

Abstract
Human coronaviruses (HCoV) are RNA viruses that cause respiratory tract infections with viral replication of limited duration. The host and viral population heterogeneity could influence clinical phenotypes. Employing long RT‐PCR with Illumina sequencing, we quantified the gene mutation load at 0.5% mutation frequency for the 4529 bp‐domain spanning the Spike gene (4086 bp) of HCoV‐OC43 in four upper respiratory clinical specimens obtained during acute illness. There were a total of 121 mutations for all four HCoV samples with the average number of mutations at 30.3 ± 10.2, which is significantly higher than that expected from the Illumina sequencing error rate. There were two mutation peaks, one at the 5′ end and the other near position 1 550 in the S1 subunit. Two coronavirus samples were genotype B and two were genotype D, clustering with HCoV‐OC43 strain AY391777 in neighbor‐joining tree phylogenetic analysis. Nonsynonymous mutations were 76.1 ± 14% of mutation load. Although lower than other RNA viruses such as hepatitis C virus, HCoV‐OC43 did exhibit quasi‐species. The rate of nonsynonymous mutations was higher in the HCoV‐OC43 isolates than in hepatitis C (HCV) virus genotype 1a isolates analyzed for comparison in this study. These characteristics of HCoV‐OC43 may affect viral replication dynamics, receptor binding, antigenicity, evolution, transmission, and clinical illness.
Keywords: coronavirus, genetic variability, genetic variation, hepatitis C virus, humoral immunity, mutation
1. INTRODUCTION
Human coronavirus‐OC43 (HCoV‐OC43) is within the Betacoronavirus genus (family Coronaviridae) and is an enveloped, positive sense, single‐stranded RNA virus.1, 2 There are five known genotypes and other betacoronaviruses include severe acute respiratory syndrome (SARS) CoV and Middle East respiratory syndrome (MERS) CoV.3 HCoV‐OC43 is prevalent among humans and genotype D has been prominent in recent years.3, 4, 5 Little is known about how HCoV‐OC43 genotypes persist in human populations, but continuous adaptation by viral antigenic genes in the Spike protein through genetic drift may be necessary. The Spike protein is the major antigenic protein and is under selection pressure by the host immune response; it is important for host range and tissue tropism. It is cleaved into S1 and S2 subunits for receptor binding and membrane fusion. The N‐terminal domain of the S1 subunit is responsible for sugar receptor binding and the S2 subunit is responsible for fusion of viral and host membranes.6 The S1 subunit is more divergent in sequence and the S2 subunit is more conserved.1, 2, 3, 7
Human coronaviruses cause the common cold and influenza‐like illnesses, but can be associated with more severe illnesses such as pneumonia, exacerbations of asthma and chronic obstructive pulmonary disease, croup, and bronchiolitis. In patients with chronic obstructive pulmonary disease studied during the 1998‐1999 influenza season, 13.5% of illnesses were associated with HCoV‐229E and HCoV‐OC43 infection, while in another study between 2009 and 2013, 19% of acute respiratory illnesses in patients with cardiopulmonary diseases and 21.5% in healthy young adults were associated with HCoV.8, 9, 10, 11 Coronavirus‐associated illness was less severe than influenza but was associated with multiple respiratory and systemic symptoms, and hospitalization.10 HCoV‐229E and HCoV‐OC43 infection rates of 2.8‐26% in healthy young and elderly adults, high‐risk adults, and hospitalized patients were reported during the winters of 1999‐2003 and they contributed to medical disease burden.12
Little is known about the degree of heterogeneity of HCoV‐OC43 viral quasi‐species present in upper respiratory secretions. If present, this may help explain persistent incidence of HCoV‐OC43 infections in human populations, if the mutational changes result in antigenic drift. This might allow escape from host immunity and contribute to virus infectivity and pathogenicity.
In the current study, we combined RT‐PCR and Illumina sequencing to measure the diversity of HCoV‐OC43 Spike gene quasi‐species through direct count of the Spike gene mutations, determination of percent nonsynonymous mutation rates and comparison of these rates to (HCV), which is in the genus Hepacivirus, family Flaviviridae. HCV is an RNA virus with a heterogeneous population of quasi‐species in chronically infected patients.13, 14, 15
2. MATERIALS AND METHODS
2.1. Patient samples
We studied nasal and oropharyngeal swab specimens that were obtained from each of four patients early during symptomatic acute respiratory illness and positive for HCoV‐OC43 nucleic acids by multiplex RT‐PCR.11 Serum and nasal wash specimens were obtained at the time of acute illness and 3‐4 weeks after illness onset. They were assayed by enzyme‐linked immunosorbent assay for serum IgG and nasal wash IgA antibodies to tissue culture‐adapted HCoV‐OC43 (American Type Culture Collection #VR‐1558, GenBank: NC_005147.1) that was inactivated by psoralen compound and long‐wavelength ultraviolet light, as described.10, 11, 16 Severity of acute respiratory illness was measured by two scores: a self‐reported visual analogue scale of overall illness severity, ranging from 1 (mildest) to 10 (most severe), and a severity of influenza‐like symptoms and signs score that was the sum of 16 symptoms and signs that were graded on a scale of 0 (absent)‐15 (most severe) with a maximum score of 240, as described.10, 11, 17 Respiratory and systemic symptoms of the acute illness were recorded. The patients gave written informed consent and the study was approved by the Institutional Review Boards at the VA St. Louis Health Care System and Saint Louis University. Two recombinant clones from a previous study, #1701 and #1709, each containing a 9022 bp HCV insert, were used to estimate potential errors associated with Illumina sequencing.14, 15 Also, 19 HCV genotype 1a samples from an earlier report3 were available for re‐analysis and comparison in the current study.
