

Abstract
Previously, it was reported that productive viral infection, viral protein synthesis, and viral RNA replication of respiratory syncytial virus (RSV) operated efficiently in two human epithelial cell lines (HEp‐2 and A549), but not in a human mast‐cell line, HMC‐1. Based on these observations, it was hypothesized that HMC‐1 cells lack the machinery required for RSV replication. To identify the host factors required for RSV replication, cDNA subtraction using A549, HEp‐2, and HMC‐1 cells was performed, and cytokeratin 18 (C18) was identified as a candidate host factor. Because C18 is generally expressed in simple epithelia with cytokeratin 8 (C8), HMC‐1 cells that constitutively express C18 and C8 (HMC‐1‐C8/18) were established to evaluate the role of C8/18 in RSV replication. In HMC‐1‐C8/18 cells, RSV RNA replication was increased, and the amount of infective virus produced was also increased in the cellular fraction after RSV spinoculation, whereas RSV production was decreased in A549 cells in which C18 expression was knocked down. These data suggest that the replication of RSV increases in the presence of C8/18. J. Med. Virol. 84:365–370, 2012. © 2011 Wiley Periodicals, Inc.
Keywords: respiratory syncytial virus (RSV), HMC‐1, cytokeratin 8, cytokeratin 18
Abbreviations:
C18, cytokeratin 18; C8, cytokeratin 8; FCS, fetal calf serum; IMDM, Iscove's modified Dulbecco's medium; PBS, phosphate‐buffered saline; RSV, respiratory syncytial virus; PCR, polymerase chain reaction.
MAIN TEXT
Human respiratory syncytial virus (RSV), of the family Paramyxoviridae, subfamily Pneumovirinae, genus Pneumovirus, is a major causative agent of respiratory tract infections in children worldwide. Preterm children or those with underlying cardiopulmonary disorders are at a particularly high risk of developing severe and lethal RSV respiratory tract infections [American Academy of Pediatrics, 2003; Girard et al., 2005].
Previously, it was reported that viral infection, viral protein synthesis, and viral RNA replication operated efficiently in two RSV‐inoculated human epithelial cell lines (HEp‐2 and A549) but not in an RSV‐inoculated human mast‐cell line, HMC‐1. To address the hypothesis that HMC‐1 lacks the machinery required for RSV replication [Shirato and Taguchi, 2009], a cDNA subtraction assay was performed using HMC‐1 cells and two human epithelial cell lines (A549 and HEp‐2). Using a polymerase chain reaction (PCR)‐Select cDNA Subtraction Kit (Clontech, Mountain View, CA), A549 or HEp‐2 cells were used as the tester, and HMC‐1 cells were used as the driver. The cDNA synthesis, adapter ligation, first hybridization, and second hybridization steps were performed following the manufacturer's protocol, with modifications. After the second hybridization step, PCR and nested PCR were performed using Advantage cDNA polymerase (Clontech) and primers supplied in the PCR‐select cDNA Subtraction Kit. The obtained PCR products were then cloned into the pT7Blue2‐vector (EMD4B biosciences, Gibbstown, NJ), and colonies were selected. The subtraction assay was performed three times, yielding a total of 49 clones for A549 versus HMC‐1 and 85 clones for HEp‐2 versus HMC‐1. The sequences of these clones were confirmed using a 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA) with SP6 and U19mer reverse primers. Using BLASTn to identify sequence homologies, three genes (Cytokeratin 18, GenBank accession no. X12883; Laminin receptor homolog, S35960; and H. sapiens CL100 mRNA for protein tyrosine phosphatase, X68277) were identified as common to both A549 versus HMC‐1 and HEp‐2 versus HMC‐1 experiments (Table I). Among them, cytokeratin 18 (C18) was detected in 2 out of 85 tests (HEp‐2 vs. HMC‐1 cells) and in 19 out of 49 tests (A549 vs. HMC‐1 cells).
Table I.
