

Abstract
A nonribosomal peptide synthetase (NRPS)-nonreducing polyketide synthase (NRPKS) hybrid enzyme (AnATPKS) from Aspergillus niger was shown to produce amino acid derived α-pyrone natural products (pyrophen and campyrone B). Biochemical characterization of the NRPS module in vitro reveals that the adenylation domain is promiscuous towards a variety of substituted phenylalanine analogues. Using precursor feeding and heterologous expression of AnATPKS and an associated O-methyltransferase (AnOMT), we were able to access a library of substituted pyrophen analogues. Our study paves the way for future combinatorial biosynthesis of diverse α-pyrone natural products using NRPS-NRPKS hybrids.
Graphical Abstract
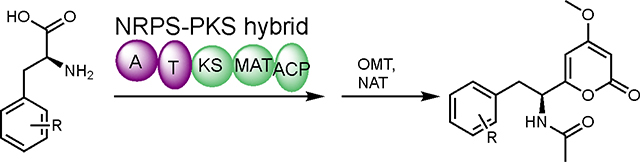
Fungal type I polyketide synthases (PKSs) are monomodular multidomain enzymes that use a single set of elongation and tailoring domains in an iterative and permutative fashion to synthesize a structurally diverse family of natural products exhibiting a wide range of biological activities.1,2 Based on the domain architecture and extent of β-keto reduction, fungal PKSs can be classified into three types: nonreducing (NR), partial reducing (PR), and highly reducing (HR). The highly reducing PKSs (HRPKSs) are often found being paired with a nonribosomal peptide synthetase (NRPS) resulting in a bi-modular PKS-NRPS hybrid, which can generate highly reduced polyketide/amino acid hybrid molecules with increased structural complexity, expanded chemical space, and unique biological activities (Figure 1A).1,2
Figure 1.

Diverse functions of fungal PKS, PKS-NRPS, and NRPS-PKS hybrids. (A) Examples of well-studied fungal PKS and PKS-NRPS hybrids. (B) Examples of fungal NRPS-PKS hybrids.
In comparison, fungal NRPS-PKS hybrids have been largely unexplored. To date, only a handful of NRPS-PKS hybrids have been characterized (Figure 1B). For example, a truncated NRPS-PKS hybrid TenA, in which only the KS domain is present in the PKS module, was shown to catalyze condensation between an activated L-isoleucine and acetoacetyl-CoA, followed by Dieckmann cyclization to release the mycotoxin tenuazonic acid.3 Here the KS catalyzed cyclization is an atypical function for this domain. In addition, an NRPS-PKS hybrid SwnK was suggested to synthesize the indolizidine backbone of the mycotoxin swainsonine.4 Although the intact PKS module of SwnK performs a complete elongation and β-reduction cycle, it does not carry out the iterative chain elongation that is the hallmark of fungal PKSs.
One could argue that the current accepted dogma of the enzymology of fungal HRPKS, i.e., that no loading module is required for chain initiation,5 is incompatible with the chemical logic of NRPS-PKS hybrids in which the NRPS acts as a loading module. Because in an NRPS-PKS hybrid, the KS domain is responsible for vectorial transfer of the starter unit from NRPS to PKS, the PKS module must resemble bacterial modular assembly line PKSs rather than fungal iterative HRPKSs.6 Indeed, phylogenetic analysis of SwnK KS domain indicated that it shares the last common ancestor with assembly line PKSs and displays highest sequence identity (55%) with the KS domain from module 6 of rapamycin polyketide synthase (Fig. S1).7 This finding also implies that for a fungal NRPS-PKS hybrid where PKS needs to carry out iterative chain elongation, NRPKS is more likely to be found. This hypothesis is supported by the recent discovery of HispS,8 a mushroom NRPS-NRPKS hybrid involved in biosynthesis of hispidin (Figure 1B). In HispS, the minimal PKS module (KS-MAT-ACP) performs two cycles of chain elongation leading to a triketide intermediate, which then spontaneously off-loads through lactonization leading to the α-pyrone hispidin. Although the enzymology of HispS has not been studied, this NRPS-NRPKS suggests that other pyrone natural products may be biosynthesized using similar logic.
