

ABSTRACT
It is widely believed that host prion protein (PrP), without nucleic acid, converts itself into an infectious form (PrP‐res) that causes transmissible encephalopathies (TSEs), such as human sporadic CJD (sCJD), endemic sheep scrapie, and epidemic BSE. There are many detailed investigations of PrP, but proteomic studies of other proteins in verified infectious TSE particles have not been pursued, even though brain homogenates without PrP retain their complete infectious titer. To define proteins that may be integral to, process, or protect an agent genome, we developed a streamlined, high‐yield purification of infectious FU‐CJD mouse brain particles with minimal PrP. Proteinase K (PK) abolished all residual particle PrP, but did not reduce infectivity, and viral‐size particles lacking PrP were ∼70S (vs. 90–120S without PK). Furthermore, over 1,500 non‐PrP proteins were still present and positively identified in high titer FU‐CJD particles without detectable PrP by mass spectrometry (LC‐MS/MS); 114 of these peptides were linked to viral motifs in the environmental–viral database, and not evident in parallel uninfected controls. Host components were also identified in both PK and non‐PK treated particles from FU‐CJD mouse brain and human sCJD brain. This abundant cellular data had several surprises, including finding Huntingtin in the sCJD but not normal human brain samples. Similarly, the neural Wiskott–Aldrich sequence and multivesicular and endosome components associated with retromer APP (Alzheimer amyloid) processing were only in sCJD. These cellular findings suggest that new therapies directed at retromer–vesicular trafficking in other neurodegenerative diseases may also counteract late‐onset sCJD PrP amyloid pathology. J. Cell. Biochem. 115: 2012–2021, 2014. © 2014 Wiley Periodicals, Inc.
Keywords: TRANSMISSIBLE ENCEPHALOPATHIES (TSEs), INFECTIOUS BRAIN PARTICLES, VIRAL PEPTIDES, AMYLOID, ALZHEIMER's DISEASE, RETROMERS, WISKOTT‐ALDRICH, HUNTINGTIN, PROTEOMICS
The infectious agents that cause transmissible encephalopathies (TSEs) such as human Creutzfeldt–Jakob disease (CJD), endemic sheep scrapie, and epidemic BSE, inevitably induce late‐onset brain degeneration with pathological changes in host‐encoded prion protein (PrP). PrP was first identified as a relatively proteinase K (PK) resistant 27–30 kDa protein (PrP‐res, PrPSc, PrPCJD, PrPTSE) in infectious fractions of scrapie infected hamster brain [Bolton et al., 1982]. Shortly thereafter, new purification methods showed that PrP‐res formed amyloid fibrils [Diringer et al., 1983], and comparable abnormal fibrils were also induced by the human sporadic CJD (sCJD) agent [Merz et al., 1983; Manuelidis et al., 1985]. These data led to two fundamentally different interpretations: first, that host PrP‐res is the causal infectious agent, versus second, that PrP‐res is a late amyloid response to infection caused by a foreign environmental pathogen. TSE agents require host PrP for infection [Fischer et al., 1996], a fact that parallels the known viral requirements for specific host proteins. Nevertheless, this PrP requirement, combined with the assertion that PrP‐res levels are proportional to infectious titers [McKinley et al., 1983; Prusiner, 1991], gave great credence to the prion hypothesis, that is, that PrP‐res, without a nucleic acid genome, is the sole component of the infectious entity [Prusiner, 1997].
Whereas the prion hypothesis has engendered an intense focus on models of PrP misfolding and amyloid formation, the major biological characteristics of infectious TSE agents are most consistent with an exogenous viral structure. These features include: (i) the epidemic spread of TSEs in the environment; (ii) host recognition of a foreign entity with demonstrable immune responses elicited before PrP misfolds [Manuelidis et al., 1997; Lu et al., 2004]; (iii) distinct agent strains that do not reflect the PrP‐res band patterns which are cell type and species‐specific [Manuelidis et al., 2009]; (iv) agent replication and latency features that are inexplicable by prion conversion models [Miyazawa et al., 2011b]; and (v) fundamental viral characteristics of the infectious particle including its ∼25 nm size [Manuelidis, 2007; Manuelidis et al., 2007]. Additionally, PrP‐res itself has failed to correlate with infectious titer, or to fulfill Koch's postulates [Manuelidis, 2007; Gonzalez et al., 2012]. Two highly publicized reports claim that recombinant PrP (rPrP) can be converted into an infectious form in vitro [Legname et al., 2004; Wang et al., 2010]. This is critical data because it excludes non‐PrP proteins in addition to nucleic acids from the infectious agent. However, these rPrP findings have never been reproduced by others using independent material [Manuelidis, 2013], and laboratory contamination remains an acknowledged problem with in vitro PrP to PrP‐res conversions [Cosseddu et al., 2011]. Indeed, numerous attempts to make rPrP infectious during the past 8 years have failed to yield any infectivity, and unfortunately only one group has published their negative results [Timmes et al., 2013]. Finally, in the living cell PrP‐res increases to enormously high levels while infectivity is reduced by 4 logs [Miyazawa et al., 2012]. This demonstrates that PrP is part of a defensive host response to eliminate the infectious agent. This build up of host prions (PrP‐res, amyloid) becomes a runaway defense mechanism against the infecting agent. Most critically, PK can destroy virtually all PrP in brain homogenates without reducing the infectious titer [Miyazawa et al., 2011a]; in these experiments PK digested all forms of PrP, including PK sensitive PrP (PrP‐sen), the recently proposed “real” infectious form [Cronier et al., 2008; Colby and Prusiner, 2011].
Because many non‐PrP proteins were visualized in highly infectious but complex brain homogenates after PrP was destroyed, it became critical to isolate and characterize a smaller subset of residual proteins that strongly bind or associate with the infectious agent. One or more of these proteins, especially in more purified agent preparations, can protect an agent with a strain‐specific genome and/or participate in pathogenesis. To better purify infectivity we first established a reproducible rapid culture system for quantitative agent titration with representative CJD and scrapie strains and these in vitro assays were verified by animal assays [Nishida et al., 2005; Liu et al., 2008; Miyazawa et al., 2011a, 2012]. We then developed a new and streamlined procedure that quantitatively recovers the FU‐CJD agent from infected brain homogenates. In contrast to this quantitative recovery, <5% of the starting titer is recovered with purifications of molecularly complex PrP‐res aggregates. Nuclei and abundant brain lipid are removed without detergents and enzymes, and sucrose step gradients that retrieve >80% of the starting brain infectivity reduce PrP to ∼6%, and also minimize other cell proteins. PK digestions of simplified particles again further demonstrated that high particle infectivity is preserved even when PrP is undetectable. In sum, non‐PrP proteins are more critical for agent integrity and infection than any form of detectable PrP.
