

Abstract
The ATP-gated P2X7 ion channel has emerging roles in amyotrophic lateral sclerosis (ALS) progression. Pharmacological blockade of P2X7 with Brilliant Blue G can ameliorate disease in SOD1G93A mice, but recent data suggests that this antagonist displays poor penetration of the central nervous system (CNS). Therefore, the current study aimed to determine whether the CNS-penetrant P2X7 antagonist, JNJ-47965567, could ameliorate ALS progression in SOD1G93A mice. A flow cytometric assay revealed that JNJ-47965567 impaired ATP-induced cation dye uptake in a concentration-dependent manner in murine J774 macrophages. Female and male SOD1G93A mice were injected intraperitoneally with JNJ-47965567 (30 mg/kg) or 2-(hydroxypropyl)-beta-cyclodextrin (vehicle control) three times a week from disease onset until end stage, when tissues were collected and studied. JNJ-47965567 did not impact weight loss, clinical score, motor (rotarod) coordination or survival compared to control mice. NanoString analysis revealed altered spinal cord gene expression in JNJ-47965567 mice compared to control mice, but such differences were not confirmed by quantitative PCR. Flow cytometric analyses revealed no differences between treatments in the frequencies or activation status of T cell or dendritic cell subsets in lymphoid tissues or in the concentrations of serum cytokines. Notably, serum IL-27, IFNβ and IL-10 were present in relatively high concentrations compared to other cytokines in both groups. In conclusion, JNJ-47965567 administered thrice weekly from disease onset did not alter disease progression or molecular and cellular parameters in SOD1G93A mice.
Electronic supplementary material
The online version of this article (10.1007/s11302-020-09692-4) contains supplementary material, which is available to authorized users.
Keywords: Amyotrophic lateral sclerosis, Motor neurone disease, SOD1G93A mice, P2X7 receptor, Purinergic receptor, Nqo1
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neurone disease [1]. ALS is characterised by the death of upper and lower motor neurones, which results in progressive muscle weakness and paralysis, culminating in death [2]. ALS remains incurable, with the only approved drug treatments being riluzole and edaravone, each of which have very limited clinical benefits [3]. Thus, there is an urgent need for new therapeutics in ALS.
Mutations in the gene encoding superoxide dismutase 1 (SOD1) are associated with familial ALS [4]. These findings led to the development of the first [5] and most commonly used [6] mouse model of ALS. In this model, mice overexpress multiple copies of a human variant of mutant SOD1 (SOD1G93A) and develop clinical signs of ALS (disease onset) at 70–140 days, depending on the strain and housing conditions [7]. These mice show many features of ALS, such as axonal and neuromuscular dysfunction, microgliosis, astrogliosis, motor neurone loss and muscle wasting [8].
The P2X7 receptor is a trimeric ligand-gated ion channel activated by extracellular ATP, which results in the rapid flux of cations, including fluorescent cation dyes, across the plasma membrane [9]. P2X7 is primarily expressed on leukocytes and plays important roles in inflammation and immunity [10]. P2X7 is also present on microglia, astrocytes and motor neurones in the central nervous system (CNS), where it plays various roles in neurological function and disorders [11]. A number of CNS-penetrant P2X7 antagonists have been developed including JNJ-47965567, which displays high amounts of brain occupancy in vivo [12], and established efficacy in neurological disorders including neuropathic pain [12] and epilepsy [13, 14]. JNJ-47965567 was originally considered to inhibit human and rat P2X7 in a competitive manner [12], but structural and activity studies of panda P2X7 revealed this compound to be a non-competitive antagonist of P2X7 [15]. Thus, the mechanism of action of JNJ-47965567 against P2X7 in different species remains to be fully elucidated.
Based on studies of rodent models of ALS and people with this disease, a role for CNS P2X7 in the progression of ALS has recently emerged. P2X7 is upregulated in post-mortem spinal cords of patients with ALS [16] and SOD1G93A rats [17]. Notably, P2X7 activation on microglia and astrocytes from SOD1G93A mice or rats, but not wild-type controls, can induce the death of motor neurones [18, 19]. Moreover, P2X7 activation on motor neurones can directly induce death of these cells [20]. This neurotoxicity potentially results in ATP release from dying cells to provide a positive feedback loop to further activate P2X7 on neighbouring neural cells and promote ALS progression. Collectively, this has led to the current idea that P2X7 activation in the CNS promotes ALS and that pharmacological blockade of P2X7 has therapeutic benefits [21, 22]. To this end, our group and others have examined the effect of pharmacological blockade of P2X7 in disease progression in SOD1G93A mice. In each study, P2X7 blockade was achieved by intraperitoneal (i.p.) injection of the P2X7 antagonist, Brilliant Blue G (BBG), into SOD1G93A mice from disease onset (defined as the first signs of clinical disease). In the first study, Cervetto et al. [23] showed that BBG (45.5 mg/kg, every 2 days) improved motor coordination in mice of both sexes, and reduced weight loss in male mice. Whilst Apolloni et al. [24] showed that BBG (250 mg/kg, thrice per week) improved motor coordination, with no differences between sexes. Finally, in our study [25], BBG (45.5 mg/kg, thrice per week) improved weight and survival in female, but not male, mice.
Combined, the above studies with BBG indicated that P2X7 blockade has a therapeutic effect in the SOD1G93A mouse model of ALS when drug administration commences at disease onset [26]. However, recent observations have questioned the ability of BBG to cross the blood-brain barrier in high concentrations. Administration of BBG into rats did not display brain P2X7 occupancy and only minimal amounts of BBG are detected within the brain [27]. Whilst in our study [25], BBG (blue) colouration was not apparent in the brains or spinal cords, despite extensive BBG colouration in systemic tissues [26]. Collectively, these studies suggest that BBG lacks the ability to cross the blood-brain barrier, thereby predominantly inhibiting systemic P2X7 rather than CNS P2X7. Therefore, the primary aim of the current study was to determine if the highly CNS-penetrant P2X7 antagonist, JNJ-47965567, administered via a regime similar to that used previously for BBG [25], could ameliorate ALS progression in SOD1G93A mice. JNJ-47965567 is a selective, potent P2X7 antagonist, and is clearly detectable in both plasma and CNS tissue [12], making it a suitable candidate to assess both systemic and CNS P2X7 blockade in SOD1G93A mice. Despite JNJ-47965567 demonstrating blockade of murine P2X7 in vitro, this compound when administered thrice weekly from disease onset did not alter disease progression or molecular and cellular parameters of ALS in SOD1G93A mice.
Materials and methods
Materials
RPMI-1640 medium, GlutaMAX and Dulbecco’s PBS (PBS) were from Life Technologies (Grand Island, USA). Foetal calf serum (FCS) was from Bovogen Biologicals (East Kellior, Australia) and was heat-inactivated before use. ATP, dimethyl sulphoxide (DMSO), 2-(hydroxypropyl)-beta-cyclodextrin (β-CD) and RNAlater were from Sigma Chemical Co (St. Louis, USA). Ethidium bromide and other reagent grade chemicals were from Amresco (Solon, USA). JNJ-47965567 was from Tocris Bioscience (Ellisville, USA). For in vitro studies, JNJ-47965567 was dissolved in 100% DMSO at 30 mM and stored at − 20 °C until required. For in vivo studies, JNJ-47965567 was dissolved in a cyclodextrin-based solvent as previously described [12]. Briefly, β-CD was solubilised in Milli-Q water (Millipore, Burlington, USA) at 0.3 mg/mL (30% w/v). JNJ-47965567 was solubilised in 30% (w/v) β-CD at 5 mg/mL by rotation at 45 °C for 2 h and sonicated in a 37 °C water bath for 15 min. Both β-CD and JNJ-47965567 were filter sterilised (0.22 μM MillexGV, Millipore) and stored at 4 °C for less than 7 days, with fresh stocks prepared weekly.
The Zombie Violet Fixable Viability Kit, and PerCP-Cy5.5-conjugated hamster anti-murine CD11c (clone N418), PE-conjugated rat anti-murine CD39 (clone Duha59), PE-Cy7-conjugated hamster anti-murine CD80 (clone 16-10A1) monoclonal antibodies (mAb) were from BioLegend (San Diego, USA). Rat anti-murine CD16/CD32 (Mouse BD Fc Block), PerCP-Cy5.5-conjugated rat anti-murine CD4 (clone RM4-5), APC-Cy7-conjugated rat anti-murine CD8 (clone 53-6.7) and PE-Cy7-conjugated rat anti-murine CD44 (clone IM7) mAb were from BD Biosciences (San Diego, USA). FITC-conjugated hamster anti-murine CD3 (clone 145-2C11), FITC-conjugated rat anti-murine CD11b (clone M1/70), APC-conjugated rat anti-murine Foxp3 (clone FJK-16s) and APC-conjugated rat anti-murine MHC Class II (clone M5/114.15.2) mAb were from eBioscience (San Diego, USA).
Murine macrophages
Murine macrophage J774 cells (American Type Culture Collection, Manasses, USA) were maintained in RPMI-1640 medium supplemented with 2 mM GlutaMAX and 10% (v/v) FCS at 37 °C and 95% air/5% CO2. Cells were passaged and harvested for experiments by mechanical scraping.
ATP-induced cation dye uptake assay
P2X7 activity and inhibition was assessed using an ATP-induced cation dye uptake assay as described [28]. Briefly, 1 × 106 cells were washed and resuspended in low-divalent medium (145 mM NaCl, 2 mM KCl, 0.2 mM CaCl2, 13 mM D-glucose, 10 mM HEPES, pH 7.4) and incubated at 37 °C for 5 min in the presence of 25 μM ethidium bromide in the absence or presence of ATP. Ethidium+ uptake was terminated by the addition of an equal volume of ice-cold stop solution (low-divalent medium containing 20 mM MgCl2) and centrifugation. Cells were washed once with low-divalent medium. In some experiments, cells were pre-incubated at 37 °C for 15 min in the presence or absence of JNJ-47965567 (as indicated) before addition of ethidium bromide and ATP. Data was acquired using a LSRFortessa X-20 (BD Biosciences) and the mean fluorescence of ethidium+ uptake determined using FlowJo software v8.7.1 (TreeStar Inc., Ashland, USA). ATP-induced ethidium+ uptake was determined as the difference in uptake in the presence and absence of ATP.