Indeed, a family of fungal amino acid-derived α-pyrone natural products, many of which exhibit antifungal and cytotoxic activity, are likely to be biosynthesized by NRPS-NRPKSs (Figure 2A).9 In 2018, Reber and Burdge reported six-step total syntheses of pyrophen and campyrones A-C, and proposed a similar biosynthetic route.10 Here, we show that an NRPS-NRPKS hybrid is responsible for constructing the α-pyrone scaffold of pyrophen from L-phenylalanine. Through precursor feeding and heterologous expression, we were able to access a library of amino acid derived α-pyrones.
Figure 2.

Amino acid derived α-pyrone fungal natural products. (A) Structures and retro-biosynthetic analysis. (B) Candidate gene cluster. AnATPKS is an NRPS-NRPKS hybrid with domain architecture indicated. AnOMT is an O-methyltransferase.
RESULTS AND DISCUSSION
To identify the biosynthetic genes involved in pyrophen or campyrone biosynthesis, we searched the genome sequences of known producers for NRPS-NRPKS hybrids.9 As a result, we found a gene cluster consisting of only an NRPS-NRPKS (named as AnATPKS hereafter) and an O-methyltransferase (named as AnOMT) in the genome of Aspergillus niger (Figure 2B). Because there is only one NRPS-NRPKS gene in the pyrophen/campyrone-producing fungi and its co-localization with an O-methyltransferase is consistent with the retro-biosynthetic analysis, we hypothesized that this is the putative pyrophen/campyrone biosynthetic gene cluster.
To test our hypothesis, we heterologously reconstituted AnATPKS and AnOMT activity in vivo. Overexpressing both atpks gene (EAH27898.1) and omt gene (EAH27897.1) in the heterologous host A. nidulans ΔEM strain11 led to the production of a dominant metabolite (1) with molecular mass (m/z = 288 for [M+H]+) and a distinct UV absorption profile (λmax = 282 nm) consistent with a pyrone functional group (Figure 3a). Isolation and structural characterization of 1 confirmed its identity as pyrophen. A minor metabolite 2 was isolated and characterized to be campyrone B. The product ratio (1:2) ranges from 100:1 to 10: 1 depending on the culture conditions (e.g., media manufacturer and fermentation method). Overexpressing atpks alone resulted in the production of a new metabolite 3 albeit at lower titer (Figure 3A). Isolation and structural characterization of 3 revealed it as desmethyl-desacetyl-pyrophen (Figure 3B). Production of 3 instead of desmethyl-pyrophen indicates that L-phenylalanine is the direct substrate for AnATPKS opposed to the N-acetyl-L-phenylalanine proposed by Reber and Burdge.12 The acetylation must occur at a later stage and must be carried out by an endogenous yet-unidentified N-acetyltransferase. It is worthwhile to mention that overexpression of atpks alone drastically affected the metabolite profile, and caused growth defects leading to lower amount of cell mass, suggesting that overproducing compound 3 may be toxic to the fungal host. Indeed, the growth and development of A. nidulans was weakly inhibited on solid medium in the presence of compound 3 (Figure S2).
Figure 3.

Heterologous expression of atpks and omt in A. nidulans. (A) HPLC analysis of A. nidulans metabolite profile. (B) Pyrophen biosynthetic intermediates. (C) Chemical hydrolysis of pyrophen 1 to obtain relevant biosynthetic intermediates.
Since the timing for the methylation or acetylation of 3 is not clear from the reconstitution study, to unambiguously assign the sequence of the modification events, in vitro activity of AnOMT was characterized. Soluble and active AnOMT was prepared as a fusion protein with an N-terminal maltose-binding protein (MBP) tag (Figure S3). To prepare the possible AnOMT substrate desmethyl-pyrophen (4) (Figure 3B), we hydrolyzed 1 in 6N HCl (Figure 3C). Incubating purified 4 with anOMT and the obligatory methyl donor S-adenosine-L-methionine (SAM) in vitro did not yield the corresponding methylated product 1. In contrast, we readily observed conversion of 3 to desacetyl-pyrophen 5 under the same condition (Figure 4). The standard 5 was obtained through chemical hydrolysis of 1 (Figure 3C). These results indicate that the free α-amino group is important for AnOMT recognition and thus methylation occurs before acetylation, while the yet-unidentified N-acetyltransferase selectively recognizes methylated pyrone 5 but not 3, which explains why the formation of 4 was not observed in vivo in the absence of AnOMT.