Although, a few proteins that purify with TSE particles can be visualized on blots, such as nucleic acid binding proteins, more sensitive, broad, and sequence‐specific analyses were needed. LC‐MS/MS, combined with sophisticated computer comparisons of parallel uninfected and infected brain, were advantageous for characterizing particle peptides. LC‐MS/MS is a powerful technique that has extraordinary sensitivity, and is oriented towards the separation, general detection and identification of molecules resolved by their mass and ionization [Fenn et al., 1989]. We used LC‐MS/MS and SWATH to compare particles from normal and FU‐CJD infected brains. We also compared control and sCJD infected human brain particle preparations. We here detail the many viral‐linked proteins that remain in highly infectious FU‐CJD particle preparations that have no detectable PrP by LC‐MS/MS. These robust results are not compatible with the belief that PrP is the major, or even an essential structural element of the infectious particle. The purification and analytic approaches shown can now be used to resolve TSE molecular components that are most fundamental for infection.
MATERIALS AND METHODS
Simplified Purification of Infectious Particles From Brains
Two grams of frozen infected or control uninfected brain, containing 2 × 109 (2e9) cell equivalents (CE), were homogenized with 20 strokes of a smooth glass B type pestle in 9 volumes of TBS (50 mM Tris–Cl pH 8.9, 100 μM PMSF 0.5 μg/ml DAPI). To remove collagen and other large debris, the homogenate was filtered through a Steriflip vacuum‐driven system (Millipore Corp, Bellierica, CA) and the filter washed with TBS to yield ∼7.7e7 CE/ml. A starting reference aliquot of the filtered homogenate was frozen for analytic and infectivity titrations (Fx0). Separation of nuclei from cytosol and myelin‐membranes was facilitated by adjusting the filtrate to 25% sucrose followed by centrifugation at 5,500g × 10 min in a Beckman HS‐4 rotor at 20°C. The small nuclear pellet was strongly DAPI positive for DNA, and the thick floating white membrane‐lipid fraction showed only a weak DAPI fluorescence. The non‐fluorescent cytosol (Fx1) was collected by syringe, adjusted to 8% sucrose with TBS, and centrifuged at 10,000g × 5 min at 22°C to clear residual membrane and nuclear contamination. This yielded a highly infectious 10 k supernatant (s10). The release of 90% of the starting FU‐CJD brain infectivity into Fx1 cytosol was achieved without any detergents, making it possible to study the effect of later added detergents and enzymes such as Triton X‐100 (final concentration of 0.1%) and RNAse, as done here. The same method was used to make p18 particles from control and sCJD cerebral cortex; the latter was from a 67‐year‐old male with typical clinical and pathologic sCJD and positive for PrP‐res by Western blot.
Enzyme Digestions and Sucrose Gradients
The s10 was digested with 120 μg/ml RNAse A (Calbiochem) for 30 min at 37°C, and infectious particles were rapidly concentrated by pelleting at 18,000g × 30 min at 20°C in a microfuge. The use of pooled 18 k pellets (p18), suspended by bath sonication in 20 mM Tris–Cl pH 8.9, enabled increased loads on sucrose step gradients [Sun et al., 2008] for further purification. Briefly, ∼2e7 CE/ml were loaded on gradients and spun in a TLA 55 rotor at 135,000g for 1 h at 20°C. A reduced centrifugation time (1 h vs. 2 h previously) was used to better assess changes in sedimentation after RNAse and Proteinase K (PK). PK digestions, detailed in the Results Section, were done prior to gradient loading and stopped by PMSF [Miyazawa et al., 2011a]. For gradients, 600 μl samples in 8% sucrose were layered over 200 μl of 15%, 150 μl of 25%, and 50 μl of 60% sucrose. Benzonase (BZ, Sigma) was tested at 240, 500, and 650 U/ml for 1 h at 37°C in reactions supplemented with 4 mM MgCl2 and BZ was terminated by 6 mM EDTA pH 7.5.
Titration of FU‐CJD Infectivity and PrP/PrP‐res in Brain Subcellular Fractions
Infectious titers were determined in GT1 neuronal cell assays by dilution [Liu et al., 2008; Miyazawa et al., 2011a,2011b] to determine tissue culture infectious doses (TCID). This bioassay is only threefold less sensitive than prolonged end‐point mouse LD50 bioassays, and is reliable for a variety of mouse‐adapted TSE agent strains including the human Kuru agent and the 22 L and RML scrapie strains [Sun et al., 2008; Miyazawa et al., 2011b]. FU‐CJD infected mouse brain typically contained 0.3 TCID per cell, equivalent to 3.3e8 (3.3 × 108) TCID per gram of brain (e9 cells). Titers done on all fractions in multiple independent experiments were averaged in Table I, as were other molecular components. Western blots for specific proteins such as PrP and tubulin, and colloidal gold staining for other proteins, were done as previously [Manuelidis et al., 2009]. Extracted nucleic acids were assayed by Nanodrop reader and Sybr gold stained agarose gels loaded with 3e6 cell equivalents (CE) of purified nucleic acids [Manuelidis, 2011]; 500 pg per band were easily detected in lanes with diluted markers; this reflects a sensitivity of six orders of magnitude from starting cell DNA (250 μg per 3e6 cells).
Table I.
Sequential Molecular Purification of FU‐CJD Particles
| Starting total (100%) | Membranes/nuclei | Fx1 cytosol | s10 | s18 | p18 | Gradient |
|---|---|---|---|---|---|---|
| A: Infectivity | 10%/2% | 90% | 83% | <5% | 80% | 80% |
| B: Protein | 6%/1% | 90% | 65% | 40% | 12% | <2% |
| C: %PrP/PrP‐res | 30%/0.55× | 70%/0.5× | 30/0.5× | 15%/0.5× | 15%/0.6× | 6%/0.4× |
| D: Nucleic acids | >99% DNA | nd | nd | nd | <0.2% | <0.2% |
Sequential purification of FU‐CJD infectious brain particles from total starting homogenate (100%). Major components in each subcellular fraction (average of six independent experiments) is shown. Infectious titers varied by less than threefold among experiments and recoveries are based on replicate TCID determinations in GT1 neuronal cells in sequential cell passages as previously for FU‐CJD [Miyazawa et al., 2011b] (and see Fig. 4). Individual preparations typically showed 3e8 (3 × 108) TCID/gram brain (e9 cells). The above data summarizes >60 independent infectivity titrations, with average PrP/PrP‐res, proteins, and nucleic acid for each preparation.
