

Abstract
Background
The worldwide epidemic of severe acute respiratory syndrome (SARS) in 2003 was caused by a novel coronavirus called SARS‐CoV. We report the use of DNAzyme (catalytic DNA) to target the 5′‐untranslated region (5′UTR) of a highly conserved fragment in the SARS genome as an approach to suppression of SARS‐CoV replication. A mono‐DNA enzyme (Dz‐104) possessing the 10–23 catalytic motif was synthesized and tested both in vitro and in cell culture.
Materials and methods
SARS‐CoV total RNA was isolated, extracted from the SARS‐CoV‐WHU strain and converted into cDNA. We designed a RNA‐cleaving 10–23 DNAzyme targeting at the loop region of the 5′UTR of SARS‐CoV. The designed DNAzyme, Dz‐104, and its mutant version, Dz‐104 (mut), as a control consist of 9 + 9 arm sequences with a 10–23 catalytic core. In vitro cleavage was performed using an in vitro transcribed 5′UTR RNA substrate. A vector containing a fused 5′UTR and enhanced green fluorescent protein (eGFP) was co‐transfected with the DNAzyme into E6 cells and the cells expressing eGFP were visualized with fluorescence microscopy and analyzed by fluorescence‐activated cell sorting (FACS).
Results and conclusions
Our results demonstrated that this DNAzyme could efficiently cleave the SARS‐CoV RNA substrate in vitro and inhibit the expression of the SARS‐CoV 5′UTR‐eGFP fusion RNA in mammalian cells. This work presents a model system to rapidly screen effective DNAzymes targeting SARS and provides a basis for potential therapeutic use of DNA enzymes to combat the SARS infection. Copyright © 2007 John Wiley & Sons, Ltd.
Keywords: DNAzyme, inhibition, SARS‐CoV, 5′UTR
Introduction
Severe acute respiratory syndrome (SARS) is a life‐threatening form of pneumonia. In the course of a few months, a total of 8422 probable cases of this highly infectious disease had been reported to the WHO by August 2003 1 including 916 deaths. The disease has been etiologically linked to a novel coronavirus named the SARS‐associated coronavirus (SARS‐CoV) 2. Genome sequences of SARS‐CoV isolates obtained from a number of index patients have been published and have provided important information on the phylogeny and variability 3, 4. The viral genome has the characteristics of a linear and positive single‐stranded RNA virus about 29.7 kb in length, containing 14 identifiable open reading frames (ORFs) and short untranslated regions at both termini 5.
There are currently no approved antiviral drugs that are highly effective against coronaviruses. Strategies exemplified by the use of antisense RNA or deoxyoligonucleotides 6 were reported to inhibit gene expression by interfering with RNA transcription or translation. Unfortunately, the administration of large amounts of antisense phosphorothioate oligonucleotides has led to a toxicity problem 7.
Recently, several RNA‐cleaving DNAzymes have been shown to be active in cleaving their substrate RNA under simulated physiological conditions. Two representative catalytic motifs, 10–23 and 8–17 (Figure 1), were reported by Santoro and Joyce through in vitro selection from a combinatorial library of DNA sequences 8. The most efficient DNAzyme, termed ‘10–23’, consists of a conserved catalytic core of 15 nucleotides, flanked by two variable hybridizing arms that can theoretically be designed to be complementary to any target RNA. It can cleave between any unpaired purine and a paired pyrimidine residue, which endow them with tremendous potential for applications in down‐regulation of targeted genes. To date, a number of groups have explored the potential of the 10–23 DNAzyme in biological systems. Banerjea and co‐workers have attempted to inhibit HIV‐1 gene expression by DNAzymes targeted at the HIV‐1 tar, gag, tat/rev RNA, and coreceptor CCR5 9, 10, 11, 12. Sun and co‐workers targeted the viral sequences from the HPV16 E6 and E7 genes 13 and the c‐myc gene in smooth muscle cells 14 by DNAzymes. The biological activity of DNAzymes that cleave RNA derived from the bcr‐abl, Egr‐1, huntingtin, the env gene of HIV and PB2 mRNA of influenza virus A have also been examined 16, 17, 18, 19, 20. All the reported work demonstrated that DNAzymes can be used as a potential tool to specifically inhibit the disease‐related genes.
Figure 1.
