

Abstract
The Notch signaling pathway plays important roles in a variety of cellular processes. Aberrant transduction of Notch signaling contributes to many diseases and cancers in humans. The Notch receptor intracellular domain, the activated form of Notch receptor, is extremely difficult to detect in normal cells. However, it can activate signaling at very low protein concentration to elicit its biological effects. In the present study, a cell based luciferase reporter gene assay was established in K562 cells to screen drugs which could modulate the endogenous CBF1‐dependent Notch signal pathway. Using this system, we found that the luciferase activity of CBF1‐dependent reporter gene was activated by baicalin and baicalein but suppressed by niclosamide in both dose‐ and time‐dependent manners. Treatment with these drugs modulated endogenous Notch signaling and affected mRNA expression levels of Notch1 receptor and Notch target genes in K562 cells. Additionally, erythroid differentiation of K562 cells was suppressed by baicalin and baicalein yet was promoted by niclosamide. Colony‐forming ability in soft agar was decreased after treatment with baicalin and baicalein, but was not affected in the presence of niclosamide. Thus, modulation of Notch signaling after treatment with any of these three drugs may affect tumorigenesis of K562 cells suggesting that these drugs may have therapeutic potential for those tumors associated with Notch signaling. Taken together, this system could be beneficial for screening of drugs with potential to treat Notch signal pathway‐associated diseases. J. Cell. Biochem. 106: 682–692, 2009. © 2009 Wiley‐Liss, Inc.
Keywords: Notch, CBF1, baicalin, baicalein, niclosamide
The Notch signaling pathway regulates several cellular processes including proliferation, differentiation, apoptosis, cell fate decision, and maintenance of stem cells [Artavanis‐Tsakonas et al., 1999; Miele and Osborne, 1999; Kopan, 2002]. Abnormalities in Notch signaling are linked to many diseases such as Alagille's syndrome [Li et al., 1997; Oda et al., 1997], CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) [Joutel et al., 1996], prion disease [Ishikura et al., 2005], rheumatoid synoviocytes [Nakazawa et al., 2001], Alzheimer's disease, and Down syndrome [Fischer et al., 2005].
The deregulated Notch signaling also participates in the tumorigenesis of hematologic and solid malignancies, including T‐cell acute lymphoblastic leukemia, pancreatic carcinoma, cervical carcinoma, prostatic carcinoma, classical Hodgkin lymphoma, anaplastic large cell lymphoma, multiple myeloma, melanoma, lung cancer, colon cancer, breast cancer, hepatocellular carcinoma, and Kaposi's sarcoma [Radtke and Raj, 2003; Leong and Karsan, 2006]. Moreover, Notch signaling pathway can be both oncogenic and tumor suppressive. Many reports documented that the cellular context and the cross‐talk with other signaling pathways determined the roles of the Notch signal pathway in tumorigenesis [Radtke and Raj, 2003; Yoo et al., 2004; Leong and Karsan, 2006; Nguyen et al., 2006].
Notch receptors, single‐span transmembrane proteins, contain several functional domains. The Notch receptor intracellular domain consists of a RAM domain, ankyrin repeats, two nuclear localization signals, a transcriptional activator domain, and a proline‐glutamate‐serine‐threonine‐rich (PEST) domain. In the canonical pathway of Notch signaling, Notch receptors are activated and cleaved in order to release and translocate the intracellular domain into the nucleus after the extracellular portions interacting with their ligands on the neighboring cells. This Notch receptor intracellular domain, the activated form of Notch receptor, regulates downstream target genes through both C promoter binding factor‐1 (CBF1)‐dependent and ‐independent pathways [Baron, 2003]. The mechanism underlying the control of Notch signaling is very complicated and not yet fully understood.
Mounting evidence indicates that the aberrant Notch signal pathway caused by the mutations of Notch receptors or their ligands is involved in human malignancies and a variety of human diseases [Radtke and Raj, 2003; Leong and Karsan, 2006]. Therefore, both genetic or pharmacological manipulation of this signaling could serve as a potential therapeutically strategy for the Notch signal pathway‐associated diseases [Zlobin et al., 2000; Nam et al., 2002; Nickoloff et al., 2003; SjÖlund et al., 2005; Grabher et al., 2006; Miele et al., 2006; Shi and Harris, 2006].
The Notch receptor intracellular domain, also the nuclear form of Notch receptor, has been detected in several tumor cells such as SUP‐T1, HeLa, and Jurkat cells [Rand et al., 2000; Yeh et al., 2003]. However, the endogenous Notch receptor intracellular domain is hard to be detected in normal cells owing to its rapid turnover and/or low level expression [Lieber et al., 1993; Lam et al., 2000]. The C‐terminal PEST domain of Notch receptor contributes to its instability and degradation by the ubiquitin‐proteasome pathway [Wu et al., 2001]. Nevertheless, the Notch receptor intracellular domain can elicit its biological effects at a very low protein concentration to prevent potentially deleterious level of transcription [Rechsteiner and Rogers, 1996; Schroeter et al., 1998; Struhl and Adachi, 1998]. Therefore, it is important to scrutinize the endogenous Notch signal pathway to prevent the possibility of fortuitous abnormalities caused by the constitutive overexpression of Notch receptor intracellular domain.
It was demonstrated that both baicalin and baicalein possess anti‐tumor activity [Kubo et al., 1984; Fukutake et al., 1998; Chan et al., 2000; Ikezoe et al., 2001; Liu et al., 2003; Huang et al., 2005; Ma et al., 2005; Miocinovic et al., 2005]. After the cleavage of the glycoside moiety, baicalin is converted to baicalein in vivo. Both baicalin and baicalein cause G1 arrest and apoptosis in prostate cancer cells [Chen et al., 2001]. In human hepatoma cells, baicalin and baicalein induce apoptosis or inhibit proliferation [Chang et al., 2002]. Baicalin and baicalein also exhibit the anti‐angiogenesis potential through down‐regulating the activity of matrix metalloproteinase 2 [Liu et al., 2003].
Niclosamide, an anti‐helmintic drug, is useful for the treatment of beef, dwarf, and dog tapeworms [Reynolds, 1989]. It is also used as an active chemical ingredient to control sea lamprey and a molluscicide against schistosomiasis [Applegate et al., 1961; Goldsmith, 1984; Reynolds, 1989]. Recently, niclosamide was shown to exhibit the activity against severe acute respiratory syndrome coronavirus [Wu et al., 2004; De Clercq, 2006].
In the present study, we established a permanent human erythroleukemia K562 cell line to monitor the endogenous Notch signaling by reporter gene assay. We found that the endogenous Notch signal pathway is activated by baicalin and baicalein, whereas it is suppressed by niclosamide. In addition to modulating the autonomous Notch signaling, the tumorigenesis of K562 cells was also evaluated after the treatment with baicalein, baicalin, and niclosamide.
MATERIALS AND METHODS
Plasmids and Plasmid Construction
The expression construct of Notch1 receptor intracellular domain (pcDNA‐HA‐N1IC) contains cDNA encoding the amino acid residues 1764–2444 of the human Notch1 receptor with an N‐terminal HA tag [Yeh et al., 2003]. Reporter plasmid pCBF1‐RE‐Luc‐neo contains four copies of the wild‐type CBF1‐response elements (GTGGGAA) in front of the simian virus 40 promoter‐driven luciferase reporter gene in pGL2‐promoter vector and neomycine resistance gene in pcDNA3 vector. To construct this reporter plasmid, the pGL2‐promoter vector with four copies of the wild‐type CBF1‐response elements was digested by Kpn I and Bam HI. Then the Kpn I–Bam HI DNA fragment (3.1 kb) containing CBF1‐response elements and luciferase reporter gene was inserted at Bgl II and Kpn I sites of pcDNA3 vector which had the Bgl II–Kpn I DNA fragment (0.9 kb) containing the CMV promoter already been deleted. All constructs were verified by sequencing.