Sample Preparation for Mass Spectrometry (MS)
Concentrated mouse and human brain p18 fractions containing ∼30 μg protein were solubilized in fresh 8 M urea/0.4 M NH4HCO3 with 4 mM DTT for 20 min at 22°C, quenched with iodoacetamide to 8.3 mM, and diluted to <2 M urea prior to digesting with 5 μg Lys C for 4 h at 37°C followed by sequential sequencing grade trypsin (Promega) digestions for ∼3 h (5 μg), and then overnight (2 μg) for 4 h. For p18 + PK samples, ∼8× the CE were treated as above, except trypsin was reduced by ½ because of the markedly reduced protein content (see the Results Section). Samples were then desalted and run for LC‐MS/MS and SWATH as parallel pairs by members of the Yale Keck facility as described (Supplemental Information).
RESULTS
We first developed procedures to better purify and expose infectious brain particles to test their inherent sensitivity to enzymatic digestions. The variety of heterogeneous and uncharacterized components of most previous infectious brain fractions, as well as TSE agent trapped by PrP amyloid, can compromise digestions. Close to quantitative yields of starting brain infectivity along with increased agent purity was achieved, whereas our previous disaggregation approaches yielded only 15% of the starting homogenate titer [Manuelidis et al., 1995]. The new streamlined procedure detailed here for brain FU‐CJD particles is representative (Fig. 1). It has been used with success for other TSE strains/species models, and comparable subcellular characteristics were obtained with 263 K hamster scrapie brain. Although, human brain sCJD titers cannot be accurately evaluated by human inoculation, the overall protein and PrP reductions in human sCJD followed the same pattern as found in FU‐CJD. Hence, more purified human sCJD particles are also compared to uninfected controls in proteomic analyses here.
Figure 1.
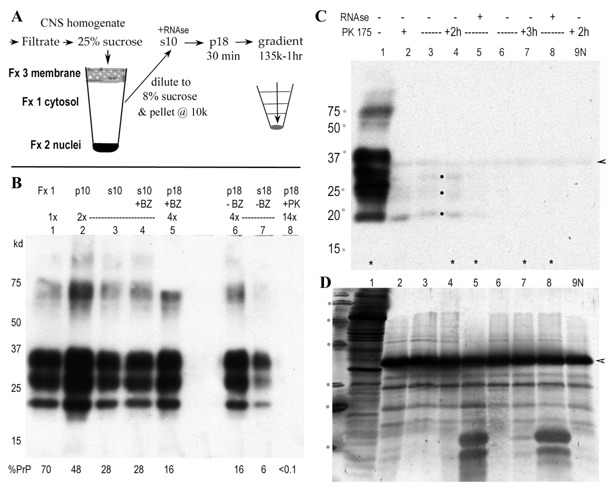
Purification and rapid concentration of FU‐CJD infectious particles from brain. A: Diagrams the new streamlined procedure with major infectious fractions (Fx). The brain cytosol (Fx1), spun at 10,000g, yields a highly infectious supernatant (s10) with reduced lysosomes, mitochondria, and pathological debris. The 18,000g pellet (p18) concentrates infectious particles, reduces soluble proteins and also allows increased loads on sucrose step gradients for further purification. B: Western blot shows typical FU‐CJD brain PrP, with the % of total PrP in each sequential fraction indicated under each lane. Note the p10 contains large amounts of PrP, but little infectivity (<10%, Table I). The addition of enzymes (BZ and PK) are indicated above each lane, as well as the relative load of each fraction. Note the 14× load of p18 in lane 5 after PK (100 μg/ml × 2 h) shows no visible PrP, with <0.1% of the starting PrP detectable by chemifluorescence. In contrast, Benzonase at 500 U/ml has no effect on p18 PrP (compare +BZ and −BZ lanes). Markers (kDa) are indicated. C: Western blot shows PrP in crude Fx1 cytosol before (lane 1) or after PK digestion (175 μg/ml) for 1.5 h (lane 2), or for 2 h (lanes 3–5) or 3 h as indicated. In addition samples in lanes 4 and 8 were treated with RNAse prior to PK. The 2 h digest with RNAse (lane 5) has less visible PrP‐res bands (dots), and little PrP‐res is seen in 3 h digests. The control lane with the same load of uninfected brain (lane 9 N) shows the PK band (at arrowhead) that binds some PrP antibody non‐specifically. Without any background subtraction the 3 h digests had as little as 0.3% starting PrP, and samples in lanes marked with an * were assayed for infectivity (see Fig. 2A). D: Gold stain of blot in C showing prominent residual protein bands after PK with the intense PK band (29 kDa at arrowhead) and smaller RNAse bands (at 12 and 14 kDa in lanes 5 and 8). RNAse was effectively removed in by p18 centrifugation.
Progressive Agent Purification With Significantly Reduced PrP and Nucleic Acids
Titrations of the FU‐CJD agent showed enhanced separation of TSE agent particles from the brain's cellular complexity, high lipid content, and secondary neurodegenerative pathology (Table I). Moreover, agent isolation here contrasts with standard sarkosyl procedures to “purify” PrP amyloid aggregates that yield only 0.1–5% of the starting titer [Hilmert and Diringer, 1984; Weber et al., 2008]. Nuclear contamination, poor separation of agent from myelin, lipid‐rich membranes, pathological debris, blood vessels, and collagen all compromised previous agent purifications. We found that: (1) filtration of homogenates removes virtually all blood vessels and collagen with no loss of starting titer; (2) addition of sucrose to 25% stabilizes nuclei and improves their separation by low speed pelleting. This high sucrose concentration also separates the majority of floating lipid‐rich membranes, and at the same time removes 30% of starting PrP and PrP‐res; (3) Omission of detergents and their impermeable micelles enhances separation of 90% of the starting brain infectivity into the cytosol without artifactual aggregation (Fx1, Fig. 1A and Table I); and (4) We also discovered that RNAse treatment facilitates concentration of essentially all infectious particles by a simple 18,000g × 30 min centrifugation. Moreover, these more purified p18 particles contain relatively low protein and nucleic acid, features necessary for detection of agent‐specific molecules. The method outlined (Fig. 1A) is straightforward, takes only 1 day, and a rapid sucrose step gradient further purifies infectious particles from residual proteins that are not required for infection.