SOD1G93A mice
The use of mice was approved by the University of Wollongong Animal Ethics Committee (Wollongong, Australia), and complied with the Australian code for the care and use of animals for scientific purposes (National Health and Medical Research Council, Canberra, Australia). SOD1G93A mice (B6.Cg-Tg(SOD1*G93A)1Gur/J), originally obtained from Jackson Laboratory (Bar Harbour, USA) and maintained on a C57BL/6J background [29], were bred at Australian BioResources (Moss Vale, Australia). Transgene copy number of SOD1G93A mice was assessed by quantitative PCR (qPCR) of mouse tail snips as described [25] and was similar between treatment groups (Table S1).
Pre-clinical drug trial in SOD1G93A mice
The pre-clinical trial with JNJ-47965567 in SOD1G93A mice was designed in accordance with recommendations of the ALS Therapy Development Institute (Cambridge, USA) [30] and the European ALS/MND Group [31]. Equal numbers of female and male mice were used in experiments and mice were matched for date of birth and sex. Each treatment group consisted of 24 mice per group, but 1 male control-treated mouse died (day 93) from causes unrelated to ALS and was excluded from all analysis. Mice were scored and weighed three times a week in a blinded fashion from day 60 using the criteria outlined by the ALS Therapy Development Institute [32]. Motor coordination of mice was assessed by rotarod performance once per week as described [25]. From disease onset (days 97–104) until end stage disease, mice were injected i.p. with 30 mg/kg JNJ-47965567 or an equivalent volume of β-CD (vehicle control) three times per week. The dose of 30 mg/kg for JNJ-47965567 was based on previous studies demonstrating efficacy of this drug at this same dose in rodent models of neuropathic pain [12] or epileptic seizure [13, 14]. Commencement and dosing frequency of JNJ-47965567 was based on our previous study using BBG which displayed efficacy in SOD1G93A mice when injected i.p. from disease onset [25]. Disease onset was defined as when mice in either group had a clinical score of 1 over two consecutive measurements. Disease end stage was defined as when a mouse lost 20% body weight compared to the initial pre-treatment maximum body weight or when a mouse was unable to right itself within 10 s of being placed on either of its sides. End stage mice were euthanised by CO2, and whole blood (via cardiac puncture), spleens and CNS draining (cervical) and non-draining (inguinal) lymph nodes were collected. Mice were then perfused transcardially with PBS, and spinal cords (lumbar region) stored in RNAlater at − 20 °C until required.
Gene expression analysis in spinal cords
RNA was isolated from spinal cords using the ISOLATE II RNA Mini Kit (Bioline, London, UK) as described [33]. Differential gene expression in spinal cords between the two treatment groups was assessed using the nCounter Mouse Neuropathology Panel (NanoString Technologies, Seattle, USA) according to manufacturer’s instructions. Briefly, a hybridisation reaction was prepared by incubating 25 ng of total RNA overnight at 65 °C in nCounter Reporter CodeSet, Capture ProbeSet and hybridisation buffer. Following hybridisation, samples were processed using the nCounter SPRINT Profiler (NanoString Technologies). Data was analysed using nSolver Advanced Analysis software version 4.0 (NanoString Technologies). The relative expression of each gene was normalised to eight housekeeping genes: Aars, Ccdc127, Cnot10, Csnk2a2, Fam104a, Lars, Mto1 and Tada2b.
Quantitative PCR of spinal cords
RNA was isolated from spinal cords (stored frozen in RNAlater) as above and cDNA synthesised using the qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, USA) as described [33]. The relative expression of Nqo1 and P2rx7 was determined by qPCR using FAM-labelled primers/probes for Nqo1 (Mm01253561_m1) or P2rx7 (Mn01199500_m1), and VIC-labelled primers/probes for the housekeeping gene Gapdh (Mm99999915_g1) with the TaqMan Universal Master Mix II (Thermo Fisher Scientific, Waltham, USA) according to manufacturer’s instructions. Reactions were conducted in triplicate for each gene and analysed using QuantStudio Design and Analysis software version 1.4.3 (Thermo Fisher Scientific). Relative gene expression was quantified using the ΔΔCt method and presented relative to a randomly selected vehicle control sample.
Immunophenotyping of lymphoid cells
Following collection, spleens and lymph nodes were mechanically dissociated in ice-cold RPMI-1640 medium containing 10% (v/v) FCS. Spleens were cleared of red blood cells by mechanical agitation in lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na4EDTA) for 5 min at room temperature. All samples were washed in ice-cold PBS containing 10% (v/v) FCS. Approximately 5 × 106 cells per sample were incubated with Zombie Violet for 30 min on ice in the dark. Cells were washed twice with PBS containing 10% (v/v) FCS, and labelled in the presence of Mouse BD Fc Block with panels of fluorochrome-conjugated mAb (T cell panel: CD3, CD4, CD8, CD39 and CD44; myeloid panel: CD11b, CD11c, CD80 and MHC Class II) for 30 min on ice in the dark. Cells in the T cell panel were washed twice and labelled with APC-conjugated Foxp3 mAb using the Foxp3/Transcription Factor Staining Buffer Set (eBiosicence) according to manufacturer’s instructions. Finally, all cells were washed with PBS and data acquired using a LSRII Fortessa X-20. Proportions of leukocyte subsets and the relative expression of CD39 or CD80 were determined using FlowJo software.
Serum cytokine measurements
Following collection, whole blood was centrifuged at 1700×g for 10 min and the resulting sera stored at − 80 °C until required. Cytokine concentrations (in duplicate) in serum were quantified using the LEGENDplex Mouse Inflammation Panel (13-plex) (BioLegend) according to the manufacturer’s instructions. Data was acquired using a LSR Fortessa X-20 and analysed using the LEGENDplex Data Analysis software version 8.0 (BioLegend). Cytokine concentrations below the detection limit were assigned a value of 0.01.
Data presentation and statistical analysis
All data were analysed using Prism 5 software (GraphPad, San Diego, USA). Data is presented as mean ± standard error of the mean (SEM). Differences between treatment groups were determined using: a two-way ANOVA (with Bonferroni post hoc test) for clinical score, body weight loss and motor coordination; the log-rank (Mantel-Cox) test for survival; and a two-tailed unpaired Student’s t test for transgene copy number, PCR, flow cytometric and cytokine analyses. Statistical differences with P < 0.05 were considered significant.
Results
JNJ-47965567 inhibits murine P2X7 in concentration-dependent manner
JNJ-47965567 is a CNS-penetrant P2X7 antagonist [12] but its mechanism of action against murine P2X7 remains to be elucidated. Therefore, flow cytometric measurements of ATP-induced ethidium+ uptake into J774 macrophages were used to determine the mechanism of action of JNJ-47965567 against murine P2X7. To determine the EC50 value for ATP against murine P2X7, J774 macrophages were incubated with increasing concentrations of ATP. ATP induced ethidium+ uptake into cells in a concentration-dependent fashion with an EC50 value of 510 ± 96 μM (Fig. 1a). J774 macrophages were pre-incubated with DMSO (vehicle control) or increasing concentrations of JNJ-47965567 and incubated with 500 μM ATP, a concentration approximate to the EC50 value. JNJ-47965567 impaired ATP-induced ethidium+ uptake into cells in a concentration-dependent fashion with an IC50 value of 54 ± 24 nM (Fig. 1b). DMSO alone had no effect on ethidium+ uptake (data not shown). Finally, J774 macrophages were pre-incubated in the presence of DMSO or 0.03 μM, 0.3 μM and 3 μM JNJ-47965567, and then incubated with increasing concentrations of ATP. ATP in the presence of DMSO alone induced ethidium+ uptake into cells in a concentration-dependent fashion with an EC50 value of 358 ± 27 μM (Fig. 1c). In the presence of 0.03 μM, 0.3 μM and 3 μM JNJ-47965567, ATP induced ethidium+ uptake with EC50 values of 519 ± 13 μM, 1021 ± 61 μM and 1850 ± 40 μM, respectively (Fig. 1c). Maximal ATP-induced ethidium+ uptake responses in the presence of JNJ-47965567 were decreased in a concentration-dependent manner, with maximal responses reduced by 37 ± 0.5%, 61 ± 4% and 89 ± 2% by 0.03 μM, 0.3 μM and 3 μM JNJ-47965567, respectively (Fig. 1c). This inhibition of maximal ATP-responses by JNJ-47965567 is consistent with a non-competitive mechanism of inhibition.
Fig. 1.