2.2. RNA extraction, RT‐PCR, and illumina sequencing
Total RNA from each nasal and oropharyngeal swab specimen sample was purified using the QIAamp Ultrasens Virus Kit (Qiagen, Valencia, CA) according to the manufacturer's procedures. RT‐PCR was then applied to amplify a 4529 bp amplicon spanning the full‐length spike gene (4086 bp). In brief, 10.6 μL of extracted RNA was mixed with 9.4 μL RT matrix consisting of 1x SuperScript III buffer, 10 mM DTT, 1 μM OC43R1 (reverse primer, 5′‐TGC CCC ACA TAC CAC ACA G‐3′, position 28 164‐28 182, numbering is according to HCoV‐OC43 strain, GenBank accession number: AY391777), 2 mM dNTPs, 20 U of RNase OUT recombinant Ribonuclease Inhibitor, and 200 U of SuperScript III Reverse transcriptase (Life Technologies). After 75 min. incubation at 50°C and subsequent inactivation, an aliquot of 5 μL of RT reaction was applied for the first round of PCR that contained 1x GC enhancer (New England Biolabs), 1x Q5 buffer (New England Biolabs), 1.6 mM dNTPs, 0.4 μM OC43F1 (forward primer, 5′‐GTA CAG GTT GTT GAT TCG CG‐3′, position 23 210‐23 229), 0.4 μM OC43R1 and 1.6 U Q5 High Fidelity DNA Polymerase (New England Biolabs). After initial heating at 94°C for 1 min, cycle parameters were programmed as the first 10 cycles of 94°C for 30 sec, 65°C for 30 sec and 68°C for 5 min followed by 20 cycles of 94°C for 30 sec, 60°C for 30 sec and 68 °C for 5 min with a 2 sec autoextension at each cycle. Two μL of the first round of PCR product was used for the second round amplification with primers OC43F2 (forward primer, 5′‐TCT GGC CTC TCT ACC CCT ATG GC‐3′, position 23 439‐23 461) and OC43R2 (reverse primer, 5′‐CTT GAT TAC GGC ACC AAG CAT GAC‐3′, position 27 944‐27 967), under the same cycle parameters as the first round of PCR. Product at expected size was gel‐purified using QIAquick PCR purification Kit (Qiagen) and quantitated. About 4‐5 μg of purified DNA product was subjected to library construction. The fragment library was constructed using Illumina Nextera XT DNA library preparation kit, and followed by Illumina sequencing on NextSeq 500 machine with 1 × 250 bp read output.
2.3. Sequence data analysis
We first estimated the error rate associated with Illumina sequencing using two recombinant HCV clones. In doing so, raw sequence reads in fastq format were first filtered in PRINSEQ (v 0.19.5) for quality control, including read length ≥70 bp, mean read quality score ≥25, low complexity with DUST score ≤7, ambiguous bases ≤1% and all duplicates.18 Filtered reads were mapped onto HCV genotype 1a prototype strain H77 (GenBank accession number AY009606) using a gapped aligner Bowtie 2.19 Mapped files were then converted into binary format (BAM), sorted and indexed in SAMtools20 followed by local realignment and base quality recalibration in Genome Analysis Toolkit (GATK).21 Next, by converting post‐alignment BAM files into mpileup format in SAMtools, the consensus sequence for each clone was called in VarScan (v 2.2.3) with the settings of ≥1,000 x coverage, ≥25 base quality at a position to count a read and ≥50% mutation frequency.22, 23 The entire pipeline was repeated using individual consensus sequences. Mutations were called at each position in VarScan under the setting of 0.5% frequency and base quality from 15 to 40, followed by manual check in the Integrative Genomics Viewer.22
Using the value of base quality to define a mutation from above analysis, similar procedures were applied to four patient samples. The HCoV‐OC43 strain (GenBank AY391777) was used as the reference at initial mapping. Over the entire coronavirus Spike gene, the mutation load, the total number of mutations at a given site, was counted through sliding windows, size = 300 bp, overlap = 100 bp. Finally, under the frame of full‐length HCoV Spike gene (4086 bp), the nature of each mutation, either synonymous or nonsynonymous, was determined using a custom script.24
2.4. Phylogenetic analysis
The consensus full‐length HCoV spike sequences from four patients and reference sequences retrieved from GenBank were used for phylogenetic analysis. The tree was constructed using neighbor‐joining approach under nucleotide substitution model of maximum composite likelihood in MEGA program (version 5.2).25
2.5. Statistical analysis
Statistical analyses were done with either two‐tailed, unpaired Students test or Chi‐square. When applicable, data were expressed as mean value and standard deviation. P < 0.05 was considered statistically significant.
