

Abstract
Summary. Detection of residual HCV in individuals with SVR after treatment of CHC can be significantly heightened by analyzing ex vivo mitogen‐activated T and B lymphocytes and applying sensitive nucleic acid amplification assays. However, it remained unknown if synergistic activation of lymphocytes and monocytes would further augment HCV detection, if viral replication becomes universally upregulated in treated cells, and if examining sequential sera and lymphoid cells would improve detection of occult infection. Using paired sera and lymphoid cells collected 1 year apart from 17 individuals with normal liver enzymes for up to 72 months after SVR, it was found that simultaneous activation of lymphocytes and monocytes enhanced identification of silent HCV infection and revealed that in some cases monocytes were the principal immune cell type where HCV persisted. Testing of serial samples further increased detection of occult infection. Ultimately, by combining the above two approaches, all individuals with SVR were found to be silent carriers of HCV. Clonal sequencing revealed HCV variations in sera and lymphoid cells and evolution of viral genomes confirming ongoing virus replication. Surprisingly, similar to those with CHC, naive lymphoid cells from some individuals carried ∼103 HCV copies/μg total RNA. HCV loads in naive lymphoid cells predetermined the outcome of ex vivo stimulation with respect to upregulation or inhibition of HCV replication. HCV RNA levels in occult infection were inversely proportional to the expression of IFNα and IFN‐inducible MxA, but not to IFNγ or tumour necrosis factor α in naive and mitogen‐treated lymphoid cells.
Keywords: hepatitis C virus lymphotropism, hepatitis C virus persistence, interferon alpha, monocytes, occult hepatitis C virus infection, T and B lymphocytes
Abbreviations:
- HCV
hepatitis C virus
- SVR
sustained virological response
- CHC
chronic hepatitis C
- PBMC
peripheral blood mononuclear cells
- IFNα
interferon α
- RT‐PCR
reverse transcription‐polymerase chain reaction
- NAH
nucleic acid hybridization
- PHA
phytohemagglutinin
- PWM
pokeweed mitogen
- LPS
lipopolysaccharide
- MMLV‐RT
Moloney murine leukemia virus reverse transcriptase
- 5′UTR
5′ untranslated region
- IFNγ
interferon γ
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- HIV
human immunodeficiency virus
- WHV
woodchuck hepatitis virus
- UT
untreated
- IL‐2
interleukin‐2
- rIFN5α
recombinant IFN5α
Introduction
Hepatitis C virus is an important human pathogen, which causes chronic infection in at least 170 million people world‐wide. Up to 85% of acutely infected patients become carriers of the virus and develop CHC, which can advance to cirrhosis and/or hepatocellular carcinoma in up to 20% of patients [1]. HCV is a positive single‐stranded RNA virus that replicates by making the so‐called ‘negative strand’. Although HCV is highly hepatotropic, evidences indicate that it also propagates in the lymphatic [2, 3, 4, 5, 6, 7, 8] and nervous [9, 10] systems. In regard to the lymphatic system, circulating PBMC from patients with CHC [2, 3, 4, 5] and from individuals with apparent complete resolution of hepatitis C, who were serum HCV RNA nonreactive by standard laboratory assays and had normal liver enzymes, have been shown to carry replicating HCV genomes [5, 6, 7, 8]. Low levels of HCV RNA have also been detected in lymphoid cells in a significant proportion of patients with persistently elevated liver enzymes of unknown aetiology [11, 12]. Furthermore, susceptibility of T‐ and B‐cell lines, as well as primary human T cells and monocyte/macrophages to HCV infection has also been shown [13, 14, 15, 16]. It is thought that by invading the immune system, HCV can conceal its presence and enhance the likelihood of persistence, as it has been observed in many other long‐term viral infections [17, 18].
The current standard treatment for CHC is a combination therapy with pegylated IFNα and Ribavirin for 24 or 48 weeks depending on the HCV genotype. This therapy results in the so‐called SVR in no more than 50% of patients [19]. The SVR is defined as serum HCV RNA negativity for at least 6 months after completion of treatment, as determined by clinical laboratory RT‐PCR assays with the current sensitivities between 50 and 3200 virus genome (copies) equivalents [vge]/mL or 50–615 IU/mL [20]. Clinically, individuals with SVR and normal liver function tests have been considered to be free of HCV. However, it has recently been documented that by employing more sensitive RT‐PCR detection methods, identifying <10 HCV copies or <5 IU/mL, the virus can be consistently found in sera, lymphoid cells and hepatic tissue for years after the apparent complete recovery from hepatitis C [5, 6, 8, 21, 22, 23]. In addition, liver biopsies from these individuals occasionally reveal histological evidence of protracted minor inflammation, frequently mild‐to‐moderate fibrosis or in rare cases, even active chronic hepatitis [21, 22, 24].