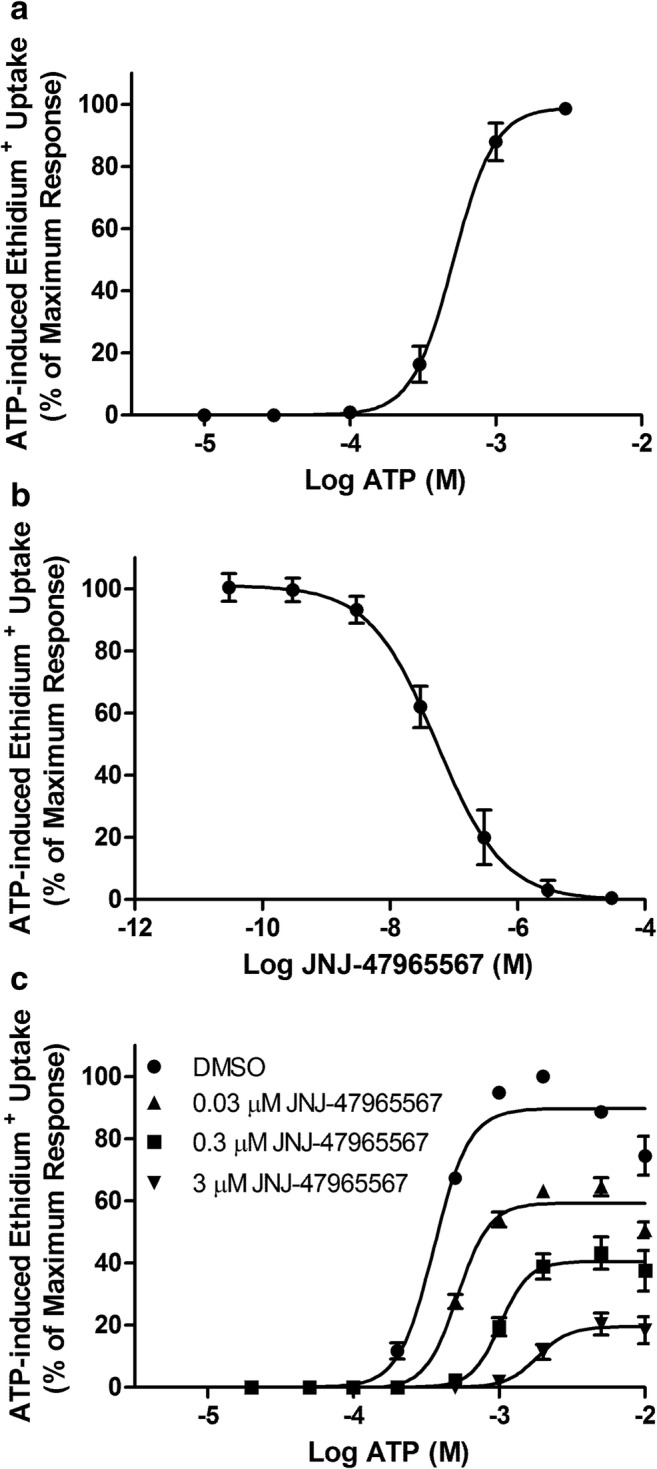
JNJ-47965567 inhibits murine P2X7 in a concentration-dependent manner. a J774 cells in low-divalent medium containing 25 μM ethidium bromide were incubated in the absence or presence of ATP (as indicated) for 5 min. b, c J774 cells in low-divalent medium were pre-incubated with DMSO or JNJ-47965567 (as indicated) for 15 min. Ethidium bromide (25 μM) was then added and cells were incubated with b 500 μM ATP or c ATP (as indicated) for 5 min. a–c ATP incubations were stopped by addition of low-divalent medium containing 20 mM MgCl2 and centrifugation. Ethidium+ uptake was assayed by flow cytometry, and ATP-induced ethidium+ uptake determined as the difference in uptake between cells incubated in the presence or absence of ATP. Data is represented as percent ethidium+ uptake relative to maximum ATP response. Results are mean ± SEM (n = 3)
JNJ-47965567 does not alter clinical disease progression in SOD1G93A mice
To determine if a CNS-penetrant P2X7 antagonist could impair ALS progression, female and male SOD1G93A mice were injected i.p. with 30 mg/kg JNJ-47965567 (n = 24 mice) or β-CD (vehicle; n = 23 mice) three times per week from disease onset (two consecutive scores of 1) until end stage disease. Clinical score and weight were assessed three times per week, and motor coordination (rotarod performance) was assessed once per week [25]. Mice in both treatment groups displayed similar trends and no significant difference (P = 0.72) in clinical scores over the course of the study (Fig. 2a). Mice in both treatment groups also exhibited similar changes in mean body weight over time, with initial signs of weight loss from day 111, followed by an accelerated decline from day 139 and no significant difference between groups (P = 0.31) (Fig. 2b). Mice in both treatment groups also displayed similar motor coordination over the course of the study, with motor coordination declining from day 139 and no significant differences between groups (P = 0.63) (Fig. 2c). Finally, the rate of survival of mice in both treatment groups was similar, with all mice reaching end stage disease and a mean lifespan of 165 ± 10 days in mice treated with JNJ-47965567 compared to 163 ± 9 days in mice treated with vehicle (P = 0.34) (Fig. 2d). Previous studies have reported sex-dependent effects on disease progression in SOD1G93A mice treated with the P2X7 antagonist BBG [23, 25]. However, separate analyses of female and male SOD1G93A mice did not reveal any significant differences in clinical score, weight loss, motor coordination or survival between treatments in mice of either sex (Fig. S1).
Fig. 2.

JNJ-47965567 does not alter clinical disease progression in SOD1G93A mice. a–d SOD1G93A mice were injected i.p. with JNJ-47965567 (30 mg/kg) or an equivalent volume of β-CD (vehicle) three times per week from disease onset until end stage disease. Mice were assessed for a ALS score, b body weight loss (percent of pre-disease maximum), c motor (rotarod) coordination and d survival. a–d Results are mean ± SEM (β-CD, n = 23; JNJ-47965567, n = 24) with differences between groups compared using a–c a two-way ANOVA or d the log-rank (Mantel-Cox) test
In addition to the above observations, several mice displayed unexpected adverse events following prolonged treatment with either JNJ-47965567 or vehicle. These adverse events were observed as ocular discharge and blepharospasm (n = 10 mice), vasculitis of the ears (n = 2 mice) and/or hair loss (n = 3 mice), and were observed in both JNJ-47965567- and vehicle-treated mice (n = 8 and 7 mice, respectively) (data not shown). Histological examination of one eye sample revealed corneal oedema, but no other changes to the eye or optic nerve (data not shown). Combined, this data suggests that these observed effects were due to the vehicle β-CD rather than JNJ-47965567.
JNJ-47965567 does not alter spinal cord gene expression in SOD1G93A mice
To determine if JNJ-47965567 altered molecular CNS parameters in SOD1G93A mice, RNA was isolated from the spinal cords of mice treated with JNJ-47965567 or vehicle and the expression of 760 genes analysed by NanoString (Table S2). Three female and three male mice treated with JNJ-47965567 were randomly chosen for this analysis and matched with vehicle-treated mice in a pair-wise manner for sex and date of birth. The data for each mouse and all 760 genes is presented in Table S2. Volcano plot analysis revealed that a large number of genes were upregulated and a smaller number of genes were downregulated by JNJ-47965567 compared to vehicle control (Fig. 3a). However, these differences were not statistically significant following Bonferroni correction for multiple comparisons (Table S2). The data for the top 20 genes altered by JNJ-47965567 are displayed in Table 1. Nqo1 showed the greatest fold change in gene expression, with a log2 fold-change of − 2.2 ± 0.3 in JNJ-47965567-treated mice compared to vehicle-treated mice (P = 0.0897). P2rx7 was partially upregulated in JNJ-47965567-treated mice with a log2 fold-change of 1.8 ± 0.7 (P = 1) and was ranked twelfth for all genes altered by JNJ-47965567.
Fig. 3.
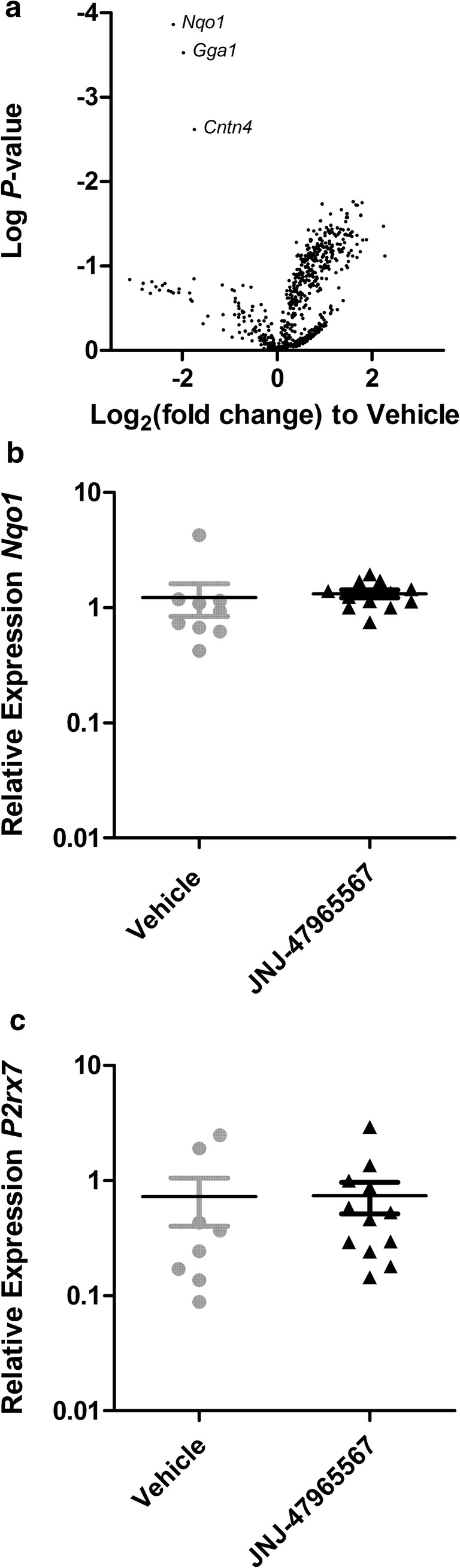
JNJ-47965567 does not alter spinal cord gene expression in SOD1G93A mice. a RNA and b, c synthesised cDNA from spinal cords from end stage β-CD- and JNJ-47965567-treated SOD1G93A mice were analysed by a NanoString and b, c qPCR. a Volcano plot showing relative change in gene expression in JNJ-47965567-treated mice compared to β-CD-treated mice; symbols represent individual genes (n = 6 mice per group); log P values were determined using nSolver Advanced Analysis software. b, c Results are mean ± SEM; symbols represent individual mice (β-CD, n = 8; JNJ-47965567, n = 12)
Table 1.
