccines 98.
Collectively, these results suggest that, although previous comparisons showed that murine macrophage cell lines behaved in a similar way to human monocytes 99, ADE of influenza A viruses is cell type‐ and host‐specific and underscores the need for a thorough characterization of the cellular model utilized with different viruses.
Pneumovirus
Human respiratory syncytial virus (RSV) is a negative‐sense, single‐stranded RNA virus of the family Paramyxoviridae that is the leading cause of lower respiratory infections in neonates and children 100. The virus is a global burden with seasonal epidemics and efforts have been ongoing to curb its incidence with a preventive vaccine. Since the initial demonstration that a formalin‐inactivated vaccine actually worsened disease severity in children, the possibility of ADE was considered, as immunized children developed sub‐neutralizing antibody titers compared to those of a comparable age who had natural RSV infections and had not been immunized 101. The first hints that ADE could be the underlying mechanism contributing to disease manifestations in some infants came from in vitro studies in which both murine (P388D1) and human (THP1‐ and U937) macrophage‐like cell lines were used. Thus, two MAbs specific to separate surface GP of RSV and human sera containing virus‐specific antibodies were found to enhance RSV infection. Of note, the ability to increase virus titer was greater for convalescent sera and correlated with the antibody titers 102, 103. This phenomenon could be reversed by blocking either the Fc segment of the MAb or the Fc receptor on the cells, suggesting that it involved opsonization of the virus followed by binding of immune complexes to Fc receptors on target cells 103. Collectively, these reports indicate that antibody‐mediated enhancement of RSV infection can occur in vitro in macrophages. Further evidence for ADE as a possible mechanism for disease manifestations following immunization with the formalin‐inactivated vaccine against RSV was obtained in a macaque model 104. However, several recent studies reporting ‘vaccine‐enhanced disease’ have exonerated ADE 105 and the mechanisms of lung immunopathology following formalin‐inactivated vaccination in mice have been ascribed to distinct CD4+ T‐cell subsets that orchestrate discrete and specific disease manifestations resulting in the deposition of immune complexes that start in the lung 106. Despite obvious differences in the generation of immunopathology, these two types of antibody‐dependent disease share the common trait of being dependent on inadequate responses of the innate immune system triggered by either natural infection or vaccination. Interestingly, a formalin‐inactivated vaccine against human metapneumovirus induced immune‐mediated disease 107, indicating that this may be a common feature shared by members of the Paramyxoviridae family, including measles virus 108.
Ebola virus
Ebola virus is a non‐segmented, negative‐strand RNA virus belonging to the Filoviridae family that is responsible for a severe hemorrhagic fever with a high fatality rate 109. The current Ebola epidemic in Liberia, Guinea, and Sierra Leone is the largest and most complex Ebola outbreak since this virus was first discovered in 1976, with almost 25 000 confirmed, probable and suspected cases and more than 10 000 deaths. With no available treatments of proven efficacy (although several trials are underway), the development of a safe vaccine has become one of the main areas of research. In this context, it is useful to review evidence that immunization with the envelope GP of the Zaire strain of Ebola virus induced an antibody response that enhanced virus infectivity in vitro 110. Although evidence for the involvement of ADE as a mechanistic explanation for the extreme virulence of Ebola virus is lacking, these observations raised concerns on the development of safe vaccines. A subsequent study revealed that sera from convalescent patients (51–135 days after onset of symptoms) enhanced infection of primate cells 12. On the other hand, two MAbs against the Zaire GP produced diametrically opposite effects, i.e. protection or ADE, and it was concluded that distinct epitopes underlie these features 86, 111. By constructing a chimeric envelope GP in which the epitopes of Zaire strain required for ADE were replaced with corresponding sequences of Reston Ebola virus, which appears to cause only asymptomatic infections in humans 109, the ability to trigger ADE was drastically reduced 111. The enhanced infectivity is mediated by a novel molecular correlate that requires antibodies directed against the envelope GP to interact with the complement factor, C1q. Thus, C1q binds to the antibodies complexed to the virus, thereby favoring interaction with its receptor and, consequently, increasing interactions at the cell surface 111. Because C1 receptors have been identified in most mammalian cells 112, this mechanism extends the range of cells that become susceptible to infection well beyond the range of Fc receptor‐expressing cells of the immune system. It has been hypothesized that C1q‐dependent ADE may be responsible for the rapid dissemination of the infection to secondary targets with ensuing exacerbation of symptoms. These observations have raised concerns about the development of Ebola virus vaccines based on the envelope GP and the usefulness of passive therapy with convalescent sera 111. However, recent studies have demonstrated that the potential risks and inherent disadvantages in using whole polyclonal serum for passive immune therapy can be overcome with specific and well‐characterized MAbs that showed protective efficacy in a non‐human primate model of Ebola virus disease 113, 114 and are being currently tested in clinical trials 115. In addition, several vaccine candidates have shown safety in primate models and have been rolled out during the last months of the current Ebola outbreak 116, 117, 118, 119. Vaccinated humans appear to achieve immune responses comparable in magnitude with those associated with protection in non‐human primates, suggesting that immunological data could be used to demonstrate vaccine efficacy, although full reports have not yet appeared in the literature. Collectively, these observations suggest that ADE of Ebola may be a complex phenomenon related to the balance between different neutralizing, non‐neutralizing, and enhancing antibodies elicited by different immunization protocols.
Characterizing ADE of virus infection: DENV as a case study
Epitopes responsible for ADE
The association between ADE of infection and severity of viral disease remains an area of intense study. Infection‐enhancing epitopes are therefore recognized for their potential to contribute to viral pathogenesis. Studies of the antigenic determinants that mediate ADE of enveloped viruses have found overwhelming evidence for the role of epitopes within surface structural proteins. Antibodies generated against the envelope proteins of multiple viruses have been shown to enhance infection in vitro. Examples of such epitopes include the spike protein of SARS‐CoV 66, 68, E2 GP of hepatitis C virus 120, GP of Ebola virus 111, transmembrane GP gp41 of HIV 121, and GP 5 of porcine reproductive and respiratory syndrome virus 122. Mutational analysis and epitope mapping has allowed further dissection of the amino acids and domains within these proteins responsible for ADE. Evidence also exists, however, of infection‐enhancing antibodies generated against internal structural proteins such as porcine reproductive and respiratory syndrome virus nucleocapsid and DENV premembrane (prM) 122, 123. It seems likely that these proteins, generally concealed by the virus envelope, may stimulate an antigenic response due to an abundant overexpression during natural infection or release of incomplete or immature virions from the cell. Notably also, antibodies against non‐structural protein 1 (NS1) can promote endothelial cell damage via antibody‐dependent complement‐mediated cytolysis, due to the high homology between NS1 of DENV and cell surface molecules on endothelium and platelets, as well as blood coagulation‐related molecules 124, 125. NS1 is not expressed on the virion surface, but is detectable on the infected cell surface 126 and in serum 127. With viral serotypes, strains, and even members of the same viral genus or family able to present common epitopes during infection, the potential to generate antibodies capable of ADE‐mediated infection enhancement is further increased. ADE of infection is therefore a significant obstacle to the development of effective vaccines for a number of viral pathogens. Understanding of the epitopes responsible for enhancing viral infection can therefore aid development of novel vaccination strategies, for example engineering vaccines that exclusively express neutralizing epitopes.
DENV comprises four serotypes that are antigenically cross‐reactive with one another. Antibodies elicited during primary infection with one serotype, although offering lifelong protection against that serotype, can be sub‐neutralizing and cause ADE upon secondary infection with other serotypes. Moreover, within a particular DENV serotype, the diversity of DENV genotypes adds further complexity to the elicited antibody response, with amino acid sequences varying by up to 6%. This is likely to be further exacerbated by the generation of quasispecies in dengue infection 128. Perhaps not surprisingly, the antibody response to DENV infection in humans is predominantly cross‐reactive; many are non‐neutralizing and have the potential for ADE. In addition, live attenuated DENV vaccine candidates produce a similar humoral response, dominated by serotype cross‐reactive weakly neutralizing antibodies, to that seen in DENV infection 129. A safe DENV vaccine must therefore stimulate protection against all DENV serotypes at once, or else risk development of severe forms of the disease, such as DHF, in a secondary infection. Multiple studies have examined the different DENV serotype structures to aid rational design of vaccines that induce neutralizing antibodies while lowering the risk of DHF and DSS. The host‐derived DENV membrane is a dynamic structure studded with multiple copies of the multidomain envelope (E) GP, in either dimeric or trimeric conformation, and membrane (M) proteins. Immature virus particles contain trimeric spikes made from heterodimers of E and prM proteins, which, during maturation undergo furin cleavage to leave the c‐terminal M transmembrane domain associated with the virus particle. Numerous studies have attempted to characterize neutralizing epitopes on DENV structural proteins, with serotype‐specific MAbs against DENV E protein reportedly having enhanced neutralizing activity 130. Indeed, potential therapeutic antibodies that recognize epitopes on domain III (DIII) of the E protein have shown neutralizing activity, not only in DENV infections but also in other flaviviruses and across heterologous DENV genotypes 130, 131, 132. DIII‐specific antibodies, however, are only part of a larger antibody repertoire and whether these antibodies contribute to DENV neutralization in vivo remains a contentious issue, only complicated by ADE. Recent studies have confirmed that highly cross‐reactive antibodies with both neutralizing and enhancing activity dominate the human immune response to DENV infection 133.
Using antibody‐depletion techniques, studies have attempted to identify the specific viral antigens and epitopes recognized by enhancing human antibodies after DENV infection. DENV‐specific antibody sub‐populations were removed from primary DENV‐immune human sera and the effect on ADE examined 123. Removal of serotype cross‐reactive antibodies ablated enhancement of heterotypic virus infection both in vitro and in vivo, confirming that cross‐reactive antibodies binding to the virion structural proteins are the main antibody component in human immune serum responsible for heterotypic DENV enhancement both in cell culture and in the ADE mouse model. Specific depletion of cross‐reactive E‐binding antibodies, antibodies that predominantly recognize the fusion loop region in E DII, only partially decreased the enhancement capacity of the immune sera against heterotypic serotypes, indicating that additional structural protein antibodies play a role in ADE of heterotypic DENV infections. Competition ADE studies using prM‐specific Fab fragments in human immune sera indicated that both E protein fusion loop and prM‐specific antibodies in primary DENV‐immune sera independently contribute to enhancement of heterotypic DENV infection.
FcγR‐mediated ADE of infection has been shown to occur with antibodies that bind both DENV E and prM proteins 123, 134, 135, 136. Recent advances in human hybridoma technology have confirmed the enhancing effect of human anti‐DENV MAbs, which in the past had largely been observed with mouse MAbs 136. These advances have allowed a better understanding of the humoral immune response to dengue virus infection in humans. Interestingly, human hybridomas could be derived from the cells of subjects infected with DENV 20 years or more prior to collection of peripheral blood. These long‐term memory B cells that predominated in the circulation were found to encode enhancing antibodies, creating the potential for ADE of DENV infection, and thus disease enhancement, for decades after the initial infection. Anti‐prM antibodies have been shown to be a major component of the human serological response to DENV infection. However, most of these antibodies exhibited limited virus neutralization capacity and were highly cross‐reactive among the dengue virus serotypes 135. Even at high concentrations, anti‐prM was unable to fully neutralize DENV infection but potently promoted ADE in FcR‐bearing cells. The inefficient cleavage of prM to M during DENV processing results in relatively high levels of immature, prM‐containing virus particles released from infected cells. It is therefore likely that ADE of immature, normally non‐infectious DENV particles by anti‐prM antibodies facilitates efficient virus binding and cell entry into Fc receptor‐expressing cells 137. Concurrently, the partial cleavage of prM in DENV exudates may reduce the density of antigen available for viral neutralization by anti‐prM antibodies. Thus, anti‐prM antibodies may enable infection by the otherwise inactive immature by‐products of infected cells via ADE of infection, an interesting proposition, given also that prM antibody responses were shown to be significantly higher in patients with secondary DENV infection than primary infection 138. It is possible that anti‐prM‐mediated ADE of DENV infection contributes to the severe disease pathologies associated with secondary infection. It has been suggested that in promoting an anti‐prM antibody response, DENV benefits not only from immune enhancement but also as an immune evasion strategy. In addition to restoring the infectivity of non‐infectious immature DENV, prM‐specific antibodies have also been found to cross‐react with a cryptic epitope on the E protein located to the DI/DII junction 139. Despite being unavailable on a native, functionally folded E protein, this cryptic epitope may impede E protein or E protein subunit‐based immunization strategies; such approaches may be unable to completely avoid generation of anti‐prM antibodies that enhance DENV infection.
Recent studies by Luo et al. 140 have analyzed the antibody response to individual epitopes of DENV prM protein. Using epitope mapping and bioinformatic analysis, five epitope peptides were chosen to immunize BALB/c mice from a library of 11 overlapping prM peptides. Mice immunized with a peptide spanning amino acids 19–34 of prM protein, named pr4, produced remarkably high titers of anti‐pr4 antibody, which recognized epitopes that overlapped with sera from DENV‐infected patients, indicating that an antibody response against the pr4 epitope was elicited during natural DENV infection. Interestingly, anti‐pr4 sera showed limited neutralizing activity but significant ADE activity toward all four DENV serotypes in Fc receptor‐positive K562 cells. Similarly, a cross‐reactive antibody that poorly neutralized but potently enhanced infection of the four DENV serotypes was mapped to amino acid residues 14–18 of DENV prM protein 141. Identification of such infection‐enhancing epitopes will further understanding of the pathogenesis associated with ADE of infection and aid rational vaccine design.
