

Abstract
In the year 2015, new Zika virus (ZIKV) broke out in Brazil and spread away in more than 80 countries. Scientists directed their efforts toward viral polymerase in attempt to find inhibitors that might interfere with its function. In this study, molecular dynamics simulation (MDS) was performed over 444 ns for a ZIKV polymerase model. Molecular docking (MD) was then performed every 10 ns during the MDS course to ensure the binding of small molecules to the polymerase over the entire time of the simulation. MD revealed the binding ability of four suggested guanosine inhibitors (GIs); (Guanosine substituted with OH and SH (phenyl) oxidanyl in the 2′ carbon of the ribose ring). The GIs were compared to guanosine triphosphate (GTP) and five anti‐hepatitis C virus drugs (either approved or under clinical trials). The mode of binding and the binding performance of GIs to ZIKV polymerase were found to be the same as GTP. Hence, these compounds were capable of competing GTP for the active site. Moreover, GIs bound to ZIKV active site more tightly compared to ribavirin, the wide‐range antiviral drug.
Keywords: guanosine inhibitors, molecular docking, molecular dynamics simulation, polymerase, protein‐ligand docking, Zika virus
1. INTRODUCTION
Zika virus (ZIKV) infection was reported for the first time 70 years ago in Uganda.1, 2 It appeared in different central and western African countries in 2007 but the spread was not as fast as the 2015 outbreak in Brazil and Caribbean countries.3, 4 By the year 2016, more than 80 countries reported ZIKV infection worldwide.5 The World Health Organization (WHO) declared it as a public health emergency of international concern in February 2016.5
ZIKV is transmitted through the bites of Ades mosquitos (Aedes africanus, Aedes aegypti, Aedes luteocephalus, and Aedes albopictus).6, 7, 8 It can be detected in body fluids like saliva, urine, and blood.9, 10 Slight fever, rash, arthralgia, and conjunctivitis are common symptoms of ZIKV infection.5, 11 Strong links between ZIKV infection and microcephaly and neuronal disorders in newborn infants of ZIKV‐infected women were confirmed in March 2016.12
Non‐structural 5 (NS5) viral protein have two domains; the helicase domain and the RNA‐dependent RNA polymerase (RdRp) domain which is vital for viral replication.13, 14 The polymerase domain has a highly conserved active site in different viruses including hepatitis C virus (HCV), West Nile virus, and Middle East Respiratory Syndrome Coronavirus.15, 16, 17, 18, 19 The antiviral activity of different nucleotide inhibitors were studied against the RdRp of HCV. Sofosbuvir is an example of a successful nucleotide inhibitor that was approved by Food and Drug Administration (FDA) in December 2013.20, 21, 22, 23 The same compound was also studied as a possible inhibitor against ZIKV.24, 25, 26
Molecular modeling is usually used to mimic the behavior of macromolecules.27, 28 Molecular docking (MD) is important in testing the binding of small molecules to protein active site and in virtual screening.18 These in silico techniques were used to study viral proteins.18, 27, 29, 30, 31, 32, 33 Molecular dynamics simulation (MDS) offers a means to study protein dynamics in silico. It is able to mimic and predict the dynamical properties of protein in solution28 in addition to suggesting possible interaction potency.34, 35, 36, 37 In this study, MD was used to mimic the interaction between 10 of nucleotide inhibitors and ZIKV RdRp domain. MDS was performed on the predicted model of the viral polymerase for more than 400 ns, while MD was carried out each 10 ns to test the binding of the inhibitors.
2. MATERIALS AND METHODS
2.1. Preparation of small molecules and protein target
ZIKV RdRp sequence was downloaded from the National Center for Biotechnology Information.38 Protein Homology/analogY Recognition Engine PHYRE 2.0 server39 was used to build the 3D structure model from ZIKV sequence. Dengue virus NS5 RdRp domain structure was used as a template (sequence identity 67%).