Top 20 genes in spinal cords altered by JNJ-47965567. RNA from spinal cords from end stage SOD1G93A mice was analysed by NanoString to determine changes in gene expression in JNJ-47965567-treated mice relative to β-CD-treated mice (n = 6 mice per group)
| Gene | Log2 (fold change ± SEM) | Pa |
|---|---|---|
| Nqo1 | − 2.2 ± 0.3 | 0.000139 |
| Gga1 | − 2.0 ± 0.3 | 0.00030 |
| Cntn4 | − 1.7 ± 0.4 | 0.00244 |
| Pllp | 1.6 ± 0.6 | 0.0175 |
| Mal | 1.8 ± 0.6 | 0.0178 |
| Plcb1 | 2.0 ± 0.3 | 0.0186 |
| Ugt8a | 1.7 ± 0.6 | 0.0190 |
| Cdkn1a | 1.7 ± 0.6 | 0.0192 |
| Bcl2l1 | 1.4 ± 0.5 | 0.0216 |
| Jam3 | 1.5 ± 0.6 | 0.0240 |
| Gnai1 | 1.1 ± 0.4 | 0.0245 |
| P2rx7 | 1.8 ± 0.7 | 0.0253 |
| Pla2g16 | 1.5 ± 0.6 | 0.0265 |
| Arhgef10 | 1.5 ± 0.6 | 0.0268 |
| Fa2h | 1.5 ± 0.6 | 0.0269 |
| Plcb4 | 0.9 ± 0.4 | 0.0295 |
| Cers2 | 1.4 ± 0.6 | 0.0299 |
| Gria3 | 1.1 ± 0.5 | 0.0333 |
| Pmp22 | 2.2 ± 0.9 | 0.0339 |
| Nf1 | 1.0 ± 0.4 | 0.0351 |
aFollowing Bonferroni correction for multiple comparisons, all P values equalled 1 except P = 0.0897 for Nqo1 and P = 0.193 for Gga1
To validate the findings of the NanoString analysis, the relative expression of Nqo1 and P2rx7 in spinal cords of SOD1G93A mice were analysed by qPCR. RNA from the same 12 mice used for the NanoString analysis, as well as RNA from an additional six JNJ-47965567-treated and two vehicle control-treated mice were examined. qPCR analysis revealed that the relative expression of Nqo1 (Fig. 3b) and P2rx7 (Fig. 3c) did not differ between treatment groups.
Finally, NanoString analysis revealed no significant differences in the expression of the microglia markers Itgam (Cd11b) or P2ry12 [34], or the astrocyte markers Aldh1l1 or Gfap [35], between the two treatment groups (Table 2; Table S2). This suggests that JNJ-47965567 did not alter microgliosis or astrogliosis in SOD1G93A mice.
Table 2.
JNJ-47965567 does not alter gene expression of microglia or astrocyte markers in SOD1G93A mice. RNA from spinal cords from end stage SOD1G93A mice was analysed by NanoString to determine the changes in gene expression in JNJ-47965567-treated mice relative to β-CD-treated mice (n = 6 mice per group)
| Gene | Log2 (fold change ± SEM) | Pa |
|---|---|---|
| Microglia | ||
| Itgam(Cd11b) | 0.6 ± 0.5 | 0.257 |
| P2ry12 | 0.2 ± 1.0 | 0.879 |
| Astrocytes | ||
| Aldh1l1 | 0.3 ± 0.5 | 0.520 |
| Gfap | 0.2 ± 0.9 | 0.816 |
aFollowing Bonferroni correction for multiple comparisons, all P values equalled 1
JNJ-47965567 does not alter proportions of lymphoid leukocytes in SOD1G93A mice
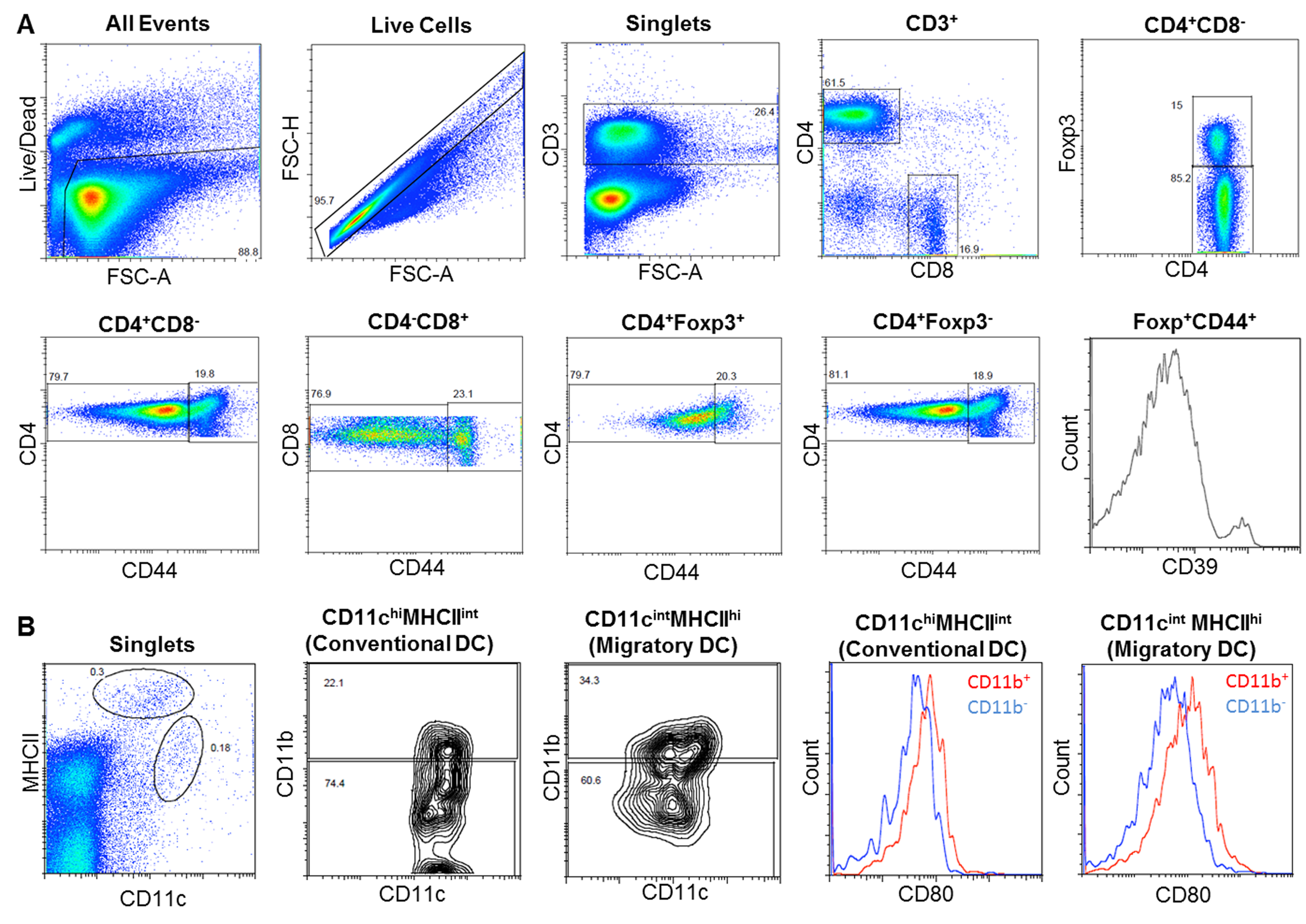
There is increasing evidence that systemic T cells [36, 37] including regulatory T (Treg) cells are altered in ALS patients and SOD1G93A mice [38, 39]. To determine if JNJ-47965567 treatment altered the proportion of systemic T cells in SOD1G93A mice, the spleens and CNS draining and non-draining lymph nodes from mice were assessed by flow cytometry (Fig. S2A) with the findings presented in Table 3. There was no significant difference in the proportion of CD4+ or CD8+ T cells amongst CD3+ T cells within spleens and draining and non-draining lymph nodes from SOD1G93A mice treated with either JNJ-47965567 or vehicle. Similarly, there was no significant difference in the proportion of conventional CD4+ T cells (Foxp3− cells) or Treg cells (Foxp3+ cells) within these three lymphoid tissues between treatment groups. There were also no significant differences in the proportion of activated (CD44+) cells amongst conventional CD4+ T cells or Treg cells between treatment groups. The relative expression of the ecto-ATPDase CD39, which can regulate P2X7 activity on T cells [40], did not differ on CD4+ T cells including conventional T cells or Treg cells, or on CD8+ T cells within the spleens or lymph nodes from mice treated with either JNJ-47965567 or vehicle. Notably, the proportion of each aforementioned T cell subset including activated T cells or the relative expression of CD39 on these cells did not differ between CNS draining and non-draining lymph nodes from SOD1G93A mice.
Table 3.