Figure 4.

HPLC analysis of AnOMT enzymatic reaction product in vitro: (i) substrate 3; (ii) boiled AnOMT + 3 + SAM; (iii) AnOMT + 3 + SAM; (iv) standard 5; (v) substrate 4; (vi) AnOMT + 4 + SAM; (vii) standard 1. Reactions were stopped after 30 min at room temperature.
Since production of both 1 and 2 are linked to AnATPKS expression, campyrone B (2) is likely produced from the same pathway. We hypothesize that AnATPKS has relaxed substrate specificity to activate and extend branched-chain amino acid L-leucine to give 2. This hypothesis is consistent with our early observation that the product ratio of 1 over 2 depends on the culture conditions, namely the availability of L-leucine relative to L-phenylalanine. The hypothesis also suggests that amino acids other than phenylalanine may be tolerated by AnATPKS to afford diverse pyrone natural products. Intrigued by this possibility, we set out to biochemically characterize the enzymatic activity of AnATPKS in vitro in order to gain more insights into its substrate specificity. Numerous attempts to purify the full-length AnATPKS, to prepare soluble and active proteins for biochemical study were unsuccessful. Instead, a soluble construct of the A-T didomain by heterologous expression in E. coli (Figure S3) was prepared. The substrate specificity of the AnATPKS adenylation (A) domain was characterized by using a hydroxylamine trapping assay to measure the adenylation activity.12 As shown in Figure 5A, the A domain prefers L-phenylalanine over the other proteinogenic amino acids, which is consistent with the observations in vivo. In addition, the substrate profiling experiment shows that A domain is not enantioselective since D-phenylalanine can be activated to the same extent as L-phenylalanine. Similar lack of stereospecificity was first reported for the archetypal phenylalanine activating A domain (PheA) in gramicidin synthetase 1.13 To test whether D-phenylalanine can be utilized as a starter unit by AnATPKS, isotope labeled D-phenylalanine (ring-d5) was fed to the A. nidulans transformants (Figure S3). However, no mass-shift was observed for 1. In contrast, feeding isotope labeled L-phenylalanine (ring-d5) led to a +5 Da mass shift of 1. These results indicate that although the A domain does not discriminate between two enantiomeric phenylalanine, the downstream KS domain must play a gatekeeping role to stereoselectively accept the L-phenylalanyl-S-phosphopantetheine (Ppant)-T domain intermediate for chain elongation. Taken together, we propose the following biosynthetic pathway for pyrophen (Figure 6). The NRPS-NRPKS hybrid AnATPKS prefers L-phenylalanine as starter unit and extends the acyl chain with two malonyl-CoA units. The resulting amino acid-derived diketides is off-loaded through lactonization to yield the α-pyrone intermediate, which is then methylated and acetylated by a dedicated O-methyltransferase and a yet-unknown endogenous acetyltransferase, respectively.
Figure 5.

Substrate specificity of AnATPKS A domain. (A) Substrate profiling of the adenylation domain. The A domain exhibits relaxed substrate specificity towards substituted phenylalanine analogs. (B) Precursor feeding led to biosynthesis of substituted pyrophen analogues. Feeding of different phenylalanine analogues to A. nidulans transformants led to production of substituted pyrophen analogues (6–11), which were isolated and structurally characterized. Note that 11 may be racemic as suggested by the optical rotation data.
Figure 6.

Proposed biosynthetic pathway for pyrophen.