Cell culture and Transfection
Human erythroleukemia K562 cells and COS‐7 cells were cultured in RPMI 1640 and DMEM containing 10% fetal bovine serum, respectively. For the establishment of stable K562 cell lines to monitor the endogenous Notch signaling pathway (K562/CBF1‐RE‐Luc cells), K562 cells (2 × 106) were transfected with a lineralized reporter plasmid of pCBF1‐RE‐Luc‐neo (5 µg) by electroporation using Bio‐Rad gene pulser electroporator. Forty‐eight hours after electroporation, cells were diluted to about 0.8 cell/well in 96‐well dishes and selected with 800 µg/ml G418. The stable clones derived from single cells suitable for monitoring the endogenous Notch signaling were screened by reporter gene assay. The stable COS‐7 cell lines expressing secreted form of human Jagged1 (COS‐7/Jagged1ext) and their control cells (COS‐7/pcDNA3.1‐myc‐His) were established as described previously [Liao et al., 2007].
After seeding onto 6‐well plates, K562 cells (1 × 106) were transiently transfected by the SuperFect transfection reagent (Qiagen) as described for the luciferase reporter assay [Yeh et al., 2003, 2004]. Then the luciferase activities were measured using the Dual‐Luciferase™ reporter assay system (Promega). To activate the endogenous Notch signal pathway, K562/CBF1‐RE‐Luc cells were co‐cultured with Jagged1‐exressing COS‐7/Jagged1ext cells or their control COS‐7/pcDNA3.1‐myc‐His cells. The COS‐7/Jagged1ext cells or their control cells (5 × 105) were seeded onto 6‐well culture plates in DMEM medium. After 24 h of seeding, K562/CBF1‐RE‐Luc cells (5 × 105) were co‐cultured with COS‐7/Jagged1ext cells or their control cells in RPMI 1640 medium. Twenty‐four hours later, the K562/CBF1‐RE‐Luc cells were harvested for luciferase reporter assay.
DAPT {N‐[N‐(3,5‐Difluorophenacetyl)‐l‐alanyl]‐S‐phenylglycine t‐butyl ester}, baicalin (baicalein 7‐β‐d‐glucopyranosiduronate), baicalein (5,6,7‐trihydroxyflavone), and niclosamide (2′,5‐dichloro‐4′‐nitrosalicylanilide) were purchased from Sigma–Aldrich. These drugs at indicated concentrations in DMSO or an equal volume of DMSO were added for 24 h, followed by washing with PBS three times and cells were harvested for reporter gene assay.
Western Blot Analysis
Whole‐cell lysates were prepared as previously described [Liao et al., 2007]. Laemmli's sample buffer was added to the cell lysates and heated at 95°C for 5 min, then analyzed by SDS–PAGE. Western blotting was performed with anti‐cleaved Notch1 (Val1744, Cell Signaling), anti‐HES‐1 (Santa Cruz), anti‐cyclin D1 (Lab Vision), anti‐c‐Myc (Santa Cruz), and anti‐GAPDH antibodies (Biogenesis).
Real‐Time PCR Analysis
Total RNA was extracted using Trizol reagent (Invitrogen) and real‐time PCR analysis was performed as described previously [Liao et al., 2007]. Briefly, total RNA (2 µg) was used to synthesize cDNA using reverse transcriptase M‐MLV (New England BioLabs) with oligo (dT)18 primer. The 85‐bp cDNA of human Notch1 receptor was amplified with primers 5′‐CACTGTGGGCGGGTCC‐3′ and 5′‐GTTGTATTGGTTCGGCACCAT‐3′. The 84‐bp cDNA of HES‐1 was amplified with 5′‐AGCGGGCGCAGATGAC‐3′and 5′‐CGTTCATGCACTCGCTGAA‐3′. The 86‐bp cDNA of cyclin D1 was amplified with 5′‐CCGTCCATGCGGAAGATC‐3′ and 5′‐ATGGCCAGCGGGAAGAC‐3′. The 478‐bp cDNA of c‐Myc was amplified with 5′‐TACCCTCTCAACGACAGCAG‐3′ and 5′‐TCTTGACATTCTCCTCGGTG‐3′. The 71‐bp cDNA of Hey‐1 was amplified with 5′‐GAAAAAGCCGAGATC‐3′ and 5′‐TAACCTTTCCCTCCT‐3′. The 51‐bp cDNA of Hey‐2 was amplified with 5′‐AGATGCTTCAGGCAACAGGG‐3′and 5′‐CAAGAGCGTGTGCGTCAAAG‐3′. The 176‐bp cDNA of internal control GAPDH was amplified with 5′‐AAATCCCATCACCATCTTCC‐3′ and 5′‐TCACACCCATGA CGAACA‐3′. The quantitative real‐time PCR was analyzed on a LightCycler system with LightCycler FastStart DNA MasterPLUS SYBR Green I (Roche). The amplification condition of PCR was as follows: 95°C for 5 min; and then 35 cycles of 95°C for 10 s, 60°C for 5 s, and 72°C for 12 s. The relative quantification of mRNA expression level was normalized to that of GAPDH and corrected to a calibrator using the RelQuant software (Roche). All data are representative of the mean values and standard deviations from three to four independent experiments.
Flow Cytometry
As described previously [Liao et al., 2007], K562 cells were washed with ice‐cold phosphate‐buffered saline (PBS) and fixed in 70% ethanol at −20°C overnight. After being washed with PBS, the cell pellets were resuspended in 1.0 ml of PBS with 100 µg/ml of RNase A and incubated at 37°C for 30 min. Then 100 µl of 200 µg/ml propidium iodide (PI) was added and further incubated on ice for 30 min. The emitted fluorescence from the PI‐DNA complex was measured using a FACSCalibur flow cytometry (Becton‐Dickinson).
Erythroid Differentiation and Benzidine‐Staining
To induce erythroid differentiation, K562 cells treated with baicalin, baicalein, and niclosamide were incubated with 40 µM hemin (Sigma–Aldrich) for 3 days. The stock solution of benzidine contains 0.2% benzidine dihydrochloride (Sigma–Aldrich) in 0.5 M glacial acetic acid. Immediately before use, hydrogen peroxide at the concentration of 0.3% was added to prepare benzidine solution. After being washed two times with ice‐cold PBS, cells (5 × 104) were resuspended in 15 µl of ice‐cold PBS. Fifteen microliters of benzidine solution were added and further incubated for 10 min at room temperature. The benzidine‐positive cells with dark blue color were quantitated under light microscopy. At least 100 cells were counted in triplicate for each experiment.
Colony‐Forming Assay
The base agar (1.5 ml/well) contains RPMI medium with 0.6% agarose in 6‐well plates. Two thousand of K562 cells were resuspended in the top agar (1.0 ml/well) containing 0.2% agarose in RPMI medium with or without baicalin, baicalein, and niclosamide. Cells were incubated at 37°C in a humidified incubator with 5% CO2 for 7 days and 200 µl of RPMI medium containing the mentioned drugs were added every 3 days to prevent desiccation. Plates were stained with 0.5 ml of 0.005% crystal violet for 1 h and colonies larger than 0.1 mm in diameter were counted under the microscope from 10 random fields.