JNJ-47965567 does not alter proportions of T cell subsets in SOD1G93A mice. Spleens and CNS draining and non-draining lymph nodes from end stage β-CD- and JNJ-47965567-treated SOD1G93A mice were labelled with fluorochrome-conjugated mAb and the percentages of T cell subsets, as well as relative expression of CD39 (MFI), assessed by flow cytometry. Results are mean ± SEM
| Cell typea | Spleen | Draining lymph node | Non-draining lymph node | |||
|---|---|---|---|---|---|---|
| Vehicle (n = 14) | JNJ-47965567 (n = 21) | Vehicle (n = 14) | JNJ-47965567 (n = 19) | Vehicle (n = 14) | JNJ-47965567 (n = 20) | |
| % CD4+ of CD3+ | 54.8 ± 0.7 | 54.2 ± 2.1 | 48.3 ± 1.0 | 50.3 ± 0.6 | 49.2 ± 1.4 | 50.2 ± 0.8 |
| % CD44+ | 18.9 ± 1.1 | 16.9 ± 0.8 | 15.8 ± 0.8 | 16.0 ± 0.8 | 14.4 ± 1.0 | 14.9 ± 0.8 |
| CD39 MFI | 17.7 ± 3.8 | 15.2 ± 2.0 | 8.9 ± 1.1 | 9.0 ± 0.9 | 10.3 ± 2.2 | 8.0 ± 0.8 |
| % Foxp3+ (Treg) | 13.1 ± 0.7 | 12.8 ± 0.5 | 17.2 ± 0.7 | 18.2 ± 1.0 | 14.4 ± 0.6 | 15.5 ± 0.7 |
| % CD44+ | 25.5 ± 1.7 | 23.3 ± 1.1 | 40.5 ± 1.7 | 38.5 ± 1.6 | 43.0 ± 2.2 | 42.6 ± 1.9 |
| CD39 MFI | 27.2 ± 6.6 | 20.6 ± 2.1 | 15.5 ± 2.1 | 14.2 ± 1.4 | 20.3 ± 4.4 | 15.9 ± 1.3 |
| % Foxp3− (Tconv) | 87.2 ± 0.7 | 87.3 ± 0.5 | 83.1 ± 0.8 | 82.0 ± 1.0 | 85.7 ± 0.6 | 84.6 ± 0.7 |
| % CD44+ | 18.3 ± 1.2 | 16.9 ± 0.9 | 11.1 ± 0.8 | 11.7 ± 0.7 | 9.3 ± 0.7 | 9.5 ± 0.6 |
| CD39 MFI | 15.8 ± 3.4 | 13.7 ± 2.0 | 5.9 ± 0.7 | 6.2 ± 0.7 | 5.9 ± 1.3 | 4.4 ± 0.5 |
| % CD8+ of CD3+ | 33.3 ± 1.7 | 35.4 ± 0.7 | 41.0 ± 2.7 | 42.3 ± 0.8 | 41.3 ± 2.1 | 40.6 ± 1.2 |
| % CD44+ | 15.2 ± 1.3 | 12.7 ± 0.8 | 15.6 ± 1.3 | 13.0 ± 0.6 | 15.7 ± 1.6 | 14.8 ± 0.5 |
| CD39 MFI | 6.4 ± 1.8 | 5.3 ± 1.3 | 3.4 ± 0.6 | 2.6 ± 0.2 | 5.4 ± 1.7 | 2.9 ± 0.3 |
aTreg regulatory T cells, Tconv conventional T cells
Dendritic cells (DCs) are antigen-presenting cells crucial in the activation and regulation of T cells [41]. Therefore, the proportions of conventional DCs (CD11chiMHC class IIint cells) and migratory DCs (CD11cintMHC class IIhi cells) in the CNS draining and non-draining lymph nodes from SOD1G93A mice were studied by flow cytometry (Fig. S2B) (Table 4). There was no significant difference in the proportion of these DC subsets in lymph nodes from mice treated with either JNJ-47965567 or vehicle. Notably, the proportion of conventional DCs, but not migratory DCs, was lower in CNS draining lymph nodes compared to non-draining lymph nodes from mice in either treatment group (P < 0.05). The proportions of CD11b+ DCs and CD11b− DCs, with the latter corresponding to CD8α+ DCs [41], amongst conventional or migratory DCs in lymph nodes from mice were not significantly different between treatment groups. Finally, the relative expression of the DC activation marker CD80 on any of these four DC subsets was not significantly different between treatment groups.
Table 4.
JNJ-47965567 does not alter proportions of DC subsets in SOD1G93A mice. CNS draining and non-draining lymph nodes from end stage β-CD- and JNJ-47965567-treated SOD1G93A mice were labelled with fluorochrome-conjugated mAb and the percentages of DC subsets, as well as relative expression of CD80 (MFI), assessed by flow cytometry. Results are mean ± SEM
| Cell typea | Draining lymph node | Non-draining lymph node | ||
|---|---|---|---|---|
| Vehicle (n = 13) | JNJ-47965567 (n = 17) | Vehicle (n = 10) | JNJ-47965567 (n = 18) | |
| % cDC (CD11chi MHCIIint) of total cells | 0.2 ± 0.3 | 0.3 ± 0 | 0.8 ± 0.2b | 0.9 ± 0.1b |
| CD80 MFI | 30.5 ± 6.7 | 44.5 ± 9.7 | 20.5 ± 3.8 | 41.5 ± 8.4 |
| % CD11b+ | 39.4 ± 3.7 | 39.3 ± 3.7 | 24.3 ± 3.1 | 30.0 ± 2.1 |
| CD80 MFI | 57.3 ± 8.5 | 82.7 ± 14.8 | 75.4 ± 18.1 | 107.2 ± 27.6 |
| % CD11b− | 57.2 ± 3.8 | 56.8 ± 3.8 | 75.7 ± 3.1 | 70.0 ± 2.1 |
| CD80 MFI | 17.4 ± 2.6 | 24.3 ± 3.3 | 13.8 ± 2.8 | 28.2 ± 5.6 |
| % mDC (CD11cint MHCIIhi) of total cells | 0.9 ± 0.2 | 0.7 ± 0.1 | 1.0 ± 0.4 | 0.8 ± 0.1 |
| CD80 MFI | 4.2 ± 1.8 | 10.6 ± 4.6 | 3.2 ± 0.7 | 13.3 ± 6.2 |
| % CD11b+ | 37.8 ± 1.6 | 39.5 ± 1.7 | 29.4 ± 3.9 | 28.6 ± 2.5 |
| CD80 MFI | 6.4 ± 3.6 | 14.6 ± 6.2 | 6.5 ± 2.3 | 19.4 ± 7.6 |
| % CD11b− | 57.9 ± 1.8 | 57.0 ± 1.8 | 70.6 ± 3.9 | 71.4 ± 2.5 |
| CD80 MFI | 3.2 ± 0.9 | 8.6 ± 4.1 | 2.7 ± 1.7 | 11.5 ± 5.9 |
acDC conventional DCs, mDC migratory DCs, MHCII MHC class II
bP < 0.05 compared to corresponding conventional DCs in draining lymph nodes
JNJ-47965567 does not alter serum cytokine concentrations in SOD1G93A mice
There is emerging evidence that systemic cytokines are altered in ALS patients [42] and SOD1G93A mice [43]. Therefore, to determine if JNJ-47965567 treatment altered the concentrations of serum cytokines in SOD1G93A mice, sera were assessed by a flow cytometric multiplex cytokine assay. Of the 13 cytokines measured, 12 were detected in both treatment groups, with mean cytokine concentrations displaying a rank order of IL-27 > IFNβ > IL-10 > IL-1β = IL-6 = IL-17A = GM-CSF > IL-23 = MCP-1 = TNFα > IL-1α = IFNγ (Fig. 4); IL-12p70 was below the detection threshold in all samples (data not shown). Serum concentrations of IL-27, IFNβ and IL-10 (Fig. 4a) were approximately one log higher than the other nine cytokines detected (Fig. 4b). However, there were no significant differences in serum concentrations of each respective cytokine between mice treated with either JNJ-47965567 or vehicle control (Fig. 4).
Fig. 4.

JNJ-47965567 does not alter serum cytokine concentrations in SOD1G93A mice. a, b Sera from end stage β-CD- and JNJ-47965567-treated SOD1G93A mice were assessed using a flow cytometric mutliplex cytokine assay. Cytokines are listed in rank order of concentration (highest to lowest). For clarity, a IL-27, IFNβ and IL-10 are presented separately from the b remaining nine cytokines detected; IL-12p70 was below the detection threshold in all samples (data not shown). a, b Bars represent group means; symbols represent individual mice (β-CD, n = 20; JNJ-47965567 n = 20)
Discussion
The current study aimed to determine whether the CNS-penetrant P2X7 antagonist, JNJ-47965567, administered via a regime similar to that used previously for the P2X7 antagonist BBG [25], could ameliorate ALS progression in SOD1G93A mice. In contrast to previous studies using BBG [23–25], JNJ-47965567 administered thrice weekly from disease onset did not reduce the clinical score, prevent loss of weight, or motor coordination or prolong survival in SOD1G93A mice. This P2X7 antagonist also had minimal impact on various molecular and cellular parameters within the CNS and periphery of these mice at end stage disease. During the course of the current study, another group [44] demonstrated that the CNS-penetrant P2X7 antagonist, A-804598, also failed to reduce the clinical score and extend survival in SOD1G93A mice. Reasons for the observed differences between BBG and the two CNS-penetrant P2X7 antagonists on ALS progression may be due to the specificity of these antagonists against P2X7. BBG is a less selective P2X7 antagonist than JNJ-47965567 and A-804598 [9], with established inhibitory actions on other channels including P2X1 [45], P2X5 [46] and the neuronal voltage-gated sodium channel [47], as well as the ATP-release channel pannexin-1 [48]. Thus, the possibility remains that BBG may be acting on a pathway other than that mediated by P2X7 in limiting disease progression in SOD1G93A mice. BBG can also inhibit prion activity [49], a feature of mutant SOD1 in ALS models [50]. However, it is questionable if BBG can penetrate the CNS of SOD1G93A mice [26, 27], so it remains unlikely that BBG reduces disease progression in these mice through its anti-prion activity.
