

Abstract
BST2/tetherin, an interferon‐inducible antiviral factor, can block the cellular release of various enveloped viruses. We previously reported that human coronavirus 229E (HCoV‐229E) infection can alleviate the BST2 tethering of HIV‐1 virions by downregulating cell surface BST2, suggesting that coronaviruses are capable of encoding anti‐BST2 factors. Here we report our new finding that severe acute respiratory syndrome coronavirus (SARS‐CoV) spike (S) glycoprotein, similar to Vpu, is capable of antagonizing the BST2 tethering of SARS‐CoV, HCoV‐229E, and HIV‐1 virus‐like particles via BST2 downregulation. However, unlike Vpu (which downmodulates BST2 by means of proteasomal and lysosomal degradation pathways), BST2 downregulation is apparently mediated by SARS‐CoV S through the lysosomal degradation pathway only. We found that SARS‐CoV S colocalized with both BST2 and reduced cell surface BST2, suggesting an association between SARS‐CoV S and BST2 that targets the lysosomal degradation pathway. According to one recent report, SARS‐CoV ORF7a antagonizes BST2 by interfering with BST2 glycosylation1. Our data provide support for the proposal that SARS‐CoV and other enveloped viruses are capable of evolving supplementary anti‐BST2 factors in a manner that requires virus replication. Further experiments are required to determine whether the BST2‐mediated restriction of authentic SARS‐CoV virions is alleviated by the SARS‐CoV spike protein.
Keywords: coronavirus, human immunodeficiency virus, immune responses, innate immunity, SARS coronavirus, virus classification
Highlight
BST2/tetherin inhibits the release of various enveloped viruses.
SARS‐CoV S antagonizes the BST2 tethering of human coronavirus and HIV‐1 virus‐like particles.
SARS‐CoV S colocalizes with BST2 and reduces cell surface BST2.
SARS‐CoV S downregulates BST2 through lysosomal degradation pathway.
1. INTRODUCTION
Bone marrow stromal antigen 2 (BST2, also designated as CD317 or tetherin) is a type II integral membrane protein containing a cytoplasmic N‐terminal region followed by a spanning transmembrane domain and a carboxy‐terminal glycosyl‐phosphatidylinositol (GPI) anchor.1 BST2 is an interferon‐inducible gene that functions as an innate defense system against virus infections. It has been described as a host restriction factor capable of impeding the release of several types of enveloped viruses, including retroviruses,2, 3, 4, 5, 6, 7 filoviruses,8, 9, 10 arenaviruses,11 influenza,12 and the Sendai virus.13
One research team has proposed that BST2 inhibits virus release by tethering nascent virions to cell surfaces via the N‐terminal transmembrane domain and C‐terminal GPI anchor.14 Most BST2‐restricted enveloped viruses bud directly from cell surfaces, but a small number of enveloped viruses (eg, herpesviruses) are subject to BST2‐related restrictions even though their final envelopment entails membranes from TGN and/or endosomal compartments and egression via exocytosis.15, 16 In a previous study we reported that the human coronavirus 229E (HCoV‐229E), whose assembly and budding occurs at the ER‐Golgi intermediate compartment and whose virions are released via vesicle exocytosis,17, 18, 19 is also subject to BST2 inhibition. Results from electron microscopy analyses indicate the presence of HCoV‐229E virions on cell surfaces or on the membranes of intracellular vesicles that tend to cluster with BST2. This suggests the BST2‐triggered tethering of budding virions to vesicle membranes that remain on cell surfaces at the plasma membrane after exocytosis.17 BST2 has been described as moderately restricting the release of the hepatitis C virus, whose assembly takes place in the ER and whose release from cells via secretory pathways occurs in a manner similar to that of coronaviruses.20, 21 Combined, these data support the assumption that enveloped virus budding and release occurring at the plasma membrane or in an intracellular compartment is subject to BST2 blocking. BST2 is a component of innate immune response in the form of restricted enveloped virion release, and many viruses have evolved specific antagonists to counteract BST2 antiviral activity: HIV‐1 Vpu, HIV‐2 Env, simian immunodeficiency virus (SIV) Nef and Env, Ebola and Sendai virus GP, Kaposi's sarcoma‐associated herpesvirus K5, and influenza virus neuraminidase are all capable of antagonizing BST2.2, 3, 4, 5, 9, 12, 13, 15, 22 Since some of these anti‐BST2 viral factors are viral envelope glycoproteins, there is speculation that SARS‐CoV spike glycoprotein may possess the property to counteract the BST2 blocking of virus release. Our work is built in part on an earlier finding by another research team that the ORF7a accessory protein (encoded by SARS‐CoV) inhibits the BST2 tethering of virions.23 We also found that the SARS‐CoV spike (S) protein is capable of downmodulating BST2, thus mitigating the BST2‐mediated restriction of virus‐like particle (VLP) release, and suggesting that SARS‐CoV and other enveloped viruses are capable of evolving additional anti‐BST2 factors.