Genes that Were Obtained in Both A549‐HMC‐1 and HEp‐2‐HMC‐1 Subtraction Experiments
C18 is an acidic cytokeratin that belongs to the cytokeratin family. This 20‐member cytokeratin family is divided into acidic type I cytokeratins (9–20) and basic type II cytokeratins (1–8). These cytokeratins are usually expressed together in a combination of acidic and basic cytokeratins, and the acidic protein C18 is generally expressed together with the basic protein cytokeratin 8 (C8) in simple epithelia [Moll et al., 1982].
To date, a few reports have suggested a relationship between RSV infection and the cytoskeleton. Jeffree et al. [2007] reported a close physical interaction between filamentous actin and the virus in RSV infection involving inclusion bodies and viral filaments. It was previously reported that RSV infection induced the up‐regulation of C17 gene expression in a NF‐kB signaling pathway‐dependent manner, and the expression of C17 was associated with the syncytia formation of RSV [Domachowske et al., 2000]. Garcia‐Barreno et al. [1988] reported that N protein of RSV required a maturation period after synthesis to associate with certain cytoskeletal intermediate filaments. They also reported that C18 in RSV‐infected HEp‐2 cells was modified in a way other than phosphorylation. Thus, a relationship between RSV replication and C18 has been suggested, but it is unclear whether C18 plays a role in RSV replication.
The expression of cytoskeleton mRNA was compared among A549, HEp‐2, and HMC‐1 cells using a quantitative real‐time PCR assay (Fig. 1). There were no marked differences in mRNA expression of vimentin or γ‐actin among the cell types except for vimentin expression in HEp‐2 cells. β‐Actin expression was slightly lower in A549 and HEp‐2 cells relative to HMC‐1 cells. By contrast, the expression of C18 mRNA in A549 and HEp‐2 cells was 100‐ to 1,000‐fold greater than in HMC‐1 cells (n = 4, P < 0.01). These findings show that the RSV‐susceptible cell lines express C18 while the unsusceptible cell line does not. Taken together, these data suggest that C18 might be involved in the replication of RSV in HEp‐2 and A549 cells. Because C18 is predominantly expressed in HEp‐2 cells [Price et al., 1988], and was identified by subtraction experiments as one of the genes expressed in both of the RSV‐sensitive cell lines but not in HMC‐1 cells, the role of C18 in RSV replication was examined further.
Figure 1.
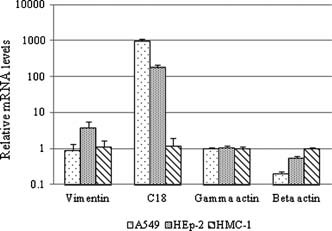
mRNA expression of cytoskeletons in A549, HEp‐2, and HMC‐1 cells. Total RNA was extracted with a Total RNA Isolation Mini Kit (Agilent, Wilmington, DE) and first‐strand cDNA was synthesized with M‐MLV Reverse Transcriptase (Takara Bio, Shiga, Japan) and Random Hexamer (ABI). Real‐time PCR analysis was performed using a LightCycler 480 with its probe master reagent (Roche, Basel, Switzerland) and specific primer and probe sets sold by Applied Biosystems Inc (ABI; C18, Hs01649353_g1; vimentin, Hs00185584_m1; β‐actin, Hs03023880_g1; and γ‐actin, Hs02340971_gH). Human GAPDH was used as an internal control. The data were analyzed using the comparative Ct method and expressed as relative to the data obtained from the HMC‐1 cells (n = 4; P < 0.01).