RESULTS
Establishment of Stable Cell Lines Expressing Luciferase Reporter Gene Modulated by CBF1‐Dependent Notch Signal Pathway
For the screening of drugs affecting endogenous Notch signaling, a cell based assay using luciferase reporter gene was established. The pCBF1‐RE‐Luc‐neo reporter plasmid containing four copies of CBF1‐response elements in front of a simian virus 40 promoter‐driven luciferase reporter gene and neomycine‐resistant gene was constructed. When K562 cells were co‐transfected with pCBF1‐RE‐Luc‐neo reporter plasmid and Notch1 receptor intracellular domain‐expressing construct, pcDNA‐HA‐N1IC, or its control vector, activity of CBF1‐dependent luciferase reporter gene was enhanced to 12.7‐fold in the presence of the exogenous Notch1 receptor intracellular domain (P < 0.001, Fig. 1A).
Figure 1.
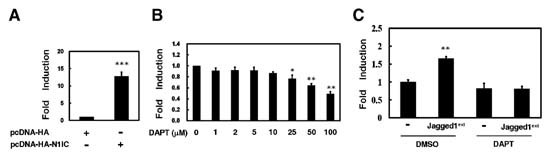
Establishment of stable K562/CBF1‐RE‐Luc cells harboring integrated luciferase reporter gene modulated by endogenous Notch signaling pathway. A: The pCBF1‐RE‐Luc‐neo reporter plasmid was co‐transfected with HA‐Notch1 receptor intracellular domain fusion protein‐expressing construct (pcDNA‐HA‐N1IC) or its control vector into K562 cells for reporter gene assay. Forty‐eight hours after transfection, luciferase activity was determined from whole‐cell extracts of the transfected cells, and basal promoter activity of the reporter construct was set to unity. Renilla luciferase activity was then used to normalize for transfection efficiency. Mean values and standard deviations from at least four independent experiments are shown. ***P < 0.001 compared with mock. B: After treatment with various concentrations of DAPT for 24 h, K562/CBF1‐RE‐Luc cells were harvested for reporter gene assay. *P < 0.05; **P < 0.01 compared with vehicle (DMSO). C: The Jagged1‐expressing COS‐7/Jagged1ext cells or their control cells were seeded onto 6‐well culture plates in DMEM medium for 24 h. Then K562/CBF1‐RE‐Luc cells were co‐cultured with COS‐7/Jagged1ext cells (Jagged1ext) or their control cells (−) in RPMI 1640 medium with 100 µM DAPT or an equal volume of DMSO for 24 h. Luciferase reporter gene activity of K562/CBF1‐RE‐Luc cells was determined and normalized with protein concentration. **P < 0.01 compared to co‐culture with control cells.
Then the pCBF1‐RE‐Luc‐neo reporter plasmid was transfected into K562 cells to establish the single cell‐derived stable clones (K562/CBF1‐RE‐Luc cells) with integrated luciferase reporter gene and its expression is regulated by CBF1‐dependent Notch signal pathway. In the K562/CBF1‐RE‐Luc cells, we also found that the expression of the exogenous Notch1 receptor intracellular domain could induce the CBF1‐dependent luciferase activity (data not shown).
K562/CBF1‐RE‐Luc Cells Can Be Used to Monitor the Activation of Endogenous Notch Signaling
The aforementioned data showed that the CBF1‐depedent reporter gene activity in K562/CBF1‐RE‐Luc cells could be induced by Notch signal pathway. We surmised that the K562/CBF1‐RE‐Luc cells might be used to assess the molecules/factors that can modulate the endogenous Notch signaling in cells. To evaluate this possibility, K562/CBF1‐RE‐Luc cells were treated with various concentrations of γ‐secretase inhibitor DAPT, for 24 h to block endogenous Notch signaling. The results showed that reporter gene activity in K562/CBF1‐RE‐Luc cells was significantly suppressed in a dose‐dependent manner after the treatment of DAPT (P < 0.01, Fig. 1B).
Previously, we had demonstrated that endogenous Notch signal pathway is activated after co‐culture of K562 cells with the single cell‐derived stable COS‐7 cells constitutively expressing the secreted form of Notch ligand Jagged1 (COS‐7/Jagged1ext cells) or after treatment with their conditioned media [Liao et al., 2007]. In this experiment, co‐culture with COS‐7/Jagged1ext cells also promoted the activity of CBF1‐dependent reporter gene in K562/CBF1‐RE‐Luc cells (P < 0.01, Fig. 1C). This enhancement of CBF1‐dependent reporter gene activity by Jagged1 was abrogated after the treatment with DAPT.
Endogenous Notch Signaling Is Activated by Baicalin and Baicalein But Suppressed by Niclosamide in K562 Cells
This cell‐based system was further used to screen drugs modulating endogenous Notch signal pathway in the present study. We found that baicalin, baicalein, and niclosamide were candidates for modulation of the endogenous Notch signal pathway. The CBF1‐dependent reporter gene activity in K562/CBF1‐RE‐Luc cells was enhanced by baicalin and baicalein in a dose‐dependent manner (P < 0.05, Fig. 2A, left and middle). In the presence of DAPT, there was no significant enhancement of CBF1‐dependent reporter gene activity after treatment with baicalin and baicalein. Furthermore, niclosamide inhibited the CBF1‐dependent reporter gene activity of K562/CBF1‐RE‐Luc cells in a dose‐dependent manner (P < 0.01, Fig. 2A, right). However, this suppression of CBF1‐dependent reporter gene activity of K562/CBF1‐RE‐Luc cells by various concentrations of niclosamide was not affected after the treatment with DAPT.
Figure 2.

Endogenous Notch signaling is activated by baicalin and baicalein but is suppressed by niclosamide in K562 cells. A: In the presence or absence of 100 µM DAPT, K562/CBF1‐RE‐Luc cells were treated with various concentrations of baicalin (left), baicalein (middle), and niclosamide (right) for 24 h. B: After seeding of K562/CBF1‐RE‐Luc cells at the same time, cells were treated with 100 µM DAPT for various periods of time and then harvested for reporter gene assay. In the presence or absence of 100 µM DAPT, K562/CBF1‐RE‐Luc cells were also treated with 100 µM baicalin, 10 µM baicalein, and 1.25 µM niclosamide for various periods of time and then harvested for reporter gene assay. Luciferase reporter gene activity was determined as described in the legend to Figure 1. *P < 0.05; **P < 0.01; ***P < 0.001 compared with vehicle.
As shown in Figure 2B, the suppression of CBF1‐dependent reporter gene activity in K562/CBF1‐RE‐Luc cells by DAPT was time‐dependent (P < 0.05). The induction of CBF1‐dependent reporter gene activity of K562/CBF1‐RE‐Luc cells by baicalin and baicalein treatment also showed a time‐dependent manner (P < 0.05). In the presence of DAPT, there was no significant enhancement of CBF1‐dependent reporter gene activity after treatment with baicalin and baicalein for various periods of time. However, niclosamide also presented a time‐dependent manner in inhibition of CBF1‐dependent reporter gene activity of K562/CBF1‐RE‐Luc cells (P < 0.05). After treatment with niclosamide for various periods of time, the suppression of CBF1‐dependent reporter gene activity in K562/CBF1‐RE‐Luc cells was not affected by DAPT significantly.