2. MATERIALS AND METHODS
2.1. Plasmid construction and expression vectors
Mammalian expression vectors encoding SARS‐CoV M, N, S, and E were provided by G. J. Nabel.24 BST2 dimerization‐defective mutant (BST2/C3A) was a gift from Klaus Strebel.25 Plasmid pTRE‐HN, kindly provided by Volker Thiel,26 served as a template to generate PCR product containing HCoV‐229E nucldocapsid coding sequence, using a forward primer 5′‐CGCAATCGATTCATGAAGGCAGTTGCT‐3′ and a reverse primer 5′‐CTTCGGATCCCGTTTACTTCATCAAT‐3. For constructing an HA‐tagged HCoV‐229E M expression vector, a plasmid containing codon optimized sequence (synthesized by Mission Biotech, Taiwan) served as a template, using a forward primer 5′‐CGCAATCGATTCATGAAGGCAGTTGCT‐3′ (forward) and a reverse primer 5′‐CTTCGGATCCCGTTTACTTCATCAAT‐3. PCR‐amplified products were digested with BamHI and ClaI and cloned into the SARS‐CoV M or N expression backbone, yielding HCoV‐229N or HA‐tagged 229M expression vectors.
2.2. Virus, cell culture, and transfection
293T, HeLa, and stable BST2‐knockdown HeLa cell lines (HeLa/BST2‐)17 were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (Invitrogen). Confluent cells were trypsinized and seeded onto 10 cm dish plates 24 hours before transfection. For each construct, cells were transfected with 20 μg of plasmid DNA by the calcium phosphate precipitation method; 50 μm chloroquine was added to enhance transfection efficiency. Unless otherwise indicated, 10 μg of each plasmid was used for cotransfection. For HeLa transfection, plasmid DNA was mixed with GenCarrier (Epoch Biolabs) at a ratio of 1 μg to 1 μL; the transfection procedure was performed according to the manufacturer's protocols. Human coronavirus 229E (HCoV‐229E) were propagated in HeLa or A549 cells as described previously.17
2.3. Protein analysis
Culture supernatant from transfected cells was collected, filtered, and centrifuged through 2 mL of 20% sucrose in TSE (10 mM Tris–HCl [pH 7.5], 100 mM NaCl, 1 mM EDTA plus 0.1 mM phenylmethylsulfonyl fluoride [PMSF]) at 4°C for 40 minutes at 274 000g. Pellets were suspended in IPB (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X‐100, 0.02% sodium azide) plus 0.1 mM PMSF. Cells were rinsed with ice‐cold phosphate‐buffered saline (PBS), collected in IPB plus 0.1 mM PMSF, and microcentrifuged at 4°C for 15 minutes at 13 700g to remove unbroken cells and debris. Either supernatant or cell samples were mixed with equal volumes of 2× sample buffer (12.5 mM Tris–HCl [pH 6.8], 2% SDS, 20% glycerol, 0.25% bromphenol blue) and 5% β‐mercaptoethanol and boiled for 5 minutes or (for the M‐containing samples) incubated at 45°C for 10 minutes. Samples were resolved by electrophoresis on SDS‐polyacrylamide gels and electroblotted onto nitrocellulose membranes. Membrane‐bound M or HA‐M proteins were immunodetected using a SARS‐CoV M rabbit antiserum or anti‐HA (LTK BioLaboratories, Taiwan) monoclonal antibody. For SARS‐CoV N or S detection, a mouse monoclonal antibody was used.27, 28 BST2 was probed with a human BST2 mouse antiserum (ab88523, Abcam) or a rabbit antiserum.29 Vpu was detected with a rabbit antiserum.30 The secondary antibody was a sheep antimouse or donkey antirabbit horseradish peroxidase‐(HRP) conjugated antibody (Invitrogen).