Viral strain‐specific differences in pathogenicity have also been linked with increased infectivity due to FcγR‐dependent ADE, as a result of variation in structural protein epitopes at the amino acid level. Investigation of Marburg virus (MARV) envelope GP from two strains of the virus, Angola and Musoke, revealed Angola GP antisera notably enhanced Angola pseudovirus infectivity, whereas Musoke GP antisera did not significantly enhance the infectivity of Angola or Musoke pseudovirus 142. The putative difference in pathogenicity between the MARV strains might therefore be partly explained by the ability to induce antibodies that enhance infectivity. Functional analysis of the Angola and Musoke GP using chimeric viruses identified that epitopes in the mucin‐like region and glycine at amino acid position 547 of Angola GP were required for maximal ADE. Curiously, while altering ADE significantly, mutation of GP amino acid 547 resulted in no significant change in reactivity of Angola and Musoke GP antisera. It is therefore possible that amino acid 547 plays a downstream role in virus entry mediated through an ADE pathway yet to be elucidated.
Although there is thought to be significant overlap between the viral epitopes responsible for both neutralization and enhancement of infection, the quantitative relationship between antibody‐mediated neutralization and ADE is less clear. Recent work by Pierson et al., using more than 100 MAbs raised against WNV and DENV, has shed light on the stoichiometry of these processes. In agreement with previous observations that ADE occurs in the presence of sub‐neutralizing quantities of antibody, analyses of neutralization and FcγR‐dependent ADE suggest that these two phenomena are related by differences in the number of antibodies bound to a single virion 45. Using MAbs that bind epitopes on the E protein, an upper and lower threshold number of MAbs was shown to dictate the stoichiometry of ADE. The neutralization threshold governs the upper limit, while the minimum number of antibodies required for ADE was found to be approximately half the number required for neutralization. The functional activity of MAbs was further determined by the location of the epitope, the antibody affinity, and epitope accessibility. For example, antibodies that recognize poorly accessible epitopes are often incapable of neutralizing heterogeneous virions, even under saturating conditions of antibody 143.
High‐resolution structural studies have further emphasized the importance of epitope availability to ADE by examining, in greater detail, the conformation and interactions of viral proteins that play a key role in binding not only antibodies but also cell receptors and potentially drugs. These studies offer comprehensive structural data with which to analyze antibody neutralization and enhancement of infection at a molecular level. Cryo‐electron microscopy of mature forms of DENV has revealed marked differences in surface charge distribution and number of contacts between the envelope proteins between serotypes 144. Crystal structures of antibody–envelope protein interactions have also highlighted the molecular determinants of infection neutralization versus enhancement and the dynamic nature of certain epitopes that may influence ADE. A MAb with varying degrees of neutralizing ability between DENV serotypes and strains was found to bind the highly conserved AB loop of DIII of the E protein of three DENV serotypes 145. Interestingly, this epitope was poorly accessible in the mature virion, suggesting that significant conformational changes in the virus take place for antibody binding. It is therefore reasonable to suppose that enhancing antibodies are also capable of recognizing concealed epitopes that, upon conformational change in the virion, are exposed to allow antibody binding and subsequent ADE. Future studies such as these to understand the structural basis for ADE will help to characterize infection‐enhancing epitopes, further aiding rational vaccine and drug design. Indeed, high‐resolution structures can be used in silico to model whole virus or epitope interactions with antibodies or molecules of interest.
Clinical data and maternal DENV‐specific antibodies
During pregnancy, mothers previously or concurrently infected with DENV transmit DENV antibodies to the developing fetus via the placenta 146 and after birth via breast milk, protecting infants from homologous infection. However, levels of protective antibodies drop from 6 months after birth, disappearing by 12 months 147, 148, 149, 150. During this period, either sub‐neutralizing levels of maternal antibodies bind to the virion with a stoichiometry below the in vivo neutralizing threshold, or non‐neutralizing maternal antibodies may enhance infectivity, increasing disease severity. This may explain why 80% of 303 serologically confirmed DENV‐infected infants in a South Vietnam study developed DHF/DSS 151.
On the other hand, a prospective, nested case–control study with 60 Filipino infants infected with DENV‐3 showed no correlation between ADE activity and DENV disease severity, although it supported the initial in vivo protective role for high level maternally derived antibody at birth and showed the decay of those protective antibodies in infants 152. ADE activity was determined by PRNT50 (plaque reduction neutralization titration by 50%) with K562 cells and this study did not attempt to correlate severity of disease with other determinants, such as cytokine levels 153. However, in other studies, the sequence of infection over time by particular DENV strains appears to modulate disease severity 154. ADE measurements in humans should thus be interpreted bearing in mind that in vitro ADE activity may vary depending on virus strain and methodology. In the mouse model, infection with serotype cross‐reactive, antibody‐opsonized DENV in AG129 mice, which lack type 1 and II interferon (IFN) receptors, also displayed a significantly higher mortality, albeit with a lower peak serum viremia than after direct virus inoculation 155. Indeed, viremia in humans is not necessarily associated with DENV disease severity 43.
Further evidence that maternally derived antibodies can worsen disease in vivo was also shown in AG129 mice. Mice that were born to DENV‐1‐infected immune mothers and infected postnatally with DENV‐2 exhibited higher levels of viremia, as well as higher levels of TNF and IL‐6, correlating with vascular leakage, compared to control DENV‐2‐infected mice born to naive mothers 156. The correlation of high systemic levels of TNF with severity of disease was also found in other in vivo experimental ADE models 134, 157. In agreement, in vitro studies either on FcR‐bearing cells, THP‐1, U937 158, 159, or human primary myeloid cells 160, 161 showed higher levels of IL‐6 and TNF. Taken together, this may also reflect the involvement of these pro‐inflammatory cytokines in the mechanism(s) of intrinsic ADE. Nevertheless, as FcγRIIa appears to be the most permissive FcγR for DENV ADE in humans 162, the question of whether a mouse model without the equivalent FcγRIIa can be reliably used to infer ADE mechanisms in humans remains unclear, although transgenic murine models in which all murine FcγR are replaced by unique human FcγR may enable some insight into this 163. Similarly, the use of mice in which the pivotal receptors for both innate cellular defense and adaptive type 1 responses are abrogated to model enhanced virus infection in vivo remains difficult to evaluate.
Molecular signaling events in ADE
Recent studies of the molecular signaling events involved in the induction of ADE have begun to shed some light on the mechanism(s) that may underlie this phenomenon. In ADE, while extrinsic ADE facilitates virus binding and entry, resulting in a significant increase in the number of infected cells, intrinsic ADE subverts the antiviral state within individual infected cells, leading to more infectious virions being produced by and released from the infected cells. The high viral load due to an increased infected cell mass and ongoing conditions permissive to virus replication are thought to presage severe disease development.
The three human FcγR classes comprise FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16). FcγRI is the only high‐affinity receptor that can bind to monomeric IgG molecules 164. All other FcγR are low‐affinity receptors and therefore require multiple interactions to produce sufficiently high‐avidity binding by IgG immune complexes to transduce the relevant signals. Of these, FcγRIIa and FcγRIIc are activating receptors that mostly signal via immunoreceptor tyrosine‐based activation motifs (ITAMs) 165, while FcγRIIb is the only known inhibitory FcγR and contains an immunoreceptor tyrosine‐based inhibitory motif (ITIM).
Among myeloid cells, monocytes express FcγRI and FcγRIIa at high levels, whereas FcγRIIb is expressed at intermediate levels. Both FcγRIIa and FcγRIIb are expressed on DCs, but FcγRIIb decreases with maturity 165. The ligation of viral immune complexes by FcγR may govern ADE through the internalization pathway, with the outcome ultimately a balance of activating versus inhibitory FcγR. Pathogen opsonization results in the formation of IgG complexes that can activate low‐affinity receptors. This leads to virus neutralization and/or phagocytosis/internalization. An investigation of 66 DENV patients and 37 healthy individuals showed that there was no correlation between serum FcγRI and severity of disease in DENV patients, despite increased levels in patients with and without vascular leakage compared to healthy subjects 166. Moreover, in vitro studies using FcγRIIb knock‐in and FcγRIIb knock‐down THP‐1 cells showed FcγRIIb ligation of immunocomplexes inhibited FcγR‐mediated phagocytosis, subsequently reducing DENV ADE 167. It is of interest to note that, in model systems, on engagement of the immunocomplex FcγRIIb dissociates and is separately directed into the recycling pathway, whereas FcγRIIa and its ligands undergo a degradation process via the lysosomal compartment in the cytoplasm. However, while these pathways may still operate during viral infection, this has not been confirmed 168.
As implied above, the susceptibility to DENV ADE appears to depend on the ratio of activating FcγRIIa and inhibitory FcγRIIb. In a study on human primary myeloid cells where mature, but not immature, DCs supported ADE 160, 161, FcγRIIa was expressed at similar levels in mature DCs and immature DCs 161. However, mature DCs had lower levels of FcγRIIb than immature DCs 165. Interestingly, immature DCs are permissive for DENV infection due to expression of DC‐SIGN, the DENV viral receptor, which is found at lower levels on mature DCs. FcγRIIb‐deficient DCs also exhibit an improved capacity for antigen presentation to CD8+ T cells, compared with wildtype DCs 169, indicating effective antigen uptake of FcγRIIa‐ligated immunocomplexes. The ratio of activating FcγRIIa and inhibitory FcγRII in favor of one or the other may explain the different ADE profiles of mature and immature DCs and may ultimately reflect a capacity to enable ADE and/or the probability of its occurrence.
Although the ligation occurs on the extracellular component of FcγR, the key element determining the behavior of FcγR isoforms is located in the cytoplasmic domain. The crucial role of the FcγR cytoplasmic tail was elegantly demonstrated by genetically exchanging ITAM and ITIM (activating/inhibitory) motifs. Notwithstanding the functionally intact extracellular component, the FcγRIIa‐ITIM significantly inhibited ADE, while the FcγRIIb‐ITAM restored ADE to wildtype levels 68, 162. Consistent with this, ITAM mutations or removal of sequences between the two tyrosines of the ITAM motif eliminate the ability of FcγRIIa to mediate ADE on K562 cells 170.
Antiviral responses under normal conditions
As mentioned above, the hallmark of ADE, especially in vitro, is increased infectivity or viral load. Under normal conditions, there are several intracellular RNA sensors, including endosomal Toll‐like receptor (TLR)‐3 and TLR‐7/8, which recognize double‐stranded RNA (dsRNA) and single‐stranded RNA, respectively 171, 172, 173, 174 (Fig. 1). Viral RNAs that escape the endosomes are likely to be recognized by TLR‐independent pathway components, such as retinoic acid‐inducible gene 1 (RIG‐1) and melanoma differentiation‐associated gene 5 (MDA‐5) 175. Recognition of dsRNA by RIG‐I/MDA‐5 or TLR‐3 is cell type‐dependent. Thus, RNA virus infection of conventional DCs or macrophages isolated from RIG‐1‐ and MDA5‐deficient mice fails to induce IFN, whereas IFN production is still observed in plasmacytoid DC 176. Upregulation of these RNA sensors has been reported following DENV infection in monocytic THP‐1 177, HUH‐7 (human hepatoma‐derived cell line) 175, and primary DCs 173, 174 (Fig. 1).
Figure 1.
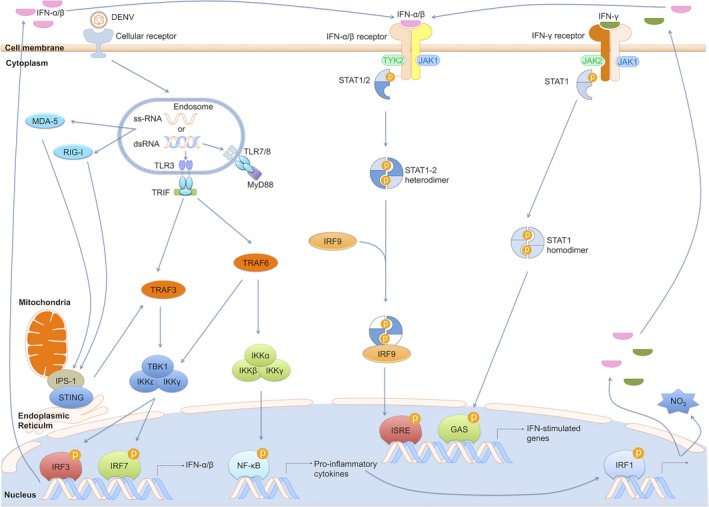
Antiviral responses under normal conditions. The TLR‐dependent, RIG‐1/MDA5 and TLR‐independent pathways induce IFN‐α/β production and the expression of ISG (interferon‐stimulated gene) to tailor the innate anti‐viral response to DENV infection. dsRNA, double‐stranded RNA; 5′‐pppRNA, 5′ triphosphate single‐stranded RNA; STING, stimulator of IFN genes; MAVS, mitochondrial antiviral signaling; TRIF, TIR‐domain‐containing adapter‐inducing interferon‐β; TRAF, TNF receptor‐associated factor; IKK, inhibitor of nuclear factor kappa‐B kinase; JAK/STAT, janus kinase/signal transducer and activator of transcription; IRF, interferon regulatory factor.