Nucleotide inhibitors (in its active triphosphate form) were sketched and optimized using SCIGRESS 3.0 software.29, 40 Optimization was done using Molecular Mechanics force field MM341 followed by quantum mechanics (B3LYP functional).42 Supplementary Figure S1 shows the structures of the inhibitors in 2D representation.43
2.2. Molecular dynamics simulation (MDS)
NAMD software34 was used to perform MDS on the Cy‐Tera super computer facility of the Cyprus Institute of Science (Project no. pro15b114s1). ZIKV model was solvated (explicitly) using TIP3P water model.34 Two chlorine ions were added to neutralize the protein charge at pH 7. The protein in the solvent system was equilibrated at room temperature (310 K) then the solvent was minimized for 100 ps. Normal pressure and temperature (NPT) ensemble was used to relax the system for a total period of 5 ns, after that normal volume and temperature (NVT) ensemble was used in the production run for 444 ns. In the production run, the box size was set to (90.32 × 90.32 × 90.32) Å3 based on the fluctuations during the 5 ns of NPT ensemble.
Every 10 ns of the production run, the structure of the protein was retrieved for docking study. Therefore, the docking study was performed for 45 times during different dynamic states (different conformations). The retrieved structures were used to check the solvent accessible surface area (SASA) and radius of gyration (R of G). These two parameters were used to check conformational changes during the course of MDS. AutoDock Vina software44 was used to perform the docking study using default parameters. The protein is treated as rigid as we need to test the contribution of dynamics of the protein into inhibitor binding. The calculated binding affinity (in kcal/mol) were used for comparison between different nucleotide inhibitors.
3. RESULTS AND DISCUSSION
As reported before anti‐HCV drugs were capable of binding to ZIKV polymerase active site.24, 25 In the current study, MDS was performed on the ZIKV RdRp model for a period of 444 ns. Despite five solved structures of ZIKV, NS5 RdRp were deposited in the Protein Data Bank in the last few months, the comparatively built model used in this study had very strong sequence and hence structural conservation (Figure 1A). Sequence comparison between the five solved structures (PDB IDs: 5WZ3, 5TFR, 5TMH, 5TIT, and 5U04) and the model used in this study is shown in Supplementary Figure S2. Figure 1B shows the root mean square deviation (RMSD) over the carbon alpha atoms of the polymerase versus time. Saturation was achieved after 200 ns with about 12 Å RMSD. During the course of MDS, two distances were recorded. These were the distance between carbon alpha of the active site D124 and the two alpha carbons of D6 and G46 (green and red curves in Figure 2A, respectively). D6 lied in the center of alpha helix (α1 in Figure 2B) near the active site. On the other hand, G46 lied at the end of the arm consisting of β1, β2, and its connecting loop (fingers tip). It was reported that this arm takes part in the interaction with the RNA during the nucleotide transfer reaction.25 Based on the structural alignment made with HCV NS5b, this arm suggested to be movable in ZIKV polymerase and this is what we found using MDS.
Figure 1.
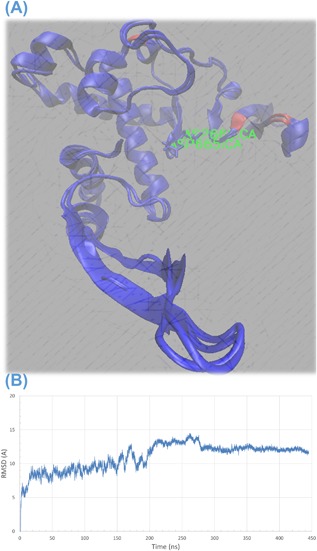
A, Structural alignment of ZIKV model built in this work in silico and a solved structure of ZIKV (PDB ID 5TFR) showing high conservation. (B) Root Mean Square Deviation (RMSD) values in Å versus simulation time in ns showing an equilibration after about 200 ns
Figure 2.