2.6. Data availability
Raw sequence data in fastq format from all four patient samples were archived in NCBI Sequence Read Archive (SRA) under SRA accession number SRP071020.
3. RESULTS
3.1. Clinical characteristics of HCoV infections and antibody responses
Samples 3 and 4, both genotype D, were collected within a month of each other in December 2010 and January 2011 from two older patients with significant acute respiratory and systemic symptoms (Table 1). The patients had underlying chronic cardiopulmonary diseases and diabetes mellitus. The two illnesses were associated with greater than a fourfold increase in nasal wash IgA antibody titers but only one with at least a fourfold increase in serum IgG antibody titer to HCoV‐OC43, comparing acute illness to convalescent specimens collected 3‐4 weeks after illness onset (Table 1).
Table 1.
Clinical characteristics of acute respiratory illnesses in patients associated with the four human sequenced coronavirus OC43 (HCoV‐OC43) clinical sample
| Anti‐HCoV‐OC43 serum/ nasal wash reciprocal antibody titers a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Study subject | Age (years) | Gender | Date of illness | Acute illness visit | Convalescent visit | Symptoms/signs of acute respiratory illness | Acute illness severity score b | Acute VAS of severity c | Medical history |
| 2 | 39 | Male | 1‐6‐2010 | 664/<5 | 1,408/36 | Sputum, rhinitis, dyspnea, headache, fatigue, sore throat | 45 | 5 | None |
| 3 | 82 | Male | 12‐17‐2010 | 1,983/59 | 6,758/1,686 | Cough, sputum, rhinitis, dyspnea, chills, headache, myalgia, fatigue, sore throat | 57 | 7 | COPD d , emphysema, ischemic heart disease, diabetes mellitus |
| 4 | 61 | Male | 1‐10‐2011 | 467/63 | 7,741/131,410 | Cough, sputum, rhinitis, dyspnea, chills, headache, myalgia, body aches and pains, fatigue, sore throat, pharyngitis | 80 | 6 | Congestive heart failure, ischemic heart disease, asbestosis, diabetes mellitus |
| 6 | 62 | Male | 3‐13‐2012 | 1,510/139 | 1,042/460 | Rhinitis, dyspnea, chills, body aches and pains, fatigue, sore throat | 27 | 6 | Ischemic heart disease, diabetes mellitus, sinusitis |
Serum antibodies were IgG and nasal wash antibodies were IgA binding to UV light and psoralen‐inactivated tissue culture‐adapted HCoV‐OC43 (ATCC#VR‐1558) measured by enzyme‐linked immunosorbent assay.
Severity of influenza‐like symptoms and signs score.
VAS is visual analogue scale score.
COPD is chronic obstructive pulmonary disease.
Samples from subjects two and 6, both genotype B, were collected about 2 years apart in January 2010 and March 2012 from a younger patient without underlying chronic illnesses and an older patient with cardiac disease and diabetes mellitus. Both had acute respiratory and systemic symptoms that may have been less severe than those reported by the two patients with genotype D isolates (Table 1). One of the two illnesses with genotype B viruses was associated with a greater than fourfold increase in nasal wash IgA antibody titer to HCoV‐OC43, but neither had a fourfold rise in serum IgG antibody titer to HCoV‐OC43, comparing acute illness to convalescent specimens collected 3‐4 weeks after illness onset (Table 1).
3.2. Quantitation of HCoV‐OC43 mutation load
The raw data output indicated 70.1% of bases read had a quality score greater than 30. Interpretation of the distribution statistics of base quality scores over read length resulted in trimming the read length at the 3′ end by 6‐10%. The final results of the quality control are shown in Supplemental Table. The large output gave a very deep base coverage for each HCoV sample, the average was 94 899 ± 21 405 (Supplemental Fig. S1).
To estimate the error rate associated with library construction and Illumina sequencing, mutations were called from two recombinant HCV clones, #1701 and #1709, under a range of base quality settings. Even if a mutation was counted under the base quality as low as 15, there were no differences in the consensus sequences derived either from Illumina or from gene‐walking Sanger sequencing (data not shown). However, the number of individual mutations had a sharp drop from the base quality 25‐30 (Supplemental Fig. S2). Under the conditions of 0.5% mutation frequency and base quality score of ≥30, there were a total of 31 mutations in the two HCV clone samples, suggesting an error rate of about 1.76 mutations per kb.