2.4. Laser scanning immunofluresecne microscopy
HeLa cells were split 1:80 onto coverslips 24 hours before transfection. Between 18 and 24 hours posttransfection, cells were washed with PBS and either directly probed with an anti‐BST2 antibody before cell membrane permeabilization. Cells then were permeablized in in acetone for 10 minutes at room temperature after fixation with 3.7% formaldehyde at 4°C for 20 minutes. Samples were incubated with the primary antibody for 1 hour and with the secondary antibody for 30 minutes. After each incubation, samples were subjected to three washes (5 to 10 minutes each) with DMEM/calf serum. BST2 was probed with a rabbit antiserum.29 SARS‐CoV S was detected with a mouse monoclonal antibody.27 A rhodamine‐conjugated or FITC‐conjugated antirabbit or antimouse antibody served as the secondary antibody (Cappel, ICN Pharmaceuticals, Aurora, OH). After a final DMEM/calf serum wash, the coverslips were washed three times with PBS and mounted in 50% glycerol in PBS for viewing. Images were analyzed and photographs taken using the inverted laser Zeiss.
3. RESULTS
3.1. BST2 restricts coronavirus VLP release
In our previous report, we determined that the coexpression of either SARS‐CoV28 or HCoV‐229E M and N (unreported results) are sufficient for VLP production. To test the ability of BST2 to inhibit coronavirus VLP release, M, and N proteins from SARS‐CoV or HC0V‐229E were coexpressed with or without BST2 in 293T cells; NL4.3delVpu (a Vpu‐deficient HIV‐1 virion‐producing expression vector) served as a control. Since SARS‐CoV M can be released into the medium as vesicles31 but N cannot be released without M coexpression,32 N detected in medium indicates VLPs formed by M and N. Accordingly, VLP release levels can be measured as N detected in medium. Data from repeat independent experiments indicate that BST2 coexpression led to significant decreases in SARS‐CoV, HCoV‐229E, and HIV‐1delVpu VLP yields ( Figures 1A‐D). The BST2 inhibitory effect on VLP release occurred dose‐dependently (Figures 1E‐G).
Figure 1.

BST2 inhibits coronavirus virus‐like particle (VLP) production. A‐D, 293T cells were cotransfected with SARS M and N (panel A), HA‐tagged HCoV‐229E M and N (panel B), or NL4.3delVpu (panel C) expression vectors, with (lane 2) or without a BST2 expression vector. Cells and supernatants were harvested and subjected to Western immunoblotting at 24 to 36 hours posttransfection. D, N proteins from medium or cell samples were quantified by scanning N band densities from immunoblots. Rations of N level in media to those in cells were determined for each sample and normalized to that of samples without BST2 coexpression. Data were derived from at least three independent experiments. *P < .05; **P < .01. (E‐G) 293T cells were transfected with SARS (panel E), HCoV‐229E (panel F), or HIV‐1 (panel G) VLP‐producing expression vectors as described for (A‐C). All tests were performed with 0.1 μg, 0.5 μg, or 2 μg of cotransfected BST2 expression plasmids (lanes 2, 3 and 4, respectively). H‐J, HeLa or BST2‐knockdown HeLa (HeLa/BST2‐) cells were transfected with the indicated SARS, HCoV‐229E, or HIV‐1 VLP‐producing plasmid. Cells and supernatants were harvested, prepared and subjected to Western immunoblotting at 48 to 72 hours posttransfection. Viral proteins were detected with anti‐SARS‐CoV M, anti‐HCoV‐229 N antiserum, or monoclonal antibodies against SARS‐CoV N, HA‐tagged HCoV‐229 M, or p24CA. BST2 was probed with a rabbit anti‐BST2 antibody. BST2, bone marrow stromal antigen 2; SARS‐CoV, severe acute respiratory syndrome coronavirus
We also used HeLa cells (which constitutively express BST2) to assess the impact of endogenous BST2 on VLP yields and found that BST2‐knockdown HeLa cells (HeLa/BST2‐) produced VLPs at higher levels compared to normal HeLa cells (Figures 1H‐J). This suggests that VLP release is also subject to restrictions associated with endogenous BST2. To determine whether reduced VLP yield is a result of VLPs being tethered to cell membranes by BST2, cells were treated with subtilisin, a nonspecific protease capable of triggering cell surface‐associated virion release via BST2 cleavage.17, 33 Our results indicate increased medium‐associated VLP quantities following subtilisin treatment (Figures 2A‐C, lane 6 vs. lane 4), confirming that BST2 trapped VLPs on cell surfaces. Combined, the data suggest that both exogenous and endogenous BST2 are capable of inhibiting SARS‐CoV and HCoV‐229E VLP release.
