

Summary
Transcription initiation requires formation of the open promoter complex, RPo. To generate RPo, RNA polymerase (RNAP) unwinds the DNA duplex to form the transcription bubble and loads the DNA into the RNAP active site. RPo formation is a multi-step process with transient intermediates of unknown structure. We used single particle cryo-electron microscopy to visualize seven intermediates containing Escherichia coli RNAP with the transcription factor TraR en route to forming RPo. The structures span the RPo formation pathway from initial recognition of the duplex promoter in a closed complex to the final RPo. The structures and supporting biochemical data define RNAP and promoter DNA conformational changes that delineate steps on the pathway, including previously undetected transient promoter-RNAP interactions that contribute to populating the intermediates but do not occur in RPo. Our work provides a structural basis for understanding RPo formation and its regulation, a major checkpoint in gene expression throughout evolution.
Graphical Abstract
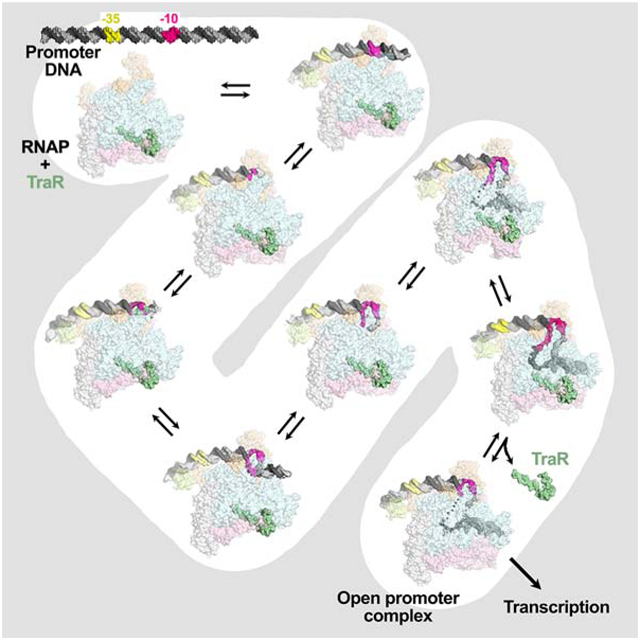
eTOC Blurb
Cryo-EM structures of RNA polymerase/promoter DNA intermediates identify stages in transcription initiation from initial recognition of double-stranded promoter DNA in RPc to final promoter melting in RPo. Structural analyses of RNA polymerase and DNA conformational changes delineate steps in the pathway. Biochemical and genetic characterization support their functional importance.
INTRODUCTION
Transcription of cellular DNA cannot begin until RNA polymerase (RNAP) locates a promoter and forms the open promoter complex (RPo). In RPo, RNAP unwinds about 13 base pairs (bps) of DNA to form the transcription bubble and loads the template-strand (t-strand) DNA into the RNAP active site located within a deep cleft (Abascal-Palacios et al., 2018; Bae et al., 2015; He et al., 2016; Nagy et al., 2015; Plaschka et al., 2016; Tafur et al., 2016; Vorländer et al., 2018; Zuo and Steitz, 2015). The vast majority of initiation events in bacteria involve the RNAP catalytic core enzyme (termed E, subunit composition α2ββ’ω) combined with the primary promoter specificity σ factor [σ70 in Escherichia coli; (Feklistov et al., 2014; Gruber and Gross, 2003)], or Eσ70. Eσ70 functions as a molecular isomerization machine, using binding free energy to generate RPo through a multi-step pathway (Ruff et al., 2015).
Structures of bacterial RPo have been well characterized (Bae et al., 2015; Boyaci et al., 2019; Hubin et al., 2017b; Narayanan et al., 2018; Zuo and Steitz, 2015), but the structural basis for RPo formation is poorly understood due to the transient nature of intermediates along the pathway from initial Eσ70 recognition of the duplex promoter in the closed complex (RPc) to the final RPo (Ruff et al., 2015). Previous kinetic analyses used salt, urea, and other perturbants to identify intermediates of RPo formation in solution (Gries et al., 2010; Kontur et al., 2010; 2008). Here, we used single particle cryo-electron microscopy (cryo-EM) to visualize RPo formation by E. coli (Eco) RNAP. To facilitate visualization of intermediates, we added TraR, an F plasmid-encoded transcription factor, and used a promoter that is inhibited by TraR but forms stable intermediates, the S20 ribosomal protein promoter rpsT P2 (Lemke et al., 2011; Gopalkrishnan et al., 2017; Chen et al., 2019b). Like its homolog DksA, TraR binds directly to RNAP rather than to promoter DNA, regulating transcription initiation in vitro and in vivo by increasing the occupancy of intermediates on the pathway (Blankschien et al., 2009; Gopalkrishnan et al., 2017; Chen et al., 2019b).
Using image classification approaches, we visualized five intermediates formed by the wild-type (wt) rpsT P2 promoter, and two additional intermediates formed by a mutant rpsT P2 promoter. Our structures span the RPo formation pathway from RPc RPo (Chen et al., 2019b). Features of the structures allow their placement in an ordered pathway that provides a structural basis for understanding RPo formation in all organisms.
RESULTS
TraR stabilizes a partially melted intermediate on the rpsT P2 promoter
Our initial studies focused on Eσ70 complexes with the well-characterized rrnB P1 promoter which forms an unstable RPo in the absence of initiating NTPs that is in rapid equilibrium with earlier intermediates (Gourse et al., 2018; Rutherford et al., 2009). However, we could not detect TraR-Eσ70-rrnB P1 complexes by native mass spectrometry (nMS) or by cryo-EM, suggesting these complexes were too unstable under cryo-EM conditions (Chen et al., 2019a). The RPo formed on rpsT P2 is more stable than on rrnB P1 but less stable than on many Eco promoters (Lemke et al., 2011), and TraR-Eσ70-rpsT P2 complexes were detected by nMS, footprinting (Figures 1, S1A, S1B) and cryo-EM (Figure 2).
Figure 1 |. Eco TraR-Eσ70 forms stable, partially melted complexes with an rpsT P2 promoter fragment.
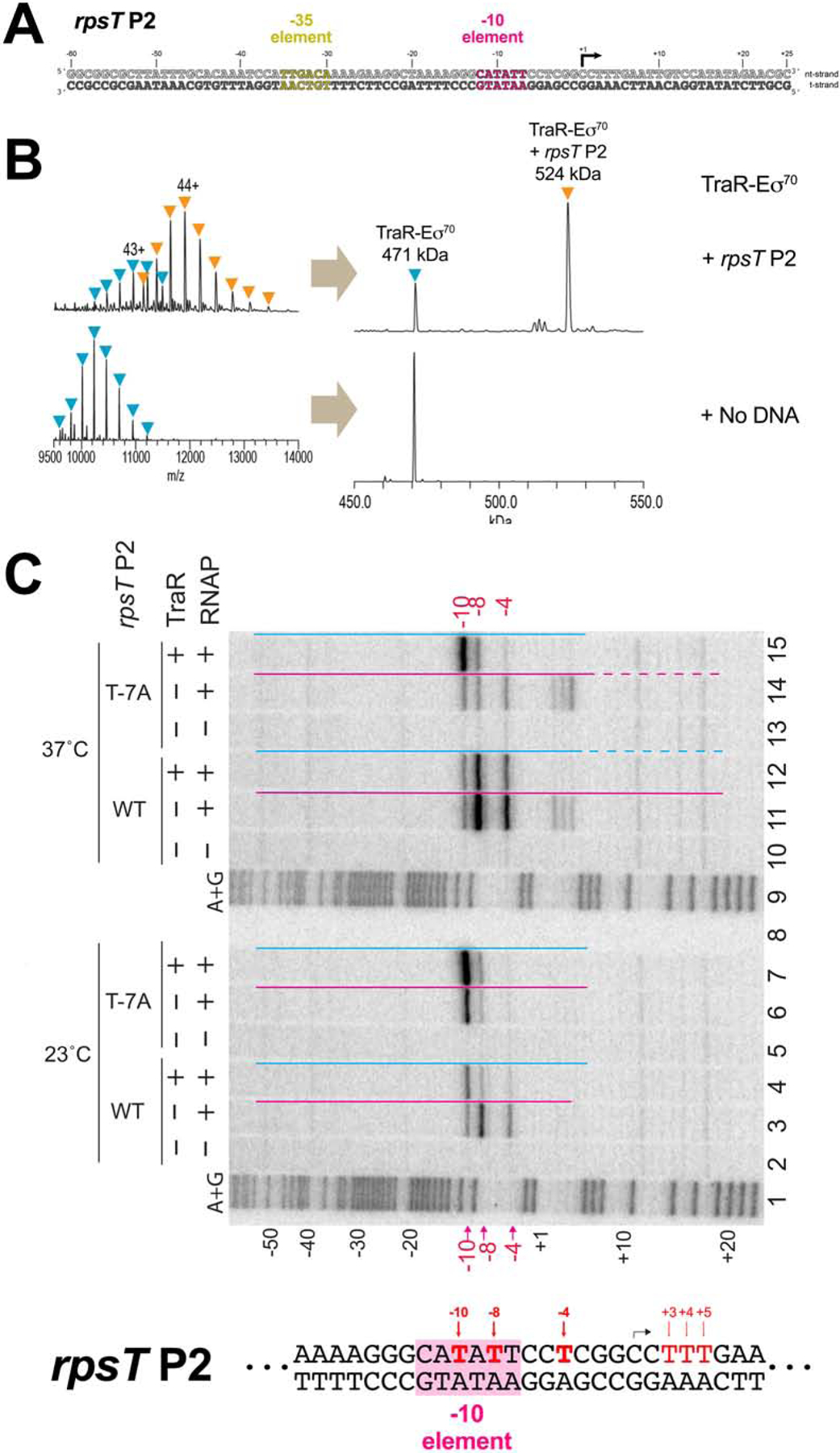
A. The wt-rpsT P2 promoter fragment (−60 to +25) used for nMS and cryo-EM.
B. nMS spectra and the corresponding deconvolved spectra for TraR-Eσ70 complexes with the rpsT P2 promoter fragment (A). TraR binds to Eσ70 in a 1:1 stoichiometry, forming a 471 kDa complex. Upon incubation of this complex with the promoter DNA (52 kDa), a predominant charge state series for the TraR-Eσ70-promoter assembly (524 kDa) was observed.
C. Detection of unpaired thymines by KMnO4 footprinting of Eσ70 complexes formed with the wt-rpsT P2 or T−7A promoters ± TraR, and DNase I footprint protection ranges, shown by red or blue lines above each lane (dahsed lines: partial protection). Strand cleavage of modified thymines at 23°C (lanes 2–7) or 37° (lanes 10–15) was detected by gel electrophoresis of DNA fragments 32P end labeled in the nt-strand. Lanes 1, 9: A+G sequence ladder. Modified thymines at −10, −8 and −4 are indicated in red above and below gel, and on the section of the wt-rpsT P2 sequence shown below the gel (−10 element shaded in pink). Black arrow: transcription start site [see Figures S1A, B for DNase I footprints at 23°C; for 37°C footprints, se e (Gopalkrishnan et al., 2017)].
See also Figure S1.
Figure 2. Eco Eσ70 promoter melting intermediates on the rpsT P2 promoter.
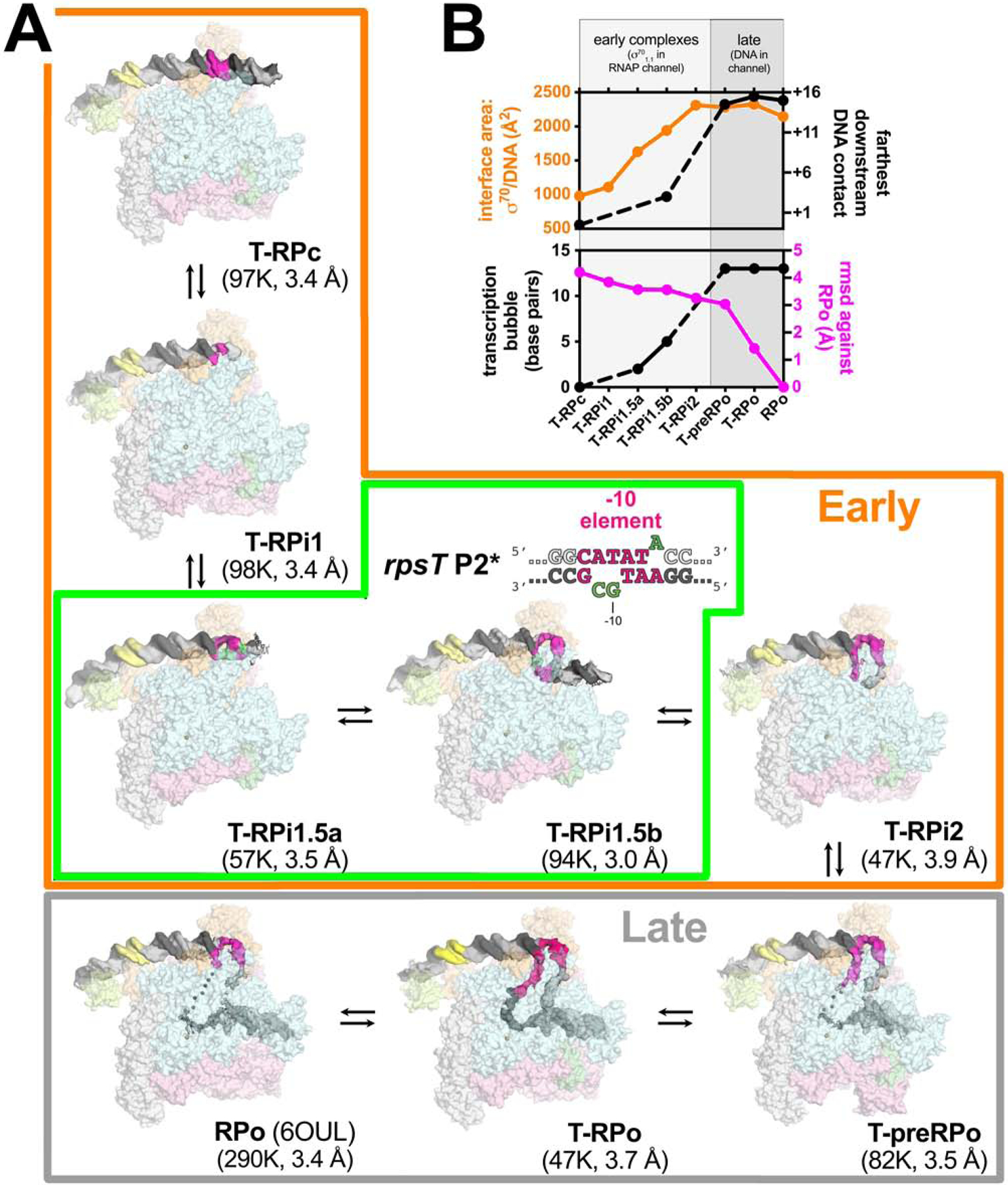
A. Overall structures of promoter melting intermediates obtained by cryo-EM. Proteins are shown as transparent surfaces (αI, αII, ω, light gray; αCTD, pale limon; β, pale cyan; β’, light pink; σ70, light orange; TraR, pale green). The Eσ70 active site Mg2+ is shown as a sand-colored sphere. The promoter DNA is shown as cryo-EM difference density (nt-strand, gray; t-strand, dark gray; −35 element, yellow; −10 element, hot pink). The eight structures were derived from three samples: Sample 1) T-RPc, T-RPi1, T-RPi2, T-preRPo, and T-RPo structures were obtained with TraR and the wt-rpsT P2 fragment (Figure 1A); Sample 2) T-RPi1.5a and T-RPi1.5b were obtained with TraR and rpsT P2* (boxed in green; nucleotide substitutions in rpsT P2* are colored green); Sample 3) RPo was determined previously with wt-rpsT P2 without TraR (Chen et al., 2019b). In the Early complexes (boxed in orange), σ701.1 occupies the Eσ70 channel. In the Late complexes (boxed in gray), downstream duplex DNA occupies the channel.