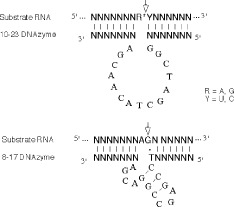
Composition of the 8–17 and 10–23 catalytic motifs. The DNAzyme (lower strand) binds the RNA substrate (top strand) through Watson–Crick pairing. Cleavage occurs at the position indicated by the arrow. R = A or G; Y = U or C
In this study, we explored the potential use of DNAzymes targeting the SARS 5′UTR fused with the green fluorescent protein (GFP) gene. The DNAzyme designed for this purpose was found to be effective both in vitro and in vivo, suggesting that the DNAzymes could be as a new class of anti‐SARS agent.
Materials and methods
Cells and virus
The African green monkey kidney (Vero E6) cells were grown and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) (Gibco Invitrogen Corporation) at 37 °C. The SARS‐CoV‐WHU strain (Accession No. AY293 850) was maintained and propagated in Vero E6 cells as described 21.
Construction of a SARS 5′UTR‐eGFP fusion vector
SARS‐CoV total RNA was isolated and extracted from the SARS‐CoV‐WHU strain. The RNA was converted into cDNA by reverse transcription. The following primers were used to amplify a 264 bp fragment of the 5′UTR of the SARS genome by polymerase chain reaction (PCR): pUTR1, 5′AGAATTCTAGGTTTTTACCTACCCAGG‐ 3′ (EcoRI digestion site underlined); pUTR2, 5′AGGATCC CTTACCTTTCGGTCACACC‐3′ (BamHI digestion site underlined).
The conditions for PCR were 94 °C, 2 min; (94 °C, 40 s; 50 °C, 40 s; 72 °C, 2 min) for 30 cycles; followed by 72 °C, 10 min. The amplified product was analyzed by electrophoresis, cloned into pMD18‐T PCR cloning vector, then subcloned into the multiple cloning site (MCS) between EcoR1 and BamH1 in peGFP‐N1 vector (BD Biosciences Clonetech), and then transformed into Escherichia coli strain DH5a. The sequence integrity of the construct was verified by restriction mapping and DNA sequencing.
DNA enzyme design
Based on Santoro and Joyce's report 8, we designed a RNA‐cleaving DNAzyme possessing the 10–23 motif. The target site was selected in the loop region of the predicted secondary structure of the 5′UTR. The designed DNAzyme, Dz‐104, consists of 9 + 9 arm sequences with a 10–23 catalytic core, and was synthesized using standard chemistry (Sangon, Shanghai, China). The details of the target sequences, the DNAzyme (Dz‐104) and the mutant control (Dz‐104 (mt)) are shown in Figure 2. As indicated, the A nucleotide of the target gene was left unpaired and cleavage is expected to take place after the A. The mutant DNA enzyme (Dz‐104 (mt)) possessed a single mutation in the catalytic motif (G to C).
Figure 2.

DNA enzyme target sites and two designed DNA enzymes. The target sequence is shown together with (A) active DNAzyme (Dz‐104) and (B) the mutated control (Dz‐104 (mut))
In vitro synthesis of substrate RNA
To generate the substrate RNA for testing DNAzyme cleavage activity in vitro, the SARS‐CoV 5′UTR was produced by PCR from the recombinant plasmid p5′UTR‐eGFP with a forward primer containing a T7 promoter sequence (5′CTGTAATACGACTCACTATAGGGCCAAGTACGCCCC‐ CTATTGA and a reverse primer (5′GTAGTTGCCGTCGT‐ CCTTGA). The amplified fragment is 1000 bp in length and the predicted cleavage site is in the 5′UTR of SARS at the midpoint of the fragment.
The conditions for PCR were 94 °C, 2 min; (94 °C, 40 s; 50 °C, 40 s; 72 °C, 5 min) for 30 cycles; followed by 72 °C, 15 min. The amplified product was analyzed by electrophoresis and then recovered from the agarose gel. Then 1 µg purified DNA template was used for in vitro transcription with a T7 transcription kit (JINGMEI BIOTECH, Beijing, China) in a volume of 20 µl as recommended by the manufacturer and incubated for 2 h at 37 °C. Upon completion of the transcription, the DNA template was removed by digestion with DNase at 1 U DNase/1 µg template DNA (TaKaRa Biotechnology, Dalian, China) for 15 min at 37 °C. The transcript RNA was further purified with TE‐saturated phenol/chloroform and chloroform/isoamyl alcohol (24 : 1).