Figure 2.

Subtilisin treatment promotes virus‐like particle release. 293T cells were cotransfected with SARS CoV (A), HCoV‐229E M and N (B), or NL4.3delVpu (C) with or without a BST2 expression vector. Cells were split equally into two dish plates at 24 hours posttransfection. After an additional 4 hours, culture medium was removed, washed twice with PBS, and incubated with PBS containing subtilisin (1 mg/mL) for 10 minutes at 37℃. Supernatants were harvested and centrifuged through 20% sucrose cushions, and pellets and cell lysates were subjected to western immunoblotting. Viral proteins were probed as described in the Figure 1 caption. BST2, bone marrow stromal antigen 2; PBS, phosphate‐buffered saline; SARS‐CoV, severe acute respiratory syndrome coronavirus
3.2. BST2 dimerization is required for coronavirus VLP release inhibition
BST2 forms stable cysteine‐linked dimers. Blocking BST2 dimerization by replacing cysteines C53, C63, and C91 with alanine in the BST2 ectodomain, in turn, blocks the BST2 inhibition of HIV‐1 release, suggesting that such dimerization is required for virion release blocking.25 To test whether BST2 dimerization is also required for restricting coronavirus VLP release, M and N proteins were coexpressed with a dimerization‐defective BST2/C3A mutant containing alanine substitutions for C53, C63, and C91. Results indicate that wild‐type BST2 was capable of inhibiting SARS‐CoV, HCoV‐229E, and HIV‐1 VLP production, but BST2/C3A was not (Figures 3A‐C, lane 2 vs. lane 3), further suggesting that BST2 dimerization is required to inhibit coronavirus VLP release.
Figure 3.

Cysteine residues in the BST2 N‐terminal ectodomain are important for inhibiting VLP release. 293T cells were cotransfected with SARS M plus N (panel A), HA‐tagged HCoV‐229E M plus N (panel B), or NL4.3delVpu (panel C) plus a wild‐type or mutant BST2 expression vector (BST2/C3A) containing alanine substitutions for three cysteine residues in the BST2 ectodomain. Cells and supernatants were harvested and subjected to western immunoblotting at 24 to 36 hours posttransfection. BST2, bone marrow stromal antigen 2; VLP, virus‐like particle
3.3. SARS‐CoV spike (S) alleviates BST2 restriction of HIV‐1 release by downregulating BST2
As stated above, several viral membrane glycoproteins such as HIV‐2 and SIV Env, as well as Ebola and Sendai virus GP proteins, exert counteractive effects on BST2. We, therefore, attempted to identify anti‐BST2 activity associated with the SARS‐CoV spike (S) protein. Since BST2 is known to restrict HIV‐1 release in the absence of Vpu, we performed tests to determine whether SARS‐CoV S counteracts BST2 and therefore supports HIV‐1 release. As shown in the upper panel of Figure 4A (lane 2 vs. lanes 3 and 4), the inhibitory effect of BST2 on NL4.3delVpu virion release decreased in the presence of either SARS‐CoV S or Vpu in step with reduced BST2 expression (Figure 4A, middle panel, lanes 2‐4), suggesting that SARS‐CoV S is capable of promoting the release of HIV‐1 virus particles from cells via BST2 downregulation. Additional experiments confirmed that SARS‐CoV S, like Vpu, is capable of reducing BST2 expression in a dose‐dependent manner (Figures 4B and C). Combined, the data suggest that SARS‐CoV S may counteract the BST2‐mediated restriction of VLP release.
Figure 4.

