

Abstract
There is a worldwide concern about the new coronavirus 2019‐nCoV as a global public health threat. In this article, we provide a preliminary evolutionary and molecular epidemiological analysis of this new virus. A phylogenetic tree has been built using the 15 available whole genome sequences of 2019‐nCoV, 12 whole genome sequences of 2019‐nCoV, and 12 highly similar whole genome sequences available in gene bank (five from the severe acute respiratory syndrome, two from Middle East respiratory syndrome, and five from bat SARS‐like coronavirus). Fast unconstrained Bayesian approximation analysis shows that the nucleocapsid and the spike glycoprotein have some sites under positive pressure, whereas homology modeling revealed some molecular and structural differences between the viruses. The phylogenetic tree showed that 2019‐nCoV significantly clustered with bat SARS‐like coronavirus sequence isolated in 2015, whereas structural analysis revealed mutation in Spike Glycoprotein and nucleocapsid protein. From these results, the new 2019‐nCoV is distinct from SARS virus, probably trasmitted from bats after mutation conferring ability to infect humans.
Keywords: coronavirus, epidemiology, macromolecular design, SARS coronavirus
Highlights
An epidemic by a new Coronavirus, the 2019‐nCoV is worrying worldwide.
The epdiemic originated in Wuhan, China causing severe infections in some cases.
The infection probably originated in bats.
A mutation in the viral spike glycoprotein could have favured the cross‐sepcies tramsnimssion from bats to human.
1. INTRODUCTION
The family Coronaviridae comprises a group of large, single, plus‐stranded RNA viruses isolated from several species, and it is previously known to cause the common cold and diarrheal illnesses in humans. 1 , 2 In 2003, a new coronavirus (severe acute respiratory syndrome coronavirus [SARS‐CoV]) was associated with the SARS outbreak. 1 , 2 Recently, a new coronavirus (2019‐nCoV) has emerged in the region of Wuhan (China) as a cause of severe respiratory infection in humans. Since December 2019, different cases of pneumonia of unknown origin associated with permanence at the Wuhan market in China have been reported. 3 , 4 A new coronavirus, named 2019‐nCoV, belonging to the Orthocoronavirinae subfamily, distinct from MERS‐CoV and SARS‐CoV, was described. 5 To date, a total of 1975 pneumonia cases have been confirmed in China (the State Council Information Office in Beijing, capital of China, 26 January 2020). 6 , 7 Animal to human transmission is considered the origin of epidemics, as many patients declared to have visited a local fish and wild animal market in Wuhan in November. Quite recently, evidence has been gathered for the animal to the human and interhuman transmission of the virus. 7 , 8
Although prompt diagnosis and patient isolation are the hallmarks for initial control of this new epidemic, molecular epidemiology, evolutionary models, and phylogenetic analysis can help estimate genetic variability and the evolutionary rate, which in turn have important implications for disease progression as well as for drug and vaccine development. In this short report, we provide a phylogenetic tree of the 2019‐nCoV and identify sites of positive or negative selection pressure in distinct regions of the virus.