B. Structural properties used to order the complexes in the RPo formation pathway. (top panel) Plotted in orange (left scale) is the σ70/DNA interface area (Å2)(Krissinel and Henrick, 2007). Plotted in black (right scale) is the most downstream protein/duplex DNA contact. For T-RPi1, T-RPi1.5a, and T-RPi2, most or all of the downstream duplex DNA was disordered so no point is included.
(bottom panel) Plotted in black (left scale) is the extent of the transcription bubble. For T-RPi1 and T-RPi2, the downstream fork of the transcription bubble was disordered so no point is included. Plotted in magenta (right scale) is the root-mean-square deviation of α-carbons (Å) for each complex superimposed with RPo.
See also Figures S2 – S5 and Table S1.
A shift in the occupancy of the rpsT P2 promoter from RPo to earlier intermediates was first detected by a shorter length of protected DNA in DNase I footprints with RNAP at 23°C vs 37°C (Figure 1C, la nes 3, 11), or upon addition of TraR at 37°C [Figure 1C, lanes 11, 12; (Gopalkrishnan et al., 2017); protection indicated by colored lines]. The downstream protection bounary was shifted from +20 (characteristic of RPo at most promoters) to +6 with respect to the transcription start site at +1 (Figure 1A) by reduction of the temperature or by inclusion of TraR. The upstream protection boundary (−54) remained unchanged, indicating that an intermediate complex is formed at rpsT P2 either by reducing the temperature or by TraR.
The pattern of unstacked thymines detected by KMnO4 footprinting (Ross and Gourse, 2009) in these complexes suggested that the transcription bubble was partially melted. At both 23°C and 37°C with RNAP alone, non- template strand (nt-strand) T’s at −10, −8, and −4 were KMnO4 reactive with RNAP alone [Figure 1C, lanes 3, 11; T’s at +3,+4,+5 in the 37°C complex were also reactive, la ne 11, indicating “scrunching” of a minor fraction of the complexes at this temperature (Winkelman et al., 2016)]. The almost totally conserved nt-strand T at the −7 position [T−7(nt); Shultzaberger et al., 2007] was not reactive, reflecting its protection from KMnO4 by binding in a pocket of σ70 subunit domain 2 (σ702)(Feklistov and Darst, 2011). At 23°C, TraR increas ed the KMnO4 signal at −10 and reduced the signals at −8 and −4, suggesting that TraR stabilized a partially melted intermediate (Figure 1C, lane 4). Lower temperature combined with use of the T−7A(nt) substituted promoter strengthened the −10 signal even further and eliminated the signal at −4, independent of TraR (Figure 1C, lanes 6, 7). We infer that all three perturbations [TraR, reduced temperature, and T−7A(nt)] shift the population to earlier DNA melting intermediates.
Two lines of evidence suggest that TraR stabilizes partially open intermediates. First, a higher percentage of Eσ70 was incorporated into complexes with rpsT P2 DNA with TraR than without, as detected by nMS (compare Figure 1B and S1C). Second, the KMnO4-reactive band at position −10 seen in the presence of TraR (Figure 1C and S1D, lanes 1,2) was also observed when the reaction was performed with RNAP containing a small deletion in the clamp module that prevents stabilizing interactions with downstream duplex DNA in RPo [β’Δ215–220-RNAP; (Bartlett et al., 1998)] but not with promoter DNA alone (Figure S1D, lanes 3–5). These results suggest that rather than populating an earlier intermediate indirectly by destabilizing RPo, TraR stabilizes the earlier intermediate directly (Galburt, 2018; Chen et al., 2019b).
Structures along the promoter melting pathway
TraR-Eσ70 complexes were incubated with the wt-rpsT P2 promoter fragment (Figure 1A) or a promoter variant (rpsT P2*; Figure 2A) engineered to trap early melting intermediates detected by KMnO4 (Figure 1C, lane 7) and Dnase I footprinting (Figure S1A). Noncomplementary base pairs at −11 to −10 were introduced in rpsT P2* to favor bubble nucleation while the T−7A(nt) substitution was made to disfavor propagation of downstream base opening. Basal transcription (without TraR) from rpsT P2* was very weak compared with wt-rpsT P2 (Figure S1E), indicating these substitutions depopulated RPo. TraR inhibited transcription from the wt-rpsT P2 promoter fragment (Figure 1A) under conditions similar to those used for cryo-EM (Figure S1F).
The TraR-Eσ70-promoter complexes were visualized by cryo-EM. Steps of maximum-likelihood classification (Scheres, 2012) revealed five TraR-Eσ70-wt-rpsT P2 structures (T-RPc, T-RPi1, T-RPi2, T-preRPo, T-RPo; Figure 2A) at 3.4 – 3.9 Å nominal resolution, and 3.0 – 3.4 Å in the central core of the structures (Figures S2–S4, Table S1). Classification of TraR-Eσ70-rpsT P2* complexes gave rise to two distinct structures (T-RPi1.5a, T-RPi1.5b; Figure 2A) at 3.5 and 3.0 Å nominal resolution, with the central core of the structures resolved to 3.0 and 2.6 Å, respectively (Figures S4, S5, Table S1). In our structural analysis, we also include a previously determined nominal 3.4 Å structure of a complex between Eσ70 and the wt-rpsT P2 promoter fragment prepared in the absence of TraR (RPo, Figure 2A; Chen et al., 2019b).
The eight complexes observed by cryo-EM with rpsT P2 and rpsT P2* were ordered in the pathway such that the DNA/σ70 interface area, the downstream boundary of the DNA/RNAP contacts, and the extent of the transcription bubble monotonically increased, while the root-mean-square deviation of α-carbon positions of each complex compared to RPo decreased monotonically, with progress along the pathway (Figure 2B). A clear demarcation between early and late complexes could be made based on the presence of the N-terminal domain of σ70, σ701.1 (early complexes) or downstream duplex DNA (late complexes) in the RNAP channel (Bae et al., 2013; Mekler et al., 2002).
In all eight structures, Eσ70 interacts with upstream promoter DNA (from −43 to −17) in the same manner: i) domain 4 of σ70 (σ704) engages specifically with the major groove of the promoter −35 element from −37 to −30 (Campbell et al., 2002), ii) an α-subunit C-terminal domain (αCTD) binds just upstream of σ704, interacting with the DNA minor groove from −43 to −38 (Benoff et al., 2002; Ross et al., 2001; 1993) and also interacting with σ704 (Ross et al., 2003), and iii) conserved residues of the β’zipper (β’Y46 and R47) interact with the DNA backbone from −18 to −17 (Bae et al., 2015; Yuzenkova et al., 2011). By contrast, Eσ70 interacts with the promoter DNA downstream of −17 in diverse configurations that we propose represent steps on the RPo formation pathway (Figure 2A).
Structure of a closed complex
Initial recognition of the duplex promoter sequence prior to melting is thought to give rise to the closed complex, RPc (Ruff et al., 2015). RPc has been enriched at some promoters by formation of the complex at 0–4°C. DNa se I or hydroxyl-radical footprinting revealed an upstream DNA protection in RPc similar to that in RPo (Kovacic, 1987; Schickor et al., 1990). However, downstream protection extended only to about −3, with weak protection sometimes extending to about +2 (Kovacic, 1987; Schickor et al., 1990), indicating that the duplex DNA downstream of the −10 element was mostly solvent exposed. The earliest complex in our pathway (T-RPc, Figures 2A, 3), which contains entirely duplex DNA and thus precedes nucleation of transcription bubble melting, forms Eσ70-promoter interactions consistent with these earlier footprinting results.
Figure 3. Structure of the TraR-Eσ70 closed promoter complex (T-RPc).
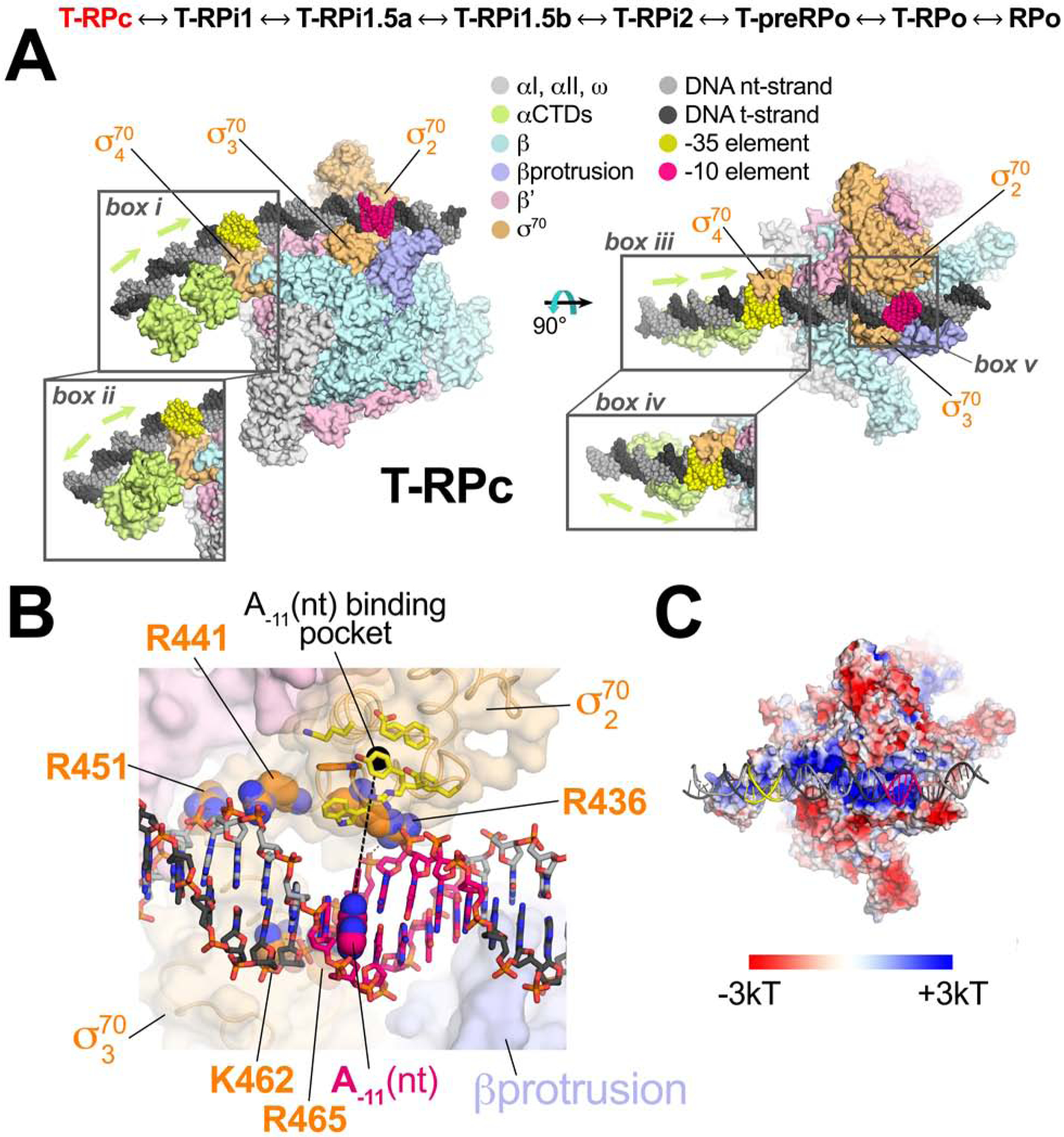
(top) The structures determined here are ordered through the RPo formation pathway (see Figure 2). T-RPc, highlighted in red, is the focus of this figure.
A., B. Color-coding is shown in the key.
A. Orthogonal views of T-RPc. Proteins are shown as molecular surfaces, DNA is shown as Corey-Pauling-Koltun (CPK) spheres. The proximal (adjacent to σ704) and distal (further upstream) αCTDs were visualized in two co-existing dispositions on the DNA upstream of the −35 element, head-to-tail (box i and iii) and head-to-head (box iiand iv). The region around the duplex −10 element (box v) is magnified in (B).
B. Magnified view of Eσ70 interactions with the duplex −10 element showing the absence of sequence-specific interactions (Feklistov and Darst, 2011). The DNA is shown as sticks with the A−11(nt) base highlighted in CPK spheres, and the location of the cognate binding pocket in σ702 (yellow side chains) occupied by A−11(nt) in subsequent intermediates indicated by a dashed black line connecting A−11(nt) to the pocket. RNAP is shown as a transparent molecular surface. The side chains shown as CPK spheres (σ702 R436, R441, R451; σ703 K462, R465), absolutely conserved among primary σ’s (Gruber and Bryant, 1997), interact with the duplex DNA phosphate backbone.
C. The electrostatic charge distribution (Baker et al., 2001) is shown on the molecular surface of the T-RPc RNAP (same view as the right view of A). The DNA is shown in cartoon format.
See also Figure S6.
In T-RPc, base-specific protein-DNA interactions do not occur within the −10 element (Figures 3B, S6A), consistent with the conclusion that recognition of the −10 element sequence is coupled with melting (Feklistov and Darst, 2011). The duplex −10 element DNA is drawn to a shallow, basic channel on the Eσ70 surface (Figure 3C) by phosphate backbone interactions with invariant basic residues of σ702 (R436, R441, R451) and σ703 (K462, R465) (Figure 3B). Sequence-specific recognition of the −35 element by σ704 fixes the register of the DNA with respect to Eσ70, positioning the critical and conserved A−11 (nt) (Shultzaberger et al., 2007) in line with the σ702 residues that ultimately capture the flipped out base to nucleate transcription bubble formation (Feklistov and Darst, 2011; yellow residues in Figure 3B). DNA downstream of the −10 element (−2 to +2) interacts with the tip of the βprotrusion, introducing an ~17° bend in the DNA helical axis centered within the −10 element (Figure 3A).