Figure 1B shows a typical Western blot of PrP in the sequential subcellular FU‐CJD brain fractions. The cytosol (Fx1) contains 70% of the starting brain PrP, including abnormal PrP‐res. Fx1 (lane 1) is further purified by a 10 kg spin for 5 min. This pellets residual membranes and nuclei (p10, lane 2) with little loss of agent. Additional PrP (30%) is removed, while the 10 k supernatant contains the remaining 28% of total PrP (s10, lane 3). Benzonase (BZ) digestion of nucleic acids does not reduce the PrP content as shown with more purified p18 particles after BZ (lane 5) compared to its undigested p18 control (lane 6). Thus BZ should not reduce infectious titer according to the prion protein‐only hypothesis. In stark contrast, digestion of p18 particles with 100 μg PK for 2 h shows no visible PrP (lane 8), even though this lane contains a >3‐fold higher load than lanes 5–7. In principle, such PK digested “prionless” samples should lose 3 logs of infectivity in accord with a loss of 99.9% of the starting total PrP but the infectious titer was not reduced (see Fig. 2B). No background was subtracted, and the 0.1% PrP value can reflect the residual 29 kDa PK band that binds PrP antibodies. For example, 3 h digests of Fx1 with very high PK (175 μg/ml, Fig. 1C, lanes 6–8) show this 29 kDa PK band (arrowhead). Other proteins, subsequently stained with gold on the same blot (Fig. 1D) are also markedly reduced in the PK+ lanes (compare undigested lane 1), while the added PK at 29 kDa is the dominant band. Although, PrP is barely detectable in these 3 h PK digests, many other gold‐stained proteins from 15 to 75 kDa clearly survive PK digestion. Large amounts of RNAse, added after PK was stopped (Fig. 1D, lanes 5 and 8, bands at 13–15 kDa) are removed by concentration of p18 particles. The above data encouraged further evaluation of agent particles in sequentially purified fractions with high infectivity.
Figure 2.

Infectivity in multiple experiments of different cell fractions. The PK conditions are noted at top and the %PrP remaining are shown at bottom for each sample, with the parallel undigested control (dark bar at left of each group). A: The crude Fx1 shows no loss of titer with either 2 or 3 h PK at a high concentration. The SEM for each is indicated (n = 8). Gel blots of these assayed samples are shown in Figure 1C,D. B: The p18, digested for 6 h with PK after RNAse, shows a low PrP that is not meaningful because a value of <0.2 cannot be distinguished from background [Miyazawa et al., 2011a]. Regardless, infectivity remains high, and indistinguishable from its parallel control with appreciable PrP (as shown in Fig. 1). C: The total infectivity recovered from sucrose step gradients where s10 was treated with 100 μg/ml for 2 h before loading gradients. Note the same high TCID is found in these recovered control, RNAse only and PK only gradients, even though no PrP could be detected by any means (“0” bar). The different gradient steps of the PK sample are shown in Figure 3, with its strong positive TCID shown in Figure 4. D: Other s10 brain fractions, treated with high BZ at concentrations indicated, show a significant 1 log loss of titer in contrast to PrP‐less PK samples. Triton X‐100 (TX) at 0.1% did not alter agent titers.
Titers of the Infectious Agent in FU‐CJD Brain Fractionation Steps
Table I also summarizes infectivity, determined by established dilution titrations (see Methods), along with the sequential stepwise reduction of total protein, PrP, and nucleic acids. These results were highly reproducible and represent averages for six independent brain preparations with >60 determinations of tissue culture infectious doses (TCID) per sample fraction. Non‐infectious proteins and lipids are visibly reduced in the p18 and sucrose gradient sediment, with low final amounts of all forms of PrP (6%) in the gradient sediment. PrP‐res is also reported for each subcellular fraction because it is the presumed infectious form, and PrP‐res represents 50–60% of the total FU‐CJD brain PrP, that is, a factor of 0.55×. PrP‐res in all the subcellular fractions was reduced in parallel with total PrP. Quantitative recovery of infectivity was routinely obtained in the p18 and sedimenting gradient particles. In contrast, the vast majority of PrP and PrP‐res are removed without any detergent. Other proteins were also reduced, and nucleic acids were substantially removed as indicated.
Digestion of PrP in Infectious Subcellular Fractions Does Not Reduce Infectivity
To minimize artifacts induced by preparative procedures, whole brain homogenates were used previously to test PK induced PrP loss on infectious brain titers [Miyazawa et al., 2011a]. For reference, maximal PrP‐res bands are visualized with very limited PK (25 μg/ml for <30 min). Although, previously all forms of PrP in whole brain homogenate were reduced to invisible levels with more extreme PK digestions (250 μg for 2 h) without loss of titer, many other homogenate proteins survived; similarly as shown above for the less complex Fx1 cytosol (Fig. 1D). PrP was again reduced and thus the more purified subcellular infectious fractions were pursued to further minimize the set of proteins most critical for agent structure and resistance. With progressive purification, less obvious pathologic host components that adhere most tightly to infectious particles should also be revealed. Whereas aggregating PrP can trap the infectious agent and also entangle other brain proteins, including ubiquitin and Apolipoprotein J [Manuelidis et al., 1997], only a few of these may be retained with increasing agent purification, especially after PK digestion.
A series of infectivity titrations of different subcellular fractions all showed that PK abrogated both PrP and PrP‐res, but did not reduce infectivity. Figure 2 graphs the TCID of complex to more purified agent preparations, with and without different additives as noted beneath each bar. Each set of experiments (bar clusters A to D) shows an independent brain preparation, with the non‐PK parallel fraction (darker bars in each cluster). In cluster A, the Fx1 cytosol was treated with high levels of PK (175 μg/ml) for 2 and 3 h, as indicated above the bars. Western blots of aliquots titered are shown in Figure 1C, with the untreated control (lane 1), and the four digested samples assayed for infectivity in lanes marked by an asterisk. The % PrP of the starting homogenate (Fx0) for each sample is also indicated beneath each bar. Reduction of PrP from 70% to 0.3% did not reduce infectivity of Fx1, and RNAse also had no effect on titer. Moreover, a more purified p18, treated with 50 μg/ml for 6 h (B cluster), resulted in even less retained PrP (0.2% of the total). Despite this 3 log reduction in PrP, the infectious titer was again completely preserved without any loss. By comparison, 8 M urea‐trypsin treatments for SWATH analysis did not alter the PrP levels, but instead reduced the starting p18 titer by >4 logs.