To elucidate the general physiological function of C18 in RSV infection, plasmids encoding C18 and C8 were co‐transfected into HMC‐1 cells and selected using 2 µg/ml puromycin to establish HMC‐1‐C8/18 cells. Although HMC‐1‐C8/18 cell growth was slower than that of the original HMC‐1 cells, the expression of these genes did not affect cell viability (Fig. 2a). C8/18 mRNA expression in HMC‐1‐C8/18 cells was 100‐ to 1,000‐fold higher than that in HMC‐1 cells (Fig. 2b). C8/18 expression was confirmed by flow cytometry (Fig. 2c). The frequency of C8/18 double‐positive cells was 5.3% in HMC‐1 cells and 64.8% in A549 cells. By contrast, 41.6% of the HMC‐1‐C8/18 cells were double‐positive using the same cut‐off, indicating that the HMC‐1‐C8/18 cells expressed C8/18 at higher levels than the original HMC‐1 cells.
Figure 2.
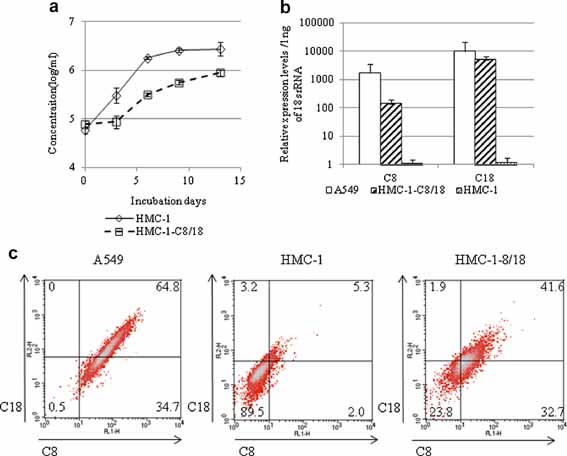
Cloning of HMC‐1‐C8/18 cells. a: Growth kinetics of HMC‐1 and HMC‐1‐C8/18 cells. First, 5 × 104 cells were incubated with 10% FCS‐IMDM in six‐well plates; the cell concentrations were determined every third day (n = 6). b: C8/18 mRNA expression in A549, HMC‐1, and HMC‐1‐C8/18 cells. Total RNA was extracted, first‐strand cDNA was synthesized, and real‐time PCR was performed with specific primer and probe sets for C8 (ABI, Hs02339473_g1) and C18 with 18s rRNA (ABI, 4352930) as an internal control. The data are expressed as relative values compared to those of parental HMC‐1 cells. c: Flow cytometric analysis of C8/18 expression. For C18 detection, cells were stained with specific chicken IgY polyclonal antibodies (GenWay, San Diego, CA) and donkey anti‐chicken IgY, biotin (Chemicon, Temecula, CA) followed by PE‐conjugated streptavidin (BD PharMingen, San Diego, CA). For C8 detection, cells were stained with a mouse monoclonal antibody (M20; GeneTex, Irvine, CA) followed by FITC‐conjugated goat anti‐mouse IgG (Zymed, South San Francisco, CA). Flow cytometry was performed using the FACSCalibur system (Becton Dickinson, Franklin Lakes, NJ).