Upstream of the αCTD proximal to σ704, all of the Eσ70-rpsT P2 structures showed cryo-EM density corresponding to a distal αCTD bound to DNA, but only in T-RPc was this density interpretable. Focused classification of this region upstream of the −35 element revealed two dispositions of the αCTDs, head-to-head (about 53% of the particles) and head-to-tail (about 47%), with altered upstream DNA trajectory (Figure 3A; compare box iii and iv). In all cases, the linkers connecting the αCTDs with the αNTDs were disordered so we could not assign which αNTD was connected to which αCTD. These structures highlight the dynamic nature of the flexibly tethered αCTDs, which tune expression via variable interactions with σ704, transcription factors, and upstream DNA sites (Ross et al., 1993; Estrem et al., 1998; Ross et al., 2003; Ross et al., 2005; Benoff et al., 2002; Lee et al., 2012).
Transcription bubble nucleation and the σ70 W-dyad
The key event in nucleation of promoter melting is thought to be flipping of the A−11(nt) base from the duplex DNA into its σ702 pocket (Chen and Helmann, 1997; Feklistov and Darst, 2011; Heyduk et al., 2006; Lim et al., 2001) and isomerization of an invariant W-dyad of σ702 (W433/W434) from an ‘edge-on’ (Figures 4B, C) to a ‘chair’-like conformation (Figure 4D). In the chair conformation, the W433 side chain rotates away from W434, fills the space vacated by the flipped-out A−11(nt), and forms a π-stack with the face of the exposed −12(nt) base (Bae et al., 2015).
Figure 4. T-RPc ↔ T-RPi1 ↔ T-RPi1.5a; transcription bubble nucleation.
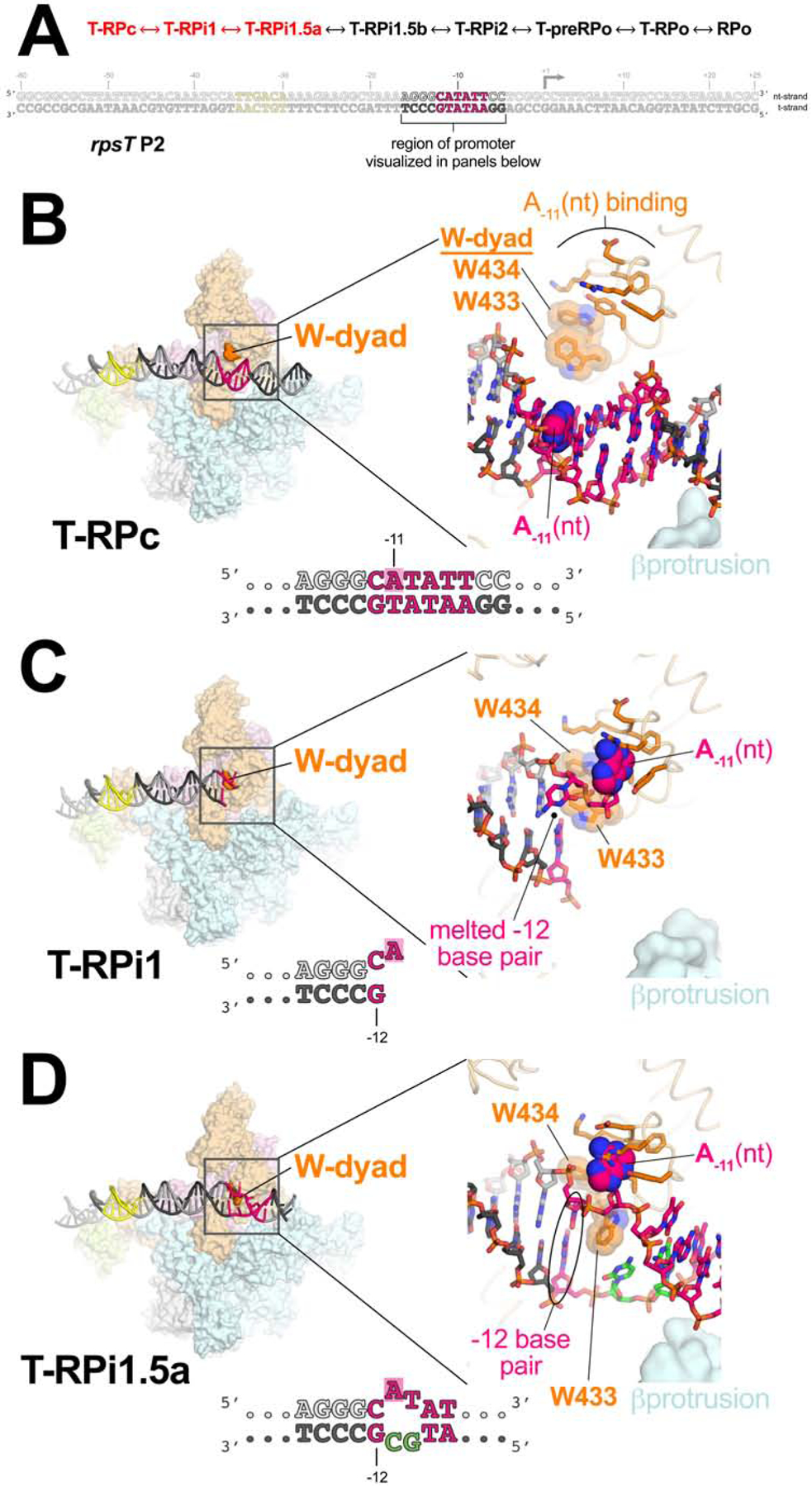
A. (top) The order of structures through the RPo formation pathway (see Figure 2). The progression from T-RPc ↔ T-RPi1 ↔ T-RPi1.5a, highlighted in red, is the focus of this figure.
(bottom) The sequence of the duplex rpsT P2 promoter fragment is shown, with the region of the promoter visualized in the panels below highlighted.
B – D. (left) Overall view of T-RPc (B), T-RPi1 (C), and T-RPi1.5a (D). Eσ70 is shown as a molecular surface with promoter DNA in cartoon format (color-coded as in Figure 2A). The σ70 W-dyad is colored dark orange. The boxed region is magnified on the right.
(right) Magnified view of promoter −10 element and W-dyad. Promoter DNA is shown in stick format with the A−11(nt) base highlighted with CPK spheres. Σ70 is shown as a backbone worm (pale orange) but with side chains of residues that interact with A−11(nt) in RPo shown (orange). The W-dyad is also shown, highlighted with transparent CPK spheres.
B. T-RPc: The −10 element is completely duplex and the W-dyad is in the edge-on conformation.
C. T-RPi1 and transcription bubble nucleation: A−11(nt) is flipped out of the duplex towards its cognate σ702 pocket, nucleating −10 element melting. Steric clash with the edge-on conformation of the W-dyad disrupts the −12 base pair. Downstream DNA lacks cryo-EM density and is presumed to be highly dynamic.
D. T-RPi1.5a: The flipped out A−11(nt) more fully engages with its cognate σ702 pocket. We modeled the W-dyad in its ‘chair’ conformation (Bae et al., 2015), allowing the −12 bp to reform. The T-RPi1.5a structure was obtained with the mutant rpsT P2* promoter (base substitutions colored green).
In T-RPi1, the A−11(nt) base is flipped and entering its cognate σ702 pocket (Figure 4C). Notably, the W-dyad remains in its edge-on conformation (Figure S6B). The edge-on orientation of the W433 side chain in T-RPi1 sterically clashes with the −12 bp, and the cryo-EM density indicates transient melting of the −12 bp in this intermediate (Figures 4C, S6B). In all subsequent structures in the pathway (T-RPi1.5a -> RPo), the flipped-out A−11(nt) base is fully engaged in its pocket and the −12 nucleotides are clearly base-paired (Figures 4D, S6C). In T-RPi1.5b -> RPo, W433 is rotated into the chair conformation and stacked with the −12(nt) base, but in the T-RPi1.5a intermediate (between T-RPi1 and T-RPi1.5b), the cryo-EM density for W433 is poorly resolved and does not unambiguously define the edge-on or chair conformations (Figure S6C). We modeled T-RPi1.5a with W433 in the chair conformation due to the strong apparent density for the −12 bp but we propose that in T-RPi1.5a, the conformation of W433 and the disposition of the −12 bp is dynamic, giving rise to the poorly resolved cryo-EM density.
Although there is fragmented cryo-EM density for the downstream duplex DNA in T-RPi1, the density is uninterpretable and we have not modeled the downstream edge of the transcription bubble nor the downstream DNA (Figure 4C). Nevertheless, T-RPi1 supports a model in which transcription bubble nucleation begins before DNA enters into the RNAP cleft. The entire T-RPc ↔ T-RPi1 ↔ T-RPi1.5a transition, illustrating transcription bubble nucleation and possible W-dyad isomerization, is shown in Movie S1.
Transcription bubble propagation and the protrusion pocket
In T-RPi1.5a (obtained with rpsT P2*; Figure 2A), the nascent transcription bubble is only two nucleotides (the engineered bubble from −11 to −10), and density for about seven bps of downstream duplex DNA is interpretable (Figure 4D). Further along the pathway, in the transition from T-RPi1.5a to T-RPi1.5b, the transcription bubble extends to 5 nucleotides (−11 to −7), the flipped-out A−11(nt) completely engages its cognate σ702 pocket, and nt-strand phosphate-backbone interactions from −10 to −8 with σ702 are established as in RPo (Figures 5A–C). Presumably because of the T−7A(nt) substitution in rpsT P2* (Figure 5A), the mutant A−7(nt) base is not engaged in the σ702 pocket normally occupied by the conserved T−7(nt) and the entire −7(nt) nucleotide is disordered (Figure 5C).
Figure 5. T-RPi1.5a ↔ T-RPi1.5b; transcription bubble propagation and the protrusion pocket.
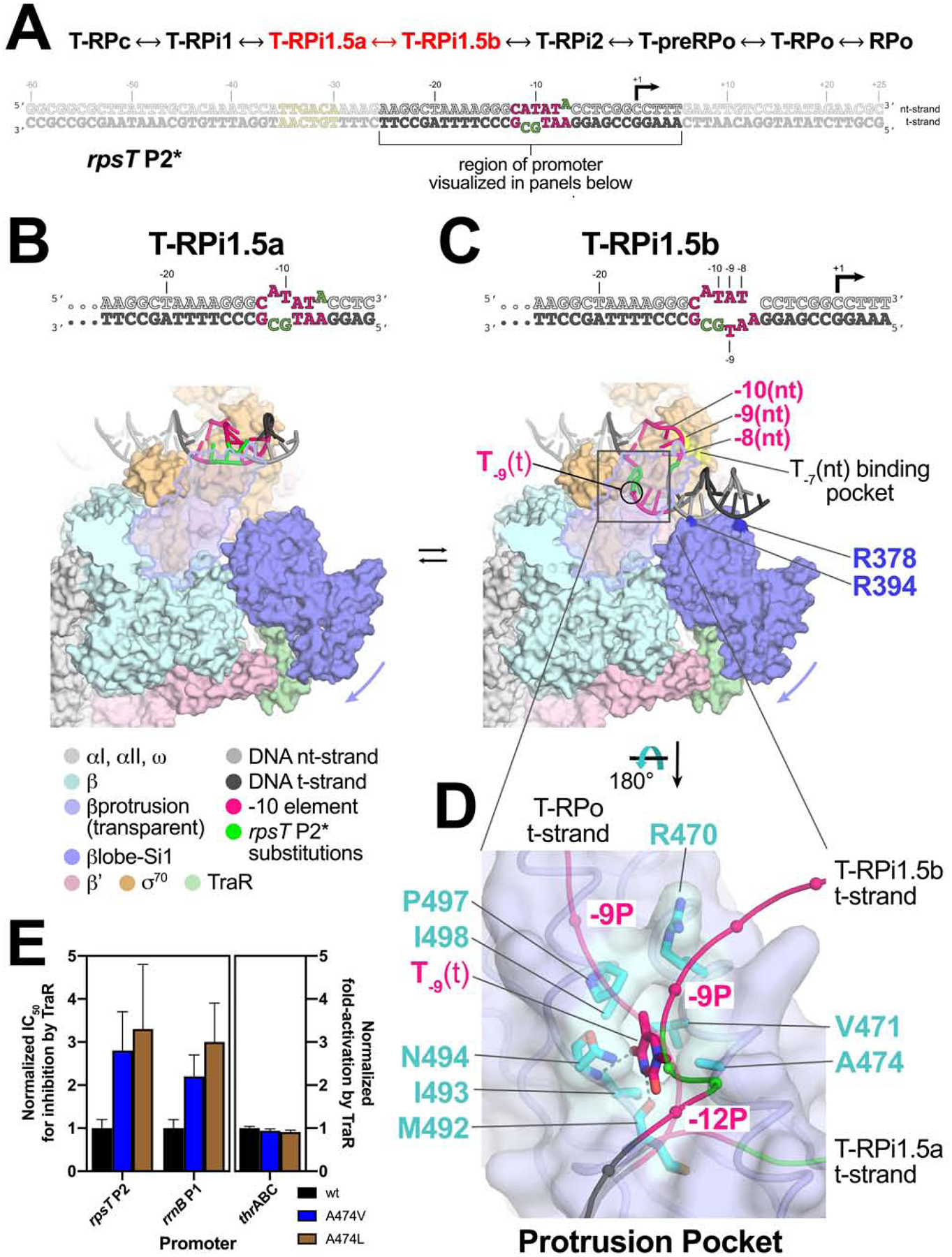
A. (top) The order of structures through the RPo formation pathway (see Figure 2). The progression from T-RPi1.5a ↔ T-RPi1.5b, highlighted in red, is the focus of this figure.
(bottom) The sequence of rpsT P2* is shown, with the region of the promoter visualized in the panels below highlighted.
B., C. Overall view of T-RPi1.5a (B) and T-RPi1.5b (C). Eσ70 is shown as a molecular surface with promoter DNA in cartoon format (color-coded as in the key). The βprotrusion (light blue) is transparent with an outline. The rotation of the βlobe-Si1 domains (slate blue) induced by TraR is indicated by the slate blue arrow.
B. T-RPi1.5a
C. In T-RPi1.5b, DNA phosphate backbone contacts between nt-strand −10 to −8 and σ70 are established as in RPo. The −7(nt) base is positioned over its cognate pocket in σ70 (highlighted in yellow) but is not bound in the pocket due to the T−7A(nt) substitution of rpsT P2*. The T−9(t) base flips up and is bound in the protrusion-pocket on the underside of the βprotrusion. βlobe residues R378 and R394, two of many residues that interact with the DNA in T-RPi1.5b but not in RPo (Table S2), are highlighted (dark blue).