It is possible that the inability of JNJ-47965567 to alter disease progression in SOD1G93A mice is a result of the drug regime used. As outlined above, the JNJ-47965567 dose used was based on previous studies demonstrating efficacy of this drug at 30 mg/kg in rodent models of neuropathic pain [12] or epileptic seizure [13, 14]. While the commencement and dosing frequency of JNJ-47965567 was based on our study using BBG which displayed efficacy in SOD1G93A mice when injected i.p. from disease onset [25]. More recent studies have also used JNJ-47965567 at 30 mg/kg i.p. to alleviate disease or symptoms in mouse models of schizophrenia [51], hypoxia-induced seizure [52] or autism [53]. However, it should be noted that these studies injected this P2X7 antagonist within 2 h prior to the disease-causing insult. Furthermore, others have reported, following a single i.p. injection of JNJ-47965567 (30 mg/kg) in C57BL/6 mice (the same background as the SOD1G93A mice used in the current study), that this drug reaches high concentrations in the brain (> 300 ng/mL) in the first 2 h of administration, but then declines towards baseline concentrations at 4–6 h post-injection [14]. Thus, i.p. injections of JNJ-47965567 (30 mg/kg) three times a week may have been insufficient to achieve continuous concentrations of this drug in the CNS to reduce disease progression in SOD1G93A mice. In support of this, a recent conference abstract (Ruiz-Ruiz C, Ceusters M, Garcia AM, P2X7 receptor antagonism with JNJ-47965567 reduced body weight loss and delayed disease onset in female amyotrophic lateral sclerosis SOD1G93A mice. 1st European Purine Meeting, September 4–6, 2019, Santiago De Compostela, Spain) reports that JNJ-47965567 (30 mg/kg) injected i.p. four times a week from pre-disease onset age to endpoint delayed disease onset in SOD1G93A mice of combined sex, and also reduced body weight loss in female SOD1G93A mice. Of note, the current study revealed a slight, but not statistically significant improvement in ALS scores of female SOD1G93A mice treated with JNJ-47965567. Collectively, this data suggests that higher doses and/or more frequent administration of JNJ-47965567 may further prevent disease progression in SOD1G93A mice.
An alternate explanation for the differences between BBG and JNJ-47965567 (or A-804598) on ALS progression in SOD1G93A mice may be due to the propensity of these antagonists to cross the blood-brain barrier. As noted above, the capacity of BBG to cross the blood-brain barrier has been recently questioned [26, 27]. Thus, at least in SOD1G93A mice, BBG can be considered a CNS-nonpenetrant P2X7 antagonist, whilst JNJ-47965567 and A-804598 remain well-established CNS-penetrant P2X7 antagonists [12, 54]. If so, this suggests that the potential therapeutic effects of BBG observed in SOD1G93A mice [23–25] may be due to blockade of systemic P2X7 rather than CNS P2X7. To reconcile why the therapeutic effects of BBG in SOD1G93A mice are not observed with either JNJ-47965567 or A-804598, which would also block systemic P2X7, it is proposed that blockade of CNS P2X7 by JNJ-47965567 or A-804598 may be detrimental in ALS and that this blockade circumvents any benefits of systemic P2X7 blockade. Consistent with this idea are observations showing that global genetic deficiency of P2X7 worsens disease progression and neural pathogenesis in SOD1G93A mice [55]; thereby suggesting that the absence of CNS P2X7 activity exacerbates disease in SOD1G93A mice despite the corresponding absence of systemic P2X7 activity.
It is also possible that the different effects between BBG and JNJ-47965567 or A-804598 on ALS progression in SOD1G93A mice may relate to the purported role of P2X7 as a phagocytic receptor within the CNS [56], which occurs independently of extracellular ATP [57]. Given that P2X7 deficiency worsens ALS in SOD1G93A mice [55], the possibility exists that microglial P2X7-mediated phagocytosis plays a protective role against ALS development as proposed for other neurological disorders [56]. As such, JNJ-47965567 or A-804598, but not BBG (due to poor CNS penetrance), may block P2X7-mediated phagocytosis in the CNS to promote ALS in SOD1G93A mice and negate any benefits of systemic P2X7 blockade in these mice. However, a caveat to this argument is that P2X7-mediated phagocytosis is not blocked by AZ10606120 [58], which docks the same non-competitive inhibitory binding site within P2X7 as JNJ-47965567 and A-804598 [15]. In this regard, in vitro characterisation of JNJ-47965567 in J774 cells in the current study revealed that this compound impairs P2X7 in a manner consistent with non-competitive mechanism of inhibition, potentially binding this same site in mouse P2X7. P2X7-mediated phagocytosis, however, is blocked by oxidised ATP [58]; therefore, it remains to be directly shown if JNJ-47965567 and A-804598 block this process or not. Finally, it should be noted that a recent study reveals that human P2X7 displays an allosteric drug binding pocket [59] similar to that identified in panda P2X7 [15]. Collectively, this supports the concept that JNJ-47965567 is a non-competitive antagonist of mouse P2X7. Thus, based on conclusions arising from in vitro observations [15], it is likely that this drug when bound to P2X7 in vivo also prevents narrowing of the allosteric drug binding pocket and the turret-like architecture of the receptor to prevent channel opening in the presence of extracellular ATP released in inflamed environments.
It is also possible that β-CD, the vehicle used in the current study, altered disease progression in SOD1G93A mice masking any potential therapeutic benefits of JNJ-47965567. The mean survival time of vehicle-treated SOD1G93A mice in the current study (163 days) was longer than that in our previous study (141 days) in which saline was used as the vehicle [25]. Although this difference may simply reflect experimental differences between these two studies, β-CD has a variety of cellular effects on the endothelium of the blood brain barrier and within the CNS [60]. Most notably, β-CD reduces amyloid plaque formation and microgliosis in a murine model of Alzheimer’s disease [61], as well as Tau pathology, microgliosis and astrogliosis in a murine model of Niemann-Pick type C disease [62]. Based on these and other observations, β-CD has been proposed as an active pharmaceutical agent in neurological disorders [60]. Thus, future studies planning the use of β-CD in SOD1G93A mice or other ALS mouse models should include a saline (or other suitable) control group to determine if β-CD alone alters disease progression and if this compound has therapeutic efficacy in ALS.
Despite the absence of significant clinical benefits of JNJ-47965567 in SOD1G93A mice, NanoString analysis revealed that this compound altered the gene expression profile within the spinal cords of end stage mice with a large number of genes upregulated and a smaller number of genes downregulated by JNJ-47965567. Of these changes, Nqo1 showed the greatest fold change (reduction) in gene expression; however, qPCR analysis did not validate this finding. This may reflect a type II error within the NanoString analysis or methodological differences between the two assays. Spinal cord P2rx7 mRNA expression was also similar in both treatment groups in the current study consistent with similar expression of spinal cord P2X7 protein in SOD1G93A mice treated with saline or BBG [24, 25].
A counterargument to the role of systemic P2X7 in promoting disease progression in SOD1G93A mice proposed above is the absence of any significant impact by JNJ-47965567 on molecular or cellular parameters within the periphery of these mice. Although this lack of difference may simply reflect analysis of tissues from mice at end stage disease (rather than earlier time points), it could be argued that JNJ-47965567 should have increased the proportion of Treg cells, given the reported increases in these cells following P2X7 blockade in murine models of muscular dystrophy [63] and renal injury [64]. Study of Treg cells, as well as other molecular or cellular parameters, within the periphery of SOD1G93A mice at earlier time points may reconcile these findings.
Finally, it should be highlighted that the circulating concentrations of IL-27, IFNβ and IL-10 were approximately one log higher than other cytokines detected in the serum of SOD1G93A mice. Thus, there is possibility that these cytokines may play important roles in ALS progression and be potential biomarkers of ALS progression. However, in people with ALS, meta-analyses of 14 cytokines from various studies revealed that serum IL-10 concentrations are similar between ALS patients and controls [42]. To the best of our knowledge, serum concentrations of IL-27 and IFNβ in ALS patients have not been reported. Moreover, analysis of 16 cytokines in the serum of SOD1G93A mice revealed higher concentrations of each cytokine in asymptomatic (day 40) SOD1G93A mice compared to age-matched control mice [65]. However, serum concentrations of IL-10 did not correlate with survival time [65], whilst serum IL-27 and IFNβ were again not assessed. Thus, it appears that serum IL-10 is unlikely to be a biomarker in people or mice with ALS, but further studies of serum IL-27 and IFNβ in ALS are warranted. It should also be noted that circulating concentrations of IL-1β in SOD1G93A mice did not differ between the two treatment groups. As discussed above for Treg cells and given the well-established role of P2X7 activation in IL-1β release [10], it could be argued that JNJ-47965567 should have decreased circulating concentrations of this cytokine. Again, this lack of difference may reflect sampling of serum from mice at end stage disease and suggests that serum IL-1β analyses at earlier time points may address this point.
In conclusion, the current study demonstrated that the CNS-penetrant P2X7 antagonist, JNJ-47965567, administered thrice weekly from disease onset does not alter disease progression in SOD1G93A mice. Furthermore, this drug regime had minimal impact on various molecular and cellular parameters within the CNS and periphery of these mice at end stage disease. Findings from this study will assist in the design of future pre-clinical drug trials using P2X7 antagonists in SOD1G93A mice and other animal models of ALS aimed at preventing this disease or its progression.
Electronic supplementary material
{kind=link}
JNJ-47965567 does not alter clinical disease progression in female or male SOD1G93A mice. β-CD- and JNJ-47965567-treated SOD1G93A mice (Fig. 1) were stratified according to sex (female, left panels; male, right panels) to compare (A) ALS score, (B) body weight loss (percent of pre-disease maximum), (C) motor (rotarod) coordination and (D) survival. (A-D) Results are mean ± SEM (β-CD, n = 12 females or 11 males; JNJ-47965567, n = 12 females or 12 males) with differences between groups compared using (A-C) a two-way ANOVA or (D) the log-rank (Mantel-Cox) test. (PNG 36 kb)
{kind=link}
Gating strategies used to determine proportions of leukocyte subsets in SOD1G93A mice. (A) For analysis of T cell subsets, sequential flow cytometric gates were selected left to right as shown (top dot plots) and used to determine the proportion of total CD4+ and CD8+ T cells, as well as conventional CD4+ T cells (Foxp3−) and regulatory T cells (Foxp3+). The proportion of activated T cells (CD44+) in each T cell subset was then determined (bottom dot plots), with the relative CD39 expression on CD44+ T cell subsets also determined (histogram). (B) For analysis of DC subsets, single live cells were gated as for T cells (not shown) and then the proportion of conventional DCs (CD11c+MHC class IIlow cells) and migratory DCs (CD11c+MHC class IIhi cells) determined (left dot plot). The proportion of CD11b+ and CD11b− DCs amongst conventional and migratory DCs was then determined (middle dot plots). Finally, the relative CD80 expression on DC subsets was determined (histograms). (PNG 1937 kb)
(DOC 32 kb)
(XLSX 100 kb)
Acknowledgements
The authors kindly acknowledge Sarah Toole (University of Wollongong) for advice regarding adverse events in mice; Kara L. Vine (University of Wollongong) and Anindya Bhattacharya (Janssen Research and Development) for advice regarding drug formulation; and Sam R. Adhikary (University of Wollongong) for assistance with necropsy. The authors also kindly acknowledge the technical staff of the Illawarra Health and Medical Research Institute and the University of Wollongong Rodent Facility.