2. MATERIAL AND METHODS
The complete genomes of 15 2019‐nCoV sequences have been downloaded from GISAID (https://www.gisaid.org/) and GenBank (http://www.ncbi.nlm.nih.gov/genbank/). A dataset has been built using five highly similar sequences for SARS, two sequences for the Middle East respiratory syndrome (MERS), and five highly similar sequences for bat SARS‐like coronavirus. The percentage of similarity has been identified using a basic local alignment search tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi); eventually duplicated sequences have been excluded from the datasets. The dataset including 27 sequences has been aligned using multiple sequence alignment online tool 9 and manually edited using BioEdit program v7.0.5. 10
Maximum likelihood (ML) methods were employed for the analyses because they allow for testing different phylogenetic hypotheses by calculating the probability of a given model of evolution generating the observed data and by comparing the probabilities of nested models by the likelihood ratio test. The best‐fitting nucleotide substitution model was chosen by jModeltest software. 11 ML tree was reconstructed using generalized time‐reversible plus gamma distribution and invariant sites (+G+I) as an evolutionary model using MEGA‐X. 12
The adaptive evolution server (http://www.datamonkey.org/) was used to find eventual sites of positive or negative selection. For this purpose, the following test has been used: fast unconstrained Bayesian approximation (FUBAR). 13 This test allowed us to infer the site‐specific pervasive selection, the episodic diversifying selection across the region of interest, and to identify episodic selection at individual sites. 14 The statistically significant positive or negative selection was based on P value less than .05. 14
Homology models have been built relying on the website SwissModel. 15 Structural templates have been searched and validated using the software available within the SwissModel environment and HHPred. 16 Homology models have been validated using the QMEAN tool. 17 Three‐dimensional structures have been analyzed and displayed using PyMOL. 18 To map the structural variability of the N, E, S, and M regions of the virus and their sites under selection pressure, homology modeling has been applied to the sequence of 2019‐nCoV.
3. RESULTS
The ML phylogenetic tree, performed on whole genome sequences, is represented in Figure 1. In the tree, MERS virus sequences formed a distinct clade (clade I) from Bat SARS‐like coronavirus, SARS virus, and the 2019‐nCoV clustering together in clade II. This clade includes two different clusters: cluster IIa with Bat SARS‐like coronavirus and the 2019‐nCoV sequences, and cluster IIb with the bat SARS‐like coronavirus and the SARS virus sequences. The 2019‐nCoV is significantly and closely related only to the specific bat SARS‐like coronavirus isolated from Rhinolophus sinicus in 2015 in China (MG772934.1) (Figure 1).
Figure 1.
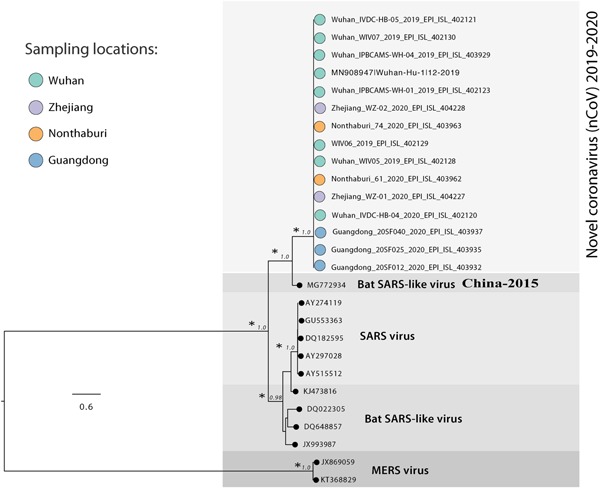
Maximum likelihood tree of 2019‐nCoV. Along the branches, an asterisk (*) indicates a statistical value from bootstrap (>97%)
Regarding the FUBAR analysis performed on the N Region, significant (P < .05) pervasive episodic selection was found in two sites (380th and 410th nucleotide position using the reference sequence; Wuhan seafood market pneumonia virus isolate labeled Wuhan‐Hu‐1 MN908947.3). On the 380th aminoacidic position in the Wuhan coronavirus sequence, there is a glutamine residue instead of an asparagine residue, whereas on the 410th aminoacidic position in Wuhan coronavirus sequence, there is a threonine residue instead of an alanine residue. A significant (P < .05) pervasive negative selection in six sites (14%) has been evidenced and confirmed by FUBAR analysis. Concerning the sequences in clade II, on the 409th aminoacidic position in the Wuhan coronavirus sequence, there is glutamine instead of an asparagine residue, whereas, on the 380th aminoacidic position in Wuhan coronavirus sequence, there is a threonine residue instead of an alanine. A significant (P < .05) pervasive negative selection in six sites (14%) has been evidenced and confirmed by FUBAR analysis.