Most TLRs activate nuclear translocation of the transcription factor NF‐κB via the MyD88‐dependent pathway to produce pro‐inflammatory cytokines. Only TLR3 acts independently of MyD88 via TRIF, activating IRF‐3, to induce IFN‐β 178, 179. Although RIG‐I and MDA‐5 recognize different ligands, they share common signaling features with the TLR‐dependent pathway. Recognition of dsRNA results in their recruitment by the adapter protein interferon‐β promoter stimulator 1 (IPS‐1) to the mitochondrial outer membrane and consequent activation of several transcription factors, including IRF‐3, IRF‐7, and NF‐κB 180 via TRAF family member‐associated NF‐κB activator (TANK)‐binding kinase 1 (TBK1), leading to expression of type I IFNs and various cytokines 181. The newly synthesized IFN, in its turn, via the JAK/STAT pathway, strongly induces antiviral nitric oxide (NO) by upregulating IRF‐1 expression. However, RIG‐I/MDA5 downstream signaling can be inhibited by DENV NS2B/3, which cleaves STING (stimulator of IFN genes) in humans and disrupts IFN production 182, 183 (Fig. 1). Moreover, IL‐1β and IL‐18 are also produced by the cleavage of pro‐IL‐1 and activation of the inflammasome, respectively, upon engagement of the virion with the C‐type lectin family 5 member A (CLEC5A) surface molecule on the macrophage 184. Recently, IL‐18 produced by CD16+ monocytes in vitro has been shown to regulate the antiviral response of primary γδ T cells against DENV infection. Co‐culture of CD16+ monocytes and γδ T cells or addition of IL‐18 to γδ T cell cultures increases their IFN‐γ production and their lysis of autologous DENV‐infected DCs 185.
Surprisingly, although these cytokines contribute to immune cell recruitment to the site of infection and promote antiviral responses 186, blockade of CLEC5A/DENV interaction in vivo improved survival rate by 50% in a lethal challenge in STAT‐1‐deficient mice 187. However, improved survival may be attributed to the deficiency of the IFN‐γ signaling pathway and was also shown in IFN‐γR‐deficient mice 188. Together, the TLR‐dependent RIG‐1/MDA5 and TLR‐independent pathways induce various pro‐inflammatory cytokines including IL‐12, IFN‐α/β, and IL‐8 to tailor the innate antiviral response and are active in the defense of various cells against DENV infection, but their regulation is also intimately linked to the activation of FcγR 189, 190, 191.
Antiviral responses during ADE
After the ligation of immunocomplexes to FcγRIIa, internalized FcγRIIa‐immunocomplex enters the endosomal compartment, where the low affinity of FcγRIIa facilitates release of the immunocomplex. By an unknown pathway, the ligation or dissociation of FcγRIIa‐immunocomplex alters the innate response from antiviral to immune‐suppressive, inducing IL‐6, TNF, and IL‐10, and suppressing the expression of type I IFN. Several possibilities may explain this intrinsic ADE. Firstly, ADE may sabotage the TLR‐independent antiviral pathway by downregulating RIG‐I and MDA5 expression and promoting dihydroxyacetone kinase (DAK) and autophagy‐related 5/12 protein (Atg5/12) production (Fig. 2 A). This inverse relationship between RIG‐1/MDA5 and DAK was observed in THP‐1 192 but not monocyte‐derived‐macrophages 158, 193. Secondly, ligation of FcγR‐immunocomplex can modulate cytokine production. Upregulated TNF, IL‐6, and IL‐10 have been reported 153, 194, 195, 196, 197, 198 in several human investigations on DENV infection 160, 161. Intriguingly, IL‐10 has recently been considered to be a reliable biomarker for predicting clinical severity 197, 199. Although data for FcγR‐mediated cytokine responses in the context of DENV ADE are variable 200, 201, increased TNF and IL‐6 production has consistently been shown in monocytes, macrophages, and DCs upon exposure to immunocomplexes 161. Differences between studies may be attributed to antibody differences. Certainly, virus‐specific memory B cell‐derived MAb isolated using recombinant antigen may exclude some potentially neutralizing antibodies that do not bind to conformational epitopes in vitro. On the other hand, neutralizing antibodies at sub‐neutralizing concentrations might also induce ADE.
Figure 2.
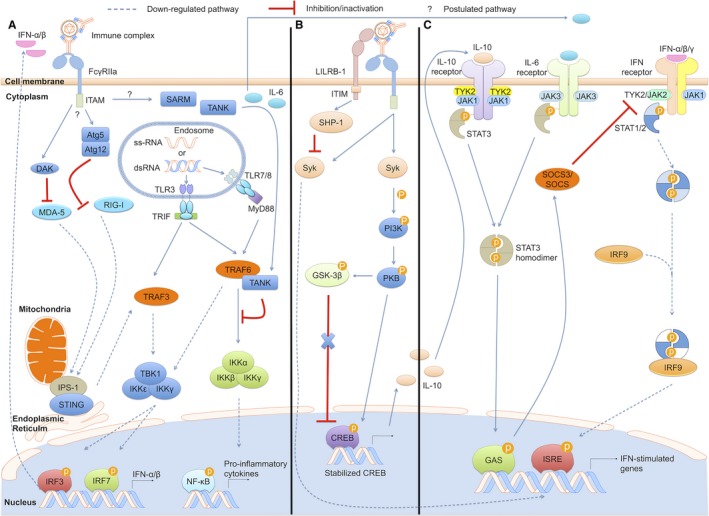
Dengue virus subversion of host innate immune responses during ADE can occur via three pathways. (A) Infectious DENV‐sub‐neutralizing antibody complexes internalized via phagocytic FcγRIIa pathway is uncoated within the endosome, where viral RNA is recognized by TLR. FcγR synergize with TLR to orchestrate the induction of cytokines: promoting DAK (dihydroxy acetone kinase), Atg5/12 (autophagy‐related 5/12 protein), SARM (Sterile‐alpha and Armadillo motif‐containing protein), and TANK (TRAF family member‐associated nuclear factor kappa‐B activator), down‐regulating RIG‐I and MDA5 expression, sabotaging the TLR‐independent antiviral pathway, subsequently suppressing production of type I IFNs. (B) Co‐ligation of FcγR and LILRB1 (leukocyte immunoglobulin‐like receptor‐B1) to DENV‐immune complexes and the DENV virion, respectively, inhibits the activation of Syk and abrogate the expression of ISGs. (C) Ligation of FcγR to immune complexes also induces upregulation of IL‐10 via Syk (spleen tyrosine kinase‐mediated PI3K/PKB (phosphoinositide 3 kinase) activation. The activation of IL‐10 as well as IL‐6 triggers SOCS (suppressor of cytokine signaling) pathway, inhibiting the JAK/STAT signaling pathway, decreasing pro‐inflammatory cytokine production. ITIM, immunoreceptor tyrosine‐based inhibitory motif; ITAM, immunoreceptor tyrosine‐based activation motif; SHP‐1, Src homology 2‐containing protein tyrosine phosphatase‐1; PKB, protein kinase B; GSK‐3β, glycogen synthase kinase; CREB, cyclic adenosine monophosphate response element‐binding; Tyk, tyrosine kinase.
In FcR‐bearing cells, FcγR‐mediated cytokine production is determined by the ratio of activating and inhibitory FcγRs. Thus, simultaneous stimulation of both activating and inhibitory FcγRs may not induce significant cytokine production without the synergizing contribution of an additional signal, e.g. from TLRs 202, 203, 204, 205. Indeed, cytokine production is likely to be due to cross‐talk of TLR3 and TLR7/8 with FcγRIIa 206, as reported for the ligation of IgG‐opsonized bacteria 207. A study using DENV ADE in THP‐1 cells showed an inverse relationship between the expression of TLR3, 4, and 7 expression and that of sterile‐alpha and Armadillo motif‐containing protein (SARM) and TANK 177. Upregulation of SARM and TANK led to downregulation of TLR signaling molecules, most significantly those using NF‐κB signaling (Fig. 2 A). Although TANK has been implicated in positive regulation of IRF3, as well as NF‐κB, it was recently shown to negatively regulate the activity of TLR signaling by directly binding to TRAF6, inhibiting TRAF6 ubiquitylation, and leading to the inhibition of NF‐κB activity 179, 208, 209. The inhibition of NF‐κB results in a reduction in IFN‐α/β, as well as IRF‐1 expression, and undermines the production of NO induced via the JAK/STAT pathway. In contrast to TNF and IL‐6, which are higher in ADE of DENV, production of NO in THP‐1 cells was significantly reduced 158, 192. Lower serum NO levels were also observed in DHF patients who had higher NS1 levels during the acute febrile phase, compared with patients with mild dengue fever 210, suggesting that DENV can suppress NO. However, the data on NO in DENV infection are conflicting. Nitric oxide synthase 2 (NOS2) expression was substantially induced in activated monocytes in a significant number of dengue patients, and in DENV‐1‐infected monocytes, high NOS2 expression was associated with reduced DENV antigen expression in vitro 211. In animal models, Nos2 −/− mice inoculated intraperitoneally with mouse‐adapted DEN‐3 showed 100% mortality compared to wildtype controls 212, suggesting that the presence of NO may help to control DENV.
NO may also be involved in immunopathogenesis. For example, the l‐arginine‐NO pathway in platelets was activated in DHF patients and this was argued to contribute to the bleeding diathesis seen in these patients 213. In line with this study, a prospective longitudinal adult study with 99 patients (11 DHF, 63 non‐DHF and 25 other febrile illnesses) showed that NO levels correlated with the development of DHF 214. This notion is supported by another study in a DEN‐3 intracranial mouse model where Nos2 −/− mice showed 100% survival, compared to the >80% mortality seen in wildtype mice 215. Thus, while NO evidently plays a role in virus control, it may also be responsible for lethality if it reaches neurotoxic levels in the brain. The discrepancy between the two studies on Nos2 −/− mice with DEN‐3 is likely explained by differences in virus strains used. In the mouse‐adapted model, the virus replicates effectively in the brain causing death in infected cells. Here, NOS2 may reduce neurovirulence by helping to control virus spread. Elevated NOS2 not only has an antiviral role in murine models of other neurotropic flaviviruses 216, 217 but also has a demonstrable immunopathological role 218.
Under ADE conditions, suppressor of cytokine signaling 3 upregulation appears to be responsible for inhibiting the JAK/STAT pathway in monocyte‐derived macrophages, with increased IL‐6 expression 193 and, in THP‐1 cells, high levels of IL‐10 192 (Fig. 2 C). This DENV ADE‐induced IL‐10 production was shown to be controlled by the activation of cyclic adenosine monophosphate response element binding (CREB) and was achieved mainly through the phosphoinositide 3‐kinase (PI3K)/PKB pathway with protein kinase A activation (Fig. 2 B). Upstream of these signaling events, ligation of FcγRIIa‐immunocomplex or engagement of CLEC5 with the virion partly induces spleen tyrosine kinase (Syk)‐mediated PI3K/PKB activation. The activation of PI3K/PKB then phosphorylates and inactivates glycogen synthase kinase (GSK)‐3b (Fig. 2 B). GSK‐3b, an important regulator for IL‐10 production following TLR signaling 219, 220, once inactivated, cannot downregulate CREB, which is stabilized by active PI3K/PKB, resulting in the observed high levels of IL‐10 221.
However, the activation of Syk via the ITAM intracellular domain of FcγRIIa also induces expression of type I IFN‐stimulated genes (ISG) in an IFN‐independent manner 222. Thus, it is not clear how DENV‐immunocomplex facilitates ADE, as the ligation of FcγR to the immunocomplex enhances the production of ISG, many of which have been identified as efficient antiviral factors against DENV, as well as WNV 223, 224.