We next explored the substrate scope of the A domain by testing different phenylalanine analogues (Figure 5A). Most para-substituted phenylalanines bearing small substitution groups (4-F, 4-Cl, 4-Br, 4-Me, 4-NH2) as well as ortho- and meta- chlorinated phenylalanine can be tolerated. The lack of enantioselectivity of A domain suggests no steric restriction is exerted to the α-position, which is consistent with the high activation efficiency (~50%) of α-methyl-substituted phenylalanine. Nevertheless, the α-amino group is still essential for enzyme recognition as no adenylation activity was observed with phenylpyruvate, phenylpropanoic acid and β-phenylalanine.
To test whether these hits can also be processed further by the downstream KS domain, 4-F, 4-Cl, 4-Br, 4-Me, 4-NH2, 2-Cl, 3-Cl substituted L-phenylalanine and α-methyl-DL-phenylalanine were fed to A. nidulans transformants. A series of substituted pyrophen analogues 6–11 were produced, and their structures confirmed by NMR (Figure 5B, Table S9–S14 and Figure S6–S46). Although KS domain also exhibits somewhat relaxed substrate specificity, it obviously does not accept DL-α-methyl-phenylalanine and 4-amino-L-phenylalanine derived thioesters. No transformation of α-substituted phenylalanine suggests that the KS active site exhibits steric restriction to the α-hydrogen in the L-configuration, which is consistent with its proposed gatekeeping role and stereoselectivity when the starter unit is transferred from NRPS module to PKS module. The intolerance of 4-amino-L-phenylalanine also indicates that the KS domain does not favor hydrophilic substitution at the para-position, which may be a safeguarding function to prevent incorporation of the isosteric L-tyrosine. To assess whether these pyrophen analogues show improved bioactivity, the in vitro antifungal activity of isolated compounds 6–11 against C. albicans SC5314 strain and S. cerevisiae JHY686 strain was tested in a microdilution assay (Figure S5). No antifungal activity of 6–11 was observed up to the concentration of 100 μg/mL. Moreover, no activity was observed for pyrophen (1), which is contradictory to the initial bioactivity characterization of 1.9c The discrepancy between these two studies is not clear. Nevertheless, it remains to be tested whether these analogues show any cytotoxic activity since pyrophen (1) was shown to inhibit the growth of T47D breast cancer cells with IC50 value of 9.2 μg/mL.
In summary, we elucidated the biosynthetic pathway of pyrophen and campyrone B, which represent a class of fungal amino acid-derived α-pyrone natural products. The relaxed substrate specificity of the essential NRPS-PKS hybrid AnATPKS enables incorporation of a series of L-phenylalanine analogues leading to the biosynthesis of pyrophen analogues. Our study demonstrated the biosynthetic potential of NRPS-NRPKS hybrid and paved the way for future combinatorial biosynthesis of pyrone-based unnatural natural products.
EXPERIMENTAL SECTION
General Experimental Procedures
LC-MS analyses were performed on a Shimadzu 2020 EV LC−MS (Phenomenex Kinetex 1.7 μm C18 100 Å, 100 × 2.1 mm LC column) using positive- and negative-mode electrospray ionization with a linear gradient of 5−95% acetonitrile MeCN−H2O supplemented with 0.5% formic acid. 1D and 2D NMR spectra were obtained on a Bruker AV500 spectrometer. High resolution mass spectra were recorded on a Waters LCT Premier equipped with ACQUITY UPLC. Phusion® high-fidelity DNA polymerase (New England Biolab) was used for all Polymerase Chain Reactions (PCRs). All phenylalanine analogues were purchased from Chem-Impex Int’l. Isopropyl-β-D-1-thio-galactopyranoside (IPTG) was purchased from Carbosynth. Tris-(2-carboxyethyl) phosphine hydrochloride (TCEP-HCl) was purchased from GoldBio Biotechnology. All other chemicals were purchased from Sigma-Aldrich. Custom oligonucleotides were synthesized by Integrated DNA Technologies. Escherichia coli strain DH10B was used for cloning procedures.