On the S region, a significant (P < .05) pervasive episodic selection was found in two different sites (536th and 644th nucleotide position using the reference sequence; the Wuhan seafood market pneumonia virus isolate labeled Wuhan‐Hu‐1 MN908947.3). For the sequences in clade II, on the 536th aminoacidic position in the Wuhan coronavirus sequence, there is an asparagine residue instead of an aspartic acid residue, whereas, on the 644th aminoacidic position in Wuhan coronavirus sequence, there is a threonine residue instead of an alanine residue. A significant (P < .05) pervasive negative selection in 1065 sites (87%) has been evidenced and confirmed by FUBAR analysis, suggesting that the S region could be highly conserved.
No sites under positive selection sites have been found in the E and M regions.
The N region of the 2019‐nCoV homology model has been built using a SARS coronavirus nucleocapsid protein structure (2jw8.1) as the statistical test has shown that this is the most stable and similar model among the other possible structures (Ramachandran favored 90.52%, GMQE 0.17, QMEAN −2.43), whereas, for the S region, the 2019‐nCoV homology model has been built using a SARS coronavirus spike glycoprotein (6acc.1) for the same reason (Ramachandran favored 87.87%, GMQE 0.83, QMEAN −3.14) (Figure 3).
Figure 3.

Cartoon model of the structural superposition between the homology model of the 2019‐nCoV in blue and the spike glycoprotein of SARS coronavirus (PDB code 6acc.1) in orange. The red circle highlights the presence of a variable region on the 2019‐nCoV at the beginning of the protein, whereas the blue square highlights the presence of two beta‐sheets on the 2019‐nCoV (401:KYR and 440:LND) that are not present on the SARS‐CoV structure; SARS, severe acute respiratory syndrome
The further structural and molecular analysis of the Nucleocapsid region of the 2019‐nCoV (MN908947.3) has highlighted that 2019‐nCoV and the bat SARS‐like coronavirus (MG772934.) share the same aminoacidic sequence near the 309th position (SKQLQQ,) whereas the SARS reference genome has a different aminoacidic sequence (SRQLQN). The same results have been found in the 380th aminoacidic position (KADET for 2019‐nCoV and bat SARS‐like coronavirus and KTDEA for the SARS reference genome). In particular, in this case, the 2019‐nCoV has a polar amino acid, whereas the SARS has a nonpolar amino acid (Figure 2).
Figure 2.

Cartoon model of the structural superposition between the homology model of the 2019‐nCoV in blue and the nucleocapsid protein of SARS coronavirus (PDB code 2jw8.1) in orange. The red circle highlights the presence of an alpha‐helix on the SARS‐CoV and the absence of the 2019‐nCoV structure, and e positional difference of the beta‐sheets. SARS, severe acute respiratory syndrome
Regarding the sites under positive selective pressure found on the spike glycoprotein, the results have shown that the 536th aminoacidic position in 2019‐nCoV has an asparagine residue, whereas the bat SARS‐like coronavirus has a glutamine residue. The SARS virus, instead, has an aspartic acid residue. On the 644th aminoacidic position in 2019‐nCoV sequence, there is a threonine residue, whereas the bat SARS‐like virus has a serine residue. The SARS virus, however, has an alanine residue.
4. DISCUSSION
The data reported above show that the new 2019‐nCoV significantly clustered with a sequence from the bat SARS‐like coronavirus isolated in 2015. Moreover, in the phylogenetic tree, these two sequences are separated from the other bat SARS‐like coronavirus sequences, suggesting that this bat SARS‐like coronavirus is homologous and genetically more similar to the 2019‐nCoV than to the other sequences of Bat SARS‐like coronavirus. This supports the hypothesis that the transmission chain began from the bat and reached the human. All other genomic sequences represented in the phylogenetic tree, also including SARS and MERS coronavirus, clustered separately, thus excluding the fact that the virus involved in the actual epidemic could belong to these subgenuses. The structural analysis of two important viral proteins, the nucleocapsid and the spike‐like nucleoprotein (protein S), confirmed the significant similarity of the new coronavirus with the bat‐like SARS coronavirus and its difference from SARS coronavirus.