Applying the same criteria for the coronavirus samples, a total of 121 mutations for all four samples were identified with an average number of mutations of 30.3 ± 10.2 (range: 20‐40 mutations per sample), which is significantly higher than that expected from the Illumina sequencing error rate (121 vs 28.76 mutations, P = 4.2 × 10−14). Nonsynonymous mutations accounted for between 61% and 90% of the total mutations (Table 2). No deletions or recombinations were detected. Of the 121 viral mutations, those with frequencies greater than 2% occurred at six positions: Spike gene position numbers 79, 81, 1229, 1859, 2244, and 2858 (Table 3).
Table 2.
Numbers of mutations in the four human coronavirus OC43 (HCoV‐OC43) samples at 0.5% mutation frequency and quality score ≥30
| No. of mutations (% of total) | |||
|---|---|---|---|
| Clinical HCoV‐OC43 samples | Synonymous | Non‐synonymous | Total |
| 2 | 9 (39%) | 14 (61%) | 23 |
| 3 | 2 (10%) | 18 (90%) | 20 |
| 4 | 5 (12.5%) | 35 (87.5%) | 40 |
| 6 | 12 (32%) | 26 (68%) | 38 |
| Total | 28 (23%) | 93 (77%) | 121 |
Table 3.
Complete listing of 121 viral mutations and population frequencies in four human coronavirus OC43 clinical samples
| Sample 2 (genotype B) | Sample 3 (genotype D) | Sample 4 (genotype D) | Sample 6 (genotype B) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide position in: | Nucleotide position in: | Nucleotide position in: | Nucleotide position in: | ||||||||
| Spike gene | Complete genome | Frequency (%) | Spike gene | Complete genome | Frequency (%) | Spike gene | Complete genome | Frequency (%) | Spike gene | Complete genome | Frequency (%) |
| 29 | 23672 | 1.26 | 41 | 23684 | 0.50 | 29 | 23672 | 1.21 | 6 | 23649 | 0.54 |
| 65 | 23708 | 0.99 | 65 | 23708 | 0.83 | 41 | 23684 | 0.84 | 29 | 23672 | 0.98 |
| 79 | 23722 | 36.92 | 81 | 23724 | 0.92 | 65 | 23708 | 0.95 | 65 | 23708 | 0.68 |
| 81 | 23724 | 7.96 | 174 | 23817 | 1.25 | 81 | 23724 | 0.82 | 79 | 23722 | 45.12 |
| 104 | 23747 | 0.56 | 259 | 23902 | 0.55 | 94 | 23737 | 0.89 | 81 | 23724 | 7.07 |
| 734 | 24377 | 0.53 | 372 | 24015 | 0.55 | 98 | 23741 | 0.56 | 104 | 23747 | 0.54 |
| 735 | 24378 | 0.65 | 997 | 24640 | 0.71 | 104 | 23747 | 0.54 | 211 | 23854 | 0.92 |
| 736 | 24379 | 0.67 | 1012 | 24655 | 0.62 | 119 | 23762 | 0.51 | 284 | 23927 | 0.95 |
| 801 | 24444 | 0.67 | 1675 | 25318 | 0.71 | 169 | 23812 | 0.59 | 320 | 23963 | 0.51 |
| 894 | 24537 | 0.50 | 1682 | 25325 | 0.53 | 242 | 23885 | 0.55 | 325 | 23968 | 0.55 |
| 979 | 24622 | 0.97 | 1691 | 25334 | 0.79 | 284 | 23927 | 0.75 | 541 | 24184 | 0.60 |
| 1036 | 24679 | 0.51 | 1709 | 25352 | 0.68 | 310 | 23953 | 0.53 | 672 | 24315 | 0.72 |
| 1431 | 25074 | 0.52 | 1712 | 25355 | 0.74 | 320 | 23963 | 0.81 | 766 | 24409 | 0.93 |
| 1773 | 25416 | 0.57 | 1716 | 25359 | 0.61 | 325 | 23968 | 0.59 | 820 | 24463 | 0.50 |
| 2172 | 25815 | 1.05 | 1730 | 25373 | 0.56 | 740 | 24383 | 0.55 | 929 | 24572 | 0.66 |
| 2244 | 25887 | 2.44 | 1732 | 25375 | 0.52 | 784 | 24427 | 0.58 | 1217 | 24860 | 0.62 |
| 2383 | 26026 | 0.51 | 1736 | 25379 | 0.60 | 1229 | 24872 | 2.03 | 1437 | 25080 | 0.50 |
| 2498 | 26141 | 0.57 | 1754 | 25397 | 0.72 | 1457 | 25100 | 0.51 | 1503 | 25146 | 0.81 |