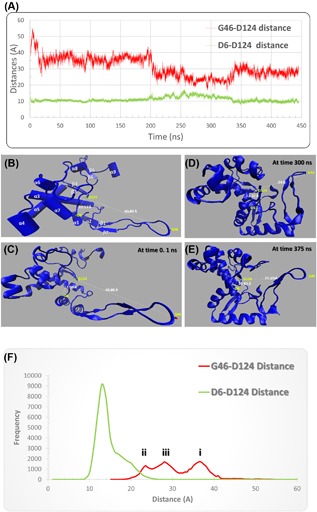
A, Two distances recorded over 444 ns of MDS. G46‐D124 (red line) and D6‐D124 (green line). (B) Secondary structural elements of the ZIKV polymerase model before the MDS run represented in carton using VMD software. (C), (D), and (E) ZIKV polymerase structure at 0.1, 300, and 375 ns, respectively of MDS run showing the differences in the two distances in the different simulation times. (F) Distances distribution showing different patterns as mentioned in the text. The color is the same as in A
As shown in Figures 2A and 2F, the D6‐D124 distance is stable with an average value of 13 Å. On the other hand, G46‐D124 distance shows a different pattern. The distribution of the values shows three maxima; in the first 200 ns of the simulation it is around 36 Å (step i in Figure 2F), the distance reduces to 24 Å (see Supplementary Video S1) in the next 150 ns of MDS (step ii in Figure 2F) and finally it is increases again to about 28 Å for the rest of the simulation time (step iii in Figure 2F). This implies a conformational change in the distal end of the arm that contains G46 and suggests its functional characteristics. Based on structural alignment made between ZIKV and HCV polymerases, this arm is a part of the fingers domain, where the fingers’ tips interact with the viral RNA during the polymerization process.45
Figure 2B illustrates the secondary structural elements of the ZIKV RdRp model before performing the MDS. It has nine alpha helices and six beta sheets. The active site aspartates are protruding from the beta turn between β3 and β4. The distance between the active site D124 and each of D6 and G46 is labeled at selected times during the MDS (Figure 2C‐E).
Figures 3A and 3B shows the calculated values of surface accessible surface area (SASA) in Å2 and the radius of gyration (R of G) in Å. These values were calculated each 10 ns of the MDS using visualizing molecular dynamics (VMD) software.46 SASA was calculated as relative exposure (0 to 1), that is, the exposure of each amino acid in the protein relative to per residue SASA values. The latter is calculated for each amino acid alone in the maximum speed molecular surface (MSMS) algorithm implemented in VMD software.47 The solvent radius was chosen to be 1.4 Å (radius of water molecule). On the other hand, radius of gyration was calculated using the following formula:
| (1) |
where ri represents atoms position, r conf represents the center of mass of the protein atoms (center of the protein molecule) and N is the number of protein atoms.
Figure 3.
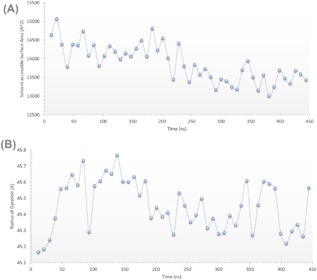
A, Solvent Accessible Surface Area (SASA) (Å2) and (B) radius of gyration (Å) of ZIKV polymerase model versus time (ns)
SASA values are in good agreement with the G46‐D124 distance distribution pattern. In the first 200 ns, SASA values are around 14 200 Å2. In the next 150 ns the values reduce to 13 500 Å2 before they rise up slightly in the rest of the simulation time. SASA values represent the compactness of the protein.47 As the arm containing G46 starts to get close to the protein core it reduces the SASA values. After the 350 ns, the arm starts to open again and SASA starts to slightly increase. Radius of gyration was also calculated every 10 ns of the MDS. One can notice that at the beginning of the MDS (in the first 70 ns), the values increase up to 45.6 Å. This can be attributed to the swallowing of the protein in the solvent.37 The R of G of the protein gradually increases up to a certain value. It then fluctuates around this value up to 200 ns before it starts to decrease again and fluctuate around 45.35 Å. This may be also attributed to the arm movement close to the protein core which reduces the R of G. After 350 ns the fluctuation in the R of G increases again, probably due to reverse movement of the arm apart from the protein core.