To avoid artifactual PrP aggregation and trapping of agent that can be induced by high g pelleting, we used the s10 for separation of particles on sucrose gradients. Three sucrose step gradients were compared in parallel. The first s10 aliquot contained only Triton X‐100, the second was additionally treated with RNAse, and the third was digested with PK at 100 μg/ml for 2 h before loading on the gradient. Both the PrP and gold stained blot for the PK digested sample are shown in Figure 3. Lane 4, after PK, has no visible PrP signal as compared with the undigested aliquot (lane 2), even though the PK lanes have a 4× higher load. Furthermore, even at 20–30× loads (where the 30× lane contains a concentrate of 107 CE), the individual sucrose fractions collected (lanes 5–8) still show no detectable PrP. Despite the lack of any detectable PrP, other visible gold stained proteins remained, in addition to the strong PK band in each gradient fraction.
Figure 3.

Blot of samples before gradient analysis and collected from the sucrose steps. The top shows the PrP and the bottom, shows subsequent colloidal gold staining for other proteins. The s10 before PK has 34% of the total cell PrP as indicated (lane 2) but only 0.4% after PK prior to loading on the gradient. No PrP is recovered in the sucrose steps (top) even with very high CE loads (107 CE in the 30× lane). Other proteins, however, are still visible by gold staining (bottom) in addition to the heavy 29 kDa PK band.
Infectivity assays showed two remarkable features of the PK digested sucrose gradient fractions. First, all s10 infectivity was recovered from the gradient, and second, there was a shift in sedimentation of the infectious agent indicating infectious particles had a smaller S value (apparent size) than control particles. Figure 4 compares the TCID assays of the PK treated (blot A) and the RNAse only control (blot B) gradient fractions. A strong PrP‐res response in indicator GT1 cells, as early as p3 post‐infection with low CE, indicates a very high titer [Miyazawa et al., 2011a,2011b]. In A, the sucrose fractions without PrP induce a vigorous PrP‐res response in GT1 cells (+PK lanes), and 85% of the recovered infectivity is in the 15% sucrose fraction. In blot B from the RNAse only gradient, the same total TCID are recovered, but the infectious particles rapidly pass through the 8–15% sucrose steps and are concentrated in the bottom 25–60% sucrose steps. Control uninfected brain (N‐CNS lane in blot B), in contrast to the FU‐CJD samples, induces no PrP‐res in this GT1 assay. The total infectivity recovered from each of the three gradients was indistinguishable and quantitative, and is graphed in Figure 2C. This result indicates a smaller TSE particle size (∼60–80S) than previously determined, but one that is still in keeping with ultrastructural ∼25 nm diameter dense particles observed in infected cells [Manuelidis et al., 2007]. Interestingly, BZ digestion of nucleic acids had a greater effect on titer than PK. Figure 2D shows BZ at two different concentrations reduced titer by 1 log. In the past, nuclease treatment of aggregated pelleted material has not caused a loss of infectivity [Manuelidis, 2011], and the nuclease sensitivity of more purified agent particles here deserve further investigation. In sum, the current results show that proteins other than PrP are essential for TSE agent infectivity.
Figure 4.

Early passage appearance of infectivity in sucrose gradient steps from (A) PK treated (shown in Fig. 3) and (B) RNAse only treatment. Identical CE of each gradient sample were inoculated into duplicate wells and indicator GT1 cell wells undigested or treated with limited PK (+) for PrP‐res are shown. The total TCID recovered in each gradient is the same (Fig. 2) and strikingly, gradient samples without PrP are highly infectious. PK pre‐treatment also reduces the apparent size of infectious particles; 85% of the infectivity in the prionless samples migrates in the 15% sucrose step. In contrast, panel B shows >78% of the TCID concentrates in the bottom small 60% cushion with <3% is in the 15% sucrose step, in accord with previous undigested particle studies [Sun et al., 2008]. Gradient recoveries were slightly higher than 100% and the arrowhead points to the FU‐CJD specific band found only in GT1 cells but not brain [Manuelidis et al., 2009]; this PrP‐res folding change in GT1 cells does not alter the agent's intrinsic characteristics as determined by re‐inoculation into mice [Arjona et al., 2004]. The standard curve for TCID assays is determined by serial dilution [Miyazawa et al., 2011b] and the end‐point titer is the most dilute CE that can induce a PrP‐res response in the GT1 indicator cells. A control normal brain sample (N CNS lanes in B) shows that no PrP‐res is provoked by uninfected homogenates.
LC‐MS/MS Analysis of Infectious Fractions
While PrP has been the singular focus of most TSE work for the last 20 years, the above data, combined with previous sucrose gradient separations of infectious particles [Sun et al., 2008] motivated a closer look at the minor set of proteins remaining in more purified agent preparations. We have detected several protein subtypes in p18 particles, such as nucleic acid binding proteins [Manuelidis, 2011], including their affinity to biotinylated recombinant Sphinx DNA sequences. We have also detected protein bands that bind antibodies to Sphinx ORFs (data not shown). However, because the total proteins in the p18, and especially in the PK treated p18 particles, had low protein levels, it was advantageous to use more sensitive and inclusive approaches for analysis. Whereas the high titer of FU‐CJD p18 particles used for MS were not reduced by PK, they lost >4 logs of infectivity with urea‐trypsin treatments (even without Lys C) prior to MS analysis. This obviated a MS biohazard risk.
As expected from Western blots, MS of both the control and infected p18 samples not subjected to PK showed many proteins including PrP. The FU‐CJD p18 contained 2.37‐fold more PrP (P = 0.03) than the uninfected control, consistent with preferential sedimentation of PrP‐res aggregates that are not present in control samples. However, after PK, neither sample had detectable PrP by MS, and it is possible that the marginal 29 kDa band we detected in a 20× overloaded Western blot of this FU‐CJD sample (0.05% of the starting PrP) reflected other peptides that can cross react with the M20 anti‐PrP polyclonal antibody. Indeed, several cellular and viral peptidases and proteases were clearly present after PK, in addition to a 29 kDa viral‐linked protein (#41, Table II). The PK sequence used for digestion was also not detectable, further solidifying the unique origins of the proteases found only in the infected sample.
Table II.