| 2858 | 26501 | 2.10 | 2000 | 25643 | 1.22 | 1566 | 25209 | 0.68 | 1523 | 25166 | 0.50 |
| 3133 | 26776 | 0.59 | 2498 | 26141 | 0.52 | 1581 | 25224 | 0.54 | 1544 | 25187 | 0.63 |
| 3480 | 27123 | 0.50 | 1622 | 25265 | 0.51 | 1553 | 25196 | 0.63 | |||
| 3958 | 27601 | 0.96 | 1641 | 25284 | 0.60 | 1596 | 25239 | 0.68 | |||
| 3970 | 27613 | 0.68 | 1670 | 25313 | 0.57 | 1600 | 25243 | 0.57 | |||
| 1675 | 25318 | 0.62 | 1601 | 25244 | 0.65 | ||||||
| 1691 | 25334 | 0.60 | 1777 | 25420 | 0.89 | ||||||
| 1913 | 25556 | 1.07 | 1841 | 25484 | 0.97 | ||||||
| 2494 | 26137 | 0.61 | 1859 | 25502 | 2.27 | ||||||
| 2738 | 26381 | 0.50 | 2078 | 25721 | 0.62 | ||||||
| 2754 | 26397 | 1.05 | 2283 | 25926 | 0.70 | ||||||
| 2775 | 26418 | 0.61 | 2383 | 26026 | 0.56 | ||||||
| 2948 | 26591 | 0.95 | 2826 | 26469 | 0.55 | ||||||
| 3079 | 26722 | 0.77 | 2862 | 26505 | 0.52 | ||||||
| 3095 | 26738 | 0.65 | 3120 | 26763 | 0.51 | ||||||
| 3137 | 26780 | 0.51 | 3151 | 26794 | 0.55 | ||||||
| 3309 | 26952 | 0.89 | 3277 | 26920 | 0.80 | ||||||
| 3462 | 27105 | 0.66 | 3284 | 26927 | 0.83 | ||||||
| 3676 | 27319 | 0.59 | 3465 | 27108 | 0.54 | ||||||
| 3763 | 27406 | 1.10 | 3620 | 27263 | 0.84 | ||||||
| 3929 | 27572 | 0.60 | |||||||||
| 4059 | 27702 | 0.57 | |||||||||
Using a sliding window analysis with window size = 300 bp and overlaps = 100 bp, there were two mutation peaks, one at the 5′ end and the other at about Spike gene position number 1550 (Fig. 1). Using consensus sequences, a Neighbor‐joining tree showed phylogenetically that two of the subjects’ samples were genotype D and two were genotype B strains of HCoV‐OC43 (Fig. 2A and B). The two genotype B samples both had high mutation frequencies at spike gene nucleotide positions 79 (36.92% and 45.12%) and 81 (7.96% and 7.07%), whereas the two genotype D samples had mutation frequencies of 0.92% and 0.82% at position 81, and less than 0.5% for position 79.
Figure 1.
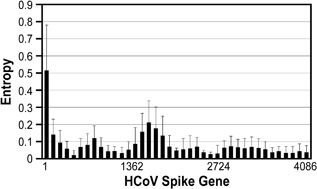
Sliding window analysis of viral mutation loads, calculated as normalized Shannon entropy, over the entire HCoV‐OC43 Spike gene (4086 bp) for the four clinical samples
Figure 2.
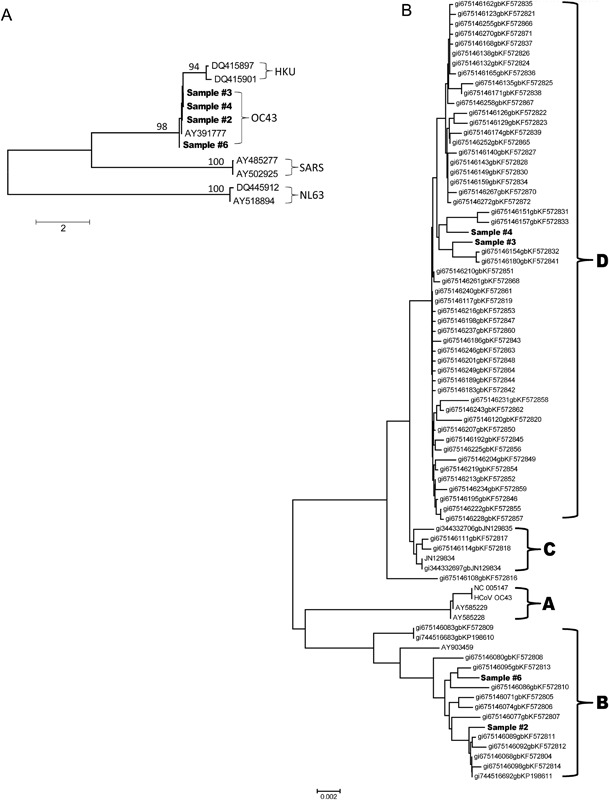
Neighbor‐joining trees constructed with HCoV consensus sequences. Panel A. All four HCoV‐OC43 consensus Spike genes with seven references showing clustering of all four HCoV‐OC43 samples with HCoV‐OC43 strain (AY391777). Panel B. Detailed analysis of the four HCoV‐OC43 samples showing two clustering in genotype D and two clustering in genotype B
3.3. Comparison of HCoV and HCV
A previously reported study found that HCV genotype 1a patients experiencing relapse after antiviral treatment (n = 19) had a higher average total mutation load measured through 454 sequencing.26 These samples were re‐analyzed for the current study using a base quality score >30 rather than 25 in the earlier report.26 The average mutation load in HCV patient isolates was significantly higher than in HCoV‐OC43 patient isolates (296.2 ± 102.2 vs 30.3 ± 10.2, P = 7.7 × 10−5). However, nonsynonymous mutations as a percentage of the total mutations were higher among the HCoV‐OC43 isolates than among the 19 HCV genotype 1a patient isolates (76.7 ± 14% vs 26 ± 8%, P = 3.5 × 10−9).