SARS‐CoV S downregulates BST2. A, SARS‐CoV S reduced the inhibition of HIV‐1 VLP production by BST2. 293T cells were transfected with NL4.3delVpu alone (lane 1) or combined with BST2 (lane 2) plus Vpu (lane 3) or SARS‐CoV S (lane 4) expression vectors. Cells and supernatants were harvested and subjected to western immunoblotting at 24 to 36 hours posttransfection. B‐C, 293T cells were transfected with 100 ng of BST2 alone (lane 1) or BST2 plus 1 μg (lane 2) or 3 μg (lane 3) of a SARS‐CoV S (panel B) or Vpu expression vector (panel C). Cells were harvested and subjected to western immunoblotting at 24 hours posttransfection. BST2, bone marrow stromal antigen 2; SARS‐CoV, severe acute respiratory syndrome coronavirus; VLP, virus‐like particle
3.4. SARS‐CoV S downregulates BST2 via a lysosomal degradation pathway
After binding to BST2, Vpu moves BST2 toward lysosomal‐ and proteasomal‐degradation pathways.2, 34, 35, 36, 37, 38 Our next task was to examine whether SARS‐CoV S mediates BST2 degradation via the same or similar pathway. Transfectants were treated with either MG132 (a proteasome inhibitor)39 or ammonium chloride (NH4Cl, a lysosome inhibitor).40 We found that BST2 downregulation mediated by SARS‐CoV S was not significantly affected when the proteasome function was inhibited, but was noticeably reduced following lysosome function inhibition (Figure 5, lanes 5‐7). Consistent with previous reports, we observed that proteasome or lysosome function inhibition resulted in markedly reduced Vpu‐mediated BST2 downregulation (Figure 5, lanes 2‐4), suggesting that BST2 downregulation as mediated by SARS‐CoV S largely occurs via the lysosomal degradation pathway.
Figure 5.
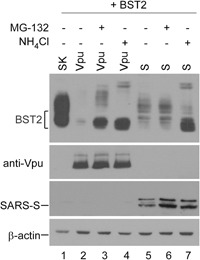
SARS‐CoV S downregulates BST2 via a lysosomal degradation pathway. 293T cells were transfected with BST2 (lane 1) or cotransfected with BST2 plus a Vpu (lanes 2‐4) or SARS‐CoV S (lanes 5‐7) expression vector. At 24 hours posttransfection, cells were either left untreated (lanes 1, 2, and 5), treated with 30 μM MG‐132 for 6 hours (lanes 3 and 6), or treated with 25 μM NH4Cl (lanes 4 and 7) for 6 hours before harvesting and immunoblotting. BST2, bone marrow stromal antigen 2; SARS‐CoV, severe acute respiratory syndrome coronavirus
3.5. SARS‐CoV spike glycoprotein colocalizes with BST2
Since BST2 largely localizes at cell surfaces (where they tether virions to prevent their release), we tested whether SARS‐CoV S antagonizes BST2 via surface BST2 downregulation. 293 cells were cotransfected with BST2 and SARS‐CoV S expression vectors. We observed that BST2 and S colocalized in perinuclear areas, but BST2 signals were barely detectable on cell surfaces. Instead, BST2 largely localized in perinuclear areas regardless of whether or not it was coexpressed with S (Figure 6A). This is consistent with a previous report that unlike HeLa‐endogenous BST2 (which localizes on cell surfaces as well as in the perinuclear area), exogenous BST2 predominantly localizes in perinuclear areas, with little distribution on cell surfaces.41 Nevertheless, flow cytometry quantification suggests that BST2 cell surface expression is noticeably reduced in 293T cells following SARS‐CoV S coexpression (Figure 6C). In the case of HeLa cells (which constitutively express BST2), we did found SARS‐CoV S colocalized with BST2 in the plasma membrane (Figure 6B). BST2 green fluorescence intensity on HeLa cell surfaces decreased slightly following SARS‐CoV S coexpression, suggesting that the capacity of SARS‐CoV S to counteract the BST2‐associated inhibition of virion release was due in part to cell surface BST2 downmodulation.
Figure 6.