3.1. Does arm movement have a contribution on small molecule binding to ZIKV polymerase?
To answer this question, MD was performed over the course of the MDS to test the binding affinities of the studied drugs to ZIKV RdRp during 444 ns of simulation. Figure 4 shows the average binding affinities of GTP, IDX‐184, MK0608, R7128, sofosbuvir, and ribavirin to ZIKV RdRp over 444 ns of MDS (error bars represent the standard deviation). The binding affinities were calculated every 10 ns of the MDS and their values were found to lie between −5.5 and −9.1 kcal/mol. GTP (red circle) is better (having more negative binding affinities) compared to ribavirin and R7128.
Figure 4.
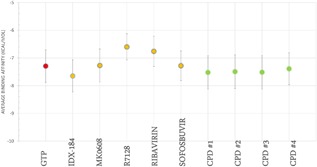
Average binding affinities calculated using AutoDock Vina for all of the small molecules upon binding ZIKV polymerase model over 444 ns of MDS. Standard Deviation values are represented by error bars. The four suggested compounds are represented in green circles, anti‐HCV drugs in orange while GTP is in red
As shown there is superposition of the GTP values with those of the compounds (CPD #1, CPD # 2, CPD # 3, and CPD # 4) (green circles in Figure 4). By examining the H‐bonds that were formed upon binding of the small molecules to ZIKV polymerase, one finds the same mode of interaction as that with HCV polymerase. H‐bonds were formed between the small molecules and ZIKV polymerase active site aspartate D124 and two amino acids in the active site cavity; R190 and E194. These results imply the ability of the suggested compounds to compete with GTP for ZIKV RdRp active site over the entire period of MDS.
Figure 5 shows an example of ligand (GTP) bound to ZIKV polymerase at 141 ns (A) and 243 ns (B) of the MDS. The former has the arm extended (G45‐D124 distance 40.6 Å) while the latter has the arm flexed (G45‐D124 distance 26.4 Å). The conformations are selected randomly before and after the arm movement and the binding affinities are recorded on the bottom of the figures. There are no dramatic changes in the binding affinities before and after arm movement. This is also reported for other inhibitors used (Supplementary Figure S3). Hence, we can conclude that the arm movement has not contributed against binding of the small molecules to ZIKV polymerase.
Figure 5.
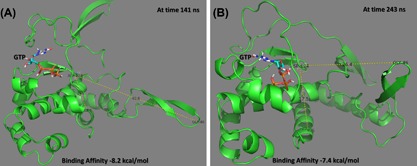
An example showing the binding of GTP to ZIKV polymerase at different dynamics states. Extended arm at 141 ns (A) and flexed arm at 243 ns (B). The binding affinities are reported in the bottom of the figures. Figures were prepared using PyMOL software
As reported by the authors in earlier studies, IDX‐184 shows promising results compared to other drugs.22, 25, 27, 48 The new suggested compounds have also promising average binding affinities to ZIKV NS5 RdRp compared to other drugs (Figure 4). Upon binding, the suggested ligands will stop the polymerization process and impair the virus life cycle.
4. CONCLUSION
In the previous studies, authors suggested the ability of anti‐HCV drugs to bind and subsequently inhibit ZIKV polymerase.25, 26 This was based on structural similarity between HCV and ZIKV polymerases. In this study, the authors simulated the dynamics of ZIKV polymerase in water over 444 ns. The results supported our previous suggestion. In addition, four new compounds were introduced as promising binders to ZIKV polymerase active site based on the calculated binding affinities. On the other hand, the arm movement that occurred after 200 ns did not affect the binding of all of the small molecules. Future work is suggested to test the arm movement effect on the viral RNA upon polymerase binding.