In vitro cleavage reaction and kinetic analysis
In cleavage reactions, the purified substrate RNA (100 nM) and the DNAzyme (various concentrations) were mixed in 10 µl of 50 mM Tris‐HCl, pH 7.5, in the presence of 10 mM MgCl2 and 1 U RNasin to prevent non‐specific RNA degradation. Prior to mixing enzyme and target RNA, both solutions were denatured separately for 2 min at 85 °C. The cleavage reaction was incubated at 37 °C. For a single turnover kinetic experiment, 10‐fold excess of DNAzyme was used in the cleavage reaction (1000 nM). In a multiple kinetic reaction, 1/10 concentration of DNAzyme (10 nM) was employed and aliquots were taken after defined intervals during the first 10% of the reaction. The reaction was stopped by adding 83 mM EDTA and cooled on ice. 10× sample buffer (37% formaldehyde and 7% 5 × MOPS buffer in formamide) was added to the cleavage reactions prior to being loaded onto an agarose gel. The gel was stained with ethidium bromide and the intensities were quantified with the Quantity One software (Gene Snap). Data were further analyzed by fitting either linearly to obtain the initial velocity v init for substrate excess experiments or with a single exponential decay function to obtain the observed cleavage rate for enzyme excess experiments using Origin (Microcal Software, Northampton, MA, USA). Values given are mean ± standard deviation (SD) of at least three independent experiments.
Transfection of Vero E6 cells with p5′UTR‐eGFP and DNA enzymes
Vero E6 cells (2 × 105) were grown to 60% confluence on a 24‐well plate and were then co‐transfected with a fixed amount of p5′UTR‐eGFP (1 µg) along with varying amounts of DNA enzymes (0.7, 3.5, 7 µg, respectively) for 12 h using Lipofectamine Plus (Invitrogen, USA) in a final volume of 500 µl. After 4 h of incubation in the presence of Lipofectamine Plus, the cells were washed with DMEM without serum and incubated for a further 12 h in the same medium with 10% FBS. Then, the cells expressing eGFP were visualized with fluorescence microscopy (Nikon, Tokyo, Japan).
Fluorescence‐activated cell sorting (FACS) analysis of the transfected Vero cells
The transfected cells were trypsinized and collected by centrifugation. The cells were further rinsed with phosphate‐buffered saline (PBS), re‐suspended in 100 µl 2% polyformaldehyde and kept at 4 °C; then 900 µl PBS were added to each sample. The cells were then analyzed on Beckman Coulter counter for the percentage of the cells expressing GFP.
Real‐time PCR assay for quantification of the GFP mRNA expression
At 24 h post‐transfection, the cells were collected for total RNA extraction using Trizol according to the manufacturer's instruction. Real‐time PCR reactions were carried out in ICycler iQ (BioRad). The primers used in the PCR were: GFP1, 5′CAAGCTGACC CTGAAGTTCA 3′ and GFP2, 5′ATGCGGTTCACCAGGGTGT3′, which gave rise to a 250 bp product of the GFP gene. The standard curve was generated using a serial of 10‐fold dilution of pEGFP‐N1 or p5′UTR‐EGFP (1010, 109, 108, 107, 106, 105copies). The amplification condition was: room temperature (RT), 70 °C for 5 min; 1 × 94 °C for 2 min; 45 × 94 °C for 45 s, 60 °C for 45 s, and 72 °C for 30 s. The mean Ct values were collected and used for analysis.
Results and discussion
Target site selection and DNAzyme design
SARS‐CoV belongs to the coronavirus family with a structure similar to the other coronaviruses, which contain S, M, E and N gene products. The virus features a discontinuous transcription of subgenomic mRNAs, regulated by the pre‐transcribed leader sequence and the specific TRSs in the 5′UTR of the genome. The 5′UTR sequence contributes to the regulation of the virus gene expression. Most strikingly, there is a high degree of sequence conservation in the 5′UTR region among all the SARS‐CoV strains identified thus far, which suggests the functional importance of the region. Targeting the 5′UTR would therefore not only present a feasible strategy, but also potentially avoid the possible emergency of escape mutant viruses.
A 10–23 DNAzyme was designed (Dz‐104) with a cleavage site at nucleotide 104. The arm length was chosen as a 9 + 9 format as previously reported, which gave rise to a much more efficient catalytic activity based on a balance of the hybridization strength (−ΔG) and kinetic turnover 14. The cleavage site was selected at an AU dinucleotide since a hierarchy of RY reactivity (where R is a purine and Y is a pyrimidine) to the 10–23 DNAzyme follows the general scheme AU = GU ≥ GC ≫ AC 15.