SARS‐CoV glycoprotein S colocalizes with BST2. 293 cells (panel A) were cotransfected with BST2 and a SRAS‐CoV expression vector. HeLa cells (panel B) were transfected with the SARS‐CoV S expression vector. At 24 to 36 hours posttransfection, cells were probed with an anti‐BST2 antibody before cell membrane permeabilization. SARS‐CoV S was probed with an anti‐S polyclonal antiserum. A rhodamine‐conjugated or FITC‐conjugated antirabbit or antimouse antibody served as a secondary antibody. C, SARS‐CoV S coexpression reduces BST2 cell surface expression. 293T cells were transfected with BST2 alone (middle panel) or together with a SARS‐CoV S expression vector (bottom panel). At 24 to 36 hours posttransfection, cells were fixed and probed with a rabbit anti‐BST2 antibody before the permeabilization of cell membranes, followed by a secondary FITC‐conjugated antirabbit antibody. Cells then were analyzed by flow cytometry. BST2, bone marrow stromal antigen 2; SARS‐CoV, severe acute respiratory syndrome coronavirus
4. DISCUSSION
BST2 is capable of inhibiting SARS‐CoV and HCoV‐229E VLP release (Figure 1). Since the BST2 inhibition of HIV‐1 release via virion tethering at cell surfaces is well documented, we used a Vpu‐deficient HIV‐1 virus‐producing vector (NL4.3delVpu) as a control in our experiments. As shown in Figure 2, coronavirus VLPs (similar to those of HIV‐1) were tethered to cell surfaces by BST2, and BST2 inhibited SARS‐CoV and HCoV‐229E VLP release in a BST2 dimerization‐dependent manner, similar to HIV‐1 (Figure 3). Further, in the same manner, as Vpu, SARS‐CoV S facilitated HIV‐1 release via BST2 downregulation (Figure 4). We determined that Vpu downmodulated BST2 via proteasomal and lysosomal degradation pathways and that the predominant lysosomal pathway was mediated by SARS‐CoV S (Figure 5). Our confocal microscopy observations suggest that SARS‐CoV S colocalized with BST2 at HeLa cell surfaces (Figure 6). SARS‐CoV S likely binds to BST2, after which it serves as a target for lysosomal degradation. Vpu is capable of trapping BST2 intracellularly and preventing its recycling back into the plasma membrane.29, 35, 37, 42, 43 Whether SARS‐CoV S similarly counteracts BST2 requires further investigation.
The transmembrane protein SARS‐CoV ORF7a has been shown to counteract BST2 tethering by interfering with BST2 glycosylation.23, 44 In addition to the likelihood that BST2‐associated restriction of SARS‐CoV virion release is mitigated by SARS‐CoV S, it is possible that a number of enveloped viruses have developed supplementary anti‐BST2 factors over time—note that in addition to Vpu, HIV‐1 Nef is capable of overcoming BST2 restrictions on virus release under certain conditions.45 SIV Nef7 and Env2 are capable of antagonizing BST2, and influenza neuraminidase12 and M246 proteins both possess anti‐BST2 capabilities. Some researchers have suggested that influenza and/or Ebola VLP release, but not virion release, is inhibited by BST2.11, 47 Due to biosafety requirements, we are currently unable to perform tests to determine whether SARS‐CoV S is capable of overcoming BST2 restrictions on SARS‐CoV virion release.
ACKNOWLEDGMENT
The anti‐BST2 reagent (cat #11722) used in this study was obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; we wish to thank Drs. Klaus Strebel and Amy Andrew for their assistance in this regard. This study was supported by grants V104C‐056 and V105C‐036 from Taipei Veterans General Hospital, and grants 104‐2320‐B‐010‐035, 105‐2320‐B‐010‐023 and 106‐2320‐B‐010‐017‐MY2 from the Taiwan Ministry of Science and Technology.