Of note, Syk activation through FcγRIIa signaling cascades appears to be tightly regulated to prevent exaggerated inflammatory responses to the few immune complexes containing harmless antigens that may be present in the circulation at any time. Regulation is accomplished by the balance between activating ITAM and inhibitory ITIM signaling. Indeed, absence of inhibitory ITIM signaling is associated with autoimmune disease 225, 226, 227. Both the inhibitory and activating signals via immunoreceptor pathways involve activation of Src family kinases following the engagement of FcγR by immunocomplex. However, the activating signals utilize the activation of Src for activating Syk, while the activation of Src in the inhibitory signaling pathway leads to recruitment of either inositol phosphatase SHIP (Src homology 2‐containing inositol polyphosphate 5‐phosphatase) or the tyrosine phosphatase SHP‐1 (Src homology 2‐containing protein tyrosine phosphatase‐1), which subsequently dampens downstream signaling responses by dephosphorylating Syk 228. In this context, DENV‐immunocomplexes may exploit inhibitory ITIM signaling to subvert the expression of ISG. As mentioned previously, FcγRIIb is one of most common ITIM‐containing transmembrane proteins on myeloid cells. However, DENV‐immunocomplex failed to bind to FcγRIIb at sub‐neutralizing levels of antibody to establish ADE 229. Additionally, ligation of DENV‐immunocomplex by FcγRIIb was only achieved at high concentrations of antibody and did not induce ADE 162, 229. A recent study, which exploited differences in inducing ADE between subclones of THP‐1 cells, found that the binding of leukocyte immunoglobulin‐like receptor‐B1 (LILRB1) to the DENV virion may inhibit the activation of Syk and abrogate the expression of ISG 230 (Fig. 2 B). LILRB1 is an ITIM‐containing transmembrane protein, expressed on T cells, NK cells, and B cells but primarily on myeloid cells 231. The inhibitory effect of LILRB1 has also been shown in the monocytic lineage in various diseases 232, 233, 234, 235. Following receptor engagement, Src family protein tyrosine kinases phosphorylate the tyrosine residue on the ITIM, which recruits either inositol phosphatase SHIP or SHP‐1. At this stage, SHP‐1 is activated by phosphorylation, then dephosphorylates and inactivates Syk 227, 230. Thus, ligation of DENV‐immunocomplexes by FcγRIIa and the binding of LILRB1 to the DENV virion can synergistically induce intrinsic ADE in THP‐1 cells. On the surface of it, these pathways controlled by Syk appear to be in conflict. Thus, it is not clear whether the inhibition of Syk by LILRB‐1 may lead to a reduction in IL‐10 production under ADE conditions, but this may involve a temporal balance of inhibition and activation of Syk yet to be elucidated. If so, based on data from patients with severe DENV infection 197, 199, this balance is presumably tuned in favor of excessive IL‐10 production.
Targeting Fc receptors with intravenous immunoglobulin infusion: a possible therapeutic avenue for treatment of viral diseases
Intravenous immunoglobulin (IVIG) is a blood product consisting of pooled IgG fractions obtained from approximately 3000 to 60 000 plasma specimens from blood donors 236. Since its discovery 60 years ago, IVIG has conventionally been used as therapeutic treatment for immuno‐compromised individuals suffering from immune deficiency disease such as hypogammaglobulinemia 237. Immuno‐compromised individuals are highly susceptible to pathogen infections; hence, infusion of well‐processed IVIG preparations that have undergone virus inactivation, depletion of blood coagulation factors, and IgG aggregates is crucial to enable protection against pathogens and avoid any deleterious effects that could be elicited by the infusion 236. To date, despite the efficacy of IVIG treatment in several autoimmune and inflammatory diseases, the mechanism of IVIG remains ill‐defined 238. Currently, the possible mechanisms underlying IVIG efficacy mainly revolve around its ability to shape the immune response 236, 239. One possible mechanism is that infusion of high‐dose IVIG prevents FcγR activation through saturated binding of these receptors with infused IgG, as well as increasing expression of inhibitory FcγRIIb 236, 240. The immunomodulation of infused IgG stems from the interaction between Fc or variable domains of IgG and FcγR 241, 242. In the earlier section of this review, we discussed the pathogenic impact of sub‐neutralizing antibody interaction with Fc receptors during viral invasion in causing ADE. Here, we assess the possible therapeutic role of IVIG infusion during some of the world's most prevalent arthropod‐transmitted virus infections and the possible complications that may arise from this treatment in the presence of ADE infection.
Flaviviruses: WNV and DENV
High‐dose IVIG is most commonly used during treatment of WNV encephalitis among all the arthropod‐borne diseases. The potential of IVIG infusion in ameliorating WNV encephalitis has been reported both experimentally and clinically. In the earliest studies conducted by Engle and Diamond, passive transfer of serum from mice that survived primary WNV infection into WT mice prevented morbidity and mortality in subsequent WNV infection. Although passive transfer of the immune sera failed to prevent mortality in immune‐compromised B cell‐deficient (μMT) mice, these mice survived for a longer period of time (range between 30 and 50 days) post infection, compared to the typical 10‐day survival in control mice 243. Subsequent work in a murine model of lethal WNV encephalitis demonstrated an increased survival rate following IVIG administration, particularly with IVIG derived from donors from a WNV endemic region – Israel – which are likely to contain anti‐WNV antibodies 244. Indeed, the presence of anti‐WNV antibodies in IVIG preparations collected from US donors protected mice from lethal dose of WNV 245. Recently, Srivastava et al. 246 shed insight into the mechanism underlying IVIG efficacy in animal models, identifying that IVIG preparations containing antibodies against WNV are capable of dampening inflammatory monocyte (Ly6chigh CD11b+) infiltration into the central nervous system, hence limiting the extent of inflammation and prolonging survival in a WNV mouse model. Consistent with this, recent studies by Getts et al. 247 demonstrated that therapeutic modulation of inflammatory monocyte immigration into the CNS using immune‐modifying microparticles significantly increased survival in a WNV mouse model. In addition to the success of IVIG treatment observed in animal studies, growing evidence pinpointing the therapeutic potential of IVIG has been reported in the clinical context. Infusion of IVIG preparations containing high titers of anti‐WNV antibodies prevented the development of neuroinvasive WNV disease in a patient who contracted WNV infection through a liver transplant obtained from an infected individual 248. The rapid efficacy of passive immunoprophylaxis with IVIG was further evidenced in a clinical study performed by Shimoni et al. 249, in which 12 WNV patients exhibiting neuroinvasive signs were given IVIG administration. Although 25% of this cohort died of neurological complications, 75% either partially or completely recovered, with 4 out of the 12 patients experiencing a rapid improvement in clinical condition as early as 48 h post IVIG treatment, suggesting a potential for IVIG infusion to treat lethal cases of WNV neuroinvasive disease.
Despite the efficacy of IVIG in treating WNV infections, the effects of IVIG treatment in DENV infection are controversial. Severe forms of DENV infection give rise to potentially lethal severe thrombocytopenia and shock syndrome 250. IVIG is widely used as treatment for immunothrombocytopenia and gives positive medical outcomes 251, 252. During the DHF outbreak in Brazil in 2002, five clinically diagnosed DHF patients were given high doses of IVIG. The rapid reduction in bleeding diathesis and immediate recovery of platelet count in these patients suggested IVIG had potential in treating DHF 253. Furthermore, a clinical study, in which intravenous anti‐D (Rh0D IgG) immune globulin was administered in children and adult patient cohorts with secondary DENV infection resulted in increased platelet counts in the children that had exhibited severe thrombocytopenic purpura 254. However, a randomized, controlled clinical study reported opposite results. In 31 patients with acute secondary DENV infection, randomized into high‐dose IVIG‐treated and untreated groups, despite early administration of IVIG, neither improvement of thrombocytopenia nor adverse side effects were observed 255. Moreover, recently, an animal study in an immunocompetent mouse model of secondary DENV infection demonstrated that increased mortality, exacerbated disease severity, and elevated virus replication were observed in IVIG‐treated mice in a dose‐dependent manner. Further examination revealed sub‐neutralizing titers of anti‐DENV antibodies recovered from serum of IVIG‐treated mice as a contributing factor to the deleterious outcome 256. Further experimental and clinical studies are warranted to examine the exact effects of IVIG on DENV infections, and the possible adverse effects of IVIG on DENV ADE infections.
Arthritogenic alphaviruses: CHIKV and RRV
The therapeutic potential of IVIG treatment for inflammatory arthritic diseases such as rheumatoid arthritis (RA) is well established. A recent study by Campbell et al. 257 demonstrated the importance of the IgG Fc portion in contributing to the efficacy of IVIG anti‐inflammatory effects in RA mouse models. In addition, reports using CHIKV and RRV murine disease models have identified bone pathologies resembling RA 15, 258, 259. The relevance of these animal studies was further supported with the emerging clinical evidence of CHIKV patients developing persistent symmetric polyarthritis, which mimics seronegative RA 260, 261. The efficacy of IVIG treatment in RA suggests it may be a possible therapeutic candidate for the treatment of alphavirus‐induced persistent arthritis.
Clinical manifestations of CHIKV infection are typically self‐limiting; however, medical complications arising from infection have been associated with occasional mortality. The recent CHIKV outbreaks in the Americas have, as of 15 July 2015, led to 1 118 763 suspected and 24 682 confirmed autochthonous transmission cases of infection, with 192 deaths reported, mainly in Latin America and the Caribbean region 262. Despite a typical 1 in 1000 low fatality ratio associated with CHIKV infection 263, a substantial increase in mortality was reported during the 2005–2006 Indian Ocean CHIKV outbreak. To date, the exact cause underlying the increase in mortality is unclear; however, medical complications such as fulminant hepatitis and neurological and cardiological manifestations have been associated with CHIKV‐induced fatalities. Previously, a clinical case study reported the outcome in a CHIKV patient who was given high‐dose IVIG treatment after being diagnosed with neurological complications. The 44‐year‐old patient exhibited febrile illness and polyarthralgia during the acute phase of infection, which rapidly subsided within 2 weeks post disease onset. However, during the convalescent phase of infection, neurological complications that led to quadriparesis and myeloneuropathy were observed. A subsequent 4‐day infusion of high‐dose IVIG rapidly ameliorated the complications and led to a complete recovery at the follow‐up after 6 months 264. This clinical case study is consistent with an earlier study by Lebrun et al. 265, which reported rapid recovery after IVIG treatment on two CHIKV patients with late‐onset neuropathy – Guillain–Barré syndrome. Surprisingly, IVIG treatment of CHIKV patients during early onset of neurological manifestations led to exacerbated disease 266, suggesting that the stage of infection and clinical manifestations exhibited should be carefully assessed prior to IVIG infusion to avoid harmful side effects from treatment. Other precautionary measures such as determining the level of acute phase innate immune protein pentraxin 3 (PTX3) in IVIG plasma preparation as well as patients’ blood specimens should also be taken into consideration 267. PTX3 is highly expressed during a panel of inflammatory diseases and disorders 268. Recently, elevated expression of PTX3 was reported in acute RRV and CHIKV patients, and subsequent experimental study characterized the pathogenic consequences of overt PTX3 expression in enhancing alphavirus replication and causing early disease onset and prolonged disease manifestation 269. Therefore, as a preventive measure for exacerbated disease, IVIG preparations with high level of PTX3 protein should not be given to CHIKV/RRV patients. In addition, high levels of PTX3 expression in patients with Kawasaki disease have been associated with unresponsiveness to IVIG treatment 270. Hence, IVIG treatment in acute RRV/CHIKV patients should also be avoided due to high levels of alphavirus‐induced PTX3 expression during the early phase of infection. With these precautionary measures in mind, studies which look into the effects of administering IVIG containing low levels of PTX3 during the convalescence phase of infection might provide new insights into the efficacy of IVIG treatment during alphavirus disease manifestation.
Conclusion
Numerous host and virus‐specific stimuli affect the course of virus infection and disease pathogenesis in the host. ADE of viral infection occurs when pre‐existing virus‐specific antibodies aid uptake of virus into cells bearing Fc receptors, drive complement‐dependent infection enhancement, or promote virus particle production early in infection. Whether FcR‐dependent ADE of infection plays a role in viral infectivity depends on a number of factors, including infecting virus strain, antibody concentration, and epitope availability. Findings to date partly explain the interaction between intracellular antiviral suppression and the extrinsic role of enhancing antibodies on viral uptake to produce the ADE phenomenon observed for several viruses, and in particular in DHF/DSS patients. Although much of our understanding is based on DENV studies, evidence of ADE of other viruses suggests this phenomenon is widely generalizable.
If we take dengue fever as the working model and assume that ADE is virus strain‐independent, it is inferred that antibodies opsonize the virion, as determined by the functional profile of the antibodies (neutralizing/cross‐reactive/non‐neutralizing and relative affinities thereof) and the accessibility of relevant viral epitopes. Immune complexes are ligated by both activating and inhibitory FcγR, and once binding occurs, the cytoplasmic ITAM or ITIM domains deliver the immunocomplex via a phagocytic or recycling pathway, respectively, into the cell. Immunocomplex internalized via the phagocytic FcγRIIa pathway is uncoated within the endosome, where viral RNA is recognized by TLRs. FcγRs synergize with TLRs to orchestrate the induction of cytokines. However, paradoxically, DENV seems also to exploit the inhibitory ITIM signaling pathway via LILRB‐1 and SHP‐1 to inhibit Syk and thus the expression of ISG, which is otherwise promoted by Syk activation following ligation of FcγRIIa with the DENV‐immunocomplex. Thus, interactions between FcγRIIa and LILRB1 with the DENV‐immunocomplex and the DENV virion, respectively, synergistically shift the immune system from an antiviral to immunosuppressive state. Thus, although still poorly defined, the outcome of ADE is evidently a balance between the activating and inhibitory elements of these pathways. Although our understanding of virus ADE has increased substantially over the last decade, important questions remain. The disparities between studies highlight the difficulty of interpreting responses in different cell types infected with different DENV strains and this will likely apply to other viruses also. It will be of significant interest to identify the factor(s) that contribute to the balance between activation and inhibition of Syk in regulating antiviral ISGs versus anti‐inflammatory and potentially immunosuppressive IL‐10. Whether ADE‐associated IL‐6 production is also controlled by Syk activation and to what extent IL‐6 and IL‐10 induction pathways are common or separate, will be of interest. There is undoubtedly a need to find an acceptable in vivo model to confirm these findings, as much of the elucidation of ADE mechanisms has come from in vitro studies using monocytic lines or primary human myeloid cells. The lack of an analogous murine equivalent of the FcγRIIa or LILR‐B1 will make these investigations more difficult.