D. The protrusion pocket, viewed from the underside of the βprotrusion. The βprotrusion is shown as a backbone worm with a transparent molecular surface. The T-RPi1.5b t-strand DNA is shown as a thin backbone worm with phosphate atom positions denoted by CPK spheres. The T−9(t) base, bound in the protrusion-pocket, is shown as sticks. The t-strand DNA backbone paths for T-RPi1.5a (precedes T-RPi1.5b in the RPo formation pathway) and T-RPo (follows T-RPi1.5b) are shown for comparison. Protrusion-pocket residues that interact with the T−9(t) base are shown as sticks and colored cyan. Thymine-specific hydrogen-bonds between RNAP and T−9(t) are denoted by dark gray dashed lines.
E. Effect of βA474 substitutions on TraR-mediated inhibition of rpsT P2 and rrnB P1 promoters (left) or activation of thrABC (right). For rpsT P2 and rrnB P1, IC50 values for TraR inhibition of wt-RNAP (black bar), βA474V-RNAP (blue bar), and βA474L-RNAP (brown bar) are plotted relative to wt-RNAP (normalized to 1.0). For thrABC, fold-activation (relative to no TraR) at 500 nM TraR is plotted relative to wt-RNAP (normalized to 1.0). Averages with standard deviation from three independent experiments are shown.
See also Figures S6, S7, Table S2 and Movie S2.
In all of the structures with TraR bound (except for T-RPo), TraR establishes a significant interface with the RNAP βlobe-Si1 domains, inducing an ~18° rotation of the βlobe-Si1 towards TraR and away from the βprotrusion (Figures 5B, C)(Chen et al., 2019b). Rotation of the βlobe-Si1 widens the gap between the βprotrusion and the βlobe by about 9 Å (Figure S5D). The new position of the βlobe alters βlobe-σ701.1 interactions (Chen et al., 2019b), but interactions between the βlobe-Gate-Loop (GL; β residues 371–376) and σ701.1 that pinch off further DNA access to the RNAP cleft are maintained. The GL-barrier hinders further DNA melting and entry into the RNAP cleft, while the widened gap between the βprotrusion and βlobe provides a channel to accommodate the downstream duplex DNA (Figure 5C). The T-RPi1.5a ↔ T-RPi1.5b transition is shown in Movie S2.
Because of the short transcription bubble and the novel disposition of the downstream duplex DNA, the single-stranded t-strand DNA in TraR1.5b follows a path between σ702 and the βprotrusion that is not as deep into the RNAP cleft as complexes later in the pathway (Figure 5D). In this intermediate path of the DNA, the −9 t-strand base [T−9(t)] flips towards the βprotrusion and binds in a distinct pocket in the underside of the βprotrusion that has not been described previously (referred to here as the protrusion pocket; Figures 5D, S7A). The T−9(t) is protected from KMnO4 reactivity in the presence of TraR compared to without TraR (Figure S7B), presumably due to protrusion pocket binding. Residues that form the protrusion-pocket and interact with T−9(t) (Figure 5D) are conserved among bacterial RNAPs, especially in proteobacteria such as Eco (Figure S7C), pointing to functional importance. The backbone carbonyl of βM492 and the backbone amide of βN494 form hydrogen-bonds with the T−9(t) base, suggesting that the protrusion pocket is thymine specific (Figures 5D, S7A). The protrusion pocket would be unable to accommodate a purine without significant rearrangement of the DNA phosphate backbone (Figure S7D).
To test whether binding of T−9(t) in the protrusion pocket has functional consequences, we constructed structure-guided RNAP protrusion pocket mutants βA474V and βA474L. These substitutions were expected to fill the pocket, excluding the thymine base (Figure 5D). There was a significant increase (2 to 3-fold) in the IC50 for inhibition by TraR of two promoters containinng T−9(t), wt-rpsT P2 and rrnB P1, with the greater effect resulting from the substitution with the larger side chain (Figures 5E, S7E–F). Thus, steric occlusion of the pocket by these RNAP substitutions reduces the efficiency of TraR inhibition. We conclude that T−9(t) binding in the protrusion pocket contributes to TraR-mediated stabilization of intermediate T-RPi1.5b, and that this stabilization is important for the mechanism of inhibition by TraR. By contrast, the protrusion pocket substitutions had no effect on activation of thrABC by TraR, as expected (Figure 5E, S7G) since this promoter has a t-strand G at −9 [G−9(t)] instead of a T, and this step is not rate limiting at activated promoters (Chen et al., 2019b). The RNAP substitutions did not affect basal transcription at the limited number of promoters tested here. We suggest that eliminating the T−9(t) interaction in the protrusion pocket would affect only promoters with the appropriate kinetic characteristics (Galburt, 2018; Chen et al., 2019b).
In T-RPi1.5b, promoter DNA establishes many Eσ70 contacts that are unique to this intermediate; these contacts are either altered or absent in the subsequent RPo-like complexes (T-RPo or RPo; Table S2). Like the conserved residues that form the protrusion pocket interaction with T−9(t) in T-RPi1.5b (Figure 5D), many other conserved residues in the βprotrusion, βlobe, and σ70 participate in promoter contacts in T-RPi1.5b but not in later complexes (Table S2). Substitution of these residues would be expected to affect RPo formation by altering the multi-step energy landscape of RPo formation even though they do not interact with promoter DNA in RPo itself. RNAPs with substitutions for βR378 and βR394, basic residues that interact with promoter DNA in T-RPi1.5b but not in RPo (Figure 5C), were defective not only in TraR-mediated inhibition but also in basal transcription (Figure S7H), consistent with a model in which Eσ70-promoter interactions that stabilize the T-RPi1.5b intermediate are important for TraR-mediated inhibition and for transcription in the absence of TraR.
σ701.1 ejection
In T-RPi2, the intermediate following T-RPi1.5b, T−7(nt) is engaged in its σ702 pocket, the single-stranded nt-strand DNA from −11 to −5 interacts with the RNAP in a similar manner as in RPo, but in contrast to RPo, σ701.1 remains in the RNAP cleft (Figures 6A, B). The five nucleotide transcription bubble of T-RPi1.5b is extended to at least 6 nucleotides, but the full extent of the T-RPi2 transcription bubble cannot be determined because the downstream ss/ds junction and the downstream duplex DNA in this complex lack cryo-EM density (Figure 6B). Masking, particle subtraction, and focused classification approaches failed to identify interpretable density for the downstream DNA, indicating that it is highly dynamic.
Figure 6. T-RPi1.5b ↔ T-RPi2 ↔ T-preRPo; transcription bubble completion and σ701.1 ejection.

(top) The order of structures through the RPo formation pathway (see Figure 2). The progression from T-RPi1.5b ↔ T-RPi2 ↔ T-preRPo, highlighted in red, is the focus of this figure.
A. – C. Overall view of T-RPi1.5b (A), T-RPi2 (B), and T-preRPo (C). Eσ70 is shown as molecular surfaces, with core RNAP transparent, revealing the RNAP active site Mg2+ (sand colored sphere), TraR in the secondary channel, and either σ701.1 (T-RPi1.5b and T-RPi2) or downstream duplex DNA (T-preRPo) in the RNAP channel. The βprotrusion (light blue) and βlobe-Si1 (slate blue) are outlined.
A. T-RPi1.5b: Downstream duplex DNA is accommodated in the gap between the βprotrusion and βlobe-Si1. The empty T−7(nt) pocket in σ702 is denoted.
B. T-RPi2: The −10 element T−7(nt) is engaged in its cognate σ70 pocket, the transcription bubble advances in the downstream direction, and the single-stranded nt-strand downstream to −4 is positioned in the complex much like RPo. The downstream edge of the transcription bubble and downstream duplex DNA are disordered and σ701.1 occupies that RNAP channel.
C. T-preRPo: The transcription bubble is fully formed (−11 to +2). The downstream duplex DNA is accommodated in the RNAP channel in place of the ejected σ701.1.
In the next intermediate (T-preRPo), the transcription bubble is fully formed (13 nucleotides, from −11 to +2) and the downstream duplex DNA occupies the RNAP channel, displacing σ701.1 (Figure 6C) and initiating the late stages of the pathway (Figure 2). We call this complex T-preRPo because the βlobe-Si1 is still in its rotated conformation, interacting with TraR, which remains bound to the complex (Figures 6C, S7I).
Next in the pathway is T-RPo, where TraR remains bound but the rest of the RNAP, including the βlobe-Si1, attains an RPo-like conformation (Figure S7I). Although the DNA is in an RPo-like state, T-preRPo and T-RPo would not be transcriptionally active because the presence of TraR sterically blocks folding of the trigger-loop (critical for efficient catalysis)(Vassylyev et al., 2007; Wang et al., 2006; Windgassen et al., 2014) and also blocks binding of the 3’-NTP substrate (Chen et al., 2019b).
DISCUSSION
We observed seven different intermediate structures that delineate changes in the conformation of both Eσ70 and the rpsT P2 promoter on the pathway to forming transcription-capable RPo. These intermediates were observed in the presence of TraR, which inhibits transcription from rpsT P2 by increasing the occupancy of intermediates earlier in the kinetic pathway (Rutherford et al., 2009; Gopalkrishnan et al., 2017), facilitating their structure determination.
Analysis of the structures of RPo intermediates provides insights into the mechanism of transcription initiation. Early intermediates reveal unanticipated transient events, including the melting of the −12 bp and capture of the T−9(t) base. We propose that these intermediate structures define steps in DNA opening at most if not all Eσ70 promoters and discuss the implications of this model for regulation. Finally we outline how these complexes inform models of DNA opening.
The RNAP clamp
Clamp dynamics play an important role in promoter melting for all cellular RNAPs (Boyaci et al., 2019; Chakraborty et al., 2012; Feklistov et al., 2017; He et al., 2013; Schulz et al., 2016). In our cryo-EM structures of TraR bound to Eσ70, the range of clamp motions in solution was narrowed by TraR binding (Chen et al., 2019b). Analysis of RNAP clamp positions in the TraR-Eσ70-promoter intermediates (relative to RPo) revealed that the initial Eσ70-promoter complex, T-RPc, has the most open clamp (7.2° open; Figure S7I). Transient closing of the c lamp in T-RPc would pinch the DNA between the βprotrusion and σ702 (Figure 3A), which might respond by untwisting, facilitating A−11(nt) flipping and capture by σ702, thereby initiating bubble nucleation (Feklistov et al., 2017). In the early intermediate complexes where A−11 capture is first detected (T-RPi1 - T-RPi1.5b), the clamp is ~5° ope n. The clamp generally closes as the pathway approaches RPo, but not monotonically (Figure S7I).
RPo formation involves transient melting of the −12 base pair
The first intermediate visualizing bubble nucleation, T-RPi1, reveals that A−11(nt) capture occurs before or concurrent with W-dyad isomerization and results in transient −12 bp melting due to steric clash with W433 (Figures 4C, S6B). Subsequently, the W433 side chain rotates into the chair conformation, relieving steric clash and stabilizing −12 bp formation by stacking on the exposed downstream face of the −12(nt) base (Figures 4D, S6C). We suggest this transient −12 bp melting may occur at most promoters and could help explain conservation of the TA bp at the −12 position (Shultzaberger et al., 2007).
T-RPi1.5b is likely on pathway during basal RPo formation and is stabilized by TraR
We designed rpsT P2* to trap an early melting intermediate detected by footprints of complexes formed on a fully duplex rpsT P2 T−7A promoter fragment. Protection of this intermediate against DNase I extends downstream to ~+6 (Figure S1A), and the transcription bubble likely extends from −11 to between −8 and −5, corresponding to a bubble of 4 to 8 nt (Figure 1C, lanes 6, 7). T-RPi1.5b, the prominent intermediate observed by cryo-EM with rpsT P2* (Figures 2A, 5C, 6A), has a transcription bubble of 5 nucleotides (−11 to −7) and downstream DNA contacts that extend to +4. Since the size of the transcription bubble detected by KMnO4 footprinting and the limits of DNase I protection of the rpsT P2 T−7A promoter are consistent with the properties of the T-RPi1.5b complex, we conclude that the structurally and biochemically detected intermediate are the same, indicating that the pre-formed bubble in rpsT P2* is not required for formation of the intermediate
Several lines of evidence support the hypothesis that the T-RPi1.5b intermediate, or a similar complex, is on the normal RPo formation pathway, even in the absence of TraR. First, at 23°C, the KMnO4 and DNase I footprints on the rpsT P2(T−7A) promoter were very similar with or without TraR (Figure 1C, lanes 6, 7), indicating that TraR is not required for its formation. Second, substitutions of Eσ70 residues that interact with the DNA in T-RPi1.5b but not in RPo affect ‘basal’ transcription (i.e. transcription in the absence of TraR) as well as inhibition by TraR (Figure S7H). Third, the rpsT P2* promoter fragment contains mismatched base pairs within the −10 element (Figure 2A) and thus would be expected to stimulate basal transcription compared to the duplex wt-rpsT P2 promoter. However, transcription from rpsT P2* in the absence of TraR is weaker than transcription from wt-rpsT P2 (35% of wt-rpsT P2; Figure S1E), suggesting that pre-melting upstream bases does not provide enough free enrgy to overcome subsequent conversions when the conserved T−7(nt) is replaced by Adenine. This demonstrates the key role of T−7(nt) binding to its cognate σ70 pocket (Feklistov and Darst, 2011) in driving subsequent opening (see below).
T-RPi2, σ701.1 ejection, and completion of the transcription bubble
The finding that rpsT P2* [with the T−7A(nt) substitution] yields the stable intermediate T-RPi1.5b with σ701.1 occupying the RNAP channel without proceeding to RPo, while the wt-rpsT P2 yields RPo-like complexes in which the downstream duplex DNA displaces σ701.1 (despite the presence of TraR), suggests that engagement of T−7(nt) with its cognate σ702 pocket is an important determinant of σ701.1 ejection. The pathway progresses from T-RPi1.5b, with its 5-nucleotide transcription bubble, to T-preRPo with its complete 13-nucleotide transcription bubble through a single intermediate (T-RPi2) in which the extent of the transcription bubble and the path of the downstream duplex DNA is highly dynamic (Figure 6). This suggests that propagation of the transcription bubble downstream to the start site (from around −4 to +2) occurs rapidly following nucleation and melting of the −10 element, consistent with kinetic analyses of RPo formation (Hubin et al., 2017a; Ruff et al., 2015; Saecker et al., 2011)
The complete RPo formation pathway and TraR binding
Movie S3 illustrates the entire RPo formation pathway, starting with the βlobe-Si1 rotation induced by TraR binding to RNAP. The βlobe-Si1 stays rotated throughout most of the pathway (until T-RPo), reflecting TraR binding, but how does this relate to basal RPo formation in the absence of TraR? Only one intermediate on the pathway, T-RPi1.5b, appears to require βlobe-Si1 rotation for its formation. We argue above that T-RPi1.5b is on the basal pathway, and by extension we suggest that βlobe-Si1 rotation occurs at this point during the basal pathway, but transiently. In early steps of the pathway preceding T-RPi1.5b (T-RPc ↔ T-RPi1 ↔ T-RPi1.5a), the promoter DNA is far from the βlobe-Si1 and it doesn’t appear that βlobe-Si1 rotation would affect these steps directly, suggesting that the salient structural features of these intermediates (binding of duplex DNA in T-RPc, transcription bubble nucleation in T-RPi1 and subsequent W-dyad isomerization) reflect steps in the standard RPo formation pathway with or without TraR. TraR stabilization of the βlobe-Si1 rotation likely increases occupancy of these early intermediates by increasing the population of T-RPi1.5b, facilitating our analysis of their structure. Movie S4 illustrates the hypothetical RPo formation pathway in the absence of TraR.