Wang S‐M, Huang K‐J, Wang C‐T. Severe acute respiratory syndrome coronavirus spike protein counteracts BST2‐mediated restriction of virus‐like particle release. J Med Virol. 2019;91:1743‐1750. 10.1002/jmv.25518
References
REFERENCES
- 1. Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. Bst‐2/HM1.24 is a raft‐associated apical membrane protein with an unusual topology. Traffic. 2003;4(10):694‐709. [DOI] [PubMed] [Google Scholar]
- 2. Gupta RK, Mlcochova P, Pelchen‐Matthews A, et al. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST‐2/CD317 by intracellular sequestration. Proceedings of the National Academy of Sciences. 2009;106:20889‐20894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jia B, Serra‐Moreno R, Neidermyer W, et al. Species‐specific activity of SIV Nef and HIV‐1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5(5):e1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Le Tortorec A, Neil SJ. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J Virol. 2009;83(22):11966‐11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV‐1 Vpu. Nature. 2008;451(7177):425‐430. [DOI] [PubMed] [Google Scholar]
- 6. Van Damme N, Goff D, Katsura C, et al. The interferon‐induced protein BST‐2 restricts HIV‐1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang F, Wilson SJ, Landford WC, et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. 2009;6(1):54‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jouvenet N, Neil SJ, Zhadina M, et al. Broad‐spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83(4):1837‐1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaletsky RL, Francica JR, Agrawal‐Gamse C, Bates P. Tetherin‐mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A. 2009;106(8):2886‐2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sakuma T, Sakurai A, Yasuda J. Dimerization of tetherin is not essential for its antiviral activity against Lassa and Marburg viruses. PLoS One. 2009;4(9):e6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Radoshitzky SR, Dong L, Chi X, et al. Infectious Lassa virus, but not filoviruses, is restricted by BST‐2/tetherin. J Virol. 2010;84(20):10569‐10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mangeat B, Cavagliotti L, Lehmann M, et al. Influenza virus partially counteracts restriction imposed by tetherin/BST‐2. J Biol Chem. 2012;287(26):22015‐22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bampi C, Rasga L, Roux L. Antagonism to human BST‐2/tetherin by Sendai virus glycoproteins. J Gen Virol. 2013;94(Pt 6):1211‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perez‐Caballero D, Zang T, Ebrahimi A, et al. Tetherin inhibits HIV‐1 release by directly tethering virions to cells. Cell. 2009;139(3):499‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mansouri M, Viswanathan K, Douglas JL, et al. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi's sarcoma‐associated herpesvirus. J Virol. 2009;83(19):9672‐9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mettenleiter TC. Herpesvirus assembly and egress. J Virol. 2002;76(4):1537‐1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang S‐M, Huang K‐J, Wang C‐T. BST2/CD317 counteracts human coronavirus 229E productive infection by tethering virions at the cell surface. Virology. 2014;449(0):287‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masters PS. The molecular biology of coronaviruses. Adv Virus Res. 2006;66:193‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hunter E. Virus assembly In: Knipe DM, Howley PM, eds. Fundamental Virology. 4th, 2001. [Google Scholar]
- 20. Jones DM, McLauchlan J. Hepatitis C virus: assembly and release of virus particles. J Biol Chem. 2010;285(30):22733‐22739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dafa‐Berger A, Kuzmina A, Fassler M, Yitzhak‐Asraf H, Shemer‐Avni Y, Taube R. Modulation of hepatitis C virus release by the interferon‐induced protein BST‐2/tetherin. Virology. 2012;428(2):98‐111. [DOI] [PubMed] [Google Scholar]
- 22. Pardieu C, Vigan R, Wilson SJ, et al. The RING‐CH ligase K5 antagonizes restriction of KSHV and HIV‐1 particle release by mediating ubiquitin‐dependent endosomal degradation of tetherin. PLoS Pathog. 2010;6(4):e1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Taylor JK, Coleman CM, Postel S, et al. Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J Virol. 2015;89(23):11820‐11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang Y, Yang Z‐y, Kong W‐p, Nabel GJ. Generation of synthetic severe acute respiratory syndrome coronavirus pseudoparticles: implications for assembly and vaccine production. J Virol. 2004;78(22):12557‐12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andrew AJ, Miyagi E, Kao S, Strebel K. The formation of cysteine‐linked dimers of BST‐2/tetherin is important for inhibition of HIV‐1 virus release but not for sensitivity to Vpu. Retrovirology. 2009;6:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schelle B, Karl N, Ludewig B, Siddell SG, Thiel V. Selective replication of coronavirus genomes that express nucleocapsid protein. J Virol. 