

Abstract
Human metapneumovirus (hMPV) has been identified previously as a cause of respiratory outbreaks in adults, including the elderly. The objective of this study was to document respiratory outbreaks that were caused by hMPV in Ontario, Canada and to identify the various circulating genotypes during April 2009–February 2012. The majority of the outbreaks that were part of this study were in adults (>65 years). Total nucleic acid extraction was done on 123 residual anonymized clinical specimens from 51 different respiratory outbreaks. Specimens were subjected to PCR amplification and Sanger sequencing targeting the F and G genes of hMPV. Phylogenetic analysis was performed to identify genotypes. HMPV accounted for 195 (8.5%) of 2,292 respiratory outbreaks. Genotype A2b was most prevalent, detected in 28 (54.9%) of 51 typed hMPV‐positive outbreaks. The genotype A2b2 that was described recently was also identified. In earlier reports, subtype A1 was reported in Canada which was absent in the specimens typed in this study. This shift in genotype may be significant in terms of disease severity, and for any future vaccine considerations. Regular testing for hMPV should be done as part of outbreak investigation. J. Med. Virol. 87:269–274, 2015. © 2014 Wiley Periodicals, Inc.
Keywords: respiratory virus, long‐term care facility, molecular typing
INTRODUCTION
Respiratory outbreaks among the elderly in long‐term care facilities are most commonly associated with rhinovirus, influenza virus, human metapneumovirus (hMPV), and respiratory syncytial viruses (RSV) [Marchand‐Austin et al., 2009]. Recently hMPV was reported to cause outbreaks in long‐term care facilities in Canada and the USA with attack rates of 18–70% [Boivin et al., 2007; Louie et al., 2007; Marchand‐Austin et al., 2009; teWierik et al., 2010]. In Ontario, the proportion of respiratory illness caused by hMPV is variable, peaking in winter months, where it causes up to 27% of outbreaks in the Greater Toronto Area [Marchand‐Austin et al., 2009].
HMPV is a negative‐sense single stranded non‐segmented RNA virus that was first reported as a causative agent of acute respiratory tract infections (ARIs) in 2001 [van den Hoogen et al., 2001]. It has been classified into two subtypes, A and B [Huck et al., 2006], based on differences in nucleotide sequences and reactivity patterns with monoclonal antibodies along with in vitro neutralization assays with subgroup‐specific antisera. These two subtypes were further divided into distinct subgroups A1, A2, B1, and B2, among which subtype A2 exhibits highest diversity [Huck et al., 2006]. Subtypes A2 and B2 have been found to have additional sublineages or subclusters or clades‐ A2a, A2b [Huck et al., 2006], A2b1, A2b2 [Regev et al., 2012], and B2a and B2b [Carr et al., 2008]. The objective of this study was to investigate the prevalence and molecular epidemiology of hMPV in respiratory outbreaks that occurred between February 1, 2009 and February 29, 2012 in Ontario, Canada.
MATERIALS AND METHODS
The study was conducted from April 1, 2009 to February 29, 2012. A standardized process exists for conducting respiratory outbreak testing in Ontario that involves coordinated efforts between Ontario's public health units, institutions, and Public Health Ontario Laboratories. When an institution or medical officer reports two or more cases of respiratory illness, an outbreak investigation may be initiated by a public health investigator and respiratory specimens are submitted to Public Health Ontario Laboratories for respiratory viral testing. Public Health Ontario Laboratories provides this outbreak testing for institutions such as daycares, special care facilities, long‐term care facilities, hospitals, psychiatric, and correctional facilities. Nasopharyngeal swabs or bronchoalveolar lavage specimens are tested for outbreak investigations based on Public Health Ontario Laboratories testing algorithm, which consists of real time reverse transcriptase polymerase reaction (rRT‐PCR) for influenza A and B followed by subtyping of at least one specimen for influenza positive outbreaks. Multiplex respiratory viral PCR that targets a number of viruses including hMPV is also performed. Molecular testing was performed on the first six specimens submitted from each outbreak during the first 2 years of the study and the first four specimens during the final influenza season (November 2011 to February 2012). Commencing in November of each influenza season (defined as October 1 to April 30), an in‐house influenza A and B PCR based on CDC protocols, and influenza subtyping based on CDC or in‐house assays was performed [Duncan et al., 2011]. In addition, MRVP [(Seeplex RV or Seeplex RV15ACE; Seegene, Seoul, Korea) or Luminex xTAG respiratory viral panel (Luminex Molecular Diagnostics, Toronto, Canada)] were used to test for adenovirus, human coronaviruses, rhinovirus/enterovirus, influenza A and B, hMPV, parainfluenza 1‐4, and RSV. During the final influenza season (November 2011–February 2012), only influenza‐negative samples were tested by Multiplex respiratory viral PCR.