The mRNA Expressions of Notch1 Receptor and Notch Target Genes Are Regulated by Baicalin, Baicalein, and Niclosamide in K562 Cells
To investigate the underlying regulatory mechanism of CBF1‐dependent reporter gene activity by baicalin, baicalein, and niclosamide in K562/CBF1‐RE‐Luc cells, mRNA expression levels of Notch1 receptor and Notch target genes were determined by real‐time PCR in K562 cells after treatment with these drugs (Fig. 3A). Data showed that mRNA expressions of Notch1 receptor, HES‐1, cyclin D1, Hey‐1 and Hey‐2 were enhanced by baicalin and baicalein whereas cyclin D1 and c‐Myc mRNA expressions were suppressed after treatment with niclosamide. However, the Hey‐2 mRNA expression was induced by niclosamide. In the presence of DAPT, the enhancement of mRNA expressions of HES‐1, cyclin D1, Hey‐1, and Hey‐2 by baicalin and baicalein were suppressed. The induction of Hey‐2 mRNA expression by niclosamide was also inhibited after treatment with DAPT.
Figure 3.

The mRNA expressions of Notch1 receptor and Notch target genes are regulated by baicalin, baicalein, and niclosamide in K562 cells. A: In the presence (bottom) or absence (top) of 100 µM DAPT, the transcript levels of Notch1 receptor, HES‐1, cyclin D1, c‐Myc, Hey‐1, and Hey‐2 in K562 cells were measured by quantitative real‐time PCR after treatment with 100 µM baicalin, 10 µM baicalein, and 1.25 µM niclosamide for 24 h. Data were compared, after being normalized to GAPDH. *P < 0.05; **P < 0.01; ***P < 0.001 compared with vehicle. B: K562 cells were treated with an equal volume of DMSO, 100 µM baicalin, 10 µM baicalein, and 1.25 µM niclosamide for 24 or 48 h. The medium containing drugs was changed every 24 h. Whole‐cell extracts were prepared for Western blot analysis using anti‐HES‐1, anti‐cyclin D1, anti‐c‐Myc, anti‐cleaved Notch1, or anti‐GAPDH antibodies as indicated.
After treatment with these drugs, the protein expressions of Notch target genes were also detected by Western blot analysis in K562 cells including HES‐1, cyclin D1, and c‐Myc (Fig. 3B). The expressions of HES‐1, cyclin D1, and c‐Myc were not significantly affected by baicalin and baicalein but suppressed by niclosamide after treatment for 24 h. However, the expression levels of HES‐1, cyclin D1, and c‐Myc were not changed after treatment with these drugs for 48 h. Analysis of expression levels of the cleaved activated Notch1 receptor showed that baicalin and baicalein elicited only a modest increase, but niclosamide exerted a decrease in K562 cells after treatment for 24 h.
Cell Cycle Progression, Erythroid Differentiation, and Colony‐Forming Ability of K562 Cells Are modulated by Baicalin, Baicalein, and Niclosamide
Based on the results described above, we further investigated effects of baicalin, baicalein, and niclosamide on the biological functions such as cell cycle progression, erythroid differentiation, and colony‐forming ability of K562 cells. After treatment with 100 µM baicalin, 10 µM baicalein, and 1.25 µM niclosamide for 24 h, K562 cells were harvested for PI‐staining and flow cytometry analysis. The cells treated with baicalin and baicalein were slightly arrested in G0/G1 phase as compared with those treated with DMSO (P < 0.05, Fig. 4). However, the cell population in S phase was increased after treatment with niclosamide.
Figure 4.
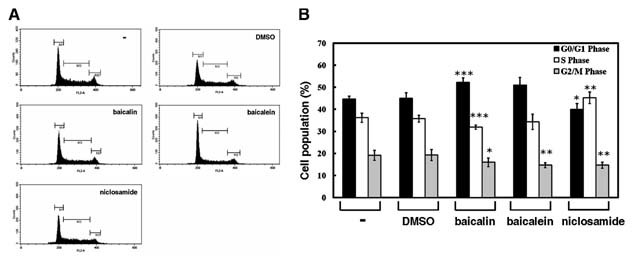
Cell cycle progression of K562 cells is slightly affected by baicalin and baicalein, but not by niclosamide. K562 cells were treated with an equal volume of PBS (−), DMSO, 100 µM baicalin, 10 µM baicalein, and 1.25 µM niclosamide for 24 h. Then these cells were stained with PI to analyze their DNA contents by flow cytometry (A). A total of 10,000 cells was assayed from each sample, and the cell proportions in G0/G1, S, and G2/M phases of cell cycle were indicated (B). Data are representative of mean values and standard deviations from three independent experiments performed in triplicate. *P < 0.05; **P < 0.01; ***P < 0.001 compared with vehicle.
Previous report has demonstrated that the activation of Notch signaling pathway inhibits erythroid differentiation of K562 cells [Lam et al., 2000]. To evaluate whether erythroid differentiation is regulated by treatment with baicalin, baicalein, and niclosamide, K562 cells were induced by 40 µM hemin for 3 days and then assessed by benzidine‐staining. The percentage of benzidine‐positive cells was suppressed by treatment with baicalin and baicalein, but promoted by niclosamide (P < 0.05, Fig. 5A,B). Furthermore, the addition of DAPT reversed the suppressed and promoted ability of erythroid differentiation by baicalin and by niclosamide, respectively (Fig. 5C).
Figure 5.
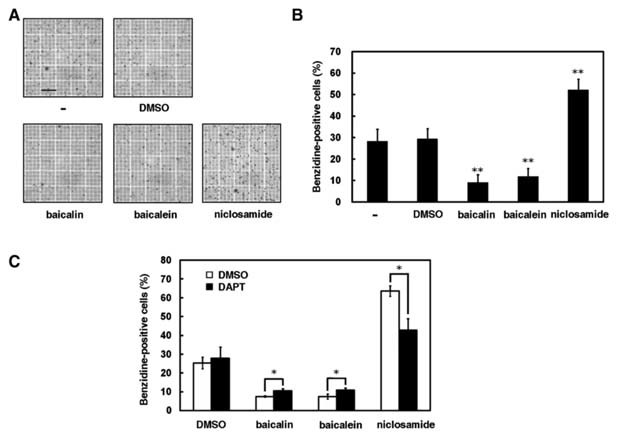
Erythroid differentiation of K562 cells is suppressed by baicalin and baicalein but enhanced by niclosamide. A: In the presence of an equal volume of PBS (−), DMSO, 100 µM baicalin, 10 µM baicalein, and 1.25 µM niclosamide, K562 cells were used to induce erythroid differentiation by 40 µM hemin for 3 days. Scale bar: 0.2 mm. B: The percentage of benzidine‐positive cells was determined using benzidine‐staining under light microscopy. Data are representative of the mean values and standard deviations from three independent experiments performed in triplicate. *P < 0.05; **P < 0.01 compared with vehicle. C: In the presence or absence of 100 µM DAPT, the percentage of benzidine‐positive K562 cells was determined after treatment with 100 µM baicalin, 10 µM baicalein, or 1.25 µM niclosamide. *P < 0.05; **P < 0.01 compared to those without DAPT treatment.
To further evaluate whether treatment with these drugs affects tumorigenesis, colony‐forming assay was also preformed. The results showed that colony numbers were decreased after treatment with baicalin and baicalein (P < 0.001, Fig. 6A,B). On the contrary, colony numbers were not dramatically affected in the presence of niclosamide. Interestingly, mean colony sizes of K562 cells treated with baicalin and baicalein but not with niclosamide were reduced as compared to the control cells treated with DMSO (Fig. 6C).
Figure 6.