Five base-specific pockets in Eσ70 modulate RPo formation
We note that RPo formation is controlled, in part, by base-specific pockets distributed throughout the Eσ70 structure. The cognate pockets for A−11(nt) and T−7(nt) in σ70 are essential for transcription bubble nucleation and −10 element melting (Feklistov and Darst, 2011), and these interactions are maintained in the final RPo. The protrusion pocket discovered here binds T−9(t) transiently (Figure 5D), contributing to regulation by TraR (Figure 5E), and may play a role in RPo formation in the absence of factors (Figure 1C, lane 6). A binding site for G−5(nt) also plays a role in modulating RPo lifetime and regulation by ppGpp/DksA (Haugen et al., 2006; 2008b). Finally, G+2(nt) binds in an RNAP β-subunit pocket (Zhang et al., 2012). These five separate pockets and the myriad possible interactions with different promoter sequences give rise to a combinatorial effect that contributes to the 10,000-fold variation in initiation rates in vivo and in vitro (McClure, 1985; Ruff et al., 2015).
While A−11(nt) and T−7(nt) are present in nearly all E σ70 promoters (Shultzaberger et al., 2007; Feklistov and Darst, 2011), G−5(nt) and G+2(nt) are not conserved but modulate RPo formation when they are present (Haugen et al., 2006; Zhang et al., 2012). T−9(t) is also not strongly conserved, but it is enriched in promoters that are negatively regulated, and underrepresented in promoters that are positively regulated, by DksA/ppGpp and TraR (Sanchez-Vazquez et al., 2019), illustrating how transcription factor promoter specificity can depend on DNA sequences that contribute to the occupancy of transient intermediates that are not represented in the initial or final steps in the mechanism.
Relationship to previously identified intermediates
It has long been appreciated that RPo formation is a multi-step process (Buc and McClure, 1985; Hawley and McClure, 1982; Kadesch et al., 1982; Roe et al., 1984; Rosenberg et al., 1982; Walter et al., 1967). RPo formation intermediates of Eco Eσ70 have been characterized on several promoters (Rogozina et al., 2009; Rutherford et al., 2009; Sclavi et al., 2005), none more extensively than λPR, where three ‘kinetically significant’ intermediates in RPo formation at λPR, I1, I2, and I3, have been identified (reviewed in Ruff et al., 2015). I1 is proposed to comprise an ensemble of closed complexes. The rate-limiting conversion from I1 to I2 involves opening of the entire transcription bubble, loading of the DNA into the RNAP cleft, and ejection of σ701.1 (Ruff et al., 2015). Thus, partially melted intermediates have not been observed at this promoter.
A study of the kinetics of RPo formation by Mycobacterium tuberculosis (Mtb) RNAP on the Mtb rrnA P3 promoter identified a minimum of two significant intermediates, termed RP1 and RP2 (Hubin et al., 2017a). The structure of a partially melted intermediate, proposed to correspond to RP2, was revealed by cryo-EM (Boyaci et al., 2019). The intermediate contained an eight-nucleotide bubble (−11 to −4). The RP2 intermediate is not structurally similar to any of the intermediates observed here, but would lie between T-RPi2 and T-preRPo. Given the differences in the nature of the N-terminal domains of Mtb σA and Eco σ70 (Hubin et al., 2017b), in lineage-specific insertions in β and β’ (Lane and Darst, 2010), and the presence of Mtb factors not found in Eco (Hubin et al., 2017a), it is unclear whether Eco Eσ70 would significantly populate an equivalent intermediate. Thus, it is not surprising that an RP2-like complex was not observed here with Eco Eσ70.
Clearly the two or three significant intermediates identified at Mtb rrnA P3 and Eco λPR cannot account for the seven intermediates observed here (Figure 2A). We suggest that ensemble footprinting or fluorescence approaches do not have the sensitivity and/or temporal resolution to distinguish some of the intermediates identified in our structures. Thus, I1, I2,, I3, RP1, RP2, and other intermediates described previously are likely ensembles of many intermediates that accumulate at kinetic bottlenecks along the RPo formation pathway.
Does DNA opening involve the same steps at every promoter or does the pathway depend on promoter sequence? We propose that RPo formation by Eσ70 proceeds through very similar conformational changes defined by these intermediates, whether assisted by transcription factors or not. Because binding free energy drives each interconversion, the overall net gain in Eσ70-DNA interactions versus the cost of duplex DNA disruption at each step determines the corresponding rate constants (Haugen et al., 2008a; Ruff et al., 2015). As DNA sequence dictates the significance of each step (i.e. whether a particular step is rate-limiting), not all intermediates are significantly populated at a given promoter, and additional intermediates not described here may be identifiable at other promoters. These kinetic differences allow regulators (as well as changes in growth conditions) to alter rates at target promoters without significantly affecting others (Haugen et al., 2008a) to generate the wide range of promoter strength in vivo.
Mechanism of promoter melting
General models for the mechanism of RPo formation by Eσ70 have been framed by two extremes that posit where duplex DNA unwinding occurs in RNAP [reviewed in (Mazumder and Kapanidis, 2019)], either outside (melt-load model) or inside the RNAP cleft (load-melt). The melt-load model arose from analysis of bacterial RNAP-holoenzyme crystal structures (the only structures available until recently) that showed a closed-clamp conformation that could not accommodate duplex DNA (Vassylyev et al., 2002). It was thus proposed that duplex DNA positioned outside the cleft could unwind and only single-stranded DNA would be allowed into the RNAP cleft (Vassylyev et al., 2002).
The load-melt model is consistent with footprinting and other kinetic studies, primarily at λPR, that suggest the ensemble of closed (i.e. KMnO4 non-reactive) complexes includes complexes in which the duplex DNA downstream of the −10 element is protected inside the RNAP cleft (Gries et al., 2010; Saecker et al., 2011). This model requires conformational changes in the RNAP to allow entry of duplex DNA into the cleft. Early crystal structures defined a mobile structural element of the RNAP termed the clamp, opening of which would allow duplex DNA entry (Gnatt et al., 2001). Multiple conformational states of the RNAP clamp have been observed in solution by cryo-EM (Boyaci et al., 2018; Chen et al., 2019b) and single-molecule FRET (Chakraborty et al., 2012), which has also been used to observe clamp opening/closing dynamics directly (Duchi et al., 2018). It should be noted that conformational changes of the βlobe could also play a role in allowing DNA access to the RNAP cleft (Boyaci et al., 2019; Chen et al., 2010).
The results of this study, combined with other available evidence, support a combination of both models. Consistent with a melt-load model, the downstream duplex DNA in T-RPc (Figure 3) is located outside the cleft, and subsequent intermediates clearly show that transcription bubble nucleation occurs outside the cleft (T-RPi1, T-RPi1.5a; Figures 4C, D).
However, consistent with a load-melt model, effects of antibiotics suggest a role for clamp dynamics in RPo formation (Boyaci et al., 2019; Feklistov et al., 2017; Lin et al., 2018; Srivastava et al., 2011). The RP2 intermediate observed with Mtb RNAP contains a partial bubble with the duplex DNA to be ultimately melted in RPo (including the transcription start site) enclosed in the RNAP cleft, indicating that final melting of the start site occurs within the RNAP cleft (Boyaci et al., 2019). Further structural characterization of RPo formation intermediates, now enabled by advances in cryo-EM, on diverse promoters and with and without transcription factors, will be required to further delineate the promoter melting mechanism.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
All unique/stable reagents generated in this study are available without restriction from the Lead Contact, Seth A. Darst (darst@rockefeller.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
RNAP core (α2ββ’ω), σ70, and TraR are proteins found in Eco. For protein expression, Eco BL21(DE3) [Eco str. B F− ompT gal dcm lon hsdSB(rB−mB−) λ(DE3 [lacI lacUV5-T7p07 ind1 sam7 nin5]) [malB+]K-12(λS)] was used.
METHOD DETAILS
Protein Expression and Purification
Eco RNAP (harboring full-length α-subunits), σ70, and TraR were purified as described previously (Chen, 2019b). A pET-based plasmid overexpressing each subunit of Eco RNAP (full-length α, β, ω) as well as β’-PPX-His10 (PPX; PreScission protease site, LEVLFQGP, GE Healthcare) was co-transformed with a pACYCDuet-1 plasmid containing Eco rpoZ (encoding ω) into Eco BL21(DE3) (Novagen). Protein expression was induced with 1 mM isopropyl ß-D-thiogalactopyranoside (IPTG) for 4 hr at 30°C. Cells were harvested and lysed with a French Press (Avestin) at 4°C. Lysate was precipitated using polyethyleneimine [PEI, 10% (w/v), pH 8.0, Acros Organics]. Pellets were washed and RNAP was eluted. The PEI elutions were precipitated with ammonium sulfate. Pellets were harvested, resuspended and loaded on to HiTrap IMAC HP columns (GE Healthcare Life Sciences) for purification by nickel affinity chromatography. Bound RNAP was washed on column, eluted and dialyzed. Dialyzed RNAP was loaded onto a Biorex-70 column (Bio-Rad) for purification by ion exchange chromatography. Eluted RNAP was concentrated by centrifugal filtration, then loaded onto a HiLoad 26/600 Superdex 200 column (GE Healthcare Life Sciences) for purification by size exclusion chromatography. Purified RNAP was supplemented with glycerol to 20% (v/v), flash frozen in liquid N2, and stored at −80°C.
Eco σ70 was purified as described previously (Chen, 2019b). Plasmid encoding Eco His10-SUMO-σ70 was transformed into Eco BL21(DE3) (Novagen). Protein expression was induced with 1 mM IPTG for 1 hr at 30°C. Cells were harvested and lysed with a French Press (Avestin) at 4°C. Lysate was loaded onto a HiTrap IMAC HP column (GE Healthcare Life Sciences) for purification by nickel affinity chromatography. Eluted σ70 was cleaved with ULPI SUMO protease (Thermo Fisher Scientific) to remove the His10-SUMO-tag from σ70, followed by dialysis. Cleaved sample was further purified on a HiTrap IMAC HP column (GE Healthcare Life Sciences). Tagless σ70 was collected in the flowthrough and concentrated by centrifugal filtration. The sample was then loaded onto a HiLoad 16/60 Superdex 200 for purification by size exclusion chromatography. Purified σ70 was supplemented with glycerol to a final concentration of 20% (v/v), flash-frozen in liquid N2, and stored at −80°
Eco TraR was purified as described previously (Chen, 2019b). His10-SUMO-TraR plasmid was transformed into Eco BL21(DE3) (Novagen). Protein expression was induced with 1 mM IPTG for 3 hr at 37°C. Cells were harvested and lysed with a French Press (Avestin) at 4°C. The supernatant was loaded onto HiTrap IMAC HP columns (GE Healthcare Life Sciences) for purification by nickel affinity chromatography. Eluted TraR was cleaved with ULPI SUMO protease (Thermo Fisher Scientific) to remove the His10-SUMO-tag, followed by dialysis. Cleaved sample was further purified on a HiTrap IMAC HP column (GE Healthcare Life Sciences). Tagless TraR was collected in the flowthrough and concentrated by centrifugal filtration. The sample was loaded onto a HiLoad 16/60 Superdex 200 column (GE Healthcare Life Sciences) and purified by size exclusion chromatography. Purified TraR was concentrated by centrifugal filtration, flash-frozen in liquid N2, and stored at −80°C.
Native mass spectrometry analysis
The RNAP holoenzyme (holo) was assembled by incubating RNAP core and σ70 (1:1.3 molar ratio) at room temperature (RT) for 10 min. TraR was then added at five-fold molar excess to an aliquot of the RNAP holo and incubated at RT for 10 min. The resulting samples (RNAP holo with and without TraR) were concentrated using Amicon Ultra 0.5-mL centrifugal filters (EMD Millipore, Burlington, MA) with a 100 kDa molecular weight cutoff (MWCO).
The samples were buffer-exchanged into native MS solution (150 mM ammonium acetate, pH 7.5, 0.01% Tween-20) using Zeba microspin desalting columns (Thermo Fisher Scientific, Waltham, MA) with a 40-kDa MWCO (Olinares et al., 2016). The promoter DNA (rpsT P2: −60 to +25) was initially desalted into HPLC-grade H2O. Prior to mixing, the concentrations of the protein complex post-buffer exchange and the DNA components were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific). To assemble the protein-DNA complexes, the promoter DNA was mixed at 3.2- to 4-fold excess with the buffer-exchanged protein sample and incubated at RT for 10 min. The ammonium acetate concentration of the sample was varied from 75 mM to 300 mM to determine optimal conditions for complex assembly.
For native MS analysis, 2–3 μL of sample was loaded into a gold-coated quartz emitter that was prepared in-house and then electrosprayed into an Exactive Plus EMR instrument (Thermo Fisher Scientific) with a static nanospray source. The typical MS parameters include: spray voltage, 1.0–1.4 kV; capillary temperature, 100 °C – 150 °C; in-source dissociation, 10 V; S-lens RF lev el, 200; resolving power, 8,750 or 17,500 at m/z of 200; AGC target, 1 × 106; maximum injection time, 200 ms; number of microscans, 5; injection flatapole, 8 V; interflatapole, 4 V; bent flatapole, 4 V; high energy collision dissociation (HCD), 200 V; ultrahigh vacuum pressure, 6–8 × 10–10 mbar; total number of scans, at least 100. Mass calibration in EMR mode was performed using cesium iodide. The acquired MS spectra were visualized using Thermo Xcalibur Qual Browser (version 3.0.63) and deconvolution was performed either manually or using UniDec v 3.2 (Marty et al., 2015; Reid et al., 2019). The deconvolved spectra from UniDec were plotted using the m/z software (Proteometrics LLC, New York, NY). Experimental masses were reported as the average mass ± standard deviation (S.D.) across all the calculated mass values obtained within the observed charge state distribution. The experimentally determined masses include: 470,745 ± 15 Da (0.02% mass error) for the TraR-Eσ70 complex; 523,900 ± 150 Da (0.16% mass error) for the TraR-Eσ70-rpsT P2 complex; 462,740 ± 25 Da (0.07% mass error) for Eσ70; 452,700 ± 20 Da (0.09% mass error) for the Eσ70-ω complex; 515,800 ± 160 Da (0.2% mass error) for the Eσ70-rpsT P2 complex.