Fungal heterologous expression
The atpks gene (EAH27898.1) including the terminator region and omt gene (EAH27897.1) including the terminator region were amplified from the genomic DNA extract of A. niger ATCC 1015 strain through PCR using primers listed in Table S1. The PCR products and the corresponding digestion-linearized fungal expression vectors were assembled through yeast homologous recombination using S. cerevisiae JHY686 strain.14 The resulting plasmids were confirmed by restriction-enzyme digestion check and DNA sequencing (Laragen Inc.). The plasmids used are listed in Table S2. Expression plasmids were transformed into A. nidulans ΔEM strain. The transformants were maintained on solid CD medium [1% (w/v) D-glucose, 1x nitrate salts, 0.1% (v/v) trace elements solution, 2% agar]. For expression, the spores were inoculated into liquid CD-ST production medium at 28 °C for 3 days. For small scale analysis, 0.5 mL of liquid culture was extracted using 1 mL of ethyl acetate. The organic fraction was dried in vacuo using a Speedvac and the residue was resuspended in methanol before injection for LC-MS analysis.
Extraction and Isolation
To purify 1 and 2 for structure characterization, the A. nidulans transformants were cultured in 4 L of liquid CD-ST production medium [2% (w/v) starch, 2% (w/v) peptone, 1x nitrate salts, 0.1% (v/v) trace elements solution] and shaken at 28 °C for 3 days. Culture was filtered to separate the supernatant and the mycelium by using a Büchner funnel. Ethyl acetate was used to extract the target compounds from the supernatant. The organic extracts were dried over Na2SO4, concentrated in vaccuo and subject to silica-gel column chromatography. Running solvent was EtOAc and fractions were checked by normal phase TLC. Pooled fractions were concentrated and purified by HPLC with a semi-preparative ARII column (Cosmosil), with solvent gradient starting at 25% B for 10 minutes and rising to 40% B over 30 minutes. Solvent A was water and solvent B was acetonitrile, both were supplemented with 0.1% v/v formic acid (FA). HPLC had the following modules from Shimadzu: CBM-20A communications bus module, DGU-20A5R degassing unit, 2xLC-20AD pumps, SPD-M20A diode array detector, SIL-20A HT auto sampler, and FRC-10A fraction collector.
To ease the purification of desmethyl-desacetyl-pyrophen 3, we derivatized 3 with di-ter-butyl decarbonate (Boc anhydride). Briefly, the pH of a 2 L cell culture medium (separated from the mycelium) was adjusted to 10 by Na2CO3 followed by addition of 1L of EtOH. The resulting insoluble materials were removed by centrifugation. To the clarified supernatant, 10 grams of (Boc)2O was added and the mixture was stirred for 24 hrs at room temperature. The EtOH was removed by rotavap and the aqueous phase was extracted by EtOAc. The organic layer was concentrated in vacuo and dried over Na2SO4. The concentrated organic extracts were subject to silica-gel chromatography and further purified by HPLC. The purified Boc-protected 3 was structurally characterized and subject to deprotection. Briefly, Boc-desmethyl-desacetyl-pyrophen (10 mg) was dissolved in 5 mL of CH2Cl2 and cooled to 0 ºC. To this solution, equal volume of trifluoroacetic acid (TFA) was added and the mixture was stirred for 2 hrs at room temperature. Excess TFA was removed by rotavap and the resulting crude oil was taken up in a minimal volume of CH2Cl2. The desired desmethyl-desacetyl-pyrophen 3 was precipitated with diethyl ether, washed with cold CH2Cl2, and subject to HPLC purification with a semi-preparative reverse-phased column. Compound 3 was eluted with a solvent gradient starting at 5% B for 10 minutes and rising to 20% B over 20 minutes. Solvent A was water and solvent B was acetonitrile, both were supplemented with 0.1% v/v formic acid (FA).
Pyrophen (1): colorless needles (acetone), [α]28D: −23.3 (c=0.1, MeOH), UV (CH3CN) λmax 281 nm, 1H NMR (CDCl3, 500 MHz), δ 7.27 (1H, m), 7.21 (1H, m), 7.13 (2H, d, J = 7.3 Hz), 6.76 (1H, d, J = 8.6 Hz), 5.82 (1H, d, J = 2.1 Hz), 5.42 (1H, d, J = 2.1 Hz), 5.00 (1H, dd, J = 8.5, 7.9 Hz), 3.74 (3H, s), 3.10 (2H, m), 1.96, (3H, s). 13C NMR (CDCl3, 125 MHz) δ 171.1, 169.9, 164.5, 161.8, 136.1, 129.0, 128.7, 127.0, 101.1, 88.4, 56.0, 52.6, 38.7, 23.0. HRESI-MS m/z 288.1243 (calcd for C16H18NO4, 288.1230).