5. ACKNOWLEDGMENTS
MDS was carried out on Cy‐Tera supercomputer facility of Cyprus Institute of Science under the production run of the project “pro15b114s1.” Ali Hassanali is appreciated for his valuable discussions and efforts that without it this work would have not been possible. Alaa M. Ismail, Wissam A. Gawad, Ahmed A. Ezat, Ahmed Gameel, Mahmoud Abdelmohsen, and Mostafa Shaban are appreciated for their helpful support and discussions.
AUTHORS’ CONTRIBUTION
AAE performed the calculations and took part in writing the manuscript. WME revised the manuscript. All authors have approved the final version of the manuscript.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Figure S1. The 2D structures of the nucleotide inhibitors used in this study.
Figure S2. Sequence alignment between the in silico built model and the 5 experimentally solved structures in the protein data bank (PDB IDs: 5WZ3, 5TFR, 5TMH, 5TIT and 5U04) for ZIKV polymerase.
Figure S3. The same as figure 5 but for other inhibitors used in this study.
Video S1. Three nanoseconds of the MDS (201‐204 ns) recorded to show the movement of the fingertip (G46) close to the active site (D124).
CONFLICTS OF INTEREST
All authors declare that there is no conflict of interest in this work.
Elfiky AA, Elshemey WM. Molecular dynamics simulation revealed binding of nucleotide inhibitors to ZIKV polymerase over 444 nanoseconds. J Med Virol. 2018;90: 13–18. 10.1002/jmv.24934
REFERENCES
- 1. Dick GWA, Kitchen SF, Haddow AJ. Zika virus (I). Isolations and serological specificity. Trans R Soc Trop Med Hyg. 1952; 46:509–520. [DOI] [PubMed] [Google Scholar]
- 2. MacNamara FN. Zika virus: a report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans R Soc Trop Med Hyg. 1954; 48:139–145. [DOI] [PubMed] [Google Scholar]
- 3. Duffy MR, Chen T‐H, Hancock WT, et al. Zika virus outbreak on Yap island, federated states of Micronesia. N Eng J Med. 2009; 360:2536–2543. [DOI] [PubMed] [Google Scholar]
- 4. Kuno G, Chang GJ. Full‐length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol. 2007; 152:687–696. [DOI] [PubMed] [Google Scholar]
- 5.WHO. Zika strategic response framework and joint operations plan. 2016.
- 6. Boorman JPT, Porterfield JS. A simple technique for infection of mosquitoes with viruses transmission of Zika virus. Trans R Soc Trop Med Hyg. 1956; 50:238–242. [DOI] [PubMed] [Google Scholar]
- 7. Haddow AJW, Williams MC, Woodall JP, Simpson DIH, Goma LKH. Twelve isolations of Zika virus from Aedes (Stegomyla) africanus (Theobald) taken in and above a Uganda forest. Bull Wid Health Org. 1964; 31:69. [PMC free article] [PubMed] [Google Scholar]
- 8. Fagbami AH. Zika virus infections in Nigeria: virological and seroepidemiological investigations in Oyo State. Epidemiol Infect. 1979; 83:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tauro LB, Bandeira AC, Ribeiro GS, et al. Potential use of saliva samples to diagnose Zika virus infection. J Med Virol. 2017; 89:1–2. [DOI] [PubMed] [Google Scholar]
- 10. Musso D, Rouault E, Teissier A, et al. Molecular detection of Zika virus in blood and RNA load determination during the French Polynesian outbreak. J Med Virol. 2016; 89:1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zumla A, Goodfellow I, Kasolo F, et al. Zika virus outbreak and the case for building effective and sustainable rapid diagnostics laboratory capacity globally. Int J Infect Dis. 2016; 45:92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mlakar J, Korva M, Tul N, et al. Zika virus associated with microcephaly. N Engl J Med. 2016; 374:951–958. [DOI] [PubMed] [Google Scholar]