Dz‐104 cleavage of SARS‐CoV 5′UTR RNA
To test the cleavage activity of Dz‐104, we conducted in vitro cleavage analyses using a 1 kb RNA transcript that was transcribed from a T7‐containing primer‐based PCR fragment of the 5′UTR. The cleavage point was in the middle of the transcript; thus the two bands of the cleavage product would appear at the same position in a gel. In a single turnover assay where 10‐fold access of Dz‐104 was used, the substrate RNA was efficiently cleaved with the expected size of the cleavage products (Figure 3A) and the observed rate was 0.064 min−1 (Figure 3B). Under a multiple turnover condition, the substrate was cleaved with an initial velocity at 0.4 nM min−1 (Figure 3C). When the mutant DNAzyme (Dz‐104 (mt)) was subjected to the cleavage assay under the same conditions, it did not show any cleavage (Figure 3A, lower panel). These results demonstrated that the designed DNAzyme was chemically active in cleaving its substrate RNA with a capacity to turnover under the in vitro conditions. It also suggested that the local RNA structures where the Dz‐104 targeted are accessible under the simulated physiological conditions.
Figure 3.

In vitro cleavage of SARS‐CoV 5′UTR by DNAzyme‐104. (A) Gel analysis of the cleaved product with Dz‐104 (upper panel) and Dz‐104 (mut) (lower panel). S, substate RNA; P, cleavage products. (B) Kinetic profile of DNAzyme‐104 under single turnover conditions with a 10‐fold enzyme excess. (C) Kinetic profile of DNAzyme‐104 under multiple turnover conditions with a 10‐fold substrate excess. Y axis represents the % of cleaved product
Establishment of a model system for DNAzyme targeting in cells
To examine the efficacy of the designed DNAzymes, it is crucial to have a sensitive and meaningful system to perform the assay. We chose to use a fusion transcript of the 5′UTR and GFP gene as a model system. In this system, the DNAzyme target was linked to the 5′ end of the GFP gene, which provided a versatile assay to monitor DNAzyme activity in cells (Figure 4). When the DNAzyme cleavage occurred, it was anticipated that the truncated fusion RNA would be subjected to the nuclease degradation; therefore, the level of the GFP expression would be reduced, which could be monitored by fluorescence microscopy.
Figure 4.

Fusion construct of SARS‐CoV 5′UTR and eGFP. The construct is based on the peGFP‐N1 vector with insertion of the 5′UTR of SARS at EcoR1 and BamH1 sites at the 5′ end of the eGFP gene. The resultant construct is designated as p5′UTR‐eGFP
When the p5′UTR‐eGFP construct was transfected into Vero E6 cells, the expression of the eGFP gene could be easily observed, but appeared to be a slightly weaker than the parental vector peGFP‐N1 (Figure 5). This minor change could be due to the effect of the extra 5′UTR sequences on the promoter activity or transcription efficiency of the GFP gene. However, the system should be sensitive enough to be used for assays of the DNAzyme activity.
Figure 5.

Dz‐104‐mediated reduction of GFP expression. Dz‐104 or Dz‐104 (mut) was co‐transfected with 1 µg of peGFP‐N1 (left panel) or p5′UTR‐eGFP (right panel) into Vero cells. Various concentrations of the DNAzyme or its control are shown as 0.7, 3.5 and 7 µg. The experiments were performed twice and one set of representative data are presented here (100×)
Effect of Dz‐104‐mediated cleavage of 5′UTR‐eGFP fusion RNA on eGFP expression in Vero E6 cells
The advantage of using GFP is that it facilities the direct observation of the target gene expression by fluorescence microscopy. To demonstrate that the designed DNAzyme could target the mRNA with the SARS 5′UTR and suppress the eGFP expression, we co‐transfected Vero E6 cells with p5′UTR‐eGFP and Dz‐104 at different ratios. The data showed that co‐transfection of p5′UTR‐EGFP/Dz‐104, compared to the Dz‐104 (mt) control, significantly decreased the fluorescent level, while it had no effect on the GFP in the cells co‐transfected with pEGFP‐N1 that contained no 5′UTR sequence (Figure 5). The effect of Dz‐104 on eGFP expression was shown to be dose‐dependent and the transfection procedure had no effect on cell growth (data not shown).