A proportion of test‐positive and test‐negative specimens are stored for possible further laboratory‐based surveillance. This study was exempt from University of Toronto's Ethic Review Board as left over anonymous specimens were analyzed further as part of a surveillance program that supports Ontario's Ministry of Health and long‐term care. Detailed clinical information is not routinely provided to Public Health Ontario Laboratories, so was not available for the patients included in this study.
A subset of 123 hMPV‐positive specimens from 51 different long‐term care facility outbreaks was retrospectively genotyped to understand the molecular epidemiology of the virus in Ontario. Typing was done on available residual stored specimens with sufficient volumes for further molecular analysis. Specimens were retrieved from 51 outbreaks; 3 outbreaks were from 2008 to 2009 influenza season (February–April 2009), 5 from 2009 spring/summer season (May–September 2009), 15 from 2009 to 2010 influenza season (October 2009–April 2010), 20 from 2010 to 2011 influenza season (October 2010–April 2011), 2 from the 2011 spring/summer season (May–September 2011), and 6 from 2011 to 2012 influenza season (October 2011–February 2012) (Supplementary Table S1). There was one fatal case and eight intensive care unit admissions from nine different outbreaks among the patients with samples genotyped.
Briefly, total nucleic acid was extracted from 200 μl of stored frozen specimen using Nuclisens easy MAG extraction system (bioMerieux Canada Incorporation, St. Laurent Quebec, Canada). RT‐PCR amplification was conducted using primers targeting a 940 bp region of the G gene [Ludewick et al., 2005] and a 780 bp region of the F gene [van den Hoogen et al., 2004]. Amplicons were sequenced using the Big Dye v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) using the same primers used for amplification. Multiple sequence alignments of the Ontario hMPV strains and hMPV sequences accessed from GenBank for the G gene and F gene were performed by BioEdit (version 7.1.19). Reference sequences were downloaded from GenBank for the various subtypes (sublineages). Reference sequences for the F gene subtype A2b2 that was described recently were kindly provided by Dr. Michal Mandelboim; Chaim Sheba Medical Center, Israel (shown as HMPVG578 and HMPV S5335 in Fig. 2). Phylogenetic analysis for the F gene was done using the reference sequences as provided by Dr. Mandelboim which have been previously used to refer to A2b2 [Regev et al., 2012]. Phylogenetic analysis was performed by constructing neighbor‐joining trees and each tree topology was statistically analyzed by bootstrapping (1,000 replicates) using Molecular Evolutionary Genetic Analysis (MEGA 5.0) software [Tamura et al., 2011]. Sequences of hMPV isolates obtained from this study were submitted to GenBank under accession numbers (KC709690‐KC709792, KC795605‐KC795682 for G‐gene and KC709690‐KC709781 for F‐gene sequences.).
Figure 2.
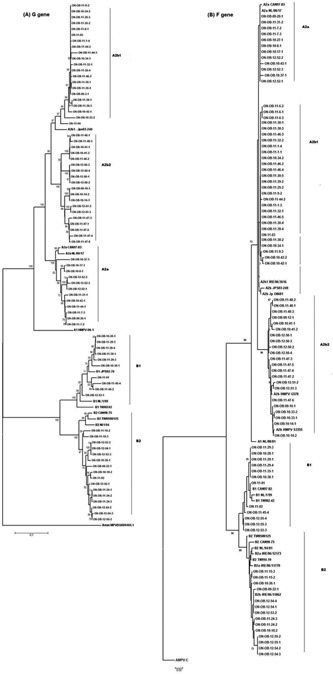
Phylogenetic trees of 80 representative Ontario hMPV strains from 51 respiratory outbreaks constructed from (A) the variable region of the G gene, and (B) partial F gene sequences. Phylogenetic sequence analysis was performed using representative sequences from different outbreaks and 100% identical sequences were removed. Multiple sequence alignments and phylogenetic trees were constructed using Clustal W and neighbour‐joining algorithm running within MEGA 5.05 software. Tree topology was supported by bootstrap analysis with 1,000 pseudo‐replicate datasets. Bootstrap values greater than 50 are shown at the branch nodes. Reference sequences are in bold text. Ontario strains have been numbered using the following format: ON‐OB‐YY‐OBN‐SS where ON = Ontario, OB = outbreak, YY = year of outbreak, OBN = representative number assigned to an outbreak and SS – representative specimen. Non‐outbreak specimens have been referred simply as ON‐YY‐SS.