Viral‐Linked Proteins in PK+ FU‐CJD Infectious Particles But Not in PK+ Uninfected Controls
| # | Protein ID | Protein name | Score | Comment |
|---|---|---|---|---|
| 1 | gi|443296517 | Envelope glycoprotein (human immunodeficiency virus 1) | 47 | Malawai; CNS child isolate, |
| 2 | gi|283467475 | rep protein (pepper leaf curl Lahore Virus—Pakistan:Lahore) | 46 | REP (replication) protein |
| 3 | gi|3335088 | Sp‐l11L (African swine fever virus) | 46 | Virulence region |
| 4 | gi|337731304 | Hypothetical protein (EBPR siphovirus 4) | 44 | Terminase domain |
| 5 | gi|2465192 | Polyprotein (bovine viral diarrhea virus 1) | 43 | Viral nucleic acid bind‐protect |
| 6 | gi|295856919 | gag protein (human immunodeficiency virus 1) | 42 | Capsid |
| 7 | gi|310831201 | Putative mRNA capping enzyme (Cafeteria roenbergensis virus) | 41 | |
| 8 | gi|402760859 | Hypothetical protein Phi87_58 (Enterobacteriaphage UAB_Phi87) | 41 | Spain |
| 9 | gi|322511129 | Hypothetical protein 162275982 (organic lake phycodnavirus 2) | 40 | Antartica, Ebola nucleoprotein family |
| 10 | gi|13447465 | DNA polymerase (caprine herpesvirus 2) | 39 | DNA polymerase |
| 11 | gi|158347774 | Envelope glycoprotein (human immunodeficiency virus 1) | 39 | China, pfam00516 |
| 12 | gi|109638628 | ORF88 (Ranid herpesvirus 2) | 38 | Capsid maturational protease |
| 13 | gi|371944963 | Putative replication factor C small subunit (Moumouvirus Monve) | 38 | REP factor C |
| 14 | gi|42716918 | pol protein (human immunodeficiency virus 1) | 36 | Argentina |
| 15 | gi|325145933 | pol protein (human immunodeficiency virus 1) | 36 | Reverse transcriptase like |
| 16 | gi|383398029 | Hypothetical protein (environmental halophage eHP‐35) | 35 | High saline metavirome |
| 17 | gi|151303250 | Unglycosylated membrane protein (equine arteritis virus) | 35 | ENV protein |
| 18 | gi|326633026 | Major tail protein (enterobacteria phage SPC35) | 35 | |
| 19 | gi|390635675 | RNA‐dependent RNA polymerase | 35 | Bat paramyxovirus |
| 20 | gi|327197952 | Putative ribonucleotide diphosphate reductase alpha | 35 | |
| 21 | gi|89515504 | orf1ab polyprotein (human coronavirus HKU1) | 34 | Conserved in bacteria |
| 22 | gi|396587254 | Polyprotein (Arracacha mottle virus) | 34 | Brazil plant |
| 23 | gi|38229178 | 13L (Yaba monkey tumor virus) | 33 | Poxvirus, lipase family |
| 24 | gi|38683765 | FirrV‐1‐C7 (Feldmannia irregularis virus a) | 33 | Algae virus, latent infections |
| 25 | gi|203454737 | gp144 (Mycobacterium phage Myrna) | 33 | |
| 26 | gi|525335124 | Tail fiber protein (Bacillus phage JL) | 33 | |
| 27 | gi|526119926 | Hypothetical protein (Pandoravirus dulcis) | 32 | Australia, ameoba virus |
| 28 | gi|326439160 | Hypothetical protein (Mavirus) | 32 | At origin large DNA transposons |
| 29 | gi|15927577 | Hypothetical protein SA1809 (Staphylococcus aureus subsp.) | 32 | |
| 30 | gi|410094842 | Protease, partial (human immunodeficiency virus 2) | 32 | |
| 31 | gi|410442733 | Polyprotein (grapevine leafroll‐associated virus 3) | 31 | |
| 32 | gi|19774246 | 7 kDa protein (tobacco necrosis virus D) | 31 | Hungary, needed for cell–cell transfer |
| 33 | gi|311993471 | Hypothetical protein Acj9p199 (Acinetobacter phage Acj9) | 31 | |
| 34 | gi|564271659 | Hypothetical protein (Oenococcus phage phiS11) | 31 | |
| 35 | gi|507866687 | Polyprotein, partial (bovine viral diarrhea virus 1) | 31 | China, capsid |
| 36 | gi|345049924 | Envelope glycoprotein, partial (human immunodeficiency virus 1) | 30 | |
| 37 | gi|167375588 | DNA polymerase catalytic subunit, partial (Equid herpesvirus 1) | 30 | Brazil, neurotropic and neuroviruent |
| 38 | gi|460838096 | Glycoprotein B (Lagenorhynchus alphaherpesvirus 1) | 30 | Pacific dolphin, glycprotein |
| 39 | gi|422936725 | Connector (Clostridium phage phi24R) | 30 | |
| 40 | gi|284504441 | Serine/threonine protein kinase (Marseillevirus) | 29 | |
| 41 | gi|15078883 | 170 L (invertebrate iridescent virus 6) | 29 | 29 kDa, insect |
| 42 | gi|559104958 | Capsid protein 2, partial (Israeli acute paralysis virus) | 29 | China, capsid picornavirus |
The PK treated p18 particles proved most informative when analyzed for sequences that could be clearly identified in FU‐CJD, but not in the parallel uninfected control. Their absence from the normal p18 does not mean that they are not present, but only that they are preferentially bound to infectious particles rather than with co‐sedimenting cellular proteins. PK treatment also allowed discovery of molecules that were insufficiently represented in the more complex undigested sample, and these numerous cellular proteins quantitatively differentiated by SWATH will be detailed separately. Table II lists 42 sequences with the highest identity viral scores that were not detectable in the parallel uninfected PK+ control. The remaining significant 72 peptides (scores >20) are listed in the supplement. First, it is notable that many different RNA and DNA viral groups are represented including retroviruses (HIV 1), coronaviruses, herpesviruses (goat and frog origin), plant viruses (pepper leaf and grapevine), insect and bacterial viruses. None of these viruses have ever been in our lab, and they are unlikely to be contaminants because infected and control samples without these sequences were always prepared in parallel with identical reagents. Secondly, a fair number of sequences in the NCBI viral database were identified in limited geographic locations unlikely to be in our environment, such as Antarctic lakes, China, Hungary, Brazil, and Australian seawater. Thirdly, the significant viral identities are limited to a few protein classes, most notably, ENV glycoproteins, viral replication and polymerase proteins, and capsid (nucleic acid binding) proteins. Together, the foregoing suggests these sequence motifs may function in several disparate viral classes. These peptides would never have been identified in high background samples, and were brought out by PK treatment that markedly reduced other proteins not required for infection. Among the replicase peptides, it is also notable that bacterial REP proteins, further confirmed DNA sequences of particle REP nucleic acids [Manuelidis, 2011]. Although these and other sequences on the list may not be integral agent components, they, as well as others that may be discovered in enriched particle preparations, deserve further study to determine their potential roles in infection and particle processing.