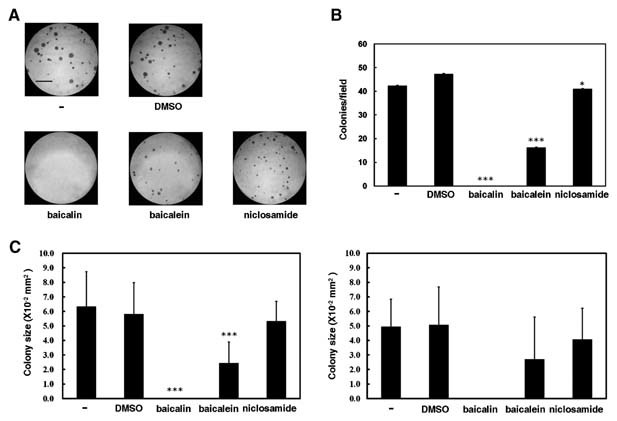
Colony‐forming ability of K562 cells is suppressed by baicalin and baicalein, but not by niclosamide. A: In the presence of an equal volume of PBS (−), DMSO, 100 µM baicalin, 10 µM baicalein, and 1.25 µM niclosamide, K562 cells were mixed with top agar in 6‐well plates. Cells were incubated for 7 days and then the plates were stained with 0.5 ml of 0.005% crystal violet for 1 h. Scale bar: 1.0 mm. Colonies larger than 0.1 mm in diameter were counted and mean colony sizes were estimated under microscope from 10 random fields. B: Data of colony numbers are representative of mean values and standard deviations from three independent experiments. C: Data of mean colony sizes are shown from one experiment (left) or mean values and standard deviations from three independent experiments (right). Because variation of colony sizes was very high, the change of mean colony sizes from three independent experiments is not statistically significant. Therefore, mean colony sizes from one experiment were also shown. *P < 0.05; ***P < 0.001 compared with vehicle.
DISCUSSION
Because the Notch signaling pathway can function as an oncogene or tumor suppressor in cancer development, targeting the steps in this signaling may have the therapeutic potential for anti‐tumor activity in the foreseeable future [Nam et al., 2002; Miele et al., 2006; Shih and Wang, 2007]. There are several potential therapeutic targeting steps for therapy in Notch signaling pathway including interaction of Notch receptors and ligands, translocation of Notch receptor intracellular domains, transactivation of downstream targets, expression of Notch components, and stability of Notch receptors. Recent studies have documented the possibility of using Notch signaling as a therapeutic target in cancer [Nam et al., 2002; Hansson et al., 2004; Li and Harris, 2005; Miele et al., 2006; Shih and Wang, 2007]. For example, there is mounting evidence that γ‐secretase inhibitors may act as a new target‐based therapy for those tumors associated with Notch signaling [Shih and Wang, 2007].
To avoid the ambiguity caused by nonphysiologic overexpression of an activated Notch receptor, it is important to study the regulatory mechanism of autonomous Notch signaling pathway. In the present study, we demonstrated that endogenous Notch signaling was modulated by treatment of baicalin, baicalein, and niclosamide in K562 cells. At least in the case of Notch1 receptor in Notch family, baicalin and baicalein affected the Notch signaling pathway through regulating mRNA expression of Notch1 receptor (Fig. 3).
Both baicalin and baicalein were shown to regulate the cell cycle progression and cell proliferation in cancer cells [Ikezoe et al., 2001; Chang et al., 2002; Liu et al., 2003]. We demonstrated herein that the progression of cell cycle in K562 cells was affected by the treatment of baicalin, baicalein, and niclosamide. Furthermore, we also presented the modulation of erythroid differentiation and colony‐forming ability in K562 cells by treatment with baicalin, baicalein, and niclosamide. Previously, it was also shown that the activation of Notch signal pathway suppresses erythroid differentiation of K562 cells [Lam et al., 2000]. Therefore, those results suggest that baicalin, baicalein, and niclosamide could control erythroid differentiation of K562 cells through affecting Notch signaling.
Clearly, the functional system of K562/CBF1‐RE‐Luc cells is suitable for screening drugs that modulate endogenous CBF1‐dependent Notch signal pathway. The identified Notch signaling‐inhibited drugs could be beneficial in treatment of cancers caused by the aberrant activation of oncogenic Notch receptors. While the identified Notch signaling‐activated drugs could be used for therapy of tumors that were contributed by the inhibition of tumor suppressive ability of Notch receptors. Nevertheless, the established screening system does not distinguish which Notch receptors participate in the regulation of luciferase activity of CBF1‐dependent reporter gene after drug‐treatment. All four Notch1‐4 receptors involve in the control of down‐stream target genes of CBF1‐dependent Notch signaling.
In addition, regulation of gene expression and functional relationship among four Notch receptors is very complicated and not yet fully understood so far. There are response elements in the upstream of Notch gene that can potentially bind with CBF1 to activate itself by Notch signal pathway [Ganapati et al., 2007]. The regulatory feedback loops of Notch signaling are present to induce expressions of Notch1 and Notch3 mRNAs [Hajdu et al., 2007]. Notch1 receptor also induces and cooperates with Notch4 receptor [Krebs et al., 2000; Weijzen et al., 2002]. Furthermore, Notch2 receptor represses Notch1 and Notch3 signaling pathways [Shimizu et al., 2002], and Notch3 receptor inhibits the Notch1‐mediated activation [Beatus et al., 1999]. Therefore, it needs further investigation to identify which Notch receptors contribute to the modulation of CBF1‐dependent reporter gene activity by identified drugs.
The control of HES‐1 expression is also very complicated and remains poorly understood so far. It was reported that transcription factor HES‐1 with a very short half‐life is a key repressor of its own promoter by a negative feedback loop [Hirata et al., 2002]. As presented in Figure 3, lack of modulation of HES‐1 mRNA by niclosamide and HES‐1 protein by baicalin and baicalein may be due to its complicated regulation of gene expression.
Induction of c‐myc expression contributes to tumorigenesis [for reviews, see Garte, 1993; Amati et al., 1998; Dang, 1999; Adhikary and Eilers, 2005]. Therefore, the moderate levels of mRNA and protein of c‐myc must be tightly regulated and maintained through a very short half‐life in cells [Levens, 2003]. Many transcription factors are involved in the control of c‐myc promoter activity such as NF‐κB [Weber et al., 2005], Notch1 intracellular domain [Rao and Kadesch, 2003; Satoh et al., 2004; Klinakis et al., 2006; Palomero et al., 2006; Sharma et al., 2006; Weng et al., 2006; Liao et al., 2007], MBP‐1, and α‐enolase [Ray and Miller, 1991; Chaudhary and Miller, 1995; Ray, 1995; Feo et al., 2000; Subramanian and Miller, 2000; Hsu et al., 2008]. Both α‐enolase and MBP‐1 could prevent the irregularity of c‐myc expression activated by cellular factors and stimuli. Notch1 receptor intracellular domain significantly enhances activity of c‐myc promoter by 30‐fold [Liao et al., 2007; Hsu et al., 2008]. However, expressions of c‐myc mRNA and protein are only enhanced about 4‐fold by Notch1 receptor intracellular domain in K562 cells. Furthermore, it was documented that Notch signaling may have different target genes in distinct cell types [Neves et al., 2006; Weerkamp et al., 2006]. Therefore, the slight induction of Notch1 expression after treatment with baicalin and baicalein did not significantly induce c‐myc expression in K562 cells (Fig. 3A).