The replication efficiency of RSV in the presence of C18 was examined using HMC‐1‐C8/18 cells. Productive RSV RNA replication in HMC‐1 cells was not detected by plaque assay in a previous study [Shirato and Taguchi, 2009]; therefore, in this study the replication of RSV in HMC‐1‐C8/18 cells was detected using real‐time PCR. The consensus RSV type A and B/RSV matrix gene probe and primer set described by Kuypers et al. [2004] was used. RSV RNA was quantified using a standard curve obtained from serially diluted virus stocks that were pre‐titrated by plaque assay; the obtained values were normalized with the 18s rRNA values from the same sample. Therefore, the RSV RNA copy number was determined as the ratio of the log10PFU/1 ng of 18s rRNA. The relationship between PFU value and RNA copy number was confirmed in the present study [Shirato et al., 2007; Shirato and Taguchi, 2009]. First, RSV was directly inoculated onto the cells and incubated at 34°C; viral RNA was detected at the indicated number of days postinfection (dpi) (Fig. 3a). Identical to the previous report, a >100‐fold productive increase in RSV RNA was seen in the A549 cells, while the copy number of RSV RNA was low in the HMC‐1 cells over the course of infection. In contrast, the amount of RSV RNA in HMC‐1C8/18 cells increased slightly but was significantly higher than that in HMC‐1 cells (n = 4; P < 0.01). As described previously, RSV was able to infect HMC‐1, but the efficiency of RSV infection in HMC‐1 was very low [Shirato and Taguchi, 2009]. It has been reported that spinoculation is a good method for forcing virion attachment to cells for retroviruses and coronaviruses [O'Doherty et al., 2000; Watanabe et al., 2006]. Therefore, spinoculation (3,000 rpm for 2 h at 4°C) was performed to increase viral infectivity. The spinoculation of RSV increased the amount of virus entering the HMC‐1 cells by two‐ to threefold compared with standard inoculation (data not shown). As a result, the amount of RSV RNA was increased to the level in A549 cells at 4 dpi, whereas that in HMC‐1 was unchanged (Fig. 3b, n = 4; P < 0.01). These results suggest that the amount of RSV RNA in the HMC‐1 cells increased in the presence of C8/18.
Figure 3.
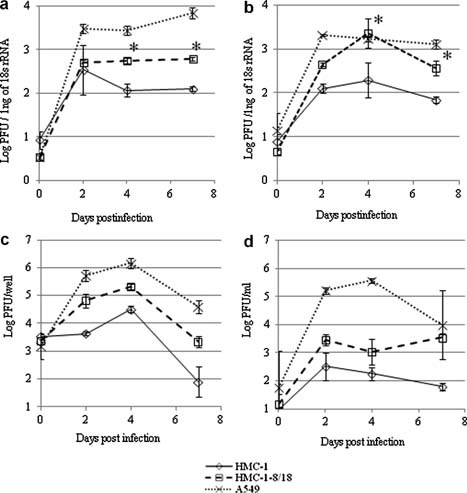
Replication kinetics of RSV genes as detected by real‐time PCR and plaque assay. a and b: Kinetics of the RSV copies upon infection by direct inoculation or spinoculation as detected by real‐time PCR. Cells were inoculated with RSV at an MOI of 1 (a) by direct inoculation or (b) spinoculation at 3,000 rpm for 2 h at 4°C. For spinoculation, the cells were mixed with 500 µl of 10% FCS‐IMDM and added to each well of type I collagen‐coated 24‐well plates (AGC Techno Glass, Chiba, Japan). At the indicated dpi, cells were collected, total RNA was extracted, and first‐strand cDNA was synthesized as described in the legend for Figure 1. Quantitative real‐time PCR was performed with an RSV‐specific primer and probe set. To quantify RSV RNA, a standard curve obtained from serially diluted virus stocks with known PFU values was used. The quantity of RNA was determined from the standard curve and normalized against 18s rRNA values from the same sample. The amount of RSV RNA is presented as the ratio of the log10PFU/1 ng of 18s rRNA (n = 4; P < 0.01). c and d: Kinetics of the infectious RSV titer upon infection by spinoculation as detected by plaque assay. Cells were inoculated with RSV at an MOI of 1 by spinoculation at 3,000 rpm for 2 h at 4°C. At the indicated dpi, (c) the cells and (d) supernatants were collected separately, and the RSV titer was determined by plaque assay using HEp‐2 cells. The titers are presented as the (c) log10PFU/well and (d) log10PFU/ml (n = 6).