To confirm the DNAzyme effect on GFP expression, the percentage of the GFP‐expressing cells was measured using flow cytometry. The results showed that there was a dose‐dependent reduction in the number of GFP+ cells that were transfected with the p5′UTR‐eGFP and Dz‐104 (Table 1). This reduction was not seen in the cells transfected with either the parental vector pEGFP‐N1 or mutant DNAzyme Dz‐104 (mut).
Table 1.
FACS analysis of the transfected Vero cells
| Vector | Dosage (µg) | Percentage of fluorescent cells (%)* | |
|---|---|---|---|
| Dz‐104 | Dz‐104 (mt) | ||
| eGFP‐N1 | 0.0 | 69.5 | 69.5 |
| 0.7 | 68.6 | 71.6 | |
| 3.5 | 66.8 | 68.5 | |
| 7.0 | 69.3 | 67.0 | |
| 5′UTR‐eGFP | 0.0 | 57.6 | 57.6 |
| 0.7 | 25.8 | 55.6 | |
| 3.5 | 17.0 | 56.8 | |
| 7.0 | 9.1 | 58.2 | |
The data were the mean of two independent experiments.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
To determine if the effect of Dz‐104 on the GFP expression was due to a cleavage of the 5′UTR‐GFP mRNA, real‐time PCR was performed to measure the level of the target RNA. It was shown that Dz‐104 treatment of the p5′UTR‐eGFP‐transfected cells led to a marked reduction in the target mRNA level, as evidenced in Table 2. This demonstrates that DNAzyme elicits the degradation of the 5′UTR‐EGFP mRNA sequence in a sequence‐specific manner, resulting in suppression of the eGFP gene expression.
Table 2.
GFP mRNA expression determined by real‐time PCR
| Vector | Dosage (µg) | Ct value | |
|---|---|---|---|
| Dz‐104 | Dz‐104 (mt) | ||
| eGFP‐N1 | 0.0 | 11.64 | 11.64 |
| 0.7 | 13.03 | 11.53 | |
| 3.5 | 11.94 | 9.59 | |
| 7.0 | 11.60 | 11.77 | |
| 5′UTR‐GFP | 0.0 | 16.66 | 16.66 |
| 0.7 | 19.64 | 13.84 | |
| 3.5 | 22.67 | 16.01 | |
| 7.0 | 23.80 | 16.64 |
Data were an average of two independent experiments.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
In the present study, the DNAzyme was co‐transfected with the target vector into Vero cells. Although this co‐transfection did not fully mimic the cells infected with the virus, it did show that the DNAzyme could enter the cells and find the target RNA. A previous study showed that the DNAzyme transfected into mammalian cells remained stable and biologically active up to 24 h 22, which supports our results, that sufficient DNAzymes had been transfected into the cells and functioned as a target‐specific gene suppressor.
Taken together, our study shows that the DNAzyme can efficiently cleave the 5′UTR of SARS‐CoV both in vitro and in cells, implying that the 5′UTR may well be a good target for molecular targeting. The study also presents a convenient system for screening other nucleic‐acid‐based agents for inhibition of SARS replication. Since DNAzymes have inherent physicochemical advantages over other nucleic‐acid‐based agents, such as higher target flexibility, minimal chemical modification, relative resistance to nuclease degradation and low cost of synthesis, they may be a clinically appealing new class of anti‐SARS agent.
Acknowledgements
This work was supported by Project 973 from the National Frontier Research Program of the Ministry of Science and Technology of the People's Republic of China (Grant No. 2005CB523001) applied by MVRC, IM, CAS. Most of the research was carried out at the P‐3 lab of MVRC, WHU. Therefore, MVRC, IM, CAS and MVRC, WHU share the first institution.
Contributor Information
Lunquan Sun, Email: lunqsun@optusnet.com.au.
Po Tien, Email: tienpo@sun.im.ac.cn.
References
- 1.Available: http://www.who.int/csr/sars/en/.