RESULTS
Public Health Ontario Laboratories tested 7,575 specimens submitted by various institutions from 2,292 Ontario outbreaks from April 1, 2009 to February 29, 2012. Four hundred thirty‐six specimens collected from 195 (8.5%) of the outbreaks were positive for hMPV. The majority of outbreaks (61.3%) occurred in long‐term care facilities followed by retirement homes (3.2%), hospitals (1.8%), psychiatric hospitals (0.75%), camps (0.2%), daycares (0.4%), and schools (0.69%). However, locations for 31.9% of outbreaks were not known as the information was not provided on the laboratory requisitions at the time of specimen submission. All patients showed signs and symptoms of acute respiratory illness, which is a prerequisite prior to submitting respiratory specimens for laboratory investigation. The mean and median age of hMPV‐positive patients in these outbreaks was 80.7 and 85 years, respectively. Most hMPV positivity was observed in winter and spring each year (Fig. 1). During the immediate period following the second wave of the 2009 influenza A (H1N1) pandemic (December 2009–April 2010), an increased prevalence of hMPV infections was observed, detected in 66 (21%) of 311 outbreaks tested. During the study the predominant viral pathogen detected in all laboratories testing at Public Health Ontario Laboratories was rhinovirus/enterovirus, followed by influenza A, RSV, hMPV, parainfluenza, human coronaviruses, and influenza B. During November 2009–March 2010, the percentage of specimens positive for influenza A or rhinovirus/enterovirus was very low, while the percent positivity for RSV, hMPV, and parainfluenza was high.
Figure 1.
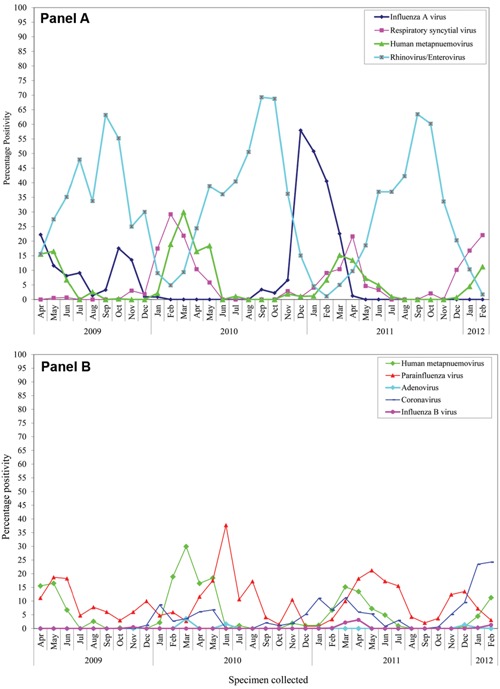
Human metapneumovirus (hMPV) percent positivity among all respiratory outbreak specimens tested at Public Health Ontario from April 2009 to February 2012. Panel A: hMPV percent positivity along with influenza A virus, respiratory syncytial virus, and rhinovirus/enterovirus percent positivities. Panel B: hMPV percent positivity along with parainfluenza virus, adenovirus, human coronavirus and influenza B percent positivities.
During the influenza season, delineated as October 1 to April 30 of the following year, hMPV was detected in 4 (2%) outbreaks during the 2008–2009 season, 60 (30.8%) during the 2009–2010 season, 77 (39.5%) during the 2010–2011 season, and 22 (11.3%) for the 2011–2012 season until February 28, 2012. During the spring/summer season, delineated as May1 to August 31, hMPV was identified in 14 (7%) outbreaks in 2009, 10 (5%) in 2010, and 8 (4%) in 2011. Table S1 gives more information on the outbreaks that were genotyped in terms of the number of the patients that were exposed, ill, and the number of hMPV‐positive cases confirmed by laboratory testing.
Phylogenetic analysis confirmed the existence of both genotypes A and B in Ontario during February 2009 to February 2012 (Fig. 2). Interestingly, genotype A2b further subdivided into A2b1 and A2b2 clades using G gene sequence data (Fig. 2). Similar phylogenetic tree topology was obtained by analyzing the F gene using the maximum likelihood method, where genotype A2b could again be further subdivided to A2b1 and A2b2. Similar observations were reported earlier by [Regev et al., 2012] using only F gene sequences.
Of the 51 hMPV associated outbreaks genotyped, 34 (67%) were type A and 17 (33%) were type B. Four outbreaks had specimens belonging to two different sublineages but belonging to the same subtype of A or B. Sublineage A2b1 was identified in 19 (34.5%) outbreaks, 9 (16.3%) were sublineage A2b2, 8 (14.3%) were sublineage A2a, 9 (16%) were sublineage B1, and 10 (17.8%) were sublineage B2 (Supplementary Table S1).