As shown in Figure 3A, Hey‐2 expression was induced by niclosamide. This may be due to the multiple control of Hey‐2 expression. Expressions of both Hey‐1 and Hey‐2 genes can be regulated through Notch‐dependent or ‐independent pathways. Hey‐1 and Hey‐2 promoters containing CBF1‐binding sites can be up‐regulated by Notch receptor intracellular domain [Maier and Gessler, 2000; Nakagawa et al., 2000; Iso et al., 2001]. It was also suggested that there are tissue‐specific factors involving in modulation of Hey‐1 and Hey‐2 expressions by CBF1 and Notch signaling [Rutenberg et al., 2006]. Alternatively, the Notch‐independent control of Hey‐1 and Hey‐2 expressions had also been found [Iso et al., 2001; Rones et al., 2002].
We show herein that treatment with baicalin and baicalein promotes mRNA expression of Notch1 receptor and activates endogenous CBF1‐dependent Notch signaling pathway. However, we did not exclude the possibility that increment of Notch1 receptor expression by baicalin and baicalein may also contribute to modulation of erythroid differentiation and colony‐forming ability through CBF1‐independent manner.
The side effect is one of the major obstacles associated with treatment of the inhibitors [Barten et al., 2006]. Because Notch signaling pathway plays an important role in several cellular processes in normal tissues, perturbation of this signaling may cause dysfunction of organs. Moreover, the identified drugs could not only target Notch signaling pathway but also affect other pathways simultaneously. Besides Notch receptors, there are several substrates of γ‐secretase including amyloid precursor protein, Notch ligands, ERBB4, syndecan‐3, CD44 [Kopan and Ilagan, 2004], ephrin‐B [Tomita et al., 2006], and MHC I proteins [Carey et al., 2007]. Although it was suggested that γ‐secretase inhibitors have a promising role in treatment of cancer, there are side effects for treatment including thymus atrophy and intestinal goblet cell hyperplasia [Katoh and Katoh, 2007].
To exclude or minimize the side effects of cytotoxicity caused by treatment of these drugs, lower concentrations of drugs were used in this study to avoid cell death. There is no apparent decrease of cell numbers in the presence of baicalin, baicalein, and niclosamide at the indicated concentrations in this study (data not shown). The activation of endogenous CBF1‐dependent Notch signaling in K562 cells is not changed dramatically, only two‐ to three‐fold, in the presence of baicalin and baicalein (Fig. 2A,B). This slight induction of Notch signal pathway is more mimic to the endogenous signaling under physiological conditions in cells.
Acknowledgements
We thank Dr. M.‐J. Tseng (National Chung Cheng University) for critical reading of the manuscript. We also thank Ms. S.‐H. Wang for the construction of pCBF1‐RE‐Luc‐neo reporter plasmid and establishment of the K562/CBF1‐RE‐Luc cells.
REFERENCES
- Adhikary S, Eilers M. 2005. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 6: 635–645. [DOI] [PubMed] [Google Scholar]
- Amati B, Alevizopoulos K, Vlach J. 1998. Myc and the cell cycle. Front Biosci 3: d250–d268. [DOI] [PubMed] [Google Scholar]
- Applegate V, Howell J, Moffett J, Johnson B, Smith M. 1961. Use of 3‐trifluoromethyl‐4‐nitrophenol as a selective sea lamprey larvicide. Technical Report 1. Ann Arbor, MI: Great Lakes Fishery Commission. pp. 1–35. [Google Scholar]
- Artavanis‐Tsakonas S, Rand MD, Lake RJ. 1999. Notch signaling: Cell fate control and signal integration in development. Science 284: 770–776. [DOI] [PubMed] [Google Scholar]
- Baron M. 2003. An overview of the Notch signalling pathway. Semin Cell Dev Biol 14: 113–119. [DOI] [PubMed] [Google Scholar]
- Barten D, Meredith JJ, Zaczek R, Houston J, Albright C. 2006. Gamma‐secretase inhibitors for Alzheimer's disease: Balancing efficacy and toxicity. Drugs R D 7: 87–97. [DOI] [PubMed] [Google Scholar]
- Beatus P, Lundkvist J, Oberg C, Lendahl U. 1999. The Notch 3 intracellular domain represses Notch 1‐mediated activation through Hairy/Enhancer of split (HES) promoters. Development 126: 3925–3935. [DOI] [PubMed] [Google Scholar]
- Carey BW, Kim DY, Kovacs DM. 2007. Presenilin/γ‐secretase and γ‐secretase‐like peptidases cleave human MHC Class I proteins. Biochem J 401: 121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan F, Choi H, Chen Z, Chan P, Huang Y. 2000. Induction of apoptosis in prostate cancer cell lines by a flavonoid, baicalin. Cancer Lett 160: 219–228. [DOI] [PubMed] [Google Scholar]
- Chang WH, Chen CH, Lu FJ. 2002. Different effects of baicalein, baicalin and wogonin on mitochondrial function, glutathione content and cell cycle progression in human hepatoma cell lines. Planta Med 68: 128–132. [DOI] [PubMed] [Google Scholar]
- Chaudhary D, Miller DM. 1995. The c‐myc promoter binding protein (MBP‐1) and TBP bind simultaneously in the minor groove of the c‐myc P2 promoter. Biochemistry 34: 3438–3445. [DOI] [PubMed] [Google Scholar]
- Chen S, Ruan Q, Bedner E, Deptala A, Wang X, Hsieh TC, Traganos F, Darzynkiewicz Z. 2001. Effects of the flavonoid baicalin and its metabolite baicalein on androgen receptor expression, cell cycle progression and apoptosis of prostate cancer cell lines. Cell Prolif 34: 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. 1999. c‐Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. 2006. Potential antivirals and antiviral strategies against SARS coronavirus infections. Expert Rev Anti Infect Ther 4: 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feo S, Arcuri D, Piddini E, Passantino R, Giallongo A. 2000. ENO1 gene product binds to the c‐myc promoter and acts as a transcriptional repressor: Relationship with Myc promoter‐binding protein 1 (MBP‐1). FEBS Lett 473: 47–52. [DOI] [PubMed] [Google Scholar]
- Fischer DF, van Dijk R, Sluijs JA, Nair SM, Racchi M, Levelt CN, van Leeuwen FW, Hol EM. 2005. Activation of the Notch pathway in Down syndrome: Cross‐talk of Notch and APP. FASEB J 19: 1451–1458. [DOI] [PubMed] [Google Scholar]
- Fukutake M, Yokota S, Kawamura H, Lizuka A, Amagaya S, Fukuda K, Komatsu Y. 1998. Inhibitory effect of Coptidis rhizoma and Scutellaria radix on azoxy‐methane induced aberrant crypt foci formation in rat colon. Biol Pharm Bull 21: 814–817. [DOI] [PubMed] [Google Scholar]
- Ganapati U, Tan HT, Lynch M, Dolezal M, de Vos S, Gasson JC. 2007. Modeling Notch signaling in normal and neoplastic hematopoiesis: Global gene expression profiling in response to activated notch expression. Stem Cells 25: 1872–1880. [DOI] [PubMed] [Google Scholar]
- Garte SJ. 1993. The c‐myc oncogene in tumor progression. Crit Rev Oncog 4: 435–449. [PubMed] [Google Scholar]
- Goldsmith RS. 1984. Basic and clinical pharmacology. Los Angeles: Lange Medical. pp. 659–660. [Google Scholar]
- Grabher C, von Boehmer H, Look AT. 2006. Notch 1 activation in the molecular pathogenesis of T‐cell acute lymphoblastic leukaemia. Nat Rev Cancer 6: 347–359. [DOI] [PubMed] [Google Scholar]