Because the human sCJD p18 could not be accurately titered, PK was not used to further purify human sCJD particles. However, a few polyproteins from diverse viruses (from onions to mammals) were identified in the non‐PK sCJD that were not detected in the control human brain. In view of the higher protein background in these non‐PK treated samples, these sequences may signify particle processing and pathologic products rather than integral agent molecules. On the other hand, the human genes identified in sCJD, but not the control human p18, began to give a strong indication of relevant pathological and neuronal repair sequences that were particle associated. High on the identity list, unexpectedly, were the neural Wiskott‐Aldridch protein and Huntintin. In addition, expected cell proteins including vesicle, endosomal, multivesicular, and chaperone proteins that are likely to be involved in agent trafficking and processing were also obvious. Expected mitochondrial proteins were also identified, consistent with the co‐sedimenting mitochondrial DNA detectable by PCR in every infectious preparation to date, including those treated with nucleases [Manuelidis, 2011]. The extensive cellular genes identified with their relevance to these and other TSE models will be detailed separately.
DISCUSSION
An exclusive focus on PrP, with the dominant belief that PrP‐res, a misfolded host protein without nucleic acid, is the causal infectious TSE agent, has obscured the unresolved nature of the real infectious particle. All infectious preparations, when tested by sensitive molecular methods, contain appreciable nucleic acids of viral size (>1 kb) as well as proteins [Manuelidis, 2011]. We are not aware of any assessments or detailed characterizations by others of non‐PrP proteins in more purified TSE agent preparations. Disruption of nucleic acid–protein complexes destroys infectivity [Manuelidis et al., 1995; Manuelidis, 2007]. In contrast, reproducible quantitative recovery of highly infectious TSE particles without detectable PrP is clear. Marked reductions in PrP and other background proteins by PK digestion led to substantial proteomic data demonstrating non‐PrP proteins strongly bind to or are essential for TSE agent infectivity. One or more of these viral and cellular proteins could protect a strain‐specific genome. The rapid purification method detailed here is reasonably universal because it has been effective for both the FU‐CJD and 22 L scrapie agents. Thus, it should be effective in other models, including high titer 263 K scrapie and sCJD agent infected hamster brains. Moreover, with this streamlined procedure, isolated FU‐CJD brain particles contained >3 logs more infectivity than found in most test tube PrP‐res conversion assays done under a variety of conditions [Castilla et al., 2005; Cosseddu et al., 2011; Klingeborn et al., 2011]. Thus, the current PK treated infectious particles are a high‐yield source for impartially identifying critical non‐PrP agent components. This approach opens the window of discovery, but it will take more work to define the integral agent sequences, including viral nucleic acids and their protective capsid proteins.
Although, it is stated that there are no viral particles in TSEs [Prusiner, 1997], virus‐like arrays of electron dense ∼25 nm particles have been identified in the cytoplasm of infected but not normal cells, and are clearly separated from the abundant intracellular PrP amyloid fibers [Manuelidis et al., 2007]. These ultrastructural particles are comparable to the biochemically isolated infectious particles of 17–27 nm that separate from abnormal PrP fibrils [Silveira et al., 2005; Manuelidis, 2007]. The ability of viruses to induce misfolded host proteins and pathological fibrillar aggregates has been increasingly realized on a detailed molecular level [Hou et al., 2011; Manuelidis, 2013], and the present studies are most consistent with PrP changes that are part of a host innate defense mechanism. Indeed, PrP misfolding can become a runaway pathological response to infection by environmental TSE agents [Miyazawa et al., 2012]. Additionally, stress induced protein aggregates, from bacterial biofilms to Alzheimer's amyloid, neurofibrillary tangles, and other neurodegenerative diseases where accumulation of abnormal protein fibrils is prominent, further underscore their pathologic unity [Manuelidis, 2013]. While others may wish to pursue the invisible PrP in infectious PK treated brain samples, study of the more abundant viral‐linked proteins that associate with TSE infectious particles would seem to be more fruitful. Viral‐linked peptide sequences enumerated here can be used immediately to synthesize corresponding cDNA oligonucleotides to identify agent probes that are positive in different TSE models. Common strain‐specific molecules can also be defined by testing cDNA probes on different human agents already propagated in a range of species and cell types [Manuelidis, 2013].
The current proteomic analyses also identified numerous cellular proteins likely to be important in new diagnostic and therapeutic approaches, although the quantitative comparative SWATH data for control and infected samples will be reported separately. However, it is already clear that the cellular genes presented here can drive the discovery of new host targets for delaying agent‐induced neurodegeneration, aggregation, and amyloid formation. The surprising finding here of several proteins associated with human sCJD particles, but not in normal control brain, included Wiskott–Aldridch protein as well as the expected vacuolar–endosomal and vesicular trafficking retromer proteins. Particular sets of these proteins in the CSF may be able to diagnostically distinguish different types of dementias at an early clinical stage. Several of these peptides also specify new pathways to target therapeutically. The above results suggest that the pharmacologic chaperones directed against retromers, currently being investigated for delaying Alzheimer's disease [Mecozzi et al., 2014], may also retard the accumulation of PrP amyloid and associated neurodegenerative changes in TSEs.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information.
ACKNOWLEDGMENTS
Preliminary particle experiments were supported by National Institute of Neurological Diseases and Stroke Grant RO1 012674‐34, while Mass Spectrometry and non‐PrP initiatives were kindly funded by the William Prusoff foundation, a Yale departmental OSHE grant, and contributions of families of CJD patients. We are especially indebted to Chris Colangelo and Thomas Abbott of the Yale Keck center for their analytic MS expertise and quantitative SWATH analyses.