DISCUSSION
This study shows the molecular epidemiology and the genetic variability of hMPV circulating in Ontario during February 2009–February 2012. The circulation pattern of hMPV is similar to previous reports, which document hMPV infections occurring throughout the year but peaking epidemiologically 1 or 2 months later than that observed for RSV as reported by Schildgen et al. [2011] and observed in this study during the 2009–2010 influenza season (Fig. 1). Though both hMPV subgroups A and B were found in the hMPV outbreaks analyzed in Ontario, the prevalence of genotype A2b (56.8%) was high, as documented in other recent reports [Li et al., 2012; Velez Rueda et al., 2013]. In addition to these reports, A2b lineage has been recently detected in Germany and Japan [Ishiguro et al., 2004], India [Agrawal et al., 2011], China [Li et al., 2012], and Argentina [Velez Rueda et al., 2013], which may reflect an increasing prevalence of genotype A2b. None of the specimens tested were found to be A1 subtype, which was the most common genotype detected in a previous Canadian study conducted during the 2001–2002 season that did not include any Ontario specimens, where it was identified in 57.5% of hMPV specimens [Bastein et al., 2003]. Recent reports from other investigators (that included partial or total F or partial or total G gene sequences) have also reported not detecting A1 subtype after 2006 in Quebec, Canada [Papenburg et al., 2013], Austria [Aberle et al., 2010], Argentina [Velez Rueda et al., 2013], New York state [Lamson et al., 2012] or Cambodia [Arnott et al., 2010]. HMPV genotype A2b were found in almost all the years covered by these studies.
A major limitation in this study was the unavailability of a substantial number of outbreak specimens for genotyping spanning the 3 years. Therefore, it cannot be concluded if any particular lineage was more prevalent during a particular season. While genotype A2 was evenly spread across the province, genotype B was mostly documented in the Greater Toronto Area, London, Hamilton, and Niagara. Vicente et al. [2006] observed greater clinical severity associated with genotype A than genotype B in children. However, in the current study correlation between disease severity and genotype could not be assessed as limited clinical data was available.
CONCLUSIONS
Through this study the presence of multiple lineages of hMPV circulating in Ontario during February 2009–February 2012 could be confirmed. Furthermore, the study established the presence of a novel sublineage A2b2 in Ontario using both F gene and G gene sequences that was reported recently [Regev et al., 2012]. These results suggest continuous monitoring of the molecular epidemiology of hMPV is needed. HMPV is difficult to grow in culture and is very fastidious [Boivin et al., 2007]; hence the present outbreak testing method at Public Health Ontario Laboratories, using multiplex PCR, was found to be useful in identifying hMPV.
This study illustrates the potential of hMPV as a cause of respiratory outbreaks in adults and seniors. The symptoms presented by hMPV are usually indistinguishable from common respiratory virus agents such as RSV, influenza and rhinovirus/enterovirus. There are presently no vaccines against hMPV and no treatment is available that is directed against this virus. Further studies dedicated to subtyping, viral evolution, phylodynamics, and how hMPV overcomes immunity selection will aid in developing more therapeutic and prophylactic treatments against hMPV. Further studies dedicated to correlating hMPV genotype and disease severity will enhance understanding of the epidemiology of this virus and may assist with outbreak management.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Table S1
REFERENCES
- Aberle JH, Aberle SW, Redlberger‐Fritz M, Sandhofer MJ, Popow‐Kraupp T. 2010. Human metapneumovirus subgroup changes and seasonality during epidemics. Pediatr Infect Dis J 29:1016–1018. [DOI] [PubMed] [Google Scholar]
- Agrawal AS, Roy T, Chosh S, Chwala‐Sarkar M. 2011. Genetic variability of attachment (G) and Fusion (F) protein gene of human metapneumovirus strains circulating during 2006–2009 in Kolkata, Eastern India. Virol J 8:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastein N, Ward D, Van Caeseele P, Brandt K, Lee SH, Mc Nabb G, Klisko B, Chan E, Li Y. 2003. Human metapnuemovirus infection in the Canadian population. J Clin Microbiol 41:4642–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin G, De Serres G, Hamelin ME, Côté S, Argouin M, Tremblay G, Maranda‐Aubut R, Sauvageau C, Ouakki M, Boulianne N, Couture C. 2007. An outbreak of severe respiratory tract infection due to human metapneumovirus in a long‐term care facility. Clin Infect Dis 44:1152–1158. [DOI] [PubMed] [Google Scholar]