Campyrone B (2): colorless needles (acetone), [α]28D: −103.3 (c=0.3, MeOH), UV (CH3CN) λmax 281 nm, 1H NMR (CDCl3, 500 MHz), δ 6.08 (1H, d, J = 8.7 Hz), 6.00 (1H, d, J = 2.3 Hz), 5.44 (1H, d, J = 2.3 Hz), 4.80 (1H, dd, J = 8.5, 7.9 Hz), 3.80 (1H, s), 2.00 (3H, s), 1.67 (1H, m), 1.63 (1H, m), 1.56 (1H, m), 0.93 (t, 6H, J = 7.0 Hz). 13C NMR (CDCl3, 125 MHz) δ 171.3, 169.7, 164.6, 162.7, 100.6, 88.6, 56.1, 49.8, 41.7, 25.0, 22.6, 22.4. HRESI-MS m/z 254.1401 (calcd for C13H20NO4, 254.1387).
Chemical preparation of 4 and 5
Purified pyrophen (20 mg, crystalline form) was dissolved in 10 ml 6N HCl. The solution was incubated at 50 ºC for 16 hrs to yield desacetyl-desmethyl-pyrophen 3, desmethyl-pyrophen 4, and desacetyl-pyrophen 5. Compounds 3-5 and unreacted 1 were separated by HPLC with a semi-preparative ARII column (Cosmosil), with solvent gradient starting at 10% B for 10 minutes and rising to 40% B over 30 minutes. Solvent A was water and solvent B was acetonitrile, both were supplemented with 0.1% v/v formic acid (FA).
Protein expression and purification
Expression plasmids for individual domains and protein were constructed by subcloning the corresponding gene into a modified pET28a (+) vector (Table S2). The resulting His-tagged proteins (AnOMT is fused with a His-tagged MBP protein to increase the solubility) were overexpressed in E. coli BL21(DE3) cells in LB medium in the presence of 50 mg/L kanamycin. Expression was induced by addition of 100 μM IPTG when OD600 reached 1.0 and the cells were incubated at 16 ºC overnight. Cells were harvested by centrifugation and resuspended in cell lysis buffer [50 mM K2HPO4 (pH 7.5), 10 mM imidazole, 300 mM NaCl, 5% glycerol]. Cells were stored at −80 ºC. For protein purification, stored cells were lysed by sonication and the cell lysate was cleared by centrifugation. The cleared supernatant was incubated with Ni2+-NTA resin for 30 min at 4 ºC and then the slurry was loaded onto a gravity column. The resin was washed and eluted with increasing concentrations of imidazole in cell lysis buffer. The fractions were examined by SDS-PAGE analysis and pooled fractions were concentrated and buffer exchanged into storage buffer [50 mM K2HPO4 (pH 7.5), 150 mM NaCl, 1 mM TCEP] using Amicon Ultra Centrifugal Filters (Millipore). Protein concentrations were determined by Bradford assay and supplemented with 10% (v/v) glycerol for storage at −80 ºC.
Protein activity assay
For methylation activity assay, substrates 4 and 5 were dissolved in DMSO to make stock solution (25 mM). In a centrifuge tube (1.5 mL), 2 μL of substrate (25 mM) was added to 45 μL of assay solution [50 mM HEPES buffer, pH 7.5), followed by addition of 2 μL of purified methyltransferase (stock solution at 125 μM concentration). The reaction was initiated by adding 1 μL of SAM (stock solution 50 mM) and incubated at room temperature for 30 min. The reaction was quenched by mixing with an equal volume of methanol and subject to HPLC analysis.