Funding information
This study was supported by grants from the Motor Neurone Disease Research Institute of Australia (R.S.), from the Australian National Health and Medical Research Council (NHMRC) Career Development Fellowship (APP1084144) (J.J.Y) and from the NHMRC Project Grant (APP1104295) (B.J.T). D.L. was a recipient of an Illawarra Health and Medical Research Institute Careers Development Grant. P.C., N.G and R.A.S were recipients of Australian Government Research Training Program (AGRTP) Scholarships.
Compliance with ethical standards
Conflict of interest
The authors declare they have no conflict of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the University of Wollongong at which the studies were conducted (Animal Ethics Committee protocols AE12/09 and AE16/18). This study does not contain any work with human participants performed by any of the authors.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Anjila Dongol and Peter Cuthbertson contributed equally to this work.
Contributor Information
Diane Ly, Email: dly@uow.edu.au.
Ronald Sluyter, Email: rsluyter@uow.edu.au.
References
- 1.Talbott EO, Malek AM, Lacomis D. The epidemiology of amyotrophic lateral sclerosis. Handb Clin Neurol. 2016;138:225–238. doi: 10.1016/b978-0-12-802973-2.00013-6. [DOI] [PubMed] [Google Scholar]
- 2.Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:162–172. doi: 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- 3.Jaiswal MK. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019;39:733–748. doi: 10.1002/med.21528. [DOI] [PubMed] [Google Scholar]
- 4.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 5.Gurney M, Pu HF, Chiu AY, Dal Canto M, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 6.Tan RH, Ke YD, Ittner LM, Halliday GM. ALS/FTLD: experimental models and reality. Acta Neuropathol. 2017;133:177–196. doi: 10.1007/s00401-016-1666-6. [DOI] [PubMed] [Google Scholar]
- 7.Pfohl SR, Halicek MT, Mitchell CS. Characterization of the contribution of genetic background and gender to disease progression in the SOD1 G93A mouse model of amyotrophic lateral sclerosis: a meta-analysis. J Neuromuscul Dis. 2015;2:137–150. doi: 10.3233/jnd-140068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGoldrick P, Joyce PI, Fisher EM, Greensmith L. Rodent models of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2013;1832:1421–1436. doi: 10.1016/j.bbadis.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Sluyter R. The P2X7 receptor. Adv Exp Med Biol. 2017;1051:17–53. doi: 10.1007/5584_2017_59. [DOI] [PubMed] [Google Scholar]
- 10.Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47:15–31. doi: 10.1016/j.immuni.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 11.Jimenez-Mateos EM, Smith J, Nicke A, Engel T (2018) Regulation of P2X7 receptor expression and function in the brain. Brain Res Bull 151:153–163. 10.1016/j.brainresbull.2018.12.008 [DOI] [PubMed]
- 12.Bhattacharya A, Wang Q, Ao H, Shoblock JR, Lord B, Aluisio L, Fraser I, Nepomuceno D, Neff RA, Welty N, Lovenberg TW, Bonaventure P, Wickenden AD, Letavic MA (2013) Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br J Pharmacol 170:624–640. 10.1111/bph.12314 [DOI] [PMC free article] [PubMed]
- 13.Fischer W, Franke H, Krugel U, Muller H, Dinkel K, Lord B, Letavic MA, Henshall DC, Engel T. Critical evaluation of P2X7 receptor antagonists in selected seizure models. PLoS One. 2016;11:e0156468. doi: 10.1371/journal.pone.0156468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimenez-Pacheco A, Diaz-Hernandez M, Arribas-Blazquez M, Sanz-Rodriguez A, Olivos-Ore LA, Artalejo AR, Alves M, Letavic M, Miras-Portugal MT, Conroy RM, Delanty N, Farrell MA, O'Brien DF, Bhattacharya A, Engel T, Henshall DC. Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and gliosis in experimental temporal lobe epilepsy. J Neurosci. 2016;36:5920–5932. doi: 10.1523/jneurosci.4009-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karasawa A, Kawate T (2016) Structural basis for subtype-specific inhibition of the P2X7 receptor. Elife 5. 10.7554/eLife.22153 [DOI] [PMC free article] [PubMed]
- 16.Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12. doi: 10.1186/1471-2377-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casanovas A, Hernandez S, Tarabal O, Rossello J, Esquerda JE. Strong P2X4 purinergic receptor-like immunoreactivity is selectively associated with degenerating neurons in transgenic rodent models of amyotrophic lateral sclerosis. J Comp Neurol. 2008;506:75–92. doi: 10.1002/cne.21527. [DOI] [PubMed] [Google Scholar]
- 18.D'Ambrosi N, Finocchi P, Apolloni S, Cozzolino M, Ferri A, Padovano V, Pietrini G, Carri MT, Volonte C. The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J Immunol. 2009;183:4648–4656. doi: 10.4049/jimmunol.0901212. [DOI] [PubMed] [Google Scholar]
- 19.Gandelman M, Peluffo H, Beckman JS, Cassina P, Barbeito L. Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis. J Neuroinflammation. 2010;7:33. doi: 10.1186/1742-2094-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gandelman M, Levy M, Cassina P, Barbeito L, Beckman JS. P2X7 receptor-induced death of motor neurons by a peroxynitrite/FAS-dependent pathway. J Neurochem. 2013;126:382–388. doi: 10.1111/jnc.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cieslak M, Roszek K, Wujak M. Purinergic implication in amyotrophic lateral sclerosis-from pathological mechanisms to therapeutic perspectives. Purinergic Signal. 2019;15:1–15. doi: 10.1007/s11302-018-9633-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volonte C, Apolloni S, Parisi C, Amadio S. Purinergic contribution to amyotrophic lateral sclerosis. Neuropharmacology. 2016;104:180–193. doi: 10.1016/j.neuropharm.2015.10.026. [DOI] [PubMed] [Google Scholar]
- 23.Cervetto C, Frattaroli D, Maura G, Marcoli M. Motor neuron dysfunction in a mouse model of ALS: gender-dependent effect of P2X7 antagonism. Toxicology. 2013;311:69–77. doi: 10.1016/j.tox.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Apolloni S, Amadio S, Parisi C, Matteucci A, Potenza RL, Armida M, Popoli P, D'Ambrosi N, Volonte C. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis Model Mech. 2014;7:1101–1109. doi: 10.1242/dmm.017038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartlett R, Sluyter V, Watson D, Sluyter R, Yerbury JJ. P2X7 antagonism using brilliant blue G reduces body weight loss and prolongs survival in female SOD1G93A amyotrophic lateral sclerosis mice. PeerJ. 2017;5:e3064. doi: 10.7717/peerj.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sluyter R, Bartlett R, Ly D, Yerbury JJ. P2X7 receptor antagonism in amyotrophic lateral sclerosis. Neural Regen Res. 2017;12:749–750. doi: 10.4103/1673-5374.206643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhattacharya A, Biber K. The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia. 2016;64:1772–1787. doi: 10.1002/glia.23001. [DOI] [PubMed] [Google Scholar]
- 28.Sluyter R, Vine KL. N-alkyl-substituted Isatins enhance P2X7 receptor-induced interleukin-1beta release from murine macrophages. Mediat Inflamm. 2016;2016:2097219. doi: 10.1155/2016/2097219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLeod VM, Lau CL, Chiam MDF, Rupasinghe TW, Roessner U, Djouma E, Boon WC, Turner BJ. Androgen receptor antagonism accelerates disease onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Br J Pharmacol. 2019;176:2111–2130. doi: 10.1111/bph.14657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, Bostrom A, Theodoss J, Al-Nakhala BM, Vieira FG, Ramasubbu J, Heywood JA. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9:4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- 31.Ludolph AC, Bendotti C, Blaugrund E, Chio A, Greensmith L, Loeffler JP, Mead R, Niessen HG, Petri S, Pradat PF, Robberecht W, Ruegg M, Schwalenstocker B, Stiller D, van den Berg L, Vieira F, von Horsten S. Guidelines for preclinical animal research in ALS/MND: a consensus meeting. Amyotroph Lateral Scler. 2010;11:38–45. doi: 10.3109/17482960903545334. [DOI] [PubMed] [Google Scholar]
- 32.Hatzipetros T, Kidd JD, Moreno AJ, Thompson K, Gill A, Vieira FG (2015) A quick phenotypic neurological scoring system for evaluating disease progression in the SOD1-G93A mouse model of ALS. J Vis Exp. 10.3791/53257 [DOI] [PMC free article] [PubMed]