Because a large amount of RSV RNA was detected in the HMC‐1‐C8/18 cells after spinoculation (Fig. 3b), the infectious virus titer was determined by plaque assay. RSV‐containing 10% fetal calf serum‐Iscove's modified Dulbecco's medium (FCS‐IMDM) was added to the cells and spinoculated at 3,000 rpm for 2 h at 4°C. Subsequently, the cells were washed with PBS and incubated at 34°C in 10% FCS‐IMDM. At the indicated dpi, the cells and supernatants were collected separately, and the amount of virus was determined by plaque assay using HEp‐2 cells. As shown in Figure 3c, a large number of infectious virus particles were detected in the HMC‐1‐C8/18 cell fraction, and the titer was similar to that in A549 cells; in contrast, a productive increase in virus was not seen in HMC‐1 over the course of infection. Conversely, the amount of infectious virus in the HMC‐1‐C8/18 cell supernatant was 100‐fold lower than that in A549 cells, although the titer was 10‐fold higher than that in HMC‐1 during the same period (Fig. 3d). These results suggest that infectious virus was produced in the HMC‐1‐C8/18 cell fraction after spinoculation and that the mean of virus release into the HMC‐1‐C8/18 culture medium was different from that into the A549 medium.
Next, to elucidate the effects of C18 on RSV replication from the opposite side, C18 was knocked down in A549 cells. A549 cells were transfected with a plasmid carrying a C18‐specific or control shRNA (Santa Cruz Biotechnology, Santa Cruz, CA). Cells were selected and cloned in the presence of 2 µg/ml puromycin. C18 expression in the knocked‐down A549 cells was decreased, and the knock down of C18 influenced C8 expression (Fig. 4a). C18 mRNA expression was decreased to 24 ± 4%, and that of C8 was decreased to 49 ± 5% compared with the shRNA‐controlled A549 cells (Fig. 4b). The sequence homology between C18 and C8 is about 50%, and it is possible that the shRNA for C18 might affect the C8 gene. Next, the cells were infected with RSV, and the titers were determined at 2 and 4 dpi by plaque assay; the results are presented as the relative titer (%); that in the shRNA‐controlled cells was 100%. As shown in Figure 4c, the RSV titer decreased to 45 ± 20% and 7 ± 4% at 2 and 4 dpi, respectively. This result suggests that the production of descendant viruses in A549 cells decreases depending on the amount of C8/18. Garcia‐Barreno et al. [1988] reported that C18 in RSV‐infected HEp‐2 cells received some modification, and they suggested a relationship between RSV replication and C18. However, it was unclear whether C18 plays a role in RSV replication. The results of the present study show that the production of RSV was altered depending on the presence or absence of C8/18 because the amount of RSV increased in C8/18‐expressing HMC‐1 cells and decreased in A549 cells expressing reduced levels of C8/18. These findings suggest that RSV replication is influenced by C8/18 and that RSV production is increased in the presence of C8/18.
Figure 4.
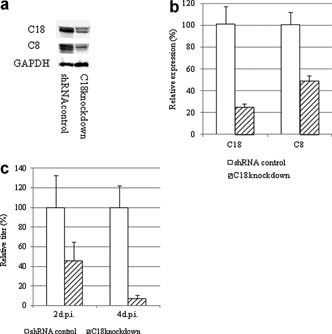
a and b: Cloning of C18 knocked‐down and shRNA control A549 cells. a: Western blot analysis of C8/C18 expression. Cells were mixed with SDS sample buffer and separated by 15% SDS–PAGE. C18 was detected with a biotin‐conjugated mouse monoclonal antibody (DC‐10; Abcam) and HRP‐conjugated streptavidin (Zymed). C8 was detected using a mouse monoclonal antibody (GeneTex) and HRP‐conjugated goat anti‐mouse antibodies (Rockland, Gilbertsville, PA). GAPDH was detected with rabbit polyclonal antibodies (Imgenex, San Diego, CA) and HRP‐conjugated guinea pig anti‐rabbit antibodies (Rockland). b: C8 and C18 mRNA expression in cloned cells. Total RNA was extracted, and first‐strand cDNA was synthesized for real‐time PCR as described in the legend for Figure 1 using GAPDH as an internal control (n = 7; *P < 0.01). c: Viral replication kinetics in C18 knocked‐down A549 cells. Cells were infected with RSV at an MOI of 1 and incubated for 2–4 days. Cells were collected and the titers were determined by plaque assay using HEp‐2 cells (n = 4).