- Carr MJ, Waters A, Fenwick F, Toms GL, Hall WW, O'Kelly E. 2008. Molecular epidemiology of human metapneumovirus in Ireland. J Med Virol 80:510–516. [DOI] [PubMed] [Google Scholar]
- Duncan C, Guthrie JL, Tijet N, Elgngihy N, Turenne C, Seah C, Lau R, McTaggart L, Mallo G, Perusini S, Rebbapragada A, Melano R, Low DE, Farrell D, Guyard C. 2011. Analytical and clinical validation of novel real‐time reverse transcriptase‐polymerase chain reaction assays for the clinical detection of swine‐origin H1N1 influenza viruses. Diagn Microbiol Infect Dis 69:167–171. [DOI] [PubMed] [Google Scholar]
- Huck B, Scharf G, Neumann‐Haefelin D, Puppe W, Weigl J, Falcone V. 2006. Novel human metapneumovirus sublineage. Emerg Infect Dis 12:147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro N, Ebihara T, Endo R, Ma X, Kikuta H, Ishiko H., et al. 2004. High genetic diversity of the attachment (G) protein of human metapneumovirus. J Clin Microbiol 42:3406–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamson DM, Griesemer S, Fuschino M, St George K. 2012. Phylogenetic analysis of human metapneumovirus from New York state patients during February through April 2010. J Clin Virol 53:256–258. [DOI] [PubMed] [Google Scholar]
- Li J, Ren L, Guo L, Xiang Z, Paranhos‐Baccalà G, Vernet G, Wang J. 2012. Evolutionary dynamics analysis of human metapneumovirus subtype A2: Genetic evidence for its dominant epidemic. PLoS ONE 7:e34544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie JK, Schnurr DP, Pan CY, Kiang D, Carter C, Tougaw S, Ventura J, Norman A, Belmusto V, Rosenburg J, Trochet G. 2007. A summer outbreak of human metapneumovirus Infection in a long‐term‐care facility. J Infect Dis 196:705–708. [DOI] [PubMed] [Google Scholar]
- Ludewick HP, Abed Y, van Niekerk N, Boivin G, Klugman KP, Madhi S. 2005. Human metapnuemovirus genetic variability, South Africa. Emerg Infect Dis 11:1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand‐Austin A, Farrell DJ, Jamieson FB, Lombardi N, Lombos E, Narang S, Akwar H, Low DE, Gubbay JB. 2009. Respiratory infections in institutions during early stages of Pandemic (H1N1) 2009, Canada. Emerg Infect Dis 15:2001–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenburg J, Carbonneau J, Isabel S, Bergeron MG, Williams JV, De Serres G, Hamelin MÈ, Boivin G. 2013. Genetic diversity and molecular evolution of the major human metapneumovirus surface glycoproteins over a decade. J Clin Virol 58:541–547. [DOI] [PubMed] [Google Scholar]
- Regev L, Meningher T, Hindiyeh M, Mendelson E, Mandelboim M. 2012. Increase human metapneumovirus mediated morbidity following pandemic influenza infection. PLoS ONE 7:e34750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildgen V, van den Hoogen B, Fouchier R, Tripp RA, Alvarez R, Manoha C, Williams J, Schildgen O. 2011. Human metapneumovirus lessons learned over the first decade. Clin Microbiol Rev 24:734–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEG A5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol Biol Evol 28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- teWierik MJ, Nguyen DT, Beersma MF, Thijsen SF, Heemstra KA. 2010. An outbreak of severe respiratory tract infection caused by human metapneumovirus in a residential care facility for elderly in Utrecht, the Netherlands, January to March 2010. Euro Surveill 17:20132. [PubMed] [Google Scholar]
- van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 7:719–71924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogen BG, Herfst S, Sprong L, Cane PA, Forleo‐Neto E, de Swart RL, Osterhaus AD, Fouchier RA. 2004. Antigenic and genetic variability of human metapneumoviruses. Emerg Infect Dis 10:658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez Rueda AJ, Mistchenko AS, Viegas M. 2013. Phylogenetic and phylodynamic analyses of human metapneumovirus in Buenos Aires (Argentina) for a three‐year period (2009‐2011). PLoS ONE 8:e63070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente D, Montes M, Cilla G, Perez‐Yarza EG, Perez‐Trallero E. 2006. Differences in clinical severity between Genotype A and Genotype B human metapneumovirus infection in children. Clin Infect Dis 42:e111–113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Table S1