Acknowledgements
Research in S. M.'s laboratory was supported by grants from the Australian National Health and Medical Research Council (NHMRC) (grants 1012292, 508600, 628011, and 1033068). Work in R. B.'s laboratory described in this review was supported by grants from Research Fund for the Control of Infectious Disease (projects 05050182 and 09080872) and Area of Excellence Scheme of the University Grants Committee (AoE/M‐12/06) of the Hong Kong Government, and the National Institute of Allergy and Infectious Diseases (contract HHSN272201400006C) to R. B. Research in NK's laboratory was supported by NHMRC grants 1030897 and 512413. A. T. is the recipient of a NHMRC Peter Doherty Early Career Fellowship (1073108). L. V. is supported by an Australia Award Scholarship and S. M. is the recipient of an NHMRC Senior Research Fellowship (1059167). We thank Dr. Michael Rolph for editorial assistance. The authors have no conflicts of interest to declare.
References
- 1. Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. Antibodies mediate intracellular immunity through tripartite motif‐containing 21 (TRIM21). Proc Natl Acad Sci USA 2010;107:19985–19990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Klasse PJ, Sattentau QJ. Occupancy and mechanism in antibody‐mediated neutralization of animal viruses. J Gen Virol 2002;83:2091–2108. [DOI] [PubMed] [Google Scholar]
- 3. Dowd KA, Jost CA, Durbin AP, Whitehead SS, Pierson TC. A dynamic landscape for antibody binding modulates antibody‐mediated neutralization of West Nile virus. PLoS Pathog 2011;7:e1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hawkes RA. Enhancement of the infectivity of arboviruses by specific antisera produced in domestic fowls. Aust J Exp Biol Med Sci 1964;42:465–482. [DOI] [PubMed] [Google Scholar]
- 5. Hawkes RA, Lafferty KJ. Enhancement of virus infectivity by antibody. Virology 1967;33:250–261. [DOI] [PubMed] [Google Scholar]
- 6. Halstead SB. Dengue hemorrhagic‐fever – a public‐health problem and a field for research. Bull World Health Organ 1980;58:1–21. [PMC free article] [PubMed] [Google Scholar]
- 7. Halstead SB, Shotwell H, Casals J. Studies on the pathogenesis of dengue infection in monkeys. II. Clinical laboratory responses to heterologous infection. J Infect Dis 1973;128:15–22. [DOI] [PubMed] [Google Scholar]
- 8. Halstead SB, Chow JS, Marchette NJ. Immunological enhancement of dengue virus replication. Nature 1973;243:24–26. [PubMed] [Google Scholar]
- 9. Cardosa MJ, Porterfield JS, Gordon S. Complement receptor mediates enhanced flavivirus replication in macrophages. J Exp Med 1983;158:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson WE Jr, Montefiori DC, Gillespie DH, Mitchell WM. Complement‐mediated, antibody‐dependent enhancement of HIV‐1 infection in vitro is characterized by increased protein and RNA syntheses and infectious virus release. J Acquir Immune Defic Syndr 1989;2:33–42. [PubMed] [Google Scholar]
- 11. Prohaszka Z, et al. Two parallel routes of the complement‐mediated antibody‐dependent enhancement of HIV‐1 infection. AIDS 1997;11:949–958. [DOI] [PubMed] [Google Scholar]
- 12. Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody‐dependent enhancement of Ebola virus infection. J Virol 2003;77:7539–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fust G, Toth FD, Kiss J, Ujhelyi E, Nagy I, Banhegyi D. Neutralizing and enhancing antibodies measured in complement‐restored serum samples from HIV‐1‐infected individuals correlate with immunosuppression and disease. AIDS 1994;8:603–609. [DOI] [PubMed] [Google Scholar]
- 14. von Kietzell K, Pozzuto T, Heilbronn R, Grossl T, Fechner H, Weger S. Antibody‐mediated enhancement of parvovirus b19 uptake into endothelial cells mediated by a receptor for complement factor C1q. J Virol 2014;88:8102–8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen W, Foo SS, Sims NA, Herrero LJ, Walsh NC, Mahalingam S. Arthritogenic alphaviruses: new insights into arthritis and bone pathology. Trends Microbiol 2015;23:35–43. [DOI] [PubMed] [Google Scholar]
- 16. Paquet C, et al. Chikungunya outbreak in Reunion: epidemiology and surveillance, 2005 to early January 2006. Euro Surveill 2006;11:E060202 060203 [DOI] [PubMed] [Google Scholar]
- 17. Laine M, Luukkainen R, Toivanen A. Sindbis viruses and other alphaviruses as cause of human arthritic disease. J Intern Med 2004;256:457–471. [DOI] [PubMed] [Google Scholar]
- 18. Suhrbier A, Jaffar‐Bandjee MC, Gasque P. Arthritogenic alphaviruses – an overview. Nat Rev Rheumatol 2012;8:420–429. [DOI] [PubMed] [Google Scholar]
- 19. Burt FJ, Rolph MS, Rulli NE, Mahalingam S, Heise MT. Chikungunya: a re‐emerging virus. Lancet 2012;379:662–671. [DOI] [PubMed] [Google Scholar]
- 20. WHO PAHO . Number of Reported Cases of Chikungunya Fever in the Americas, by Country or Territory 2013–2015. (Updated as of 1 May 2015), 2015. [Google Scholar]
- 21. Bhatt S, et al. The global distribution and burden of dengue. Nature 2013;496:504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guzman MG, Harris E. Dengue. Lancet 2015;385:453–465. [DOI] [PubMed] [Google Scholar]
- 23. WHO TDR . Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control: New Edition. Geneva, 2009. [PubMed] [Google Scholar]
- 24. Suthar MS, Diamond MS, Gale M Jr. West Nile virus infection and immunity. Nat Rev Microbiol 2013;11:115–128. [DOI] [PubMed] [Google Scholar]
- 25. Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA 2013;310:308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peiris JS, Porterfield JS. Antibody‐dependent plaque enhancement: its antigenic specificity in relation to Togaviridae. J Gen Virol 1982;58:291–296. [DOI] [PubMed] [Google Scholar]
- 27. Chanas AC, Gould EA, Clegg JC, Varma MG. Monoclonal antibodies to Sindbis virus glycoprotein E1 can neutralize, enhance infectivity, and independently inhibit haemagglutination or haemolysis. J Gen Virol 1982;58(Pt 1):37–46. [DOI] [PubMed] [Google Scholar]
- 28. Lidbury BA, et al. Macrophage‐derived proinflammatory factors contribute to the development of arthritis and myositis after infection with an arthrogenic alphavirus. J Infect Dis 2008;197:1585–1593. [DOI] [PubMed] [Google Scholar]
- 29. Assuncao‐Miranda I, Bozza MT, Da Poian AT. Pro‐inflammatory response resulting from sindbis virus infection of human macrophages: implications for the pathogenesis of viral arthritis. J Med Virol 2010;82:164–174. [DOI] [PubMed] [Google Scholar]
- 30. Labadie K, et al. Chikungunya disease in nonhuman primates involves long‐term viral persistence in macrophages. J Clin Invest 2010;120:894–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Way SJ, Lidbury BA, Banyer JL. Persistent Ross River virus infection of murine macrophages: an in vitro model for the study of viral relapse and immune modulation during long‐term infection. Virology 2002;301:281–292. [DOI] [PubMed] [Google Scholar]
- 32. Linn ML, Aaskov JG, Suhrbier A. Antibody‐dependent enhancement and persistence in macrophages of an arbovirus associated with arthritis. J Gen Virol 1996;77(Pt 3):407–411. [DOI] [PubMed] [Google Scholar]
- 33. Lidbury BA, Mahalingam S. Specific ablation of antiviral gene expression in macrophages by antibody‐dependent enhancement of Ross River virus infection. J Virol 2000;74:8376–8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahalingam S, Lidbury BA. Suppression of lipopolysaccharide‐induced antiviral transcription factor (STAT‐1 and NF‐kappa B) complexes by antibody‐dependent enhancement of macrophage infection by Ross River virus. Proc Natl Acad Sci USA 2002;99:13819–13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hallengard D, et al. Prime‐boost immunization strategies against Chikungunya virus. J Virol 2014;88:13333–13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hallengard D, et al. Novel attenuated Chikungunya vaccine candidates elicit protective immunity in C57BL/6 mice. J Virol 2014;88:2858–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Halstead SB, O'Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non‐neutralizing antibody. J Exp Med 1977;146:201–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morens DM, Venkateshan CN, Halstead SB. Dengue 4 virus monoclonal antibodies identify epitopes that mediate immune infection enhancement of dengue 2 viruses. J Gen Virol 1987;68(Pt 1):91–98. [DOI] [PubMed] [Google Scholar]
- 39. Gonzalez D, et al. Classical dengue hemorrhagic fever resulting from two dengue infections spaced 20 years or more apart: Havana, Dengue 3 epidemic, 2001‐2002. Int J Infect Dis 2005;9:280–285. [DOI] [PubMed] [Google Scholar]
- 40. Guzman MG, Kouri G, Valdes L, Bravo J, Vazquez S, Halstead SB. Enhanced severity of secondary dengue‐2 infections: death rates in 1981 and 1997 Cuban outbreaks. Rev Panam Salud Publica 2002;11:223–227. [DOI] [PubMed] [Google Scholar]
- 41. Fried JR, et al. Serotype‐specific differences in the risk of dengue hemorrhagic fever: an analysis of data collected in Bangkok, Thailand from 1994 to 2006. PLoS Negl Trop Dis 2010;4:e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Halstead SB, et al. Dengue hemorrhagic fever in infants: research opportunities ignored. Emerg Infect Dis 2002;8:1474–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simmons CP, et al. Maternal antibody and viral factors in the pathogenesis of dengue virus in infants. J Infect Dis 2007;196:416–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moi ML, Lim CK, Kotaki A, Takasaki T, Kurane I. Detection of higher levels of dengue viremia using FcgammaR‐expressing BHK‐21 cells than FcgammaR‐negative cells in secondary infection but not in primary infection. J Infect Dis 2011;203:1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pierson TC, et al. The stoichiometry of antibody‐mediated neutralization and enhancement of west nile virus infection. Cell Host Microbe 2007;1:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cardosa MJ, Gordon S, Hirsch S, Springer TA, Porterfield JS. Interaction of West Nile virus with primary murine macrophages: role of cell activation and receptors for antibody and complement. J Virol 1986;57:952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peiris JS, Porterfield JS, Roehrig JT. Monoclonal antibodies against the flavivirus West Nile. J Gen Virol 1982;58:283–289. [DOI] [PubMed] [Google Scholar]
- 48. Drosten C, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003;348:1967–1976. [DOI] [PubMed] [Google Scholar]
- 49. Peiris JS, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003;361:1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sims LD, et al. Avian influenza in Hong Kong 1997‐2002. Avian Dis 2003;47:832–838. [DOI] [PubMed] [Google Scholar]
- 51. Horimoto T, Kawaoka Y. Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 2005;3:591–600. [DOI] [PubMed] [Google Scholar]
- 52. Gao R, et al. Human infection with a novel avian‐origin influenza A (H7N9) virus. N Engl J Med 2013;368:1888–1897. [DOI] [PubMed] [Google Scholar]
- 53. Peiris JS, Poon LL, Guan Y. Emergence of a novel swine‐origin influenza A virus (S‐OIV) H1N1 virus in humans. J Clin Virol 2009;45:169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012;367:1814–1820. [DOI] [PubMed] [Google Scholar]
- 55. Cheung CY, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 2002;360:1831–1837. [DOI] [PubMed] [Google Scholar]
- 56. Kobasa D, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 2007;445:319–323. [DOI] [PubMed] [Google Scholar]
- 57. Morrison J, et al. H7N9 and other pathogenic avian influenza viruses elicit a three‐pronged transcriptomic signature that is reminiscent of 1918 influenza virus and is associated with lethal outcome in mice. J Virol 2014;88:10556–10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peiris JS, Hui KP, Yen HL. Host response to influenza virus: protection versus immunopathology. Curr Opin Immunol 2010;22:475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol 2012;2:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lai MM, Perlman S, Anderson LJ. Coronaviridae In: Fields BN, Knipe DM, Howley PM. eds. Fields Virology. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2007:1306–1335. [Google Scholar]
- 61. Li L, et al. SARS‐coronavirus replicates in mononuclear cells of peripheral blood (PBMCs) from SARS patients. J Clin Virol 2003;28:239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gu J, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med 2005;202:415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bisht H, et al. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc Natl Acad Sci USA 2004;101:6641–6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Buchholz UJ, et al. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc Natl Acad Sci USA 2004;101:9804–9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kam YW, et al. Antibodies against trimeric S glycoprotein protect hamsters against SARS‐CoV challenge despite their capacity to mediate FcgammaRII‐dependent entry into B cells in vitro. Vaccine 2007;25:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jaume M, et al. Anti‐severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH‐ and cysteine protease‐independent FcgammaR pathway. J Virol 2011;85:10582–10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang SF, et al. Antibody‐dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem Biophys Res Commun 2014;451:208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yip MS, et al. Antibody‐dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J 2014;11:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sullivan NJ. Antibody‐mediated enhancement of viral disease. Curr Top Microbiol Immunol 2001;260:145–169. [DOI] [PubMed] [Google Scholar]