From the selective pressure and structural analysis, mutations of surface proteins, as the spike protein S, and of nucleocapsid N protein conferring stability to the viral particle have been shown. The viral spike protein is responsible for virus entry into the cell after by binding to a cell receptor and membrane fusion, two key steps in viral infection and pathogenesis. The N protein is a structural protein involved in virion assembly, playing a pivotal role in virus transcription and assembly efficiency. Mutation of these proteins could determine two important characteristics of the coronavirus isolated during the 2019‐nCoV epidemic: a higher ability to infect and enhanced pathogenicity than the bat‐like SARS coronavirus but lower pathogenicity than SARS coronavirus. These features can explain the 2019‐nCoV zoonotic transmission and its initial lower severity than SARS epidemic. These results do not exclude the fact that further mutation due to positive selective pressure, led by the epidemic evolution, could favor an enhancement of pathogenicity and transmission of this novel virus.
Recently, Ji et al 8 described homologous recombination within the spike glycoprotein of 2019‐nCoV favoring cross‐species transmission and suggested snake as probable virus reservoir for human infection because its resampling similarity codon usage bias is more similar to Bungarus multicinctus snake compared with other animals and humans. In a previous article, it has been proven that compositional properties, mutation pressure, natural selection, gene expression, and dinucleotides, affect the codon usage bias of Bungarus species. 19
These data, along with analysis from our study, enforcing bat origin of infection, could clarify the transmission dynamics of the 2019‐nCoV, supporting infection control policy during the ongoing epidemic.
Benvenuto D, Giovanetti M, Ciccozzi A, Spoto S, Angeletti S, Ciccozzi M. The 2019‐new coronavirus epidemic: Evidence for virus evolution. J Med Virol. 2020;92:455–459. 10.1002/jmv.25688
Silvia Angeletti and Massimo Ciccozzi contributed equally to this study.
REFERENCES
- 1. Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967‐1976. [DOI] [PubMed] [Google Scholar]
- 2. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020. 10.1002/jmv.25681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan JF‐W, Yuan S, Kok K‐H, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person‐to‐person transmission: a study of a family cluster. Lancet. 2020. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hui DS, Azhar EI, Madani TA, et al. The continuing 2019‐nCoV epidemic threat of novel coronaviruses to global health ‐ The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;2020(91):264‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu H, Stratton CW, Tang YW. Outbreak of pneumonia of unknown etiology in Wuhan China: the mystery and the miracle. J Med Virol. 2020. 10.1002/jmv.25678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ji W, Wang W, Zhao X, Zai J, Li X. Homologous recombination within the spike glycoprotein of the newly identified coronavirus may boost cross‐species transmission from snake to human. J Med Virol. 2020. 10.1002/jmv.25682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20(4):1160‐1166. 10.1093/bib/bbx108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hall TA. BioEditA user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95‐98. [Google Scholar]
- 11. Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547‐1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murrell B, Moola S, Mabona A, et al. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol. 2013;30(5):1196‐1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012;8(7):e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Waterhouse A, Bertoni M, Bienert S. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296‐W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zimmermann L, Stephens A, Nam SZ. A completely reimplemented MPI Bioinformatics Toolkit with a new HHpred server at its core. J Mol Biol. 2018;430(15):2237‐2243. [DOI] [PubMed] [Google Scholar]
- 17. Benkert P, Künzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37(W):W510‐W514. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schrödinger LLC . The {PyMOL} Molecular Graphics System, Version~1.8, 2015.
- 19. Chakraborty S, Nag D, Mazumder TH, Uddin A. Codon usage pattern and prediction of gene expression level in Bungarus species. Gene. 2016;604:48‐60. 10.1016/j.gene.2016.11.023 [DOI] [PubMed] [Google Scholar]