- Hajdu M, Luttun A, Pelacho B, Burns T, Chase L, Gutiérrez‐Pérez M, Jiang Y, Lenvik T, Vas V, Uher F, ebestyén A, Verfaillie C. 2007. Transcriptional characterization of the Notch signaling pathway in rodent multipotent adult progenitor cells. Pathol Oncol Res 13: 302–310. [DOI] [PubMed] [Google Scholar]
- Hansson EM, Lendahl U, Chapman G. 2004. Notch signaling in development and disease. Semin Cancer Biol 14: 320–328. [DOI] [PubMed] [Google Scholar]
- Hirata H, Yoshiura S, Ohtsuka T, Bessho Y, Harada T, Yoshikawa K, Kageyama R. 2002. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science 298: 840–843. [DOI] [PubMed] [Google Scholar]
- Hsu KW, Hsieh RH, Lee YHW, Chao CH, Wu KJ, Tseng MJ, Yeh TS. 2008. The activated Notch1 receptor cooperates with α‐enolase and MBP‐1 in modulating c‐myc activity. Mol Cell Biol 28: 4829–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Tsang SY, Yao X, Chen ZY. 2005. Biological properties of baicalein in cardiovascular system. Curr Drug Targets Cardiovasc Haematol Disord 5: 177–184. [DOI] [PubMed] [Google Scholar]
- Ikezoe T, Chen SS, Heber D, Taguchi H, Koeffler HP. 2001. Baicalin is a major component of PC‐SPES which inhibits the proliferation of human cancer cells via apoptosis and cell cycle arrest. Prostate 49: 285–292. [DOI] [PubMed] [Google Scholar]
- Ishikura N, Clever JL, Bouzamondo‐Bernstein E, Samayoa E, Prusiner SB, Huang EJ, DeArmond SJ. 2005. Notch‐1 activation and dendritic atrophy in prion disease. Proc Natl Acad Sci USA 102: 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T, Sartorelli V, Chung G, Shichinohe T, Kedes L, Hamamori Y. 2001. HERP, a new primary target of Notch regulated by ligand binding. Mol Cell Biol 21: 6071–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier‐Lasserve E. 1996. Notch3 mutations in CADASIL, a hereditary adult‐onset condition causing stroke and dementia. Nature 383: 707–710. [DOI] [PubMed] [Google Scholar]
- Katoh M, Katoh M. 2007. Notch signaling in gastrointestinal tract (review). Int J Oncol 30: 247–251. [PubMed] [Google Scholar]
- Klinakis A, Szabolcs M, Politi K, Kiaris H, Artavanis‐Tsakonas S, Efstratiadis A. 2006. Myc is a Notch1 transcriptional target and a requisite for Notch1‐induced mammary tumorigenesis in mice. Proc Natl Acad Sci USA 103: 9262–9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R. 2002. Notch: A membrane‐bound transcription factor. J Cell Sci 115: 1095–1097. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MXG. 2004. γ‐Secretase: Proteasome of the membrane? Nat Rev Mol Cell Biol 5: 499–504. [DOI] [PubMed] [Google Scholar]
- Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. 2000. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev 14: 1343–1352. [PMC free article] [PubMed] [Google Scholar]
- Kubo M, Matsuda H, Tanaka M, Kimura Y, Okuda H, Higashino M, Tani T, Namba K, Arichi S. 1984. Studies on Scutellariae radix. VII. Anti‐arthritic and anti‐inflammatory actions of methanolic extract and flavonoid components from Scutellariae radix. Chem Pharm Bull 32: 2724–2729. [DOI] [PubMed] [Google Scholar]
- Lam LT, Ronchini C, Norton J, Capobianco AJ, Bresnick EH. 2000. Suppression of erythroid but not megakaryocytic differentiation of human K562 erythroleukemic cells by Notch‐1. J Biol Chem 275: 19676–19684. [DOI] [PubMed] [Google Scholar]
- Leong KG, Karsan A. 2006. Recent insights into the role of Notch signaling in tumorigenesis. Blood 107: 2223–2233. [DOI] [PubMed] [Google Scholar]
- Levens DL. 2003. Reconstructing Myc. Genes Dev 17: 1071–1077. [DOI] [PubMed] [Google Scholar]
- Li JL, Harris AL. 2005. Notch signaling from tumor cells: A new mechanism of angiogenesis. Cancer Cell 8: 1–3. [DOI] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont MEM, Rand EB, Piccoli DA, Hood L, Spinner NB. 1997. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 16: 243–251. [DOI] [PubMed] [Google Scholar]
- Liao WR, Hsieh RH, Hsu KW, Wu MZ, Tseng MJ, Mai RT, Lee YHW, Yeh TS. 2007. The CBF1‐independent Notch1 signal pathway activates human c‐myc expression partially via transcription factor YY1. Carcinogensis 28: 1867–1876. [DOI] [PubMed] [Google Scholar]
- Lieber T, Kidd S, Alcamo E, Corbin V, Young M. 1993. Antineurogenic phenotypes induced by truncated Notch proteins indicate a role in signal transduction and may point to a novel function for Notch in nuclei. Genes Dev 7: 1949–1965. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Huang TS, Cheng WF, Lu FJ. 2003. Baicalein and baicalin are potent inhibitors of angiogenesis: Inhibition of endothelial cell proliferation, migration and differentiation. Int J Cancer 106: 559–565. [DOI] [PubMed] [Google Scholar]
- Ma Z, Otsuyama KI, Liu S, Abroun S, Ishikawa H, Tsuyama N, Obata M, Li FJ, Zheng X, Maki Y, Miyamoto K, Kawano MM. 2005. Baicalein, a component of Scutellaria radix from Huang‐Lian‐Jie‐Du‐Tang (HLJDT), leads to suppression of proliferation and induction of apoptosis in human myeloma cells. Blood 105: 3312–3318. [DOI] [PubMed] [Google Scholar]
- Maier MM, Gessler M. 2000. Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem Biophys Res Commun 275: 652–660. [DOI] [PubMed] [Google Scholar]
- Miele L, Osborne B. 1999. Arbiter of differentiation and death: Notch signaling meets apoptosis. J Cell Physiol 181: 393–409. [DOI] [PubMed] [Google Scholar]
- Miele L, Miao H, Nickoloff B. 2006. Notch signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets 6: 313–323. [DOI] [PubMed] [Google Scholar]
- Miocinovic R, McCabe N, Keck R, Jankun J, Hampton J, Selman S. 2005. In vivo and in vitro effect of baicalein on human prostate cancer cells. Int J Oncol 26: 241–246. [PubMed] [Google Scholar]
- Nakagawa O, McFadden DG, Nakagawa M, Yanagisawa H, Hu T, Srivastava D, Olson EN. 2000. Members of the HRT family of basic helix‐loop‐helix proteins act as transcriptional repressors downstream of Notch signaling. Proc Natl Acad Sci USA 97: 13655–13660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa M, Ishii H, Aono H, Takai M, Honda T, Aratani S, Fukamizu A, Nakamura H, Yoshino S, Kobata T, Nishioka K, Nakajima T. 2001. Role of Notch‐1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum 44: 1545–1554. [DOI] [PubMed] [Google Scholar]
- Nam Y, Aster JC, Blacklow SC. 2002. Notch signaling as a therapeutic target. Curr Opin Chem Biol 6: 501–509. [DOI] [PubMed] [Google Scholar]
- Neves H, Weerkamp F, Gomes AC, Naber BAE, Gameiro P, Becker JD, Lucio P, Clode N, Van Dongen JJM, Staal FJT, Parreira L. 2006. Effects of Delta1 and Jagged1 on early human hematopoiesis: Correlation with expression of Notch signaling‐related genes in CD34+ cells. Stem Cells 24: 1328–1337. [DOI] [PubMed] [Google Scholar]