3.4. Consensus amino acid sequences for HCoV‐OC43
The alignment of Spike genes of consensus amino acid sequences show differences between the four clinical strains and the prototype AY391777 strain particularly at the N and C terminal ends of the S1 subunit, and at the S1 and S2 subunit cleavage site, (a.a. 762‐766) with a smaller number of amino acid differences in the S2 subunit (Fig. 3). The high genetic mutation rates at sites 79 and 81 correspond to several amino acid changes compared to the prototype strain between amino acid positions 20 and 31.
Figure 3.

Tight alignment of consensus amino acid sequences for the Spike genes of the four HCoV‐OC43 clinical samples and the prototype strain, AY391777. Dots indicated the identity and the asterisk donated stop codons
4. DISCUSSION
Through Illumina sequencing, we have introduced high‐resolution HCoV‐OC43 Spike gene mutational load to quantify viral quasi‐species population diversity. The experimental method allows the role of HCoV‐OC43 Spike gene heterogeneity to be investigated in a manner not previously reported. The HCoV‐OC43 Spike gene does have quasi‐species, but the magnitude is almost 10 times lower than the quasi‐species found in HCV, while the nonsynonymous mutations take a higher percentage of the total mutation load for HCoV‐OC43 than HCV genotype 1a. It should be noted that the Spike gene was amplified with the gene‐specific primers. Although these primers are located in the conserved HCoV HE and NS2 domains, the missing of potentially heterogeneous viral variants during the amplification cannot be excluded. As a consequence, quasi‐species diversity of HCoV may be underestimated in the current study in comparison to the use of degenerate primers or primer‐independent approaches. Viral replicative dynamics, population size, and host immune responses may contribute to this observation, and the finding has implications in terms of HCoV evolution and treatment. High mutation load facilitates the evolution of viral populations in response to external pressure. Given their slightly deleterious nature;27 however, excessive nonsynonymous mutations can be detrimental to such adaptive evolution.28 Consequently, accumulation of deleterious mutation load during viral infection may contribute to self‐limited active infection. In the case of HCoV‐OC43, this could contribute to short, self‐limited respiratory tract infections compared to HCV, which is a chronic, systemic viral infection with ongoing viral replication. However, genetic diversity manifested by a cloud of quasi‐species in some viral systems can be linked to pathogenicity and shown to allow adaptation to new environments such as infection of new hosts.29 If clonal virus populations are present in human infections with MERS‐CoV,30, 31 this could increase the chances of epidemic spread of a particularly virulent strain.32 Infections with a cloud of viral strains may enhance spread, for instance, by increasing the chance of a virulent strain being transmitted, or reduce it due to defective and less virulent strains being present. MERS‐CoV genome sequences from dromedary camels indicate presence of quasi‐species in single samples.30 Recent deep sequencing analyses reported intra‐patient viral heterogeneity during a 2015 outbreak of MERS‐CoV and the possibility that host immune response provided selection pressure to favor genetic heterogeneity.33, 34 Intra‐patient viral heterogeneity may have contributed to transmissibility.33
Our four clinical isolates were of the genotypes B and D which have been described as circulating in recent years rather than genotype A.4 Alignment of spike gene consensus amino acid sequences compared to the AY391777 prototype strain sequence (genotype A) indicates the majority of changes were in the S1 subunit. The proteolytic cleavage site of S1 and S2 subunits for the four clinical strains in our study had the RRSRR motif rather than the RRSRG motif of the prototype strain. This G to R substitution at amino acid 766 may result in increased cleavability and the cleavage process may play a part in fusion activity and viral infectivity.4, 35
There were minimal amino acid changes at Spike gene nucleotide positions 448‐459 (whole genome positions 24 091‐24 102) in the glomerular part of the Spike gene, encoding the TQDG (a.a. positions 150‐153), and low mutation frequencies in that region for our four clinical isolates, although all four had a Y154V substitution. This is in the lectin domain of the S1 subunit involved in attachment of the virus to the cellular receptor which is a derivative of neuraminic acid.36 The TQDG sequence may have evolutionary and functional importance, is not present in the closely related bovine coronaviruses and not inserted in some strains of HCoV‐OC43.36 Four critical sugar‐binding residues Y168, E188, W190, and H191 were present in our HCoV‐OC43 isolates corresponding to Y162, E182, W184, and H185 in the bovine coronavirus sequence.37