- 13. Wang B, Tan X‐F, Thurmond S, et al. The structure of Zika virus NS5 reveals a conserved domain conformation. Nat Commun. 2017; 8:14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Upadhyay AK, Cyr M, Longenecker K, Tripathi R, Sun C, Kempf DJ. Crystal structure of full‐length Zika virus NS5 protein reveals a conformation similar to Japanese encephalitis virus NS5. Acta Crystallogr F Struct Biol Commun. 2017; 73:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elfiky AA, Mahdy SM, Elshemey WM. Quantitative structure‐activity relationship and molecular docking revealed a potency of anti‐hepatitis C virus drugs against human corona viruses. J Med Virol. 2017; 89:1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elfiky AA, Elshemey WM, Gawad WA, Desoky OS. Molecular modeling comparison of the performance of NS5b polymerase inhibitor (PSI‐7977) on prevalent HCV genotypes. Protein J. 2013; 32:75–80. [DOI] [PubMed] [Google Scholar]
- 17. Ferrer‐Orta C, Ferrero D, Verdaguer N. RNA‐dependent RNA polymerases of picornaviruses: from the structure to regulatory mechanisms. Viruses. 2015; 7:4438–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ganesan A, Barakat K. Applications of computer‐aided approaches in the development of hepatitis C antiviral agents. Expert Opin Drug Discov. 2017; 12:407–425. [DOI] [PubMed] [Google Scholar]
- 19. Elfiky AA, Elshemey WM. IDX‐184 is a superior HCV direct‐acting antiviral drug: a QSAR study. Med Chem Res. 2016; 25:1005–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elfiky AA, Elshemey WM, Gawad WA. 2′‐Methylguanosine prodrug (IDX‐184), phosphoramidate prodrug (Sofosbuvir), diisobutyryl prodrug (R7128) are better than their parent nucleotides and ribavirin in hepatitis C virus inhibition: a molecular modeling study. J Comput Theoretical Nanosci. 2015; 12:376–386. [Google Scholar]
- 21. Asselah T. Daclatasvir plus sofosbuvir for HCV infection: an oral combination therapy with high antiviral efficacy. J Hepatol. 2014; 61:435–438. [DOI] [PubMed] [Google Scholar]
- 22. Elfiky AA, Gawad WA, Elshemey WM. Hepatitis C viral polymerase inhibition using directly acting antivirals, a computational approach. In: A M, ed. Software and Techniques for Bio‐Molecular Modeling. Jersey city, NJ, USA: Austin Publishing group; 2016: 197. [Google Scholar]
- 23. Asselah T. Sofosbuvir for the treatment of hepatitis C virus. Expert Opin Pharmacother. 2014; 15:121–130. [DOI] [PubMed] [Google Scholar]
- 24. Bullard‐Feibelman KM, Govero J, Zhu Z, et al. The FDA‐approved drug sofosbuvir inhibits Zika virus infection. Antivir Res. 2017; 137:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elfiky AA. Zika viral polymerase inhibition using anti‐HCV drugs both in market and under clinical trials. J Med Virol. 2016; 88:2044–2051. [DOI] [PubMed] [Google Scholar]
- 26. Sacramento CQ, de Melo GR, Rocha N, et al. The clinically approved antiviral drug sofosbuvir impairs Brazilian Zika virus replication. bioRxiv. 2016; 7:1–12. Article no. 40920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elfiky AA, Ismail AM. Molecular modeling and docking revealed superiority of IDX‐184 as HCV polymerase inhibitor. Future Virol. 2017; 12:339–347. [Google Scholar]
- 28. Leach A. 2001. Molecular dynamics simulation methods. 2nd ed. Molecular Modelling: Principles and Applications. England: Prentice Hall; 353–406. [Google Scholar]
- 29. Saleh NA, Ezat AA, Elfiky AA, Elshemey WM, Ibrahim M. Theoretical study on modified boceprevir compounds as NS3 protease inhibitors. J Comput Theoretical Nanosci. 2015; 12:371–375. [Google Scholar]