- 70. Halstead SB, Mahalingam S, Marovich MA, Ubol S, Mosser DM. Intrinsic antibody‐dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect Dis 2010;10:712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hohdatsu T, Nakamura M, Ishizuka Y, Yamada H, Koyama H. A study on the mechanism of antibody‐dependent enhancement of feline infectious peritonitis virus infection in feline macrophages by monoclonal antibodies. Arch Virol 1991;120:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Olsen CW, Corapi WV, Ngichabe CK, Baines JD, Scott FW. Monoclonal antibodies to the spike protein of feline infectious peritonitis virus mediate antibody‐dependent enhancement of infection of feline macrophages. J Virol 1992;66:956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hofmann H, et al. S protein of severe acute respiratory syndrome‐associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J Virol 2004;78:6134–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome‐associated coronavirus (SARS‐CoV) spike glycoprotein‐mediated viral entry. Proc Natl Acad Sci USA 2004;101:4240–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chang SC, et al. Longitudinal analysis of Severe Acute Respiratory Syndrome (SARS) coronavirus‐specific antibody in SARS patients. Clin Diagn Lab Immunol 2005;12:1455–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang L, et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J Med Virol 2006;78:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ho MS, et al. Neutralizing antibody response and SARS severity. Emerg Infect Dis 2005;11:1730–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee N, et al. Anti‐SARS‐CoV IgG response in relation to disease severity of severe acute respiratory syndrome. J Clin Virol 2006;35:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Webster RG, Askonas BA. Cross‐protection and cross‐reactive cytotoxic T cells induced by influenza virus vaccines in mice. Eur J Immunol 1980;10:396–401. [DOI] [PubMed] [Google Scholar]
- 80. Ochiai H, Kurokawa M, Kuroki Y, Niwayama S. Infection enhancement of influenza A H1 subtype viruses in macrophage‐like P388D1 cells by cross‐reactive antibodies. J Med Virol 1990;30:258–265. [DOI] [PubMed] [Google Scholar]
- 81. Tamura M, Webster RG, Ennis FA. Antibodies to HA and NA augment uptake of influenza A viruses into cells via Fc receptor entry. Virology 1991;182:211–219. [DOI] [PubMed] [Google Scholar]
- 82. Tong S, et al. New world bats harbor diverse influenza A viruses. PLoS Pathog 2013;9:e1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ochiai H, et al. Infection enhancement of influenza A NWS virus in primary murine macrophages by anti‐hemagglutinin monoclonal antibody. J Med Virol 1992;36:217–221. [DOI] [PubMed] [Google Scholar]
- 84. Tamura M, Webster RG, Ennis FA. Neutralization and infection‐enhancement epitopes of influenza A virus hemagglutinin. J Immunol 1993;151:1731–1738. [PubMed] [Google Scholar]
- 85. Tamura M, Webster RG, Ennis FA. Subtype cross‐reactive, infection‐enhancing antibody responses to influenza A viruses. J Virol 1994;68:3499–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Takada A, Kawaoka Y. Antibody‐dependent enhancement of viral infection: molecular mechanisms and in vivo implications. Rev Med Virol 2003;13:387–398. [DOI] [PubMed] [Google Scholar]
- 87. Vincent AL, Lager KM, Janke BH, Gramer MR, Richt JA. Failure of protection and enhanced pneumonia with a US H1N2 swine influenza virus in pigs vaccinated with an inactivated classical swine H1N1 vaccine. Vet Microbiol 2008;126:310–323. [DOI] [PubMed] [Google Scholar]
- 88. Kobinger GP, et al. Assessment of the efficacy of commercially available and candidate vaccines against a pandemic H1N1 2009 virus. J Infect Dis 2010;201:1000–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Skowronski DM, et al. Randomized controlled ferret study to assess the direct impact of 2008‐09 trivalent inactivated influenza vaccine on A(H1N1)pdm09 disease risk. PLoS ONE 2014;9:e86555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Belser JA, Katz JM, Tumpey TM. The ferret as a model organism to study influenza A virus infection. Dis Model Mech 2011;4:575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gotoff R, Tamura M, Janus J, Thompson J, Wright P, Ennis FA. Primary influenza A virus infection induces cross‐reactive antibodies that enhance uptake of virus into Fc receptor‐bearing cells. J Infect Dis 1994;169:200–203. [DOI] [PubMed] [Google Scholar]
- 92. Crum‐Cianflone NF, et al. Clinical and epidemiologic characteristics of an outbreak of novel H1N1 (swine origin) influenza A virus among United States military beneficiaries. Clin Infect Dis 2009;49:1801–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Skowronski DM, et al. Association between the 2008‐09 seasonal influenza vaccine and pandemic H1N1 illness during Spring‐Summer 2009: four observational studies from Canada. PLoS Med 2010;7:e1000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dutry I, Yen HL, Lee H, Peiris M, Jaume M. Antibody‐Dependent Enhancement (ADE) of infection and its possible role in the pathogenesis of influenza. Institut Pasteur International Network Annual Scientific Meeting. Hong Kong: BMC Proceedings, 2011:62. [Google Scholar]
- 95. Dutry I. Susceptibility of human macrophages to influenza A infection. Hong Kong: The University of Hong Kong, 2012. [Google Scholar]
- 96. Cowling BJ, et al. Protective efficacy against pandemic influenza of seasonal influenza vaccination in children in Hong Kong: a randomized controlled trial. Clin Infect Dis 2012;55:695–702. [DOI] [PubMed] [Google Scholar]
- 97. Khurana S, et al. Vaccine‐induced anti‐HA2 antibodies promote virus fusion and enhance influenza virus respiratory disease. Sci Transl Med 2013;5:200ra114. [DOI] [PubMed] [Google Scholar]
- 98. Skowronski DM, Janjua NZ, Hottes TS, De Serres G. Mechanism for seasonal vaccine effect on pandemic H1N1 risk remains uncertain. Clin Infect Dis 2011;52:831–832; author reply 832–833. [DOI] [PubMed] [Google Scholar]
- 99. Halstead SB, Larsen K, Kliks S, Peiris JS, Cardosa J, Porterfield JS. Comparison of P388D1 mouse macrophage cell line and human monocytes for assay of dengue‐2 infection‐enhancing antibodies. Am J Trop Med Hyg 1983;32:157–163. [DOI] [PubMed] [Google Scholar]
- 100. Nair H, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta‐analysis. Lancet 2010;375:1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kim HW, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol 1969;89:422–434. [DOI] [PubMed] [Google Scholar]
- 102. Gimenez HB, Keir HM, Cash P. In vitro enhancement of respiratory syncytial virus infection of U937 cells by human sera. J Gen Virol 1989;70(Pt 1):89–96. [DOI] [PubMed] [Google Scholar]
- 103. Krilov LR, Anderson LJ, Marcoux L, Bonagura VR, Wedgwood JF. Antibody‐mediated enhancement of respiratory syncytial virus infection in two monocyte/macrophage cell lines. J Infect Dis 1989;160:777–782. [DOI] [PubMed] [Google Scholar]
- 104. De Swart RL, et al. Immunization of macaques with formalin‐inactivated respiratory syncytial virus (RSV) induces interleukin‐13‐associated hypersensitivity to subsequent RSV infection. J Virol 2002;76:11561–11569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Openshaw PJ, Culley FJ, Olszewska W. Immunopathogenesis of vaccine‐enhanced RSV disease. Vaccine 2001;20(Suppl):S27–S31. [DOI] [PubMed] [Google Scholar]
- 106. Knudson CJ, Hartwig SM, Meyerholz DK, Varga SM. RSV vaccine‐enhanced disease is orchestrated by the combined actions of distinct CD4 T cell subsets. PLoS Pathog 2015;11:e1004757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. de Swart RL, et al. Immunization of macaques with formalin‐inactivated human metapneumovirus induces hypersensitivity to hMPV infection. Vaccine 2007;25:8518–8528. [DOI] [PubMed] [Google Scholar]
- 108. Huisman W, Martina BE, Rimmelzwaan GF, Gruters RA, Osterhaus AD. Vaccine‐induced enhancement of viral infections. Vaccine 2009;27:505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kortepeter MG, Bausch DG, Bray M. Basic clinical and laboratory features of filoviral hemorrhagic fever. J Infect Dis 2011;204(Suppl):S810–S816. [DOI] [PubMed] [Google Scholar]
- 110. Takada A, Watanabe S, Okazaki K, Kida H, Kawaoka Y. Infectivity‐enhancing antibodies to Ebola virus glycoprotein. J Virol 2001;75:2324–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Takada A, Ebihara H, Feldmann H, Geisbert TW, Kawaoka Y. Epitopes required for antibody‐dependent enhancement of Ebola virus infection. J Infect Dis 2007;196(Suppl):S347–S356. [DOI] [PubMed] [Google Scholar]
- 112. Eggleton P, Reid KB, Tenner AJ. C1q–how many functions? How many receptors? Trends Cell Biol 1998;8:428–431. [DOI] [PubMed] [Google Scholar]
- 113. Marzi A, et al. Protective efficacy of neutralizing monoclonal antibodies in a nonhuman primate model of Ebola hemorrhagic fever. PLoS ONE 2012;7:e36192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Qiu X, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014;514:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kupferschmidt K, Cohen J. Infectious diseases. Ebola drug trials lurch ahead. Science 2015;347:701–702. [DOI] [PubMed] [Google Scholar]
- 116. Richardson JS, et al. Enhanced protection against Ebola virus mediated by an improved adenovirus‐based vaccine. PLoS ONE 2009;4:e5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. de Wit E, et al. Safety of recombinant VSV‐Ebola virus vaccine vector in pigs. Emerg Infect Dis 2015;21:702–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Marzi A, et al. Vaccines. An Ebola whole‐virus vaccine is protective in nonhuman primates. Science 2015;348:439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Regules JA, et al. A recombinant vesicular stomatitis virus Ebola vaccine – preliminary report. N Engl J Med 2015. doi: 10.1056/NEJMoa1414216 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Meyer K, Ait‐Goughoulte M, Keck ZY, Foung S, Ray R. Antibody‐dependent enhancement of hepatitis C virus infection. J Virol 2008;82:2140–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Robinson WE Jr, et al. Human monoclonal antibodies to the human immunodeficiency virus type 1 (HIV‐1) transmembrane glycoprotein gp41 enhance HIV‐1 infection in vitro. Proc Natl Acad Sci USA 1990;87:3185–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Cancel‐Tirado SM, Evans RB, Yoon KJ. Monoclonal antibody analysis of porcine reproductive and respiratory syndrome virus epitopes associated with antibody‐dependent enhancement and neutralization of virus infection. Vet Immunol Immunopathol 2004;102:249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. de Alwis R, et al. Dengue viruses are enhanced by distinct populations of serotype cross‐reactive antibodies in human immune sera. PLoS Pathog 2014;10:e1004386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rulli NE, Rolph MS, Srikiatkhachorn A, Anantapreecha S, Guglielmotti A, Mahalingam S. Protection from arthritis and myositis in a mouse model of acute chikungunya virus disease by bindarit, an inhibitor of monocyte chemotactic protein‐1 synthesis. J Infect Dis 2011;204:1026–1030. [DOI] [PubMed] [Google Scholar]
- 125. Chuang Y‐CC, Lei H‐YY, Lin Y‐SS, Liu H‐SS, Wu H‐LL, Yeh T‐MM. Dengue virus‐induced autoantibodies bind to plasminogen and enhance its activation. J Immunol 2011;187:6483–6490. [DOI] [PubMed] [Google Scholar]
- 126. Noisakran S, Dechtawewat T. Association of dengue virus NS1 protein with lipid rafts. J Gen Virol 2008;89:2492–2500. [DOI] [PubMed] [Google Scholar]
- 127. Hermann LL, et al. Evaluation of a dengue NS1 antigen detection assay sensitivity and specificity for the diagnosis of acute dengue virus infection. PLoS Negl Trop Dis 2014;8:e3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lauring AS, Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 2010;6:e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Smith SA, et al. Human monoclonal antibodies derived from memory B cells following live attenuated dengue virus vaccination or natural infection exhibit similar characteristics. J Infect Dis 2013;207:1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shrestha B, et al. The development of therapeutic antibodies that neutralize homologous and heterologous genotypes of dengue virus type 1. PLoS Pathog 2010;6:e1000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wahala WMPB, Kraus AA, Haymore LB, Accavitti‐Loper MA, de Silva AM. Dengue virus neutralization by human immune sera: role of envelope protein domain III‐reactive antibody. Virology 2009;392:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Matsui K, Gromowski GD, Li L, Schuh AJ, Lee JC, Barrett ADT. Characterization of dengue complex‐reactive epitopes on dengue 3 virus envelope protein domain III. Virology 2009;384:16–20. [DOI] [PubMed] [Google Scholar]