For adenylation activity assay, 0.1 mM carboxylic acid substrates were added to a solution containing 100 mM Tris (pH 8.0), 15 mM MgCl2, 3 mM ATP, and15 mM hydroxylamine. The reaction was initiated by addition of 5 μM AnATPKS-A and incubated at room temperature for 12 hrs. The reaction was quenched by addition of an equal volume (150 μL) of stopping solution [10% (w/v) FeCl3 and 3.3% (w/v) trichloroacetic acid (TCA) dissolved in 0.7 M HCl]. The precipitated enzyme was removed by centrifugation and the supernatant was transferred to a 96-well plate and its absorbance at 540 nm was measured by using a TECAN M200 Pro plate reader.
Precursor feeding and isolation
A. nidulans transformants overexpression atpks and omt were cultured in liquid CD-starch medium [2% (w/v) starch, 1% (w/v) amino acid mix dropping out phenylalanine, 1x nitrate salts, 0.1% (v/v) trace elements solution]. Phenylalanine analogues (25 mg/L) were fed into the cell culture every 24 hrs for 3 days. The production of pyrophen analogues were analyzed by LC-MS and the corresponding analogues were isolated and structurally characterized using the procedure similar to pyrophen. The purity of compound 11 was also evaluated with chiral-HPLC analysis using two different chiral columns at room temperature: a LUX® Cellulose-3 (250 × 4.6 mm, 5 μm) column [mobile phase: 88/12/0.1/ H2O/acetonitrile/TFA, flow-rate: 1.0 mL/min] and a CHIRALPAK® IA-3 (150 × 4.6 mm, 3 μm) column [mobile phase: 91/9/0.1/0.1 hexanes/ethanol/TFA/diethylamine, flow-rate:1.0 mL/min].
Antifungal activity assay
To qualitatively evaluate the growth inhibition of A. nidulans by compound 3, solid CD medium containing compound 3 various concentrations (from 0 to 300 μg/mL) and other essential nutrients [0.125 μg/mL riboflavin; 0.5 μg/ml pyridoxine, 10 mM uridine and 5 mM uracil] was spotted with 5000 spores/5 μL and incubated at 37 °C for 4 days. Amphotericin B (Research Product International) and fluconazole (Sigma) were used as two positive controls.
To evaluate the antifungal activity of compound 1 and its analogues 6–11 against C. albicans SC5314 strain, a microdilution assay was employed. Briefly, C. albicans maintained in YPEG medium [2% w/v peptone, 1% w/v yeast extract, 2% v/v glycerol] were inoculated to fresh YPEG medium, or YPD medium [2% w/v peptone, 1% w/v yeast extract, 1.5% dextrose], or glycerol-based synthetic medium consisting of Difco Yeast Nitrogen base supplemented with 2% glycerol and amino acids mixture16 at a final OD600 value of 0.15 per well in 96-well microdilution plates. The resuspended cells were added with 2-fold serially diluted compounds dissolved in DMSO to a final volume of 190 μL containing 3% v/v DMSO. Amphotericin B (Research Product International) and ilicicolin H (isolated in-house)16,17 were used as two positive controls. The microplates were further incubated at 30 °C at 250 rpm for 48 hrs. The final OD600 for each well was measured with a TECAN M200 Pro plate reader.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH 1R35GM118056 to YT. YH is a Life Sciences Research Foundation postdoctoral fellow sponsored by the Mark Foundation for Cancer Research. AH is supported by the Lorraine H. and Masuo Toji Summer Research fellowship. We thank Daisy Ma for assistance with compound purification and Zhuan Zhang for isolation of ilicicolin H.
Footnotes
No competing financial interests have been declared.
ASSOCIATED CONTENT
Supporting Information.
Spectroscopic Data
Dedicated to Dr. Jon Clardy of Harvard Medical School for his pioneering work on natural products.
Contributor Information
Yi Tang, Department of Chemical and Biomolecular Engineering and Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Yang Hai, Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, California 90095, United States.