KMnO4 and DNase I footprinting
Wt or T−7A rpsT P2 promoter fragments were 32P-3’ end labeled in the nt-strand by linearizing 15 μg of plasmid DNA by NheI digestion [pRLG11272, wt, (Gopalkrishnan et al., 2017); or pRLG12844, T−7A], followed by incubation with α32P-dCTP (Perkin Elmer, Waltham, MA) and Sequenase Version 2.0 (Thermo Fisher Scientific). Promoter fragments were then generated by digestion with NcoI, purified by 5% acrylamide gel electrophoresis, eluted by diffusion and concentrated using a PCR Purification Kit (Qiagen, Hilden, Germany) as described (Gopalkrishnan et al., 2017). For 3’-labeling of the t-strand, plasmid DNA was digested at the Nco I site, labeled, and fragments were generated by Nhe I digestion. Promoter complexes were formed by incubation for 10 min at the indicated temperatures with RNAP (20 nM) and TraR (1 μM), where indicated, in 10 mM Tris-Cl, pH 8.0, 10 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA and 30 mM KCl. For KMnO4 footprinting, complexes were incubated with 2 mM KMnO4 for 30 sec, then samples were ethanol precipitated twice, incubated with 1 M piperidine at 90°C for 30 min, ethanol precipitated and run on 9.5% acrylamide, 7 M urea gels as described (Winkelman et al., 2015). For DNase I footprinting, complexes were digested with DNase I (Worthington, Columbus, OH; 10 μg/ml) for 30 sec, phenol extracted, ethanol precipitated, resuspended and analyzed by gel electrophoresis as for KMnO4 samples. Gels were dried, visualized by phosphorimaging and quantified using ImageQuant 5.2 (GE Healthcare, Pittsburgh PA). RNAPs, wt or variant, were purified by overexpression in Eco BL21(DE3) from derivatives of the multisubunit RNAP plasmid pIA900 (Svetlov and Artsimovitch, 2015), or derivatives containing β or β’ (β’Δ215–220: pRLG10030; βA474V: pRLG15444; β474L, pRLG15445).
In vitro transcription assays
In vitro transcription was carried out on supercoiled templates as described (Gopalkrishnan et al., 2017). Multiple-round in vitro transcription assays were performed on linear rpsT 2 fragments with −60/+25 endpoints (Figures S1E, F). Transcription reactions (25 μL) containing 40 nM DNA, TraR (0 – 2 μM), 60 nM RNAP and NTPs (500 μM CTP, 200 μM GTP, 200 μM ATP, 10 μM UTP, 1 μCi [α−32P] UTP) were incubated in buffer (10 mM Tris-HCl pH 7.9, 170 mM NaCl, 10 mM MgCl2, 1 mM DTT and 0.1 μg/μl BSA) at room temperature (~23 °C) for 15 minutes and were terminated by addition of equal volume of stop solution. Transcripts were separated on 8% acrylamide-7M urea denaturing gels and analyzed by phosphoimaging.
Preparation of TraR-RNAP-DNA complexes for Cryo-EM
RNAP holo was formed by mixing RNAP core and a 2-fold molar excess of σ70 and incubating for 15 minutes at RT. RNAP holo was purified over a Superose 6 Increase 10/300 GL column (GE Healthcare, Pittsburgh, PA) in gel filtration buffer (10 mM Tris-HCl, pH 8.0, 200 mM KCl, 5 mM MgCl2, 10 μM ZnCl2, 2.5 mM DTT). The eluted RNAP holo was concentrated to ~10.0 mg/mL (~20 μM) by centrifugal filtration (Amicon Ultra). TraR was added (5-fold molar excess over RNAP) and the sample was incubated for 15 min at RT. Duplex rpsT P2 promoter fragment (−60 to +25, Integrated DNA Technologies, Coralville, IA), either wild-type (Figure 1A) or rpsT P2* (Figure 2A), was added to the concentrated TraR-RNAP to 3-fold molar excess. The sample was incubated for 20 min at RT prior to cryo-EM grid preparation.
Cryo-EM grid preparation
CHAPSO {3-([3-cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate} (Anatrace, Maumee, OH) was added to the samples to a final concentration of 8 mM (Chen et al., 2019). The final buffer condition for all the cryo-EM samples was 10 mM Tris-HCl, pH 8.0, 100 mM KCl, 5 mM MgCl2, 10 μM ZnCl2, 2.5 mM DTT, 8 mM CHAPSO. C-flat holey carbon grids (CF-1.2/1.3–4Au, Protochips, Morrisville, NC) were glow-discharged for 20 sec prior to the application of 3.5 μL of the samples. Using a Vitrobot Mark IV (Thermo Fisher Scientific Electron Microscopy, Hillsboro, OR), grids were blotted and plunge-froze into liquid ethane with 100% chamber humidity at 22°C.
Cryo-EM data acquisition and processing
TraR-RNAP-wt-rpsT P2 complexes.
Grids were imaged using a 300 keV Titan Krios (Thermo Fisher Scientific Electron Microscopy) equipped with a K2 Summit direct electron detector (Gatan, Pleasanton, CA). Images were recorded with Serial EM (Mastronarde, 2005) with a pixel size of 1.3 Å over a defocus range of −0.5 μm to −3.0 μm. Movies were recorded in super-resolution mode at 8 electrons/physical pixel/s in dose-fractionation mode with subframes of 0.2 s over a 10 s exposure (50 frames) to give a total dose of 80 electrons/physical pixel. Dose-fractionated movies were gain-normalized, drift-corrected, binned, summed, and dose-weighted using MotionCor2 (Zheng et al., 2017). The contrast transfer function was estimated for each summed image using Gctf (Zhang, 2016). Gautomatch (developed by K. Zhang, MRC Laboratory of Molecular Biology, Cambridge, UK, http://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch) was used to pick particles with an auto-generated template. Picked particles were extracted from the dose-weighted images in RELION (Scheres, 2012) using a box size of 256 pixels. The TraR-RNAP-wt-rpsT P2 dataset consisted of 5,330 motion-corrected images with 1,189,185 particles (Figure S2). A subset of the particles were used to generate an initial model of the complex in cryoSPARC (ab initio reconstruction) (Punjani et al., 2017) to generate a 3D template for RELION. In RELION, a consensus refinement was performed using the extracted particles and the cryoSPARC-generated initial model resulting in a 5.5 Å map (Figure S2). Using the refinement parameters, 3D classification (N=2) was performed on the particles without alignment, revealing a high resolution class with 370,441 particles (nominal resolution 3.9 Å) after RELION 3D auto-refinement and a low-resolution ‘junk’ class that could not be classified further. Using the refinement parameters, a subsequent 3D classification (N=2) was performed on the high-resolution particles without alignment, revealing distinct classes with different DNA configurations: Class 1a contained duplex DNA bound to RNAP while class 1b contained a transcription bubble. Subsequent 3D masked classification (N=2, without alignment) was performed on particles from class 1a using a mask around the downstream DNA, βprotrusion, and σ702. Classification revealed two distinct classes: TRPc and TRPi1 (Figure S2). Using the refinement parameters, subtractive 3D classification (N=3) was performed on the particles from class 1b by subtracting density outside of TraR, βlobe-Si1, β’Si3, and the downstream channel, followed by classifying the remaining density with a mask. Classification revealed three distinct classes: TRPi2, TpreRPo and TRPo (Figure S2). After 3D classifications, the particles within each class were further processed using RELION CTF refinement and Bayesian Polishing. RELION 3D auto-refinement and post-processing of the polished particles resulted in structures with the following nominal resolutions: TRPc (3.4 Å), TRPi1 (3.4 Å), TRPi2 (3.9 Å), TpreRPo (3.5 Å), TRPo (3.7 Å). Local resolution calculations were generated using blocres and blocfilt from the Bsoft package (Cardone et al., 2013).
TraR-RNAP-rpsT P2* complexes.
Grids were imaged as for the TraR-RNAP-wt-rpsT P2 dataset with the following exceptions: 1) The defocus range was −0.5 μm to −2.0 μm. Data were collected with a dose of 5.6 electrons/pixelx/s. Images were recorded over a 15 s exposure using 0.3 s subframes (50 total frames) to give a total dose of 84 electrons/physical pixel. Dose-fractionated subframes were gain-normalized, drift-corrected, binned, summed, and dose-weighted using MotionCor2 (Zheng et al., 2017) in RELION 3.0 (Zivanov et al., 2018). The contrast transfer function was estimated for each summed image using CTFFIND4 (Rohou and Grigorieff, 2015). The TraR-RNAP-rpsT P2* dataset consisted of 1,500 motion-corrected images with 523,503 particles (Figure S5A). A subset of the particles was subjected to cryoSPARC ab initio reconstruction (Punjani et al., 2017) to generate a 3D template for RELION refinements and classifications. In RELION, 3D classification (N=2) was performed on the extracted particles with alignment to the cryoSPARC ab initio reconstruction. Classification revealed a low- resolution class and a high-resolution class containing 150,387 particles with nominal resolution of 4.6 Å after RELION 3D auto-refinement. Refinement metadata and post-processing were used as inputs for RELION CTF refinement and RELION Bayesian Polishing (Zivanov et al., 2018). Polishing improved the map to a nominal resolution of 3.1 Å after RELION 3D auto-refinement. Using the refinement parameters, subtractive 3D classification (N=3) was performed on the polished particles by subtracting density outside of σ701.1, σ702, β-lobe, β-protrusion, and downstream DNA, followed by a 3D classification of the remaining density with a mask. This classification revealed two distinct classes: TRPi1.5a (class2a) and TRPi1.5b (class 2b and class 2c combined; Figure S5A). Particles from the TRPi1.5a class were further processed using RELION CTF refinement and RELION Bayesian Polishing, resulting in an improved map with a nominal resolution of 3.5 Å after RELION 3D auto-refinement and post-processing. RELION CTF refinement and RELION Bayesian Polishing did not improve the resolution of the TRPi1.5b class (nominal resolution of 3.0 Å after RELION 3D auto-refinement and post-processing).
Model building and refinement
For initial models of the complexes, the TraR-RNAP structure (PDB ID 6N57) (Chen et al., 2019b) was manually fit into the cryo-EM density maps using Chimera (Pettersen et al., 2004) and real-space refined using Phenix (Adams et al., 2010). The DNAs were mostly built de novo based on the density maps. For real-space refinement, rigid body refinement with sixteen manually-defined mobile domains was followed by all-atom and B-factor refinement with Ramachandran and secondary structure restraints. Refined models were inspected and modified in Coot (Emsley and Cowtan, 2004).
Quantification and statistical analysis
The nMS spectra were visualized using Thermo Xcalibur Qual Browser (version 3.0.63), deconvolved using UniDec v 3.2 (Marty et al., 2015; Reid et al., 2019) and plotted using the m/z software (Proteometrics LLC, New York, NY). Experimental masses (Figures 1B, S1B and S1C) were reported as the average mass ± standard deviation across all the calculated mass values obtained within the observed charge state distribution.
ImageQuant 5.2 (GE Healthcare, Pittsburgh PA) was used to visualize and quantify gels. To quantify the transcription assays (Figures 5E, S1F, S1E, S7E–H), mean values and the standard error of the mean from at least three independent measurements were calculated.
Structural biology software was accessed through the SBGrid consortium (Morin et al., 2013). The local resolution of the cryo-EM maps (Figures S3C, S3D, S3E, S3F, S3G, S5G, S5H) was estimated using blocres (Cardone et al., 2013) with the following parameters: box size 15, verbose 7, sampling 1.3, and cutoff 0.5. The quantification and statistical analyses for model refinement and validation were generated using MolProbity (Chen et al., 2010) and PHENIX (Adams et al., 2010).
Data and code availability
The cryo-EM density maps have been deposited in the EMDataBank under accession codes EMD-20460 (TRPc), EMD-20461 (TPRi1), EMD_20462 (TRPi1.5a), EMD-20463 (TRPi1.5b), EMD-20464 (TRPi2), EMD-20465 (TpreRPo), and EMD-20466 (TRPo). The atomic coordinates have been deposited in the Protein Data Bank under accession codes 6PSQ (TRPc), 6PSR (TRPi1), 6PSS (TRPi1.5a), 6PST (TRPi1.5b), 6PSU (TRPi2), 6PSV (TpreRPo), 6PSW (TRPo).
Supplementary Material
Movie S1. Morph illustrating the T-RPc ↔ T-RPi1 ↔ T-RPi1.5a transition. Related to Figure 4. The views correspond to the views of Figure 4(right), focusing on the promoter −10 element and the σ70 W-dyad. The progress of the movie through the structures is denoted at the top.
Movie S2. Morph illustrating the T-RPi1.5a ↔ TRPi1.5b transition. Related to Figure 5. The views correspond roughly to the views of Figure 5B and 5C. The progress of the movie through the structures is denoted at the top.
Movie S3. Morph illustrating the entire R ↔ T-R ↔ T-RPc ↔ T-RPi1 ↔ T-RPi1.5a ↔ T-RPi1.5b ↔ T-RPi2 ↔ T-preRPo ↔ T-RPo ↔ RPo pathway. Related to Figure 6. The views correspond roughly to the views of Figure 2A. The progress of the movie through the structures is denoted at the top.