- 30. Saleh NA, Elfiky AA, Ezat AA, Elshemey WM, Ibrahim M. The electronic and quantitative structure activity relationship properties of modified telaprevir compounds as HCV NS3 protease inhibitors. J Comput Theoretical Nanosci. 2014; 11:544–548. [Google Scholar]
- 31. Ibrahim M, Saleh NA, Hameed AJ, Elshemey WM, Elsayed AA. Structural and electronic properties of new fullerene derivatives and their possible application as HIV‐1 protease inhibitors. Spectrochim Acta A Mol Biomol Spectrosc. 2010; 75:702–709. [DOI] [PubMed] [Google Scholar]
- 32. Saleh NA. The QSAR and docking calculations of fullerene derivatives as HIV‐1 protease inhibitors. Spectrochim Acta A Mol Biomol Spectrosc. 2014; 136PC:1523–1529. [DOI] [PubMed] [Google Scholar]
- 33. Ibrahim M, Saleh NA, Elshemey WM, Elsayed AA. Hexapeptide functionality of cellulose as NS3 protease inhibitors. Med Chem. 2012; 8:826–830. [DOI] [PubMed] [Google Scholar]
- 34. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Dijk AD, Bonvin AM. Solvated docking: introducing water into the modelling of biomolecular complexes. Bioinformatics. 2006; 22:2340–2347. [DOI] [PubMed] [Google Scholar]
- 36. Noorbatcha IA, Khan AM, Salleh HM. Molecular dynamics studies of human?‐Glucuronidase. Am J Appl Sci. 2010; 7:823–828. [Google Scholar]
- 37. Bellissent‐Funel M‐C, Hassanali A, Havenith M, et al. Water determines the structure and dynamics of proteins. Chem Rev. 2016; 116:7673–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.NCBI. National Center of Biotechnology Informatics (NCBI) database website http://www.ncbi.nlm.nih.gov/; 2017.
- 39. Kelley LA, Sternberg MJ. Protein structure prediction on the web: a case study using the Phyre server. Nat Protoc. 2009; 4:363–371. [DOI] [PubMed] [Google Scholar]
- 40. Summers KL, Mahrok AK, Dryden MD, Stillman MJ. Structural properties of metal‐free apometallothioneins. Biochem Biophys Res Commun. 2012; 425:485–492. [DOI] [PubMed] [Google Scholar]
- 41. Lii JH, Allinger NL. Molecular mechanics. The MM3 force field for hydrocarbons. 3. The van der Waals’ potentials and crystal data for aliphatic and aromatic hydrocarbons. J Am Chem Soc. 1989; 111:8576–8582. [Google Scholar]
- 42. Becke AD. Density‐functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993; 98:5648–5652. [Google Scholar]
- 43. Elfiky AA. Zika virus: novel guanosine derivatives revealed strong binding and possible inhibition of the polymerase. Future Virol. 2017; In press. [Google Scholar]
- 44. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010; 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu G, Gong P. A structural view of the RNA‐dependent RNA polymerases from the Flavivirus genus. Virus Res. 2017; 234:34–43. [DOI] [PubMed] [Google Scholar]
- 46. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996; 14:33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- 47. Durham E, Dorr B, Woetzel N, Staritzbichler R, Meiler J. Solvent accessible surface area approximations for rapid and accurate protein structure prediction. J Mol Model. 2009; 15:1093–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Elfiky AA, Elshemey WM. IDX‐184 is a superior HCV direct‐acting antiviral drug: a QSAR study. Med Chem Res. 2016;90: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Figure S1. The 2D structures of the nucleotide inhibitors used in this study.
Figure S2. Sequence alignment between the in silico built model and the 5 experimentally solved structures in the protein data bank (PDB IDs: 5WZ3, 5TFR, 5TMH, 5TIT and 5U04) for ZIKV polymerase.
Figure S3. The same as figure 5 but for other inhibitors used in this study.
Video S1. Three nanoseconds of the MDS (201‐204 ns) recorded to show the movement of the fingertip (G46) close to the active site (D124).