- 33.Geraghty NJ, Mansfield KJ, Fuller SJ, Watson D, Sluyter R. The P2X7 receptor is not essential for development of imiquimod-induced psoriasis-like inflammation in mice. Purinergic Signal. 2017;13:405–415. doi: 10.1007/s11302-017-9569-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon H, Walters G, Paulsen AR, Scarisbrick IA. Astrocyte heterogeneity across the brain and spinal cord occurs developmentally, in adulthood and in response to demyelination. PLoS One. 2017;12:e0180697. doi: 10.1371/journal.pone.0180697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gustafson MP, Staff NP. Bornschlegl S, Butler GW, Maas ML, Kazamel M, Zubair A, Gastineau DA, Windebank AJ, Dietz AB. Comprehensive immune profiling reveals substantial immune system alterations in a subset of patients with amyotrophic lateral sclerosis. PLoS One. 2017;12:e0182002. doi: 10.1371/journal.pone.0182002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murdock BJ, Zhou T, Kashlan SR, Little RJ, Goutman SA, Feldman EL. Correlation of peripheral immunity with rapid amyotrophic lateral sclerosis progression. JAMA Neurol. 2017;74:1446–1454. doi: 10.1001/jamaneurol.2017.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, Zhao W, Moore DH, Powell SZ, Appel SH. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5:64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheean RK, McKay FC, Cretney E, Bye CR, Perera ND, Tomas D, Weston RA, Scheller KJ, Djouma E, Menon P, Schibeci SD, Marmash N, Yerbury JJ, Nutt SL, Booth DR, Stewart GJ, Kiernan MC, Vucic S, Turner BJ. Association of Regulatory T-cell expansion with progression of amyotrophic lateral sclerosis: a study of humans and a transgenic mouse model. JAMA Neurol. 2018;75:681–689. doi: 10.1001/jamaneurol.2018.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Purvis HA, Anderson AE, Young DA, Isaacs JD, Hilkens CM. A negative feedback loop mediated by STAT3 limits human Th17 responses. J Immunol. 2014;193:1142–1150. doi: 10.4049/jimmunol.1302467. [DOI] [PubMed] [Google Scholar]
- 41.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Y, Cao C, Qin XY, Yu Y, Yuan J, Zhao Y, Cheng Y. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: a meta-analysis study. Sci Rep. 2017;7:9094. doi: 10.1038/s41598-017-09097-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeyachandran A, Mertens B, McKissick EA, Mitchell CS. Type I Vs. type II cytokine levels as a function of SOD1 G93A mouse amyotrophic lateral sclerosis disease progression. Front Cell Neurosci. 2015;9:462. doi: 10.3389/fncel.2015.00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fabbrizio P, Amadio S, Apolloni S, Volonte C. P2X7 receptor activation modulates autophagy in SOD1-G93A mouse microglia. Front Cell Neurosci. 2017;11:249. doi: 10.3389/fncel.2017.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seyffert C, Schmalzing G, Markwardt F. Dissecting individual current components of co-expressed human P2X1 and P2X7 receptors. Curr Top Med Chem. 2004;4:1719–1730. doi: 10.2174/1568026043387160. [DOI] [PubMed] [Google Scholar]
- 46.Bo X, Jiang LH, Wilson HL, Kim M, Burnstock G, Surprenant A, North RA. Pharmacological and biophysical properties of the human P2X5 receptor. Mol Pharmacol. 2003;63:1407–1416. doi: 10.1124/mol.63.6.1407. [DOI] [PubMed] [Google Scholar]
- 47.Jo S, Bean BP. Inhibition of neuronal voltage-gated sodium channels by brilliant blue G. Mol Pharmacol. 2011;80:247–257. doi: 10.1124/mol.110.070276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiu F, Dahl G. A permeant regulating its permeation pore: inhibition of pannexin 1 channels by ATP. Am J Phys Cell Physiol. 2009;296:C250–C255. doi: 10.1152/ajpcell.00433.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwamaru Y, Takenouchi T, Murayama Y, Okada H, Imamura M, Shimizu Y, Hashimoto M, Mohri S, Yokoyama T, Kitani H. Anti-prion activity of brilliant blue G. PLoS One. 2012;7:e37896. doi: 10.1371/journal.pone.0037896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O'Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR. Intercellular propagated misfolding of wild-type cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 2014;111:3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kovanyi B, Csolle C, Calovi S, Hanuska A, Kato E, Koles L, Bhattacharya A, Haller J, Sperlagh B. The role of P2X7 receptors in a rodent PCP-induced schizophrenia model. Sci Rep. 2016;6:36680. doi: 10.1038/srep36680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Alvarez N, Jimenez-Mateos EM, Engel T, Quinlan S, Reschke CR, Conroy RM, Bhattacharya A, Boylan GB, Henshall DC. Effects of P2X7 receptor antagonists on hypoxia-induced neonatal seizures in mice. Neuropharmacology. 2017;116:351–363. doi: 10.1016/j.neuropharm.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Horvath G, Otrokocsi L, Beko K, Baranyi M, Kittel A, Fritz-Ruenes PA, Sperlagh B. P2X7 receptors drive poly(I:C) induced autism-like behavior in mice. J Neurosci. 2019;39:2542–2561. doi: 10.1523/JNEUROSCI.1895-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donnelly-Roberts DL, Namovic MT, Surber B, Vaidyanathan SX, Perez-Medrano A, Wang Y, Carroll WA, Jarvis MF. [3H]A-804598 ([3H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacology. 2009;56:223–229. doi: 10.1016/j.neuropharm.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 55.Apolloni S, Amadio S, Montilli C, Volonte C, D'Ambrosi N. Ablation of P2X7 receptor exacerbates gliosis and motoneuron death in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:4102–4116. doi: 10.1093/hmg/ddt259. [DOI] [PubMed] [Google Scholar]
- 56.Gu BJ, Wiley JS. P2X7 as a scavenger receptor for innate phagocytosis in the brain. Br J Pharmacol. 2018;175:4195–4208. doi: 10.1111/bph.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gu BJ, Saunders BM, Jursik C, Wiley JS. The P2X7-nonmuscle myosin membrane complex regulates phagocytosis of nonopsonized particles and bacteria by a pathway attenuated by extracellular ATP. Blood. 2010;115:1621–1631. doi: 10.1182/blood-2009-11-251744. [DOI] [PubMed] [Google Scholar]
- 58.Ou A, Gu BJ, Wiley JS. The scavenger activity of the human P2X7 receptor differs from P2X7 pore function by insensitivity to antagonists, genetic variation and sodium concentration: relevance to inflammatory brain diseases. Biochim Biophys Acta Mol basis Dis. 2018;1864:1051–1059. doi: 10.1016/j.bbadis.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 59.Bin Dayel A, Evans RJ, Schmid R. Mapping the site of action of human P2X7 receptor antagonists AZ11645373, brilliant blue G, KN-62, Calmidazolium, and ZINC58368839 to the Intersubunit allosteric pocket. Mol Pharmacol. 2019;96:355–363. doi: 10.1124/mol.119.116715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vecsernyes M, Fenyvesi F, Bacskay I, Deli MA, Szente L, Fenyvesi E. Cyclodextrins, blood-brain barrier, and treatment of neurological diseases. Arch Med Res. 2014;45:711–729. doi: 10.1016/j.arcmed.2014.11.020. [DOI] [PubMed] [Google Scholar]
- 61.Yao J, Ho D, Calingasan NY, Pipalia NH, Lin MT, Beal MF. Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. J Exp Med. 2012;209:2501–2513. doi: 10.1084/jem.20121239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maulik M, Ghoshal B, Kim J, Wang Y, Yang J, Westaway D, Kar S. Mutant human APP exacerbates pathology in a mouse model of NPC and its reversal by a beta-cyclodextrin. Hum Mol Genet. 2012;21:4857–4875. doi: 10.1093/hmg/dds322. [DOI] [PubMed] [Google Scholar]
- 63.Gazzerro E, Baldassari S, Assereto S, Fruscione F, Pistorio A, Panicucci C, Volpi S, Perruzza L, Fiorillo C, Minetti C, Traggiai E, Grassi F, Bruno C. Enhancement of muscle T regulatory cells and improvement of muscular dystrophic process in mdx mice by blockade of extracellular ATP/P2X Axis. Am J Pathol. 2015;185:3349–3360. doi: 10.1016/j.ajpath.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 64.Koo TY, Lee JG, Yan JJ, Jang JY, Ju KD, Han M, Oh KH, Ahn C, Yang J. The P2X7 receptor antagonist, oxidized adenosine triphosphate, ameliorates renal ischemia-reperfusion injury by expansion of regulatory T cells. Kidney Int. 2017;92:415–431. doi: 10.1016/j.kint.2017.01.031. [DOI] [PubMed] [Google Scholar]
- 65.Moreno-Martinez L, de la Torre M, Toivonen JM, Zaragoza P, Garcia-Redondo A, Calvo AC, Osta R. Circulating cytokines could not be good prognostic biomarkers in a mouse model of amyotrophic lateral sclerosis. Front Immunol. 2019;10:801. doi: 10.3389/fimmu.2019.00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
JNJ-47965567 does not alter clinical disease progression in female or male SOD1G93A mice. β-CD- and JNJ-47965567-treated SOD1G93A mice (Fig. 1) were stratified according to sex (female, left panels; male, right panels) to compare (A) ALS score, (B) body weight loss (percent of pre-disease maximum), (C) motor (rotarod) coordination and (D) survival. (A-D) Results are mean ± SEM (β-CD, n = 12 females or 11 males; JNJ-47965567, n = 12 females or 12 males) with differences between groups compared using (A-C) a two-way ANOVA or (D) the log-rank (Mantel-Cox) test. (PNG 36 kb)
Gating strategies used to determine proportions of leukocyte subsets in SOD1G93A mice. (A) For analysis of T cell subsets, sequential flow cytometric gates were selected left to right as shown (top dot plots) and used to determine the proportion of total CD4+ and CD8+ T cells, as well as conventional CD4+ T cells (Foxp3−) and regulatory T cells (Foxp3+). The proportion of activated T cells (CD44+) in each T cell subset was then determined (bottom dot plots), with the relative CD39 expression on CD44+ T cell subsets also determined (histogram). (B) For analysis of DC subsets, single live cells were gated as for T cells (not shown) and then the proportion of conventional DCs (CD11c+MHC class IIlow cells) and migratory DCs (CD11c+MHC class IIhi cells) determined (left dot plot). The proportion of CD11b+ and CD11b− DCs amongst conventional and migratory DCs was then determined (middle dot plots). Finally, the relative CD80 expression on DC subsets was determined (histograms). (PNG 1937 kb)
(DOC 32 kb)
(XLSX 100 kb)