Movie S4. Morph illustrating the hypothetical entire R ↔ RPc ↔ RPi1 ↔ RPi1.5a ↔ RPi1.5b ↔ RPi2 ↔ RPo pathway. Related to Figure 6. The views correspond roughly to the views of Figure 2A. The progress of the movie through the structures is denoted at the top.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) | EMD Millipore | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3-([3-Cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate (CHAPSO) | Anatrace | Cat# C317 |
| E. coli RNAP (cryo-EM samples) | Chen et al., 2019b | N/A |
| E. coli RNAP, WT (Biochemistry) | Svetlov and Artsimovitch, 2015 | |
| E. coli RNAP, βA474L | This paper | |
| E. coli RNAP, βA474V | This paper | |
| E. coli RNAP, βR378A | This paper | |
| E. coli RNAP, βR378E | This paper | |
| E. coli RNAP, βR378E βR394E | This paper | |
| E. coli RNAP, βR394A | This paper | |
| E. coli RNAP, βR394E | This paper | |
| Polyethyleneimine | Fisher Scientific | Cat# AC178572500 |
| Deposited Data | ||
| Coordinates of E. coli Eσ70 | Chen et al., 2019b | PDB: 6P1K |
| Coordinates of E. coli Eσ70/Wt rpsT P2 | Chen et al., 2019b | PDB: 6OUL |
| Coordinates of E. coli TraR/Eσ70 (I) | Chen et al., 2019b | PDB: 6N57 |
| Coordinates of E. coli TraR/Eσ70 (II) | Chen et al., 2019b | PDB: 6N58 |
| Coordinates of E. coli TraR/Eσ70/Wt rpsT P2 (T-preRPo) | This paper | PDB: 6PSV |
| Coordinates of E. coli TraR/Eσ70/Wt rpsT P2 (T-RPc) | This paper | PDB: 6PSQ |
| Coordinates of E. coli TraR/Eσ70/Wt rpsT P2 (T-RPi1) | This paper | PDB: 6PSR |
| Coordinates of E. coli TraR/Eσ70/Wt rpsT P2 (T-RPi2) | This paper | PDB: 6PSU |
| Coordinates of E. coli TraR/Eσ70/Wt rpsT P2 (T-RPo) | This paper | PDB: 6PSW |
| Coordinates of E. coli TraR/Eσ70/Wt rpsT P2* (T-RPi1.5a) | This paper | PDB: 6PSS |
| Coordinates of E. coli TraR/Eσ70/Wt rpsT P2* (T-RPi1.5b) | This paper | PDB: 6PST |
| Cryo-EM map of E. coli TraR/Eσ70/rpsT P2* (T-RPi1.5a) | This paper | EMD_20462 |
| Cryo-EM map of E. coli TraR/Eσ70/Wt rpsT P2 (T-preRPo) | This paper | EMD-20465 |
| Cryo-EM map of E. coli TraR/Eσ70/Wt rpsT P2 (T-RPc) | This paper | EMD-20460 |
| Cryo-EM map of E. coli TraR/Eσ70/Wt rpsT P2 (T-RPi1) | This paper | EMD-20461 |
| Cryo-EM map of E. coli TraR/Eσ70/Wt rpsT P2 (T-RPi2) | This paper | EMD-20464 |
| Cryo-EM map of E. coli TraR/Eσ70/Wt rpsT P2 (T-RPo) | This paper | EMD-20466 |
| Cryo-EM map of E. coli TraR/Eσ70/Wt rpsT P2* (T-RPi1.5b) | This paper | EMD-20463 |
| Experimental Models: Organisms/Strains | ||
| Escherichia coli | ||
| Oligonucleotides | ||
| pIA900 βA474L: GTGTAGAGCGTCTGGTGAAAGAGCGTC | This paper, IDT | |
| pIA900 βA474V: GTGTAGAGCGTGTGGTGAAAGAGCGTC | This paper, IDT | |
| pIA900 βR378A: GAGCCGCCGACTGCCGAAGCAGCTGAAAGCCTG | This paper, IDT | |
| pIA900 βR378E: GAGCCGCCGACTGAAGAAGCAGCTGAAAGCCTG | This paper, IDT | |
| pIA900 βR394A: CTTCTCCGAAGACGCTTATGACTTGTCTGC | This paper, IDT | |
| pIA900 βR394E: CTTCTCCGAAGACGAATATGACTTGTCTGC | This paper, IDT | |
| rpsTP2(−60to+25)_−11/−10CG_bot: 5’-GCG TTC TAT ATG GAC AAT TCA AAG GCC GAG GAA TGC GCC CTT TTA GCC TTC TTT TGT CAA TGG ATT TGT GCA AAT AAG CGC CGC C-3’ |
This paper, IDT | |
| rpsTP2(−60to+25)_(T-7A)_top: 5’-GGC GGC GCT TAT TTG CAC AAA TCC ATT GAC AAA AGA AGG CTA AAA GGG CAT ATA CCT CGG CCT TTG AAT TGT CCA TAT AGA ACG C-3’ |
This paper, IDT | |
| rpsTP2(−60to+25)_bot: 5’-GCG TTC TAT ATG GAC AAT TCA AAG GCC GAG GAA TAT GCC CTT TTA GCC TTC TTT TGT CAA TGG ATT TGT GCA AAT AAG CGC CGC C-3’ |
This paper, IDT | |
| rpsTP2(−60to+25)_top: 5’-GGC GGC GCT TAT TTG CAC AAA TCC ATT GAC AAA AGA AGG CTA AAA GGG CAT ATT CCT CGG CCT TTG AAT TGT CCA TAT AGA ACG C-3’ |
This paper, IDT | |
| Recombinant DNA | ||
| E. coli RNAP, pIA900 WT | Svetlov and Artsimovitch, 2015 | |
| E. coli RNAP, pIA900 βA474L | This paper | pRLG15445 |
| E. coli RNAP, pIA900 βA474V | This paper | pRLG15444 |
| E. coli RNAP, pIA900 βR378A | This paper | pRLG15446 |
| E. coli RNAP, pIA900 βR378E | This paper | pRLG15447 |
| E. coli RNAP, pIA900 βR378E βR394E | This paper | pRLG15450 |
| E. coli RNAP, pIA900 βR394A | This paper | pRLG15448 |
| E. coli RNAP, pIA900 βR394E | This paper | pRLG15449 |
| p770 | Ross, et al., 1990 | pRLG770 |
| p770-rpsT P2 (−89 to +50) | Lemke et al., 2011 | pRLG14658 |
| p770-rrnB P1 (−88 to +50) | Ross et al., 2016 | pRLG13065 |
| p770-thrABC (−72 to +16) | Barker, 2001 | pRLG15276 |
| pACYCDuet-1_Ec_rpoZ | Twist et al., 2011 | |
| pEcrpoABC(-XH)Z | Twist et al., 2011 | |
| pET28a | EMD Millipore | |
| pET28a-His10-SUMO rpoD | Chen et al., 2017 | |
| pET28a-His10-SUMO traR | Chen et al. 2019b | pRLG15142 |
| pIA900, multisubunit RNAP plasmid | Svetlov and Artsimovitch, 2015 | |
| pSL6-rpsT P2 (−68 to +50) | Gopalkrishnan et al., 2017 | pRLG11272 |
| pSL6-rpsT P2 (−68 to +50) (T-7A) | This paper | pRLG12844 |
| Software and Algorithms | ||
| Bayesian Polishing | Zivanov et al., 2018 | https://github.com/3dem/relion |
| blocfilt | Cardone et al., 2013 | https://lsbr.niams.nih.gov/bsoft/programs/blocres.html |
| blocres | Cardone et al., 2013 | https://lsbr.niams.nih.gov/bsoft/programs/blocres.html |
| Coot | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| cryoSPARC | Punjani et al., 2017 | https://cryosparc.com/ |
| CTFFIND4 | Rohou and Grigorieff, 2015 | http://grigoriefflab.janelia.org/ctffind4 |
| Gautomatch | N/A | http://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch/ |
| Gctf | Zhang, 2016 | https://www.mrc-lmb.cam.ac.uk/kzhang/Gctf/ |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism |
| ImageQuant 5.2 | GE Healthcare, Pittsburgh PA | |
| m/z- Knexus edition | Proteometrics, LLC | |
| Molprobity | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
| MotionCor2 | Zheng et al., 2017 | |
| MTRIAGE | Afonine et al., 2018 | https://www.phenix-online.org/documentation/reference/mtriage.html |
| PDBePISA | Krissinel and Henrick, 2007 | https://www.ebi.ac.uk/pdbe/pisa/ |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org/documentation/index.html |
| Qual Browser Thermo Xcalibur version 3.0.63 | Thermo Fisher Scientific Inc. | Thermo Scientific MS instruments |
| RELION | Scheres, 2012 | https://github.com/3dem/relion |
| SBGrid | Morin et al., 2013 | https://sbgrid.org/ |
| SerialEM | Mastronarde, 2005 | http://bio3d.colorado.edu/SerialEM |
| The PyMOL Molecular Graphics System | Schrödinger, LLC | http://www.pymol.org |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera |
| UniDec version 3.2 | Marty et. al., 2015 | https://github.com/michaelmarty/UniDec/releases |
| Other | ||
| Bio-Rex 70 cation exchange resin, analytical grade, 100–200 mesh | Bio-Rad | Cat# 1425842 |
| C-flat CF-1.2/1.3 400 mesh gold grids | Electron Microscopy Sciences | Cat# CF413–100-Au |
| HiLoad 26/600 Superdex 200 pg | GE Healthcare Life Sciences | Cat# 28989336 |
| HiTrap IMAC HP | GE Healthcare Life Sciences | Cat# 17092003 |
| Isotope [α−32P]UTP | Perkin Elmer | Cat # BLU507H500UCI |
| Isotope [α32P]-dCTP | Perkin Elmer | Cat # BLU013H250UCI |
| Superose 6 INCREASE 10/300 GL | GE Healthcare Life Sciences | Cat# 29091596 |
| Zeba Micro Spin Desalting Columns, 40K MWCO | Thermo Pierce | Cat. # 87765 |
HIGHLIGHTS.
Cryo-EM structures of 7 intermediates in promoter opening pathway from RPc to RPo
Intermediates populated by using an inhibitor and a promoter with unstable RPo
RNAP and DNA conformational changes in mobile regions mark the steps in the pathway
Transient interactions identified in intermediates are not found in RPc or RPo
Acknowledgments
We thank M. Ebrahim and J. Sotiris at The Rockefeller University Evelyn Gruss Lipper Cryo-electron Microscopy Resource Center for help with cryo-EM data collection, and members of the Darst/Campbell laboratory for helpful discussion on the manuscript. This work was supported by NIH grants P41 GM109824 and P41 GM103314 to B.T.C., R01 GM37048 to R.L.G., R01 GM114450 to E.A.C., and R35 GM118130 to S.A.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests The authors declare there are no competing interests.
REFERENCES
- Abascal-Palacios G, Ramsay EP, Beuron F, Morris E, and Vannini A (2018). Structural basis of RNA polymerase III transcription initiation. Nature 553, 301–306. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Klaholz BP, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, Adams PD, and Urzhumtsev A (2018). New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr D Struct Biol 74, 814–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Davis E, Brown D, Campbell EA, Wigneshweraraj SR, and Darst SA (2013). Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ70 domain 1.1. Proc Natl Acad Sci USA 110, 19772–19777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Feklistov A, Lass-Napiorkowska A, Landick R, and Darst SA (2015). Structure of a bacterial RNA polymerase holoenzyme open promoter complex. Elife 4, e08504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett MS, Gaal T, Ross W, and Gourse RL (1998). RNA polymerase mutants that destabilize RNA polymerase-promoter complexes alter NTP-sensing by rrn P1 promoters. J Mol Biol 279, 331–345. [DOI] [PubMed] [Google Scholar]
- Benoff B, Yang H, Lawson CL, Parkinson G, Liu J, Blatter E, Ebright YW, Berman HM, and Ebright RH (2002). Structural basis of transcription activation: the CAP-alpha CTD-DNA complex. Science 297, 1562–1566. [DOI] [PubMed] [Google Scholar]
- Blankschien MD, Potrykus K, Grace E, Choudhary A, Vinella D, Cashel M, and Herman C (2009). TraR, a homolog of a RNAP secondary channel interactor, modulates transcription. PLoS Genet. 5(1):e1000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyaci H, Chen J, Jansen R, Darst SA, and Campbell EA (2019). Structures of an RNA polymerase promoter melting intermediate elucidate DNA unwinding. Nature 565, 382–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyaci H, Chen J, Lilic M, Palka M, Mooney RA, Landick R, Darst SA, and Campbell EA (2018). Fidaxomicin jams Mycobacterium tuberculosis RNA polymerase motions needed for initiation via RbpA contacts. Elife 7, e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buc H, and McClure WR (1985). Kinetics of open complex formation between Escherichia coli RNA polymerase and the lac UV5 promoter. Evidence for a sequential mechanism involving three steps. Biochemistry 24, 2712–2723. [DOI] [PubMed] [Google Scholar]
- Campbell EA, Muzzin O, Chlenov M, Sun JL, Olson CA, Weinman O, Trester-Zedlitz ML, and Darst SA (2002). Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Mol Cell 9, 527–539. [DOI] [PubMed] [Google Scholar]
- Cardone G, Heymann JB, and Steven AC (2013). One number does not fit all: mapping local variations in resolution in cryo-EM reconstructions. J. Struct. Biol 184, 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Wang D, Ebright YW, Korlann Y, Kortkhonjia E, Kim T, Chowdhury S, Wigneshweraraj SR, Irschik H, Jansen R, et al. (2012). Opening and Closing of the Bacterial RNA Polymerase Clamp. Science 337, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Noble AJ, Kang JY, and Darst SA (2019a). Eliminating effects of particle adsorption to the air/water interface in single-particle cryo-electron microscopy - Bacterial RNA polymerase and CHAPSO. J Struct Biol: X 1, 100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Gopalkrishnan S, Chiu C, Chen AY, Campbell EA, Gourse RL, Ross W, and Darst SA (2019b) E. coli TraR allosterically regulates transcription initiation by altering RNA polymerase conformation and dynamcs. eLife 8, e49375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Darst SA, and Thirumalai D (2010). Promoter melting triggered by bacterial RNA polymerase occurs in three steps. Proc Natl Acad Sci USA 107, 12523–12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YF, and Helmann JD (1997). DNA-melting at the Bacillus subtilis flagellin promoter nucleates near −10 and expands unidirectionally. J Mol Biol 267, 47–59. [DOI] [PubMed] [Google Scholar]
- Duchi D, Mazumder A, Malinen AM, Ebright RH, and Kapanidis AN (2018). The RNA polymerase clamp interconverts dynamically among three states and is stabilized in a partly closed state by ppGpp. Nucl Acids Res. 46, 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Estrem ST, Gaal T, Ross W, and Gourse RL (1998). Identification of an UP element consensus sequence for bacterial promoters. Proc Natl Acad Sci USA 95, 9761–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, and Darst SA (2011). Structural Basis for Promoter −10 Element Recognition by the Bacterial RNA Polymerase σ Subunit. Cell 147, 1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, Bae B, Hauver J, Lass-Napiorkowska A, Kalesse M, Glaus F, Altmann K-H, Heyduk T, Landick R, and Darst SA (2017). RNA polymerase motions during promoter melting. Science 356, 863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A, Sharon BD, Darst SA, and Gross CA (2014). Bacterial sigma factors: a historical, structural, and genomic perspective. Annu. Rev. Microbiol 68, 357–376. [DOI] [PubMed] [Google Scholar]
- Galburt EA (2018) The calculation of transcript flux ratios reveals single regulatory mechanisms capable of activation and repression. Proc Natl Acad Sci USA 115, E11604–E11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnatt AL, Cramer P, Fu J, Bushnell DA, and Kornberg RD (2001). Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science 292, 1876–1882. [DOI] [PubMed] [Google Scholar]
- Gopalkrishnan S, Ross W, Chen AY, and Gourse RL (2017). TraR directly regulates transcription initiation by mimicking the combined effects of the global regulators DksA and ppGpp. Proc Natl Acad Sci USA 114, E5539–E5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourse RL, Chen AY, Gopalkrishnan S, Sanchez-Vazquez P, Myers A, and Ross W (2018). Transcriptional Responses to ppGpp and DksA. Annu. Rev. Microbiol 72, 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gries TJ, Kontur WS, Capp MW, Saecker RM, and Record MT Jr, (2010). One-step DNA melting in the RNA polymerase cleft opens the initiation bubble to form an unstable open complex. Proc Natl Acad Sci USA 107, 10418–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber TM, and Bryant DA (1997). Molecular systematic studies of eubacteria, using sigma70-type sigma factors of group 1 and group 2. J Bact 179, 1734–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber TM, and Gross CA (2003). Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol 57, 441–466. [DOI] [PubMed] [Google Scholar]