- 2. Peiris J, Lai S, Poon L, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003; 361: 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marra MA, Jones SJ, Astell CR, et al. The genome sequence of the SARS‐associated coronavirus. Science 2003; 300: 1399–1404. [DOI] [PubMed] [Google Scholar]
- 4. Ruan YJ, Wei CL, Ee AL, et al. Comparative full‐length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet 2003; 361: 1779–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thiel V, Ivanov KA, Putics A, et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol 2003; 84: 2305–2315. [DOI] [PubMed] [Google Scholar]
- 6. Milligan JF, Matteucci MD, Martin JC. Current concepts in antisense drug design. J Med Chem 1993; 36: 1923–1937. [DOI] [PubMed] [Google Scholar]
- 7. Akhtar S, Agrawal S. In vivo studies with antisense oligonucleotides. Trends Pharmaceut Sci 1997; 18: 12–18. [DOI] [PubMed] [Google Scholar]
- 8. Santoro SW, Joyce GF. A general purpose RNA‐cleaving DNA enzyme. Proc Natl Acad Sci U S A 1997; 94: 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chakraborti S, Banerjea AC. Inhibition of HIV‐1 gene expression by novel DNA enzymes targeted to cleave HIV‐1 TAR RNA: potential effectiveness against all HIV‐1 isolates. Mol Ther 2003; 6: 817–826. [DOI] [PubMed] [Google Scholar]
- 10. Goila R, Banerjea AC. Sequence specific cleavage of the HIV‐1 coreceptor CCR5 gene by a hammer‐head ribozyme and a DNA‐enzyme: inhibition of the coreceptor function by DNA‐enzyme. FEBS Lett 1998; 436: 233–238. [DOI] [PubMed] [Google Scholar]
- 11. Unwalla H, Banerjea AC. Inhibition of HIV‐1 gene expression by novel macrophage‐tropic DNA enzymes targeted to cleave HIV‐1 TAT/Rev RNA. Biochem J 2001; 357: 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Unwalla H, Banerjea AC. Novel mono‐ and di‐DNA‐enzymes targeted to cleave TAT or TAT‐Rev RNA inhibit HIV‐1 gene expression. Antiviral Res 2001; 51: 127–139. [DOI] [PubMed] [Google Scholar]
- 13. Cairns MJ, Hopkins TM, Withering TC, Wang L, Sun LQ. Target site selection for an RNA‐cleaving catalytic DNA. Nat Biotechnol 1999; 17: 480–486. [DOI] [PubMed] [Google Scholar]
- 14. Sun LQ, Cairns MJ, Gerlach WL, Witherington C, Wang L, King A. Suppression of smooth muscle cell proliferation by a c‐myc RNA‐cleaving deoxyribozyme. J Biol Chem 1999; 274: 17236–17241. [DOI] [PubMed] [Google Scholar]
- 15. Cairns MJ, King AK, Sun LQ. Optimization of the 10–23 DNAzyme‐substrate paring interactions enhanced RNA cleavage activity at purine–cytosine target sites. Nucleic Acid Res 2003; 31: 2883–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dash BC, Harikrishnan TA, Goila R, et al. Targeted cleavage of HIV‐1 envelope gene by a DNA enzyme and inhibition of HIV‐1 envelope‐CD4 mediated cell fusion. FEBS Lett 1998; 431: 395–399. [DOI] [PubMed] [Google Scholar]
- 17. Santiago FS, Kavurma MM, Lowe HC, et al. New DNA enzyme targeting Egr‐1 mRNA inhibits vascular smooth muscle proliferation and regrowth after injury. Nat Med 1999; 5: 1264–1269. [DOI] [PubMed] [Google Scholar]
- 18. Toyoda T, Imamura Y, Takaku H. Inhibition of influenza virus replication in cultured cells by RNA‐cleaving DNA enzyme. FEBS Lett 1999; 458: 151–156. [DOI] [PubMed] [Google Scholar]
- 19. Warashina M, Kuwabara T, Nakamatsu Y, Taira K. Extremely high and specific activity of DNA enzymes in cells with a Philadelphia chromosome. Chem Biol 1999; 6: 237–250. [DOI] [PubMed] [Google Scholar]
- 20. Yen L, Strittmatter SM, Kalb RG. Sequence‐specific cleavage of Huntington mRNA by catalytic DNA. Ann Neurol 1999; 46: 366–373. [DOI] [PubMed] [Google Scholar]
- 21. Zhu Y, Liu M, Zhao W, et al. Isolation of virus from a SARS patient and genome‐wide analysis of genetic mutations related to pathogenesis and epidemiology from 47 SARS‐CoV isolates. Virus Genes 2005; 30: 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dass CR, Saravolac EG, Li Y, Sun LQ. Cellular uptake, distribution, and stability of 10–23 deoxyribozymes. Antisense Res Dev 2002; 12: 289–299. [DOI] [PubMed] [Google Scholar]