- 133. Beltramello M, et al. The human immune response to dengue virus is dominated by highly cross‐reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 2010;8:271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Balsitis SJ, et al. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog 2010;6:e1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Dejnirattisai W, et al. Cross‐reacting antibodies enhance dengue virus infection in humans. Science 2010;328:745–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Smith SA, Zhou Y, Olivarez NP, Broadwater AH, de Silva AM, Crowe JE. Persistence of circulating memory B cell clones with potential for dengue virus disease enhancement for decades following infection. J Virol 2012;86:2665–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Rodenhuis‐Zybert IA, et al. Immature dengue virus: a veiled pathogen? PLoS Pathog 2010;6:e1000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Lai CY, et al. Antibodies to envelope glycoprotein of dengue virus during the natural course of infection are predominantly cross‐reactive and recognize epitopes containing highly conserved residues at the fusion loop of domain II. J Virol 2008;82:6631–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Chan AHY, et al. A Human PrM antibody that recognizes a novel cryptic epitope on dengue E glycoprotein. PLoS ONE 2012;7:e1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Luo Y, et al. Comprehensive mapping infection‐enhancing epitopes of dengue pr protein using polyclonal antibody against prM. Appl Microbiol Biotechnol 2015;99:5917–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Luo YY, et al. Identification of a novel infection‐enhancing epitope on dengue prM using a dengue cross‐reacting monoclonal antibody. BMC Microbiol 2013;13:194. doi: 10.1186/1471-2180-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Nakayama E, et al. Antibody‐dependent enhancement of marburg virus infection. J Infect Dis 2011;204:S978–S985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Nelson S, et al. Maturation of West Nile virus modulates sensitivity to antibody‐mediated neutralization. PLoS Pathog 2008;4:e1000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kostyuchenko VA, Chew PL, Ng TS, Lok SM. Near‐atomic resolution cryo‐electron microscopic structure of dengue serotype 4 virus. J Virol 2014;88:477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Midgey CM, et al. Structural analysis of a dengue cross‐reactive antibody complexed with envelope domain III reveals the molecular basis of cross‐reactivity. J Immunol 2012;188:4971–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Khamim KHW, et al. Neutralizing dengue antibody in pregnant Thai women and cord blood. PLoS Negl Trop Dis 2015;9:e0003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Maroun SLC, Marliere RCC, Barcellus RC, Barbosa CN, Ramos JRM, Moreira MEL. Case report: vertical dengue infection. J Pediatr 2008;84:556–559. [DOI] [PubMed] [Google Scholar]
- 148. Tsai H‐CC, et al. Dengue virus infection in early gestation with delivery of an unaffected fetus and no vertical transmission. Taiwan J Obstet Gynecol 2010;49:112–114. [DOI] [PubMed] [Google Scholar]
- 149. Ribeiro CF, Lopes VG, Brasil P, Coelho J, Muniz AG, Nogueira RM. Perinatal transmission of dengue: a report of 7 cases. J Pediatr 2013;163:1514–1516. [DOI] [PubMed] [Google Scholar]
- 150. Sinhabahu VP, Sathananthan R, Malavige GN. Perinatal transmission of dengue: a case report. BMC Res Notes 2014;7:795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Chau TNB, et al. Clinical and virological features of dengue in vietnamese infants. PLoS Negl Trop Dis 2010;4:e657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Libraty DH, et al. A prospective nested case‐control study of Dengue in infants: rethinking and refining the antibody‐dependent enhancement dengue hemorrhagic fever model. PLoS Med 2009;6:e1000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Chau TN, et al. Dengue in Vietnamese infants–results of infection‐enhancement assays correlate with age‐related disease epidemiology, and cellular immune responses correlate with disease severity. J Infect Dis 2008;198:516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Alvarez M, Rodriguez‐Roche R, Bernardo L. Dengue hemorrhagic fever caused by sequential dengue 1–3 virus infections over a long time interval: Havana epidemic, 2001–2002. Am J Trop Med Hyg 2006;75:1113–1117. [PubMed] [Google Scholar]
- 155. Watanabe S, Chan KWK, Wang J, Rivino L, Lok S‐M, Vasudevan SG. Dengue virus infection with highly‐neutralizing levels of cross‐reactive antibodies cause acute lethal small intestinal pathology without high level of viremia in mice. J Virol 2015;89:5847–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Jowin Kai Wei N, et al. First experimental in vivo model of enhanced dengue disease severity through maternally acquired heterotypic dengue antibodies. PLoS Pathog 2014;10:e1004031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Zellweger RM, Prestwood TR, Shresta S. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody‐induced severe dengue disease. Cell Host Microbe 2010;7:128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Chareonsirisuthigul T, Kalayanarooj S, Ubol S. Dengue virus (DENV) antibody‐dependent enhancement of infection upregulates the production of anti‐inflammatory cytokines, but suppresses anti‐DENV free radical and pro‐inflammatory cytokine production, in THP‐1 cells. J Gen Virol 2007;88:365–375. [DOI] [PubMed] [Google Scholar]
- 159. Puerta‐Guardo H, et al. The cytokine response of U937‐derived macrophages infected through antibody‐dependent enhancement of dengue virus disrupts cell apical‐junction complexes and increases vascular permeability. J Virol 2013;87:7486–7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Boonnak K, et al. Role of dendritic cells in antibody‐dependent enhancement of dengue virus infection. J Virol 2008;82:3939–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Boonnak K, Dambach KM, Donofrio GC, Tassaneetrithep B, Marovich MA. Cell type specificity and host genetic polymorphisms influence antibody‐dependent enhancement of dengue virus infection. J Virol 2011;85:1671–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Boonnak K, Slike BM, Donofrio GC, Marovich MA. Human FcγRII cytoplasmic domains differentially influence antibody‐mediated dengue virus infection. J Immunol 2013;190:5659–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc Natl Acad Sci 2012;109:6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Vogelpoel LT, Baeten DL, de Jong EC, den Dunnen J. Control of cytokine production by human Fc gamma receptors: implications for pathogen defense and autoimmunity. Front Immunol 2015;6:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol 2014;14:94–108. [DOI] [PubMed] [Google Scholar]
- 166. Mohamad Zamberi Z, et al. The high‐affinity human IgG receptor Fc gamma receptor I (FcγRI) is not associated with vascular leakage of dengue. J Negat Results Biomed 2015;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Chan KR, et al. Ligation of Fc gamma receptor IIB inhibits antibody‐dependent enhancement of dengue virus infection. Proc Natl Acad Sci USA 2011;108:12479–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Zhang CY, Booth JW. Divergent intracellular sorting of Fc{gamma}RIIA and Fc{gamma}RIIB2. J Biol Chem 2010;285:34250–34258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. van Montfoort N, et al. Fcgamma receptor IIb strongly regulates Fcgamma receptor‐facilitated T cell activation by dendritic cells. J Immunol 2012;189:92–101. [DOI] [PubMed] [Google Scholar]
- 170. Moi ML, Lim C‐K, Takasaki T, Kurane I. Involvement of the Fcγ receptor IIA cytoplasmic domain in antibody‐dependent enhancement of dengue virus infection. J Gen Virol 2010;91:103–111. [DOI] [PubMed] [Google Scholar]
- 171. Liang Z, et al. Activation of Toll‐like receptor 3 impairs the dengue virus serotype 2 replication through induction of IFN‐beta in cultured hepatoma cells. PLoS ONE 2011;6:e23346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Djamiatun K, Ferwerda B, Netea MG, van der Ven AJ, Dolmans WM, Faradz SM. Toll‐like receptor 4 polymorphisms in dengue virus‐infected children. Am J Trop Med Hyg 2011;85:352–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Torres S, et al. Differential expression of Toll‐like receptors in dendritic cells of patients with dengue during early and late acute phases of the disease. PLoS Negl Trop Dis 2013;7:e2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Sariol CA, et al. Decreased dengue replication and an increased anti‐viral humoral response with the use of combined toll‐like receptor 3 and 7/8 agonists in macaques. PLoS ONE 2011;6:e19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Nasirudeen AM, Wong HH, Thien P, Xu S. Lam K‐PP, Liu DX. RIG‐I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl Trop Dis 2011;5:e926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kato H, et al. Cell type‐specific involvement of RIG‐I in antiviral response. Immunity 2005;23:19–28. [DOI] [PubMed] [Google Scholar]
- 177. Modhiran N, Kalayanarooj S, Ubol S. Subversion of innate defenses by the interplay between DENV and pre‐existing enhancing antibodies: TLRs signaling collapse. PLoS Negl Trop Dis 2010;4:e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 2012;86:2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Qian C, Cao X. Regulation of Toll‐like receptor signaling pathways in innate immune responses. Ann N Y Acad Sci 2013;1283:67–74. [DOI] [PubMed] [Google Scholar]
- 180. Loo Y‐M, Gale M Jr. Immune signaling by RIG‐I‐like receptors. Immunity 2011;34:680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kumar H, Kawai T, Akira S. Toll‐like receptors and innate immunity. Biochem Biophys Res Commun 2009;388:621–625. [DOI] [PubMed] [Google Scholar]
- 182. Rodriguez‐Madoz JR, Belicha‐Villanueva A, Bernal‐Rubio D, Ashour J, Ayllon J, Fernandez‐Sesma A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J Virol 2010;84:9760–9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Angleró‐Rodríguez YI, Pantoja P, Sariol CA. Dengue virus subverts the interferon induction pathway via NS2B/3 protease‐IκB kinase ε interaction. Clin Vaccine Immunol 2014;21:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Wu MF, et al. CLEC5A is critical for dengue virus‐induced inflammasome activation in human macrophages. Blood 2013;121:95–106. [DOI] [PubMed] [Google Scholar]
- 185. Tsai C‐Y, et al. Type I IFNs and IL‐18 regulate the antiviral response of primary human γδ T cells against dendritic cells infected with dengue virus. J Immunol 2015;194:3890–3900. [DOI] [PubMed] [Google Scholar]
- 186. van de Veerdonk FL, Netea MG. New insights in the immunobiology of IL‐1 family members. Front Immunol 2013;4:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Chen ST, et al. CLEC5A is critical for dengue‐virus‐induced lethal disease. Nature 2008;453:672–676. [DOI] [PubMed] [Google Scholar]
- 188. Prestwood TR, et al. Gamma interferon (IFN‐gamma) receptor restricts systemic dengue virus replication and prevents paralysis in IFN‐alpha/beta receptor‐deficient mice. J Virol 2012;86:12561–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Medin CL, Rothman AL. Cell type‐specific mechanisms of interleukin‐8 induction by dengue virus and differential response to drug treatment. J Infect Dis 2006;193:1070–1077. [DOI] [PubMed] [Google Scholar]
- 190. Yi‐Ting T, Sui‐Yuan C, Chun‐Nan L, Chuan‐Liang K. Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cell Microbiol 2009;11:604–615. [DOI] [PubMed] [Google Scholar]
- 191. Chen R‐F, Wang L, Cheng J‐T, Yang K. Induction of IFNα or IL‐12 depends on differentiation of THP‐1 cells in dengue infections without and with antibody enhancement. BMC Infect Dis 2012;12:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Ubol S, Phuklia W, Kalayanarooj S, Modhiran N. Mechanisms of immune evasion induced by a complex of dengue virus and preexisting enhancing antibodies. J Infect Dis 2010;201:923–935. [DOI] [PubMed] [Google Scholar]
- 193. Rolph MS, Zaid A, Rulli NE, Mahalingam S. Downregulation of interferon‐β in antibody‐dependent enhancement of dengue viral infections of human macrophages is dependent on interleukin‐6. J Infect Dis 2011;204:489–491. [DOI] [PubMed] [Google Scholar]
- 194. Tsai TT, Chuang YJ, Lin YS, Wan SW, Chen CL, Lin CF. An emerging role for the anti‐inflammatory cytokine interleukin‐10 in dengue virus infection. J Biomed Sci 2013;20:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Malavige GN, et al. Serum IL‐10 as a marker of severe dengue infection. BMC Infect Dis 2013;13:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Soundravally R, et al. Association between proinflammatory cytokines and lipid peroxidation in patients with severe dengue disease around defervescence. Int J Infect Dis 2014;18:68–72. [DOI] [PubMed] [Google Scholar]
- 197. Brasier AR, et al. A three‐component biomarker panel for prediction of dengue hemorrhagic fever. Am J Trop Med Hyg 2012;86:341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Perez AB, et al. Tumor necrosis factor‐alpha, transforming growth factor‐β1, and interleukin‐10 gene polymorphisms: implication in protection or susceptibility to dengue hemorrhagic fever. Hum Immunol 2010;71:1135–1140. [DOI] [PubMed] [Google Scholar]
- 199. Ju H, Brasier A. Variable selection methods for developing a biomarker panel for prediction of dengue hemorrhagic fever. BMC Res Notes 2013;6:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Kou Z, et al. Human antibodies against dengue enhance dengue viral infectivity without suppressing type I interferon secretion in primary human monocytes. Virology 2011;410:240–247. [DOI] [PubMed] [Google Scholar]
- 201. Sun P, et al. Infection and activation of human peripheral blood monocytes by dengue viruses through the mechanism of antibody‐dependent enhancement. Virology 2011;421:245–252. [DOI] [PubMed] [Google Scholar]