REFERENCES
- Arjona A, Simarro L, Islinger F, Nishida N, Manuelidis L. 2004. Two Creutzfeldt‐Jakob disease agents reproduce prion protein‐independent identities in cell cultures. Proc Natl Acad Sci USA 101:8768–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton DC, McKinley MP, Prusiner SB. 1982. Identification of a protein that purifies with the scrapie prion. Science 218:1309–1311. [DOI] [PubMed] [Google Scholar]
- Castilla J, Saa P, Hetz C, Soto C. 2005. In vitro generation of infectious scrapie prions. Cell 121:195–206. [DOI] [PubMed] [Google Scholar]
- Colby D, Prusiner SB. 2011. Prions. Cold Spring Harb Perspect Biol 3:epub a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosseddu G, Nonno R, Vaccari G, Bucalossi C, Fernandez‐Borges N, Di Bari M, Castilla J, Agrimi U. 2011. Ultra‐efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog 7(11):e1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronier S, Gros N, Tattum M, Jackson G, Clarke A, Collinge J, Wadsworth J. 2008. Detection and characterization of proteinase K‐sensitive disease‐related prion protein with thermolysin. Biochem J 416:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diringer H, Gelderblom H, Hilmert H, Ozel M, Edelbluth C, Kimberlin RH. 1983. Scrapie infectivity, fibrils and low molecular weight protein. Nature 306:476–478. [DOI] [PubMed] [Google Scholar]
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. 1989. Electrospray ionization for the mass spectrometry of large biomolecules. Science 246:64–71. [DOI] [PubMed] [Google Scholar]
- Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. 1996. Prion protein (PrP) with amino‐proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Gonzalez L, Thorne L, Jeffrey M, Martin S, Spiropoulos J, Beck KE, Lockey RW, Vickery CM, Holder T, Terry L. 2012. Infectious titres of sheep scrapie and bovine spongiform encephalopathy agents cannot be accurately predicted from quantitative laboratory test results. J Gen Virol 93:2518–2527. [DOI] [PubMed] [Google Scholar]
- Hilmert H, Diringer H. 1984. A rapid and efficient method to enrich SAF‐protein from scrapie brains of hamsters. Biosci Rep 4:165–170. [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. 2011. MAVS forms functional prion‐like aggregates to activate and propagate antiviral innate immune response. Cell 146:448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingeborn M, Race B, Meade‐White K, Chesebro B. 2011. Lower specific infectivity of protease‐resistant prion protein generated in cell‐free reactions. Proc Natl Acad Sci 108:1244–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legname G, Baskakov I, Nguyen H, Riesner D, Cohen F, DeArmond S, Prusiner S. 2004. Synthetic mammalian prions. Science 305:673–676. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sun R, Chakrabarty T, Manuelidis L. 2008. A rapid accurate culture assay for infectivity in transmissible encephalopathies. J Neurovirol 14:352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZH, Baker C, Manuelidis L. 2004. New molecular markers of early and progressive CJD brain infection. J Cell Biochem 93:644–652. [DOI] [PubMed] [Google Scholar]
- Manuelidis L. 2007. A 25 nm virion is the likely cause of transmissible spongiform encephalopathies. J Cell Biochem 100:897–915. [DOI] [PubMed] [Google Scholar]
- Manuelidis L. 2011. Nuclease resistant circular DNAs copurify with infectivity in scrapie and CJD. J Neurovirol 17:131–145. [DOI] [PubMed] [Google Scholar]
- Manuelidis L. 2013. Infectious particles, stress, and induced prion amyloids: A unifying perspective. Virulence 4:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L, Chakrabarty T, Miyazawa K, Nduom NA, Emmerling K. 2009. The kuru infectious agent is a unique geographic isolate distinct from Creutzfeldt‐Jakob disease and scrapie agents. Proc Natl Acad Sci USA 106:13529–13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L, Fritch W, Xi YG. 1997. Evolution of a strain of CJD that induces BSE‐like plaques. Science 277:94–98. [DOI] [PubMed] [Google Scholar]
- Manuelidis L, Sklaviadis T, Akowitz A, Fritch W. 1995. Viral particles are required for infection in neurodegenerative Creutzfeldt‐Jakob disease. Proc Natl Acad Sci USA 92:5124–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L, Valley S, Manuelidis EE. 1985. Specific proteins in Creutzfeldt‐Jakob disease and scrapie share antigenic and carbohydrate determinants. Proc Natl Acad Sci USA 82:4263–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L, Yu Z‐X, Barquero N, Mullins B. 2007. Cells infected with scrapie and Creutzfeldt‐Jakob disease agents produce intracellular 25‐nm virus‐like particles. Proc Natl Acad Sci USA 104:1965–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley MP, Bolton DC, Prusiner SB. 1983. A protease‐resistant protein is a structural component of the scrapie prion. Cell 35:57–62. [DOI] [PubMed] [Google Scholar]
- Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, Small SA. 2014. Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol 10:443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz PA, Somerville RA, Wisniewski HM, Manuelidis L, Manuelidis EE. 1983. Scrapie associated fibrils in Creutzfeldt‐Jakob disease. Nature 306:474–476. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Emmerling K, Manuelidis L. 2011a. High CJD infectivity remains after prion protein is destroyed. J Cell Biochem 112:3630–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K, Emmerling K, Manuelidis L. 2011b. Replication and spread of CJD, kuru and scrapie agents in vivo and in cell culture. Virulence 2:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K, Kipkorir T, Tittman S, Manuelidis L. 2012. Continuous production of prions after infectious particles are eliminated: implications for Alzheimer's disease. PLoS ONE 7:e35471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N, Katamine S, Manuelidis L. 2005. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science 310:493–496. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. 1999. Probability‐based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567. [DOI] [PubMed] [Google Scholar]
- Prusiner S. 1997. The Nobel Lecture: Prions. Karolinska Institutet, Stockholm: Nobelprize.org. [Google Scholar]
- Prusiner SB. 1991. Molecular biology of prion diseases. Science 252:1515–1522. [DOI] [PubMed] [Google Scholar]
- Shifman MA, Li Y, Colangelo CM, Stone KL, Wu TL, Cheung KH, Miller PL, Williams KR. 2007. YPED: A web‐accessible database system for protein expression analysis. J Proteome Res 6:4019–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira J, Raymond G, Hughson A, Race R, Sim V, Hayes S, Caughey B. 2005. The most infectious prion protein particles. Nature 437:257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Liu Y, Zhang H, Manuelidis L. 2008. Quantitative recovery of scrapie agent with minimal protein from highly infectious cultures. Viral Immunol 21:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmes AG, Moore RA, Fischer ER, Priola SA. 2013. Recombinant prion protein refolded with lipid and RNA has the biochemical hallmarks of a prion but lacks in vivo infectivity. PLoS ONE 8:e71081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Wang X, Yuan C‐G, Ma J. 2010. Generating a prion with bacterially expressed recombinant prion protein. Science 327:1095–9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber P, Reznicek L, Mitteregger G, Kretzschmar H, Giese A. 2008. Differential effects of prion particle size on infectivity in vivo and in vitro. Biochem Biophys Res Commun 369:924–928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information.