Arthur Huang, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
REFERENCES
- (1).Chooi Y-H & Tang YJ Org. Chem 2012, 77, 9933–9953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cox RJ Org. Biol. Chem 2007, 5, 2010–2026. [DOI] [PubMed] [Google Scholar]
- (3).Yun C-S; Motoyama T; Osada H Nat. Commun 2015, 6, 8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cook D; Donzelli BGG; Creamer R; Baucom DL; Gardner DR; Pan J; Moore N; Krasnoff SB; Jaromczyk JW; Schardi CL G3 (Bethesda) 2017, 7, 1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ma SM; Li JW-H; Choi JW; Zhou H; Lee KKM; Moorthie VA; Xie X; Kealey JT; Da Silva NA; Vederas JC; Tang Y Science 2009, 326, 589–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Khosla C; Herschlag D; Cane DE; Walsh CT Biochemistry 2014, 53, 2875–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Schwecke T; Aparicio JF, Molnár I; König A; Khaw LE; Haydock SF; Oliynyk M; Caffrey P; Cortés J; Lester JB; Böhm GA; Staunton J; Leadlay PF Proc. Natl. Acad. Sci. USA 1995, 92, 7839–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kotlobay AA; Sarkisyan KS; Mokrushina YA; Marcet-Houben M; Serebrovskaya EO; Markina NM; Somermeyer LG; Gorokhovatsky AY; Vvedensky A; Purtov KV; Petushkov VN; Rodionova NS; Chepurnyh TV; Fakhranurova LI; Guglya EB; Ziganshin R; Tsarkova AS; Kaskova ZM; Shender V; Abakumov M; Abakumova TO; Povolotskaya IS; Eroshkin FM; Zaraisky AG; Mishin AS; Dolgov SV; Mitiouchkina TY; Kopantzev EP; Waldenmaier HE; Oliveira AG; Oba Y; Barsova E; Bogdanova EA; Gabaldón T; Stevani CV; Lukyanov S; Smirnov IV; Gitelson JI; Kondrashov FA; Yampolsky IV Proc. Natl. Acad. Sci. USA 2018, 115, 12728–12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Barnes CL; Steiner JR; Torres E; Pacheco R; Marquez H Int. J. Pept. Protein Res 1990, 36, 292–296. [DOI] [PubMed] [Google Scholar]; (b) Laatsch H; Talontsi FM; Kongue Tatong MD; Dittrich B; Douanla-Meli C Tetrahedron 2013, 69, 7147–7151. [Google Scholar]; (c) Shaaban M; Shaaban KA; Abdel-Aziz MS Org. Med. Chem. Lett 2012, 2, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Reber KP & Burdge HE Total synthesis of pyrophen and campyrones A-C. J. Nat. Prod 2018, 81, 292–297. [DOI] [PubMed] [Google Scholar]
- (11).Liu N; Hung Y-S; Gao S-S; Hang L; Zou Y; Chooi Y-H; Tang Y Org. Lett 2017. 19, 3560–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kadi N; Challis GL Methods Enzymol. 2009, 458, 431–457. [DOI] [PubMed] [Google Scholar]
- (13).Conti E; Stachelhaus T; Marahiel MA; Brick P EMBO J. 1997, 16, 4174–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Harvey CJB; Tang M; Schlecht U; Horecka J; Fischer CR; Lin HC; Naughton B; Cherry J; Miranda M; Li YF; Chu AM; Hennessy JR; Vandova GA; Inglis D; Aiyar RS; Steinmetz LM; Davis RW; Medema MH; Sattely E; Khosla C; St. Onge RP; Tang Y; Hillenmeyer ME Sci. Adv 2018, 4, No. eaar5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Puji A; Erden W; Wahyono; Wahyuono S; Hertiani T; Asian Pac. J. Cancer Prev 2016, 17, 615–618. [DOI] [PubMed] [Google Scholar]
- (16).Singh SB; Liu W; Li X; Chen T; Shafiee A; Card D; Abruzzo G; Flattery A; Gill C; Thompson JR; Rosenbach M; Dreikorn S; Hornak V; Meinz M; Kurtz M; Kelly R; Onishi JC ACS Med. Chem. Lett 2012, 3, 10, 814–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang Z; Jamieson CS; Zhao Y-L; Li D; Ohashi M; Houk KN; Tang YJ Am. Chem. Soc 2019, 141, 14, 5659–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.