The receptor‐binding of SARS‐CoV in the S1 carboxy‐terminal domain around the receptor binding motif involving amino acids 479 and 487 are areas where nonsynonymous substitutions and genetic diversity occurred for our HCoV‐OC43 isolate sequences. These areas are important for binding affinity to human angiotensin‐converting enzyme two for SARS‐CoV, and host immune responses.38 Our isolates had nonsynonymous substitutions at amino acid residues 259 and 260‐264 in the N‐terminal domain of S1, and, in particular, had nonsynonymous amino acid substitutions within the region between amino acids 471 and 550 at a putative receptor binding domain of HCoV‐OC43 (a.a. positions 339‐549).4 In other HCoV strains, for instance HCoV‐229E, antibody neutralization of the virus is dependent on the antigenic phenotype of the S1 subunit region.39 Hence, this may also be a factor for HCoV‐OC43 needing further study. Our patients all had acute upper respiratory and systemic signs and symptoms. The small number evaluated here precludes conclusions about pathogenicity and viral mutation rate patterns, but this approach opens up pathways to future study.
In conclusion, quasi‐species were present in our HCoV‐OC43 strains involving areas of the S1 subunit of the Spike gene that may affect evolution of the viral binding process and antigenicity, as well as host specificity. Further studies are needed to more fully characterize the extent of quasi‐species in more clinical isolates and their relation to disease characteristics and host immune responses. Nonsynonymous mutations were more frequent than synonymous ones, contributing to the hypothesis that the mutations are important to viral persistence in the human population over time and to disease pathogenesis.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Legends S1.
Supporting Figure S1.
Supporting Figure S2.
Supporting Table S1.
ACKNOWLEDGMENTS
This work was supported by Veterans Affairs (VA) Research, Department of Veterans Affairs Office of Research and Development. The sponsor had no role in the design, conduct, data analysis, and reporting of the study. The authors thank Kiana Wilder for secretarial assistance, Mary Margaret Donovan, MSN for clinical nursing assistance, and the Center for Vaccine Development at Saint Louis University.
Gorse GJ, Patel GB, Fan X. Interpatient mutational spectrum of human coronavirus‐OC43 revealed by illumina sequencing. J Med Virol. 2017; 89: 1330–1338. 10.1002/jmv.24780
REFERENCES
- 1. Belouzard S, Millet JK, Licitra BN, Whittaker GR. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012; 4:1011–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bosch BJ, van der Zee R, de Haan CAM, Rottier PJM. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol. 2003; 77:8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ren L, Zhang Y, Li J, et al. Genetic drift of human coronavirus OC43 spike gene during adaptive evolution. Sci Rep. 2015; 5:11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lau SKP, Lee P, Tsang AKL, et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J Virol. 2011; 85:11325–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y, Li J, Xiao Y, et al. Genotype shift in human coronavirus and emergence of a novel genotype by natural recombination. J Infect. 2015; 70:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016; 3:237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masters PS, Perlman S. 2013. Coronaviridae. In: Knipe DM, Howley PM, editors. Fields Virology 6th Ed. Philadelphia: Lippincott Williams & Wilkins, pp 825–854. [Google Scholar]
- 8. Gorse GJ, O'Connor TZ, Young SL, et al. Efficacy trial of live, cold‐adapted and inactivated influenza virus vaccines in older adults with chronic obstructive pulmonary disease: a VA cooperative study. Vaccine. 2003; 21:2133–2144. [DOI] [PubMed] [Google Scholar]
- 9. Gorse GJ, O'Connor TZ, Young SL, et al. Impact of a winter respiratory virus season on patients with COPD and association with influenza vaccination. Chest. 2006; 130:1109–1116. [DOI] [PubMed] [Google Scholar]
- 10. Gorse GJ, O'Connor TZ, Hall SL, Vitale JN, Nichol KL. Human coronavirus and acute respiratory illness in older adults with chronic obstructive pulmonary disease. J Infect Dis. 2009; 199:847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gorse GJ, Donovan MM, Patel GB, Balasubramanian S, Lusk RH. Coronavirus and other respiratory illnesses comparing older with younger adults. Am J Med. 2015; 128:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walsh EE, Shin JH, Falsey AR. Clinical impact of human coronavirus 229E and OC43 infection in diverse adult populations. J Infect Dis. 2013; 208:1634–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan X, Mao Q, Zhou D, et al. High diversity of hepatitis C viral quasispecies is associated with early virological response in patients undergoing antiviral therapy. Hepatology. 2009; 50:1765–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fan X, DiBisceglie AM. RT‐PCR amplification and cloning of large viral sequences. Methods Mol Biol. 2010; 630:139–149. [DOI] [PubMed] [Google Scholar]