2005;79(11):6620‐6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shih Y‐P, Chen C‐Y, Liu S‐J, et al. Identifying epitopes responsible for neutralizing antibody and DC‐SIGN binding on the spike glycoprotein of the severe acute respiratory syndrome coronavirus. J Virol. 2006;80(21):10315‐10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang S‐M, Wang C‐T. APOBEC3G cytidine deaminase association with coronavirus nucleocapsid protein. Virology. 2009;388(1):112‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miyagi E, Andrew AJ, Kao S, Strebel K. Vpu enhances HIV‐1 virus release in the absence of Bst‐2 cell surface down‐modulation and intracellular depletion. Proceedings of the National Academy of Sciences. 2009;106:2868‐2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maldarelli F, Chen MY, Willey RL, Strebel K. Human immunodeficiency virus type 1 Vpu protein is an oligomeric type I integral membrane protein. J Virol. 1993;67(8):5056‐5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tseng Y‐T, Wang S‐M, Huang K‐J, Lee AI‐R, Chiang C‐C, Wang C‐T. Self‐assembly of severe acute respiratory syndrome coronavirus membrane protein. J Biol Chem. 2010;285(17):12862‐12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tseng Y‐T, Wang S‐M, Huang K‐J, Wang C‐T. SARS‐CoV envelope protein palmitoylation or nucleocapid association is not required for promoting virus‐like particle production. J Biomed Sci. 2014;21(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neil SJD, Sandrin V, Sundquist WI, Bieniasz PD. An interferon‐α‐induced tethering mechanism inhibits HIV‐1 and Ebola virus particle release but is counteracted by the HIV‐1 Vpu protein. Cell Host Microbe. 2007;2(3):193‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Fruh K, Moses AV. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST‐2/Tetherin via a {beta}TrCP‐dependent mechanism. J Virol. 2009;83(16):7931‐7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mitchell RS, Katsura C, Skasko MA, et al. Vpu antagonizes BST‐2‐mediated restriction of HIV‐1 release via beta‐TrCP and endo‐lysosomal trafficking. PLoS Pathog. 2009;5:e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goffinet C, Allespach I, Homann S, et al. HIV‐1 antagonism of CD317 is species specific and involves Vpu‐mediated proteasomal degradation of the restriction factor. Cell Host Microbe. 2009;5:285‐297. [DOI] [PubMed] [Google Scholar]
- 37. Dubé M, Bhusan Roy B, Guiot‐Guillain P, et al. Antagonism of tetherin restriction of HIV‐1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010;6(4):e1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mangeat B, Gers‐Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. HIV‐1 Vpu neutralizes the antiviral factor Tetherin/BST‐2 by binding it and directing its beta‐TrCP2‐dependent degradation. PLoS Pathog. 2009;5(9):e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di‐leucine and tri‐leucine. The Journal of Biochemistry. 1996;119(3):572‐576. [DOI] [PubMed] [Google Scholar]
- 40. Hart PD, Young MR. Ammonium chloride, an inhibitor of phagosome‐lysosome fusion in macrophages, concurrently induces phagosome‐endosome fusion, and opens a novel pathway: studies of a pathogenic mycobacterium and a nonpathogenic yeast. J Exp Med. 1991;174(4):881‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hammonds J, Ding L, Chu H, et al. The tetherin/BST‐2 coiled‐coil ectodomain mediates plasma membrane microdomain localization and restriction of particle release. J Virol. 2012;86(4):2259‐2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lewinski MK, Jafari M, Zhang H, Opella SJ, Guatelli J. Membrane anchoring by a C‐terminal tryptophan enables HIV‐1 Vpu to displace bone marrow stromal antigen 2 (BST2) from sites of viral assembly. J Biol Chem. 2015;290(17):10919‐10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt S, Fritz JV, Bitzegeio J, Fackler OT, Keppler OT. HIV‐1 Vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/tetherin to overcome the virion release restriction. mBio. 2011;2(3):e0036‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nelson CA, Pekosz A, Lee CA, Diamond MS, Fremont DH. Structure and intracellular targeting of the SARS‐coronavirus Orf7a accessory protein. Structure. 2005;13(1):75‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arias JF, Colomer‐Lluch M, von Bredow B, et al. Tetherin antagonism by HIV‐1 group M Nef proteins. J Virol. 2016;90(23):10701‐10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu S, Yin L, Mei S, et al. BST‐2 restricts IAV release and is countered by the viral M2 protein. Biochem J. 2017;474:BCJ20160861. [DOI] [PubMed] [Google Scholar]
- 47. Watanabe R, Leser GP, Lamb RA. Influenza virus is not restricted by tetherin whereas influenza VLP production is restricted by tetherin. Virology. 2011;417(1):50‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]