Domachowske et al. [2000] reported that while constitutive expression of C17 was normally restricted in basal epithelial cells in the lung, RSV infection increased the expression of C17 in lung epithelial cells. This increase was associated with fusion formation, and blocking the F protein by a specific antibody prevented it. Moreover, the localization of C17 was consistent with cell fusion [Domachowske et al., 2000]. Thus, RSV infection itself could alter the expression profile of the intracellular filament network in the infected cell and could induce the expression of cell components needed for the virus to spread via cell‐to‐cell transmission.
The expression level of C8/18 may likewise be changed after RSV infection and may be involved in cell fusion formation and the cell‐to‐cell spread of RSV. HMC‐1 cells are derived from mesenchymal cells [Butterfield et al., 1988] and as described in this study, HMC‐1 cells do not express the C18 gene constitutively. However, this study showed that external expression of C8/18 could allow the replication of RSV even in HMC‐1 cells. The present data also showed that viral load continued at high levels in the presence of C8/18 but decreased in the absence of C8/18. These results suggest that the presence of C8/18 intracellularly before RSV infection is important for the later RSV replication cycle, although further studies are needed to elucidate the direct effect of C8/18 on RSV replication.
As for other viruses, several reports have shown a relationship between viral infections and C8/18. For example, it was reported that C18 was phosphorylated [Liao et al., 1995] and that C18 rearrangement [Brunet et al., 2000] occurred after rotavirus infection without cell death; moreover, it was suggested that these virus‐induced structural alterations caused functional perturbations in the intestine. In adenovirus infection, the cleavage of C18 by adenovirus proteinase causes destruction of the cytoskeletal network, which is thought to promote cell lysis and nascent virion release [Staufenbiel et al., 1986; Chen et al., 1993]. These reports suggest a role of C18 in the later stages of viral infection (e.g., virus release).
The results of this study showed that infectious virus production increased in the HMC‐1‐C8/18 cell fraction after spinoculation, although the titer in the supernatant was low compared with that in A549 cells. This result implies the possibility that C8/18 may function not in virus release into the culture medium, but in replication within host cells. RSV is assembled in lipid rafts at the apical membrane, and budding is controlled by the apical recycling endosome‐associated protein Rab11 family interacting protein 2 [Brock et al., 2003; McCurdy and Graham, 2003; Brown et al., 2004; Utley et al., 2008]; notably, HMC‐1 may lack this type of mechanism for releasing virions into the cell‐culture medium.
In summary, this paper demonstrates that RSV production increases in the presence of C8/18 in a cell‐culture system. Additional studies are needed to elucidate the direct effect of C8/18 on RSV replication and to determine the involvement of C18 in cell fusion formation.
Acknowledgements
We thank Dr. J. Butterfield (Mayo Clinic, Rochester, MN) for supplying the human mast‐cell line, HMC‐1. We thank Dr. F. Taguchi (Nippon Veterinary and Life Science University, Tokyo, Japan) for his valuable advice.