- 15. Wang W, Zhang X, Xu Y, Weinstock GM, Di Bisceglie AM, Fan X. High resolution quantification of hepatitis C virus genome‐wide mutation load and its correlation with the outcome of peginterferon‐alpha 2a and ribavirin combination therapy. PLoS ONE. 2014; 9:e100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorse GJ, Patel GB, Vitale JN, O'Connor TZ. Prevalence of antibodies to four human coronaviruses is lower in nasal secretions than in serum. Clin Vaccine Immunol. 2010; 17:1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arden NH, Patriarca PA, Fasano MB, et al. The roles of vaccination and amantadine prophylaxis in controlling an outbreak of influenza A (H3N2) in a nursing home. Arch Intern Med. 1988; 148:865–868. [PubMed] [Google Scholar]
- 18. Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011; 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Langmead B, Salzberg SL. Fast grapped‐read alignment with bowtie 2. Nat Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011; 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Belleghem SM, Roelofs D, Van Houdt J, Hendrickx F. De Novo transcriptome assembly and SNP discovery in the wing polymorphic salt marsh beetle Pogonus chalceus (ColeopteraCarabidae). PLoS ONE. 2012; 7:e42605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar A. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol Biol Evol. 2011; 28:2731–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ren Y, Wang W, Zhang X, Xu Y, Di Bisceglie AM, Fan X. Evidence for deleterious hepatitis C virus quasispecies mutation loads that differentiate the response patterns in interferon‐based antiviral therapy. J Gen Virol. 2016; 97:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eyre‐Walker A, Keightley PD, Smith NGC, Gaffney D. Quantifying the slightly deleterious mutation model of molecular evolution. Mol Biol Evol. 2002; 191:2142–2149. [DOI] [PubMed] [Google Scholar]
- 28. Peck JR. A ruby in the rubbish: beneficial mutations, deleterious mutations and the evolution of sex. Genetics. 1994; 137:597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006; 439:344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Briese T, Mishra N, Jain K, et al. Middle East Respiratory Syndrome coronavirus quasispecies that include homologues of human isolates revealed through whole‐genome analysis and virus cultured from dromedary camels in Saudi Arabia. mBio. 2014; 5:e01146–e01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cotten M, Lam TT, Watson SJ, et al. Full‐genome deep sequencing and phylogenetic analysis of novel human betacoronavirus. Emerg Infect Dis. 2013; 19:736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gardner LM, McIntyre CR. Unanswered questions about the Middle East respiratory syndrome coronavirus (MERS‐CoV). BMC Research Notes. 2014; 7:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park D, Huh HJ, Kim YJ, et al. Analysis of intra‐patient heterogeneity uncovers the microevolution of Middle East respiratory syndrome coronavirus. Cold Spring Harb Mol Case Stud. 2016; 2:a001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim D‐W, Kim Y‐J, Park SH, et al. Variations in spike glycoprotein gene of MERS‐CoV, South Korea, 2015. Emerg Infect Dis. 2016; 22:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vijgen L, Keyaerts E, Leme P, et al. Circulation of genetically distinct contemporary human coronavirus OC43 strains. Virology. 2005; 337:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kin N, Miszczak F, Lin W, Gouilh MA, Vabret A, Consortium Epicorem. Genomic analysis of 15 human coronaviruses OC43 (HCoV‐OC43s) circulating in France from 2001 to 2013 reveals a high intra‐specific diversity with new recombinant genotypes. Viruses. 2015; 7:7052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peng G, Xu L, Lin Y‐L, et al. Crystal structure of bovine coronavirus spike protein lectin domain. J Biol Chem. 2012; 287:41931–41938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li F. Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol. 2015; 89:1954–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shirato K, Kawase M, Watanabe O, et al. Differences in neutralizing antigenicity between laboratory and clinical isolates of HCoV‐229E isolated in Japan in 2004‐2008 depend on the S1 region sequence of the spike protein. J Gen Virol. 2012; 93:1908–1917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Legends S1.
Supporting Figure S1.
Supporting Figure S2.
Supporting Table S1.
Data Availability Statement
Raw sequence data in fastq format from all four patient samples were archived in NCBI Sequence Read Archive (SRA) under SRA accession number SRP071020.