- 202. Banki Z, et al. Cross‐linking of CD32 induces maturation of human monocyte‐derived dendritic cells via NF‐kappa B signaling pathway. J Immunol 2003;170:3963–3970. [DOI] [PubMed] [Google Scholar]
- 203. Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest 2005;115:2914–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Dhodapkar KM, et al. Selective blockade of the inhibitory Fcgamma receptor (FcgammaRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J Exp Med 2007;204:1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Dai X, et al. Differential signal transduction, membrane trafficking, and immune effector functions mediated by FcgammaRI versus FcgammaRIIa. Blood 2009;114:318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Vogelpoel LTC, et al. Fc gamma receptor‐TLR cross‐talk elicits pro‐inflammatory cytokine production by human M2 macrophages. Nat Commun 2014;5:5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. den Dunnen J, et al. IgG opsonization of bacteria promotes Th17 responses via synergy between TLRs and FcγRIIa in human dendritic cells. Blood 2012;120:112–121. [DOI] [PubMed] [Google Scholar]
- 208. Kawagoe T, et al. TANK is a negative regulator of Toll‐like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol 2009;10:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Clark K, Takeuchi O, Akira S, Cohen P. The TRAF‐associated protein TANK facilitates cross‐talk within the IκB kinase family during Toll‐like receptor signaling. Proc Natl Acad Sci 2011;108:17093–17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Carvalho DM, et al. Elevated dengue virus nonstructural protein 1 serum levels and altered Toll‐like receptor 4 expression, nitric oxide, and tumor necrosis factor alpha production in dengue hemorrhagic fever patients. J Trop Med 2014;2014:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Neves‐Souza P, et al. Inducible nitric oxide synthase (iNOS) expression in monocytes during acute Dengue Fever in patients and during in vitro infection. BMC Infect Dis 2005;5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Costa VV, et al. A model of DENV‐3 infection that recapitulates severe disease and highlights the importance of IFN‐γ in host resistance to infection. PLoS Negl Trop Dis 2012;6:e1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Matsuura C, et al. Nitric oxide activity in platelets of dengue haemorrhagic fever patients: the apparent paradoxical role of ADMA and l‐NMMA. Trans R Soc Trop Med Hyg 2012;106:174–179. [DOI] [PubMed] [Google Scholar]
- 214. Thein T‐L, Wong J, Leo Y‐S, Ooi E‐E, Lye D, Yeo TW. Increased vascular nitric oxide bioavailability is associated with progression to dengue hemorrhagic fever in adults. J Infect Dis 2015;212:711–714. [DOI] [PubMed] [Google Scholar]
- 215. de Souza KP, et al. Nitric oxide synthase expression correlates with death in an experimental mouse model of dengue with CNS involvement. Virol J 2013;10:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Saxena SK, Singh A, Mathur A. Antiviral effect of nitric oxide during Japanese encephalitis virus infection. Int J Exp Pathol 2000;81:165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Saxena SK, Mathur A, Srivastava RC. Induction of nitric oxide synthase during Japanese encephalitis virus infection: evidence of protective role. Arch Biochem Biophys 2001;391:1–7. [DOI] [PubMed] [Google Scholar]
- 218. Getts DR, et al. Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)‐4‐dependent recruitment of nitric oxide‐producing macrophages. J Neuroinflammation 2012;9:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Woodgett JR, Ohashi PS. GSK3: an in‐Toll‐erant protein kinase? Nat Immunol 2005;6:751–752. [DOI] [PubMed] [Google Scholar]
- 220. Martin M, Rehani K, Jope RS, Michalek SM. Toll‐like receptor‐mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 2005;6:777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Tsai TT, et al. Antibody‐dependent enhancement infection facilitates dengue virus‐regulated signaling of IL‐10 production in monocytes. PLoS Negl Trop Dis 2014;8:e3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Nimmerjahn F, Ravetch JV. Fc[gamma] receptors as regulators of immune responses. Nat Rev Immunol 2008;8:34–47. [DOI] [PubMed] [Google Scholar]
- 223. Dai J, Pan W, Wang P. ISG15 facilitates cellular antiviral response to dengue and west nile virus infection in vitro. Virol J 2011;8:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Jiang D, et al. Identification of five interferon‐induced cellular proteins that inhibit West Nile virus and dengue virus infections. J Virol 2010;84:8332–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Joller N, et al. Cutting edge: TIGIT has T cell‐intrinsic inhibitory functions. J Immunol 2011;186:1338–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Karra L, Levi‐Schaffer F. Down‐regulation of mast cell responses through ITIM containing inhibitory receptors. Adv Exp Med Biol, 2011;716:143–159. [DOI] [PubMed] [Google Scholar]
- 227. Smith KGC, Clatworthy MR. Fc[gamma]RIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol 2010;10:328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Lowell CA. Src‐family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harbor Perspect Biol 2011;3:a002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Chan KR, et al. Ligation of Fc gamma receptor IIB inhibits antibody‐dependent enhancement of dengue virus infection. Proc Natl Acad Sci USA 2011;108:12479–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Chan KR, Ong EZ, Tan HC. Leukocyte immunoglobulin‐like receptor B1 is critical for antibody‐dependent dengue. Proc Natl Acad Sci USA 2014;111:2722–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Lamar DL, Weyand CM, Goronzy JJ. Promoter choice and translational repression determine cell type–specific cell surface density of the inhibitory receptor CD85j expressed on different hematopoietic lineages. Blood 2010;115:3278–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Pilsbury LE, Allen RL, Vordermeier M. Modulation of Toll‐like receptor activity by leukocyte Ig‐like receptors and their effects during bacterial infection. Mediators Inflamm 2010;2010:536478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Yang Z, Bjorkman PJ. Structure of UL18, a peptide‐binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci USA 2008;105:10095–10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Schultz HS, et al. Collagen induces maturation of human monocyte‐derived dendritic cells by signaling through osteoclast‐associated receptor. J Immunol 2015;194:3169–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Lichterfeld M, Yu XG. The emerging role of leukocyte immunoglobulin‐like receptors (LILRs) in HIV‐1 infection. J Leukoc Biol 2012;91:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol 2013;13:176–189. [DOI] [PubMed] [Google Scholar]
- 237. Ballow M. Primary immunodeficiency disorders: antibody deficiency. J Allergy Clin Immunol 2002;109:581–591. [DOI] [PubMed] [Google Scholar]
- 238. Ballow M. The IgG molecule as a biological immune response modifier: mechanisms of action of intravenous immune serum globulin in autoimmune and inflammatory disorders. J Allergy Clin Immunol 2011;127:315–323; quiz 324–315. [DOI] [PubMed] [Google Scholar]
- 239. Nagelkerke SQ, Kuijpers TW. Immunomodulation by IVIg and the role of Fc‐gamma receptors: classic mechanisms of action after all? Front Immunol 2014;5:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Imbach P, et al. High‐dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1981;1:1228–1231. [DOI] [PubMed] [Google Scholar]
- 241. Bruhns P, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009;113:3716–3725. [DOI] [PubMed] [Google Scholar]
- 242. van Mirre E, Teeling JL, van der Meer JW, Bleeker WK, Hack CE. Monomeric IgG in intravenous Ig preparations is a functional antagonist of FcgammaRII and FcgammaRIIIb. J Immunol 2004;173:332–339. [DOI] [PubMed] [Google Scholar]
- 243. Engle MJ, Diamond MS. Antibody prophylaxis and therapy against West Nile virus infection in wild‐type and immunodeficient mice. J Virol 2003;77:12941–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Ben‐Nathan D, et al. Using high titer West Nile intravenous immunoglobulin from selected Israeli donors for treatment of West Nile virus infection. BMC Infect Dis 2009;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Planitzer CB, Modrof J, Kreil TR. West Nile virus neutralization by US plasma‐derived immunoglobulin products. J Infect Dis 2007;196:435–440. [DOI] [PubMed] [Google Scholar]
- 246. Srivastava R, Ramakrishna C, Cantin E. The anti‐inflammatory activity of IVIG protects against West Nile virus encephalitis. J Gen Virol 2015;96:1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Getts DR, et al. Therapeutic inflammatory monocyte modulation using immune‐modifying microparticles. Sci Transl Med 2014;6:219ra217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Morelli MC, et al. Absence of neuroinvasive disease in a liver transplant recipient who acquired West Nile virus (WNV) infection from the organ donor and who received WNV antibodies prophylactically. Clin Infect Dis 2010;51:e34–e37. [DOI] [PubMed] [Google Scholar]
- 249. Shimoni Z, et al. The clinical response of West Nile virus neuroinvasive disease to intravenous immunoglobulin therapy. Clin Pract 2012;2:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Simmons CP, Farrar JJ, Nguyen v, Wills B. Dengue. N Engl J Med 2012;366:1423–1432. [DOI] [PubMed] [Google Scholar]
- 251. Hogarth PM, Pietersz GA. Fc receptor‐targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov 2012;11:311–331. [DOI] [PubMed] [Google Scholar]
- 252. Crow AR, Song S, Siragam V, Lazarus AH. Mechanisms of action of intravenous immunoglobulin in the treatment of immune thrombocytopenia. Pediatr Blood Cancer 2006;47:710–713. [DOI] [PubMed] [Google Scholar]
- 253. Ostronoff M, et al. Serious thrombocytopenia due to dengue hemorrhagic fever treated with high dosages of immunoglobulin. Clin Infect Dis 2003;36:1623–1624. [DOI] [PubMed] [Google Scholar]
- 254. de Castro RA, de Castro JA, Barez MY, Frias MV, Dixit J, Genereux M. Thrombocytopenia associated with dengue hemorrhagic fever responds to intravenous administration of anti‐D (Rh(0)‐D) immune globulin. Am J Trop Med Hyg 2007;76:737–742. [PubMed] [Google Scholar]
- 255. Dimaano EM, et al. Lack of efficacy of high‐dose intravenous immunoglobulin treatment of severe thrombocytopenia in patients with secondary dengue virus infection. Am J Trop Med Hyg 2007;77:1135–1138. [PubMed] [Google Scholar]
- 256. Costa VV, et al. Subversion of early innate antiviral responses during antibody‐dependent enhancement of Dengue virus infection induces severe disease in immunocompetent mice. Med Microbiol Immunol 2014;203:231–250. [DOI] [PubMed] [Google Scholar]
- 257. Campbell IK, et al. Therapeutic effect of IVIG on inflammatory arthritis in mice is dependent on the Fc portion and independent of sialylation or basophils. J Immunol 2014;192:5031–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Chen W, et al. Arthritogenic alphaviral infection perturbs osteoblast function and triggers pathologic bone loss. Proc Natl Acad Sci USA 2014;111:6040–6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Chen W, et al. Bindarit, an inhibitor of monocyte chemotactic protein synthesis, protects against bone loss induced by chikungunya virus infection. J Virol 2015;89:581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Miner JJ, et al. Brief report: chikungunya viral arthritis in the United States: a mimic of seronegative rheumatoid arthritis. Arthritis Rheumatol 2015;67:1214–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Manimunda SP, et al. Clinical progression of chikungunya fever during acute and chronic arthritic stages and the changes in joint morphology as revealed by imaging. Trans R Soc Trop Med Hyg 2010;104:392–399. [DOI] [PubMed] [Google Scholar]
- 262. Pan American Health Organization WHO . Number of Reported Cases of Chikungunya Fever in the Americas, by Country or Territory 2013‐2015 (Updated 1 May 2015), 2015. [Google Scholar]
- 263. Morens DM, Fauci AS. Chikungunya at the door–deja vu all over again? N Engl J Med 2014;371:885–887. [DOI] [PubMed] [Google Scholar]
- 264. Chusri S, Siripaitoon P, Hirunpat S, Silpapojakul K. Case reports of neuro‐Chikungunya in southern Thailand. Am J Trop Med Hyg 2011;85:386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Lebrun G, Chadda K, Reboux AH, Martinet O, Gauzere BA. Guillain‐Barre syndrome after chikungunya infection. Emerg Infect Dis 2009;15:495–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Wadia R. A neurotropic virus (chikungunya) and a neuropathic aminoacid (homocysteine). Ann Indian Acad Neurol 2007;10:198. [Google Scholar]
- 267. Daigo K, Mantovani A, Bottazzi B. The yin‐yang of long pentraxin PTX3 in inflammation and immunity. Immunol Lett 2014;161:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Daigo K, Hamakubo T. Host‐protective effect of circulating pentraxin 3 (PTX3) and complex formation with neutrophil extracellular traps. Front Immunol 2012;3:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Foo SS, et al. Role of pentraxin 3 in shaping arthritogenic alphaviral disease: from enhanced viral replication to immunomodulation. PLoS Pathog 2015;11:e1004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Katsube Y, et al. Ptx3, a new biomarker for vasculitis, predicts intravenous immunoglobulin unresponsiveness in patients with kawasaki disease. J Am Coll Cardiol 2011;57:E2038. [Google Scholar]