- Haugen SP, Berkmen MB, Ross W, Gaal T, Ward C, and Gourse RL (2006). rRNA Promoter Regulation by Nonoptimal Binding of σ Region 1.2: An Additional Recognition Element for RNA Polymerase. Cell 125, 1069–1082. [DOI] [PubMed] [Google Scholar]
- Haugen SP, Ross W, and Gourse RL (2008a). Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Micro 6, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen SP, Ross W, Manrique M, and Gourse RL (2008b). Fine structure of the promoter-sigma region 1.2 interaction. Proc Natl Acad Sci USA 105, 3292–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley DK, and McClure WR (1982). Mechanism of activation of transcription initiation from the lambda PRM promoter. J Mol Biol 157, 493–525. [DOI] [PubMed] [Google Scholar]
- He Y, Fang J, Taatjes DJ, and Nogales E (2013). Structural visualization of key steps in human transcription initiation. Nature 495, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Yan C, Fang J, Inouye C, Tjian R, Ivanov I, and Nogales E (2016). Near-atomic resolution visualization of human transcription promoter opening. Nature 533, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyduk E, Kuznedelov K, Severinov K, and Heyduk T (2006). A consensus adenine at position −11 of the nontemplate strand of bacterial promoter is important for nucleation of promoter melting. J. Biol. Chem 281, 12362–12369. [DOI] [PubMed] [Google Scholar]
- Hubin EA, Fay A, Xu C, Bean JM, Saecker RM, Glickman MS, Darst SA, and Campbell EA (2017a). Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. Elife 6, e22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubin EA, Lilic M, Darst SA, and Campbell EA (2017b). Structural insights into the mycobacteria transcription initiation complex from analysis of X-ray crystal structures. Nature Com 8, 16072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadesch TR, Rosenberg S, and Chamberlin MJ (1982). Binding of Escherichia coli RNA polymerase holoenzyme to bacteriophage T7 DNA. Measurements of binding at bacteriophage T7 promoter A1 using a template competition assay. J Mol Biol 155, 1–29. [DOI] [PubMed] [Google Scholar]
- Kontur WS, Capp MW, Gries TJ, Saecker RM, and Record MT Jr, (2010). Probing DNA Binding, DNA Opening, and Assembly of a Downstream Clamp/Jaw in Escherichia coliRNA Polymerase–λP RPromoter Complexes Using Salt and the Physiological Anion Glutamate. Biochemistry 49, 4361–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontur WS, Saecker RM, Capp MW, and Record MT Jr, (2008). Late steps in the formation of E. coli RNA polymerase-lambda P R promoter open complexes: characterization of conformational changes by rapid [perturbant] upshift experiments. J Mol Biol 376, 1034–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic RT (1987). The 0 degree C closed complexes between Escherichia coli RNA polymerase and two promoters, T7–A3 and lacUV5. J. Biol. Chem 262, 13654–13661. [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Lane WJ, and Darst SA (2010). Molecular Evolution of Multisubunit RNA Polymerases: Sequence Analysis. J Mol Biol 395, 671–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM, IUCr (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26, 283–291. [Google Scholar]
- Lee DJ, Minchen SD, and Busby SJW (2012). Activating Transcription in Bacteria. Ann. Rev Microbiol 66, 125–152. [DOI] [PubMed] [Google Scholar]
- Lemke JJ, Sanchez-Vazquez P, Burgos HL, Hedberg G, Ross W, and Gourse RL (2011). Direct regulation of Escherichia coli ribosomal protein promoters by the transcription factors ppGpp and DksA. Proc Natl Acad Sci USA 108, 5712–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HM, Lee HJ, Roy S, and Adhya S (2001). A “‘master’” in base unpairing during isomerization of a promoter upon RNA polymerase binding. Proc Natl Acad Sci USA 98, 14849–14852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Das K, Degen D, Mazumder A, Duchi D, Wang D, Ebright YW, Sineva E, Gigliotti M, Srivastava A, et al. (2018). Structural Basis of Transcription Inhibition by Fidaxomicin (Lipiarmycin A3). Mol Cell 70, 60–71.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty MT, Baldwin AJ, Marklund EG, Hochberg GKA, Benesch JLP, and Robinson CV (2015). Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Mazumder A, and Kapanidis AN (2019). Recent Advances in Understanding σ70-Dependent Transcription Initiation Mechanisms. J Mol Biol. doi: 10.1016/j.jmb.2019.04.046. Epub May 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure WR (1985). Mechanism and control of transcription initiation in prokaryotes. Annu. Rev. Biochem 54, 171–204. [DOI] [PubMed] [Google Scholar]
- Mekler V, Kortkhonjia E, Mukhopadhyay J, Knight J, Revyakin A, Kapanidis AN, Niu W, Ebright YW, Levy R, and Ebright RH (2002). Structural organization of bacterial RNA polymerase holoenzyme and the RNA polymerase-promoter open complex. Cell 108, 599–614. [DOI] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, and Sliz P (2013). Collaboration gets the most out of software. Elife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy J, Grohmann D, Cheung ACM, Schulz S, Smollett K, Werner F, and Michaelis J (2015). Complete architecture of the archaeal RNA polymerase open complex from single-molecule FRET and NPS. Nature Com 6, 6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A, Vago FS, Li K, Qayyum MZ, Yernool D, Jiang W, and Murakami KS (2018). Cryo-EM structure of Escherichia coli σ70 RNA polymerase and promoter DNA complex revealed a role of σ non-conserved region during the open complex formation. J. Biol. Chem 293, 7367–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olinares PDB, Dunn AD, Padovan JC, Fernandez-Martinez J, Rout MP, and Chait BT (2016). A Robust Workflow for Native Mass Spectrometric Analysis of Affinity-Isolated Endogenous Protein Assemblies. Anal. Chem 88, 2799–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Plaschka C, Hantsche M, Dienemann C, Burzinski C, Plitzko J, and Cramer P (2016). Transcription initiation complex structures elucidate DNA opening. Nature 533, 353–358. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Reid DJ, Diesing JM, Miller MA, Perry SM, Wales JA, Montfort WR, and Marty MT (2019). High-Throughput Deconvolution of Native Mass Spectra. J Am Soc Mass Spectrom 30, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe JH, Burgess RR, and Record MT Jr, (1984). Kinetics and mechanism of the interaction of Escherichia coli RNA polymerase with the lambda PR promoter. J Mol Biol 176, 495–522. [DOI] [PubMed] [Google Scholar]
- Rogozina A, Zaychikov E, Buckle M, Heumann H, and Sclavi B (2009). DNA melting by RNA polymerase at the T7A1 promoter precedes the rate-limiting step at 37 C and results in the accumulation of an off-pathway intermediate. Nucl Acids Res 37, 5390–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S, Kadesch TR, and Chamberlin MJ (1982). Binding of Escherichia coli RNA polymerase holoenzyme to bacteriophage T7 DNA. Measurements of the rate of open complex formation at T7 promoter A. J Mol Biol 155, 31–51. [DOI] [PubMed] [Google Scholar]
- Ross W, and Gourse RL (2005). Sequence-independent upstream DNA-alphaCTD interactions strongly stimulate Escherichia coli RNA polymerase-lacUV5 promoter association. Proc. Natl., Acad. Sci USA 102, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, and Gourse RL (2009). Analysis of RNA polymerase-promoter complex formation. Methods 47, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, Ernst A, and Gourse RL (2001). Fine structure of E. coli RNA polymerase-promoter interactions: alpha subunit binding to the UP element minor groove. Genes Dev 15, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, Gosink KK, Salomon J, Igarashi K, Zou C, Ishihama A, Severinov K, and Gourse RL (1993). A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science 262, 1407–1413. [DOI] [PubMed] [Google Scholar]
- Ross W, Schneider DA, Paul BJ, Mertens A, and Gourse RL (2003). An intersubunit contact stimulating transcription initiation by E coli RNA polymerase: interaction of the alpha C-terminal domain and sigma region 4. Genes Dev 17, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff EF, Record MT Jr, and Artsimovitch I (2015). Initial events in bacterial transcription initiation. Biomolecules 5, 1035–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford ST, Villers CL, Lee J-H, Ross W, and Gourse RL (2009). Allosteric control of Escherichia coli rRNA promoter complexes by DksA. Genes Dev 23, 236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saecker RM, Record MT Jr, and deHaseth PL (2011). Mechanism of Bacterial Transcription Initiation: RNA Polymerase - Promoter Binding, Isomerization to Initiation-Competent Open Complexes, and Initiation of RNA Synthesis. J Mol Biol 412, 754–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vazquez P, Dewey CN, Kitten N, Ross W, Gourse RL (2019). Genome-wide effects on Escherichia coli transcription from ppGpp binding to its two sites on RNA polymerase. Proc Natl Acad Sci USA 116, 8310–8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schickor P, Metzger W, Werel W, Lederer H, and Heumann H (1990). Topography of intermediates in transcription initiation of E.coli. EMBO J 9, 2215–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Gietl A, Smollett K, Tinnefeld P, Werner F, and Grohmann D (2016). TFE and Spt4/5 open and close the RNA polymerase clamp during the transcription cycle. Proc Natl Acad Sci USA 113, E1816–E1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclavi B, Zaychikov E, Rogozina A, Walther F, Buckle M, and Heumann H (2005). Real-time characterization of intermediates in the pathway to open complex formation by Escherichia coli RNA polymerase at the T7A1 promoter. Proc Natl Acad Sci USA 102, 4706–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultzaberger RK, Chen Z, Lewis KA, and Schneider TD (2007). Anatomy of Escherichia coli 70 promoters. Nucl Acids Res 35, 771–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Talaue M, Liu S, Degen D, Sineva E, Chakraborty A, Druzhinin SY, Chatterjee S, Mukhopadhyay J, Ebright YW, et al. (2011). New target for inhibition of bacterial RNA polymerase: ‘switch region’. Cuur Op Microbiol 14, 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetlov V, and Artsimovitch I (2015). Purification of bacterial RNA polymerase: tools and protocols. Methods Mol. Biol 1276, 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafur L, Sadian Y, Hoffmann NA, Jakobi AJ, Wetzel R, Hagen WJH, Sachse C, and Müller CW (2016). Molecular Structures of Transcribing RNA Polymerase I. Mol Cell 64, 1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev DG, Sekine S-I, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, and Yokoyama S (2002). Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature 417, 712–719. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Vassylyeva MN, Zhang J, Palangat M, Artsimovitch I, and Landick R (2007). Structural basis for substrate loading in bacterial RNA polymerase. Nature 448, 163–168. [DOI] [PubMed] [Google Scholar]
- Vorländer MK, Khatter H, Wetzel R, Hagen WJH, and Müller CW (2018). Molecular mechanism of promoter opening by RNA polymerase III. Nature 553, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter G, Zillig W, Palm P, and Fuchs E (1967). Initiation of DNA-dependent RNA synthesis and the effect of heparin on RNA polymerase. Eur J Biochem 3, 194–201. [DOI] [PubMed] [Google Scholar]
- Wang D, Bushnell DA, Westover KD, Kaplan CD, and Kornberg RD (2006). Structural Basis of Transcription: Role of the Trigger Loop in Substrate Specificity and Catalysis. Cell 127, 941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windgassen TA, Mooney RA, Nayak D, Palangat M, Zhang J, and Landick R (2014). Trigger-helix folding pathway and SI3 mediate catalysis and hairpin-stabilized pausing by Escherichia coli RNA polymerase. Nucl Acids Res 42, 12707–12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman JT, Chandrangsu P, Ross W, and Gourse RL (2016). Open complex scrunching before nucleotide addition accounts for the unusual transcription start site of E. coli ribosomal RNA promoters. Proc Natl Acad Sci USA 113, E1787–E1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman JT, Winkelman BT, Boyce J, Maloney MF, Chen AY, Ross W, and Gourse RL (2015). Crosslink Mapping at Amino Acid-Base Resolution Reveals the Path of Scrunched DNA in Initial Transcribing Complexes. Mol Cell 59, 768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzenkova Y, Tadigotla VR, Severinov K, and Zenkin N (2011). A new basal promoter element recognized by RNA polymerase core enzyme. EMBO J 30, 3766–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho MX, Arnold E, and Ebright RH (2012). Structural basis of transcription initiation. Science 338, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SHW (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, and Steitz TA (2015). Crystal Structures of the E. coli Transcription Initiation Complexes with a Complete Bubble. Mol Cell 58, 534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Morph illustrating the T-RPc ↔ T-RPi1 ↔ T-RPi1.5a transition. Related to Figure 4. The views correspond to the views of Figure 4(right), focusing on the promoter −10 element and the σ70 W-dyad. The progress of the movie through the structures is denoted at the top.
Movie S2. Morph illustrating the T-RPi1.5a ↔ TRPi1.5b transition. Related to Figure 5. The views correspond roughly to the views of Figure 5B and 5C. The progress of the movie through the structures is denoted at the top.
Movie S3. Morph illustrating the entire R ↔ T-R ↔ T-RPc ↔ T-RPi1 ↔ T-RPi1.5a ↔ T-RPi1.5b ↔ T-RPi2 ↔ T-preRPo ↔ T-RPo ↔ RPo pathway. Related to Figure 6. The views correspond roughly to the views of Figure 2A. The progress of the movie through the structures is denoted at the top.
Movie S4. Morph illustrating the hypothetical entire R ↔ RPc ↔ RPi1 ↔ RPi1.5a ↔ RPi1.5b ↔ RPi2 ↔ RPo pathway. Related to Figure 6. The views correspond roughly to the views of Figure 2A. The progress of the movie through the structures is denoted at the top.
Data Availability Statement
The cryo-EM density maps have been deposited in the EMDataBank under accession codes EMD-20460 (TRPc), EMD-20461 (TPRi1), EMD_20462 (TRPi1.5a), EMD-20463 (TRPi1.5b), EMD-20464 (TRPi2), EMD-20465 (TpreRPo), and EMD-20466 (TRPo). The atomic coordinates have been deposited in the Protein Data Bank under accession codes 6PSQ (TRPc), 6PSR (TRPi1), 6PSS (TRPi1.5a), 6PST (TRPi1.5b), 6PSU (TRPi2), 6PSV (TpreRPo), 6PSW (TRPo).