REFERENCES
- American Academy of Pediatrics . Committee on Infectious Diseases and Committee on Fetus and Newborn Pediatrics. 2003. Revised indications for the use of palivizumab and respiratory syncytial virus immune globulin intravenous for the prevention of respiratory syncytial virus infections. Pediatrics 112:1442–1446. [PubMed] [Google Scholar]
- Brock SC, Goldenring JR, Crowe JE Jr. 2003. Apical recycling systems regulate directional budding of respiratory syncytial virus from polarized epithelial cells. Proc Natl Acad Sci USA 100:15143–15148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G, Jeffree CE, McDonald T, Rixon HW, Aitken JD, Sugrue RJ. 2004. Analysis of the interaction between respiratory syncytial virus and lipid‐rafts in Hep2cells during infection. Virology 327:175–185. [DOI] [PubMed] [Google Scholar]
- Brunet JP, Jourdan N, Cotte‐Laffitte J, Linxe C, Geniteau‐Legendre M, Servin A, Quero AM. 2000. Rotavirus infection induces cytoskeleton disorganization in human intestinal epithelial cells: Implication of an increase in intracellular calcium concentration. J Virol 74:10801–10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield JH, Weiler D, Dewald G, Gleich GJ. 1988. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res 12:345–355. [DOI] [PubMed] [Google Scholar]
- Chen PH, Ornelles DA, Shenk T. 1993. The adenovirus L3 23‐kilodalton proteinase cleaves the amino‐terminal head domain from cytokeratin 18 and disrupts the cytokeratin network of HeLa cells. J Virol 67:3507–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domachowske JB, Bonville CA, Rosenberg HF. 2000. Cytokeratin 17 is expressed in cells infected with respiratory syncytial virus via NF‐kappaB activation and is associated with the formation of cytopathic syncytia. J Infect Dis 182:1022–1028. [DOI] [PubMed] [Google Scholar]
- Garcia‐Barreno B, Jorcano JL, Aukenbauer T, Lopez‐Galindez C, Melero JA. 1988. Participation of cytoskeletal intermediate filaments in the infectious cycle of human respiratory syncytial virus (RSV). Virus Res 9:307–321. [DOI] [PubMed] [Google Scholar]
- Girard MP, Cherian T, Pervikov Y, Kieny MP. 2005. A review of vaccine research and development: Human acute respiratory infections. Vaccine 23:5708–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffree CE, Brown G, Aitken J, Su‐Yin DY, Tan BH, Sugrue RJ. 2007. Ultrastructural analysis of the interaction between F‐actin and respiratory syncytial virus during virus assembly. Virology 369:309–323. [DOI] [PubMed] [Google Scholar]
- Kuypers J, Wright N, Morrow R. 2004. Evaluation of quantitative and type‐specific real‐time RT‐PCR assays for detection of respiratory syncytial virus in respiratory specimens from children. J Clin Virol 31:123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Lowthert LA, Omary MB. 1995. Heat stress or rotavirus infection of human epithelial cells generates a distinct hyperphosphorylated form of keratin 8. Exp Cell Res 219:348–357. [DOI] [PubMed] [Google Scholar]
- McCurdy LH, Graham BS. 2003. Role of plasma membrane lipid microdomains in respiratory syncytial virus filament formation. J Virol 77:1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. 1982. The catalog of human cytokeratins: Patterns of expression in normal epithelia, tumors and cultured cells. Cell 31:11–24. [DOI] [PubMed] [Google Scholar]
- O'Doherty U, Swiggard WJ, Malim MH. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol 74:10074–10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AS, Keil LB, DeBari VA. 1988. Immunocytochemical localization of cytoskeletal antigens in KB and HEp‐2cells. Diagn Clin Immunol 5:400–413. [PubMed] [Google Scholar]
- Shirato K, Taguchi F. 2009. Mast cell degranulation is induced by A549 airway epithelial cell infected with respiratory syncytial virus. Virology 386:88–93. [DOI] [PubMed] [Google Scholar]
- Shirato K, Nishimura H, Saijo M, Okamoto M, Noda M, Tashiro M, Taguchi F. 2007. Diagnosis of human respiratory syncytial virus infection using reverse transcription loop‐mediated isothermal amplification. J Virol Methods 139:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staufenbiel M, Epple P, Deppert W. 1986. Progressive reorganization of the host cell cytoskeleton during adenovirus infection. J Virol 60:1186–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, Goldenring JR, Crowe JE Jr. 2008. Respiratory syncytial virus uses a Vps4‐independent budding mechanism controlled by Rab11‐FIP2. Proc Natl Acad Sci USA 105:10209–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Matsuyama S, Taguchi F. 2006. Receptor‐independent infection of murine coronavirus: Analysis by spinoculation. J Virol 80:4901–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]