- Nguyen BC, Lefort K, Mandinova A, Antonini D, Devgan V, Della Gatta G, Koster MI, Zhang Z, Wang J, di Vignano AT, Kitajewski J, Chiorino G, Roop DR, Missero C, Dotto GP. 2006. Cross‐regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev 20: 1028–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff B, Osborne B, Miele L. 2003. Notch signaling as a therapeutic target in cancer: A new approach to the development of cell fate modifying agents. Oncogene 22: 6598–6608. [DOI] [PubMed] [Google Scholar]
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. 1997. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 16: 235–242. [DOI] [PubMed] [Google Scholar]
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O'Neil J, Neuberg D, Weng AP, Aster JC, Sigaux F, Soulier J, Look AT, Young RA, Califano A, Ferrando AA. 2006. Notch1 directly regulates c‐Myc and activates a feed‐forward‐loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci USA 103: 18261–18266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke F, Raj K. 2003. The role of Notch in tumorigenesis: Oncogene or tumour suppressor? Nat Rev Cancer 3: 756–767. [DOI] [PubMed] [Google Scholar]
- Rand MD, Grimm LM, Artavanis‐Tsakonas S, Patriub V, Blacklow SC, Sklar J, Aster JC. 2000. Calcium depletion dissociates and activates heterodimeric Notch receptors. Mol Cell Biol 20: 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P, Kadesch T. 2003. The intracellular form of Notch blocks transforming growth factor β‐mediated growth arrest in Mv1Lu epithelial cells. Mol Cell Biol 23: 6694–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray R. 1995. Induction of cell death in murine fibroblasts by a c‐myc promoter binding protein. Cell Growth Differ 6: 1089–1096. [PubMed] [Google Scholar]
- Ray R, Miller D. 1991. Cloning and characterization of a human c‐myc promoter‐binding protein. Mol Cell Biol 11: 2154–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers S. 1996. PEST sequences and regulation by proteolysis. Trends Biochem Sci 21: 267–271. [PubMed] [Google Scholar]
- Reynolds JEF. 1989. Martindale: The extra pharmacopoeia. London: The Pharmaceutical Press. pp. 987–991. [Google Scholar]
- Rones M, Woda J, Mercola M, McLaughlin K. 2002. Isolation and characterization of Xenopus Hey‐1: A downstream mediator of Notch sign. Dev Dyn 225: 554–560. [DOI] [PubMed] [Google Scholar]
- Rutenberg JB, Fischer A, Jia H, Gessler M, Zhong TP, Mercola M. 2006. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy‐related transcription factors. Development 133: 4381–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Matsumura I, Tanaka H, Ezoe S, Sugahara H, Mizuki M, Shibayama H, Ishiko E, Ishiko J, Nakajima K, Kanakura Y. 2004. Roles for c‐Myc in self‐renewal of hematopoietic stem cells. J Biol Chem 279: 24986–24993. [DOI] [PubMed] [Google Scholar]
- Schroeter EH, Kisslinger JA, Kopan R. 1998. Notch‐1 signalling requires ligand‐induced proteolytic release of intracellular domain. Nature 393: 382–386. [DOI] [PubMed] [Google Scholar]
- Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L, Krishnamoorthy V, Bhasin M, Capobianco AJ, Kelliher MA. 2006. Notch1 contributes to mouse T‐cell leukemia by directly inducing the expression of c‐myc. Mol Cell Biol 26: 8022–8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Harris A. 2006. Notch signaling in breast cancer and tumor angiogenesis: Cross‐talk and therapeutic potentials. J Mammary Gland Biol Neoplasia 11: 41–52. [DOI] [PubMed] [Google Scholar]
- Shih IM, Wang TL. 2007. Notch signaling, γ‐secretase inhibitors, and cancer therapy. Cancer Res 67: 1879–1882. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Chiba S, Saito T, Kumano K, Hamada Y, Hirai H. 2002. Functional diversity among Notch1, Notch2, and Notch3 receptors. Biochem Biophys Res Commun 291: 775–779. [DOI] [PubMed] [Google Scholar]
- SjÖlund J, Manetopoulos C, Stockhausen M, Axelson H. 2005. The Notch pathway in cancer: Differentiation gone awry. Eur J Cancer 41: 2620–2629. [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A. 1998. Nuclear access and action of Notch in vivo. Cell 93: 649–660. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Miller DM. 2000. Structural analysis of alpha‐enolase. mapping the functional domains involved in down‐regulation of the c‐myc protooncogene. J Biol Chem 275: 5958–5965. [DOI] [PubMed] [Google Scholar]
- Tomita T, Tanaka S, Morohashi Y, Iwatsubo T. 2006. Presenilin‐dependent intramembrane cleavage of ephrin‐B1. Mol Neurodegener 1: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Liu J, Collins I, Levens D. 2005. TFIIH operates through an expanded proximal promoter to fine‐tune c‐myc expression. Mol Cell Biol 25: 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerkamp F, Luis TC, Naber BAE, Koster EEL, Jeannotte L, van Dongen JJM, Staal FJT. 2006. Identification of Notch target genes in uncommitted T‐cell progenitors: No direct induction of a T‐cell specific gene program. Leukemia 20: 1967–1977. [DOI] [PubMed] [Google Scholar]
- Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC, Rudolf M, Siziopikou K, Kast WM, Miele L. 2002. Activation of Notch‐1 signaling maintains the neoplastic phenotype in human Ras‐transformed cells. Nat Med 8: 979–986. [DOI] [PubMed] [Google Scholar]
- Weng AP, Millholland JM, Yashiro‐Ohtani Y, Arcangeli ML, Lau A, Wai C, del Bianco C, Rodriguez CG, Sai H, Tobias J, Li Y, Wolfe MS, Shachaf C, Felsher D, Blacklow SC, Pear WS, Aster JC. 2006. c‐Myc is an important direct target of Notch1 in T‐cell acute lymphoblastic leukemia/lymphoma. Genes Dev 20: 2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, Chui I, Deshaies R, Kitajewski J. 2001. SEL‐10 is an inhibitor of notch signaling that targets notch for ubiquitin‐mediated protein degradation. Mol Cell Biol 21: 7403–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CJ, Jan JT, Chen CM, Hsieh HP, Hwang DR, Liu HW, Liu CY, Huang HW, Chen SC, Hong CF, Lin RK, Chao YS, Hsu JTA. 2004. Inhibition of severe acute respiratory syndrome coronavirus replication by niclosamide. Antimicrob Agents Chemother 48: 2693–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh TS, Lin YM, Hsieh RH, Tseng MJ. 2003. Association of transcription factor YY1 with the high molecular weight Notch complex suppresses the transactivation activity of Notch. J Biol Chem 278: 41963–41969. [DOI] [PubMed] [Google Scholar]
- Yeh TS, Hsieh RH, Shen SC, Wang SH, Tseng MJ, Shih CM, Lin JJ. 2004. Nuclear βII‐tubulin associates with the activated Notch receptor to modulate Notch signaling. Cancer Res 64: 8334–8340. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Bais C, Greenwald I. 2004. Crosstalk between the EGFR and LIN‐12/Notch pathways in C. elegans vulval development. Science 303: 663–666. [DOI] [PubMed] [Google Scholar]
- Zlobin A, Jang M, M L. 2000. Toward the rational design of cell fate modifiers: Notch signaling as a target for novel biopharmaceuticals. Curr Pharm Biotechnol 1: 83–106. [DOI] [PubMed] [Google Scholar]