In previous studies, it is reported that ex vivo treatment of lymphocytes from persons who achieved SVR with nonspecific mitogens led to an upregulation of viral replication, allowing detection of the virus in the cells which were apparently nonreactive [5, 6]. It became evident that such an approach facilitates a more accurate detection of HCV than by testing sera or naive PBMC. This finding also reaffirmed the notion that lymphotropism is an intrinsic property of HCV. Nevertheless, it remained unknown whether HCV replication is universally upregulated following stimulation of lymphoid cells or is augmented only in specific situations dictated by not as yet determined viral or cellular factors. Investigation of this issue is important to not only better understand HCV‐lymphoid cell interactions, but also to assess whether testing of ex vivo stimulated lymphoid cells can be suitable for routine diagnosis of occult HCV infection.
In the present study, it is found that the level of HCV expression in lymphoid cells in some cases of occult long‐term infection can be as high as that encountered in PBMC of those with CHC. Importantly, HCV replication in lymphoid cells during occult infection was inversely proportional to the level of transcription of IFNα. Moreover, sequence analysis of HCV clones derived from lymphoid cells and serum collected in the course of occult infection suggested lymphoid cell‐specific compartmentalization of viral variants providing further evidence for the existence of active viral replication in the host's immune system after apparently complete resolution of hepatitis C.
Materials and Methods
Patients and samples
Twenty randomly selected individuals (12 men and eight women) with a mean age of 36.5 ± 14.5 years who, except Patient 11 with spontaneous recovery of symptomatic HCV infection (Table 1), had recovered from CHC based on the clinical, standard virological and biochemical criteria were included in this study. All patients resolved CHC after achieving SVR following the therapy with IFNα and/or Ribavirin. Specifically, eight individuals received a 24‐ or 48‐week treatment with IFNα‐2a (Roferon A; Roche Pharmaceuticals, Warsaw, Poland) or 2b (Intron A; Schering‐Plough, Warsaw, Poland), while another six were on a combination therapy of IFNα and Ribavirin for 48 weeks (Schering‐Plough) (Table 1). The remaining five patients were treated with IFNα for 12, 24 or 48 weeks and then with IFNα and Ribavirin for 48 weeks. At the time of the first sample collection for this study, all individuals had been followed for 12–60 months after achieving the SVR (Table 1). In all of them, HCV RNA and liver function tests were evaluated every 6–12 months. During the follow‐up, serum samples from all patients were found to be negative for HCV RNA by the Roche Cobas Amplicor HCV v2.0 assay (sensitivity 1000 virus copies/mL) and liver function tests deemed normal.
Table 1.
Clinical characteristics and detection of hepatitis C virus (HCV) RNA in two serum and lymphoid cell samples obtained a year apart from individuals with occult HCV infection continuing after resolution of hepatitis C†
| Patient | Age/ sex | HCV genotype | Antiviral treatment/ duration (wk) | Follow‐up after SVR (mo) | First sample | Second sample | Overall positivity§ | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serum | Untreated peripheral blood mononuclear cells (PBMC) | Serum | Untreated PBMC | Mitogen‐treated PBMC | ||||||||||
| HCV RNA+ | HCV RNA+ | HCV RNA− | HCV RNA+ | HCV RNA+ | HCV RNA− | C5, HCV RNA+ | C5L, HCV RNA+ | C5 or C5L, HCV RNA− | ||||||
| Group 1 | ||||||||||||||
| 1 | 18/M | NA | IFN 24 and IFN/R 48 | 24 | NA | NA | NA | NA | − | NT | 100 | 350 | + | + |
| 2 | 49/F | 1a | IFN 48 and IFN/R 48 | 24 | <40 | 100 | + | <40 | − | NT | 100 | NA | NA | + |
| 3 | 24/M | NA | IFN/R 48 | 30 | NA | NA | NA | − | − | NT | 10 | 50 | + | + |
| 4 | 9/M | 1b | IFN/R 48 | 36 | <40 | <50 | + | − | − | NT | − | 50 | + | + |
| 5 | 23/F | 1a | IFN 24 and IFN/R 48 | 36 | <40 | − | NT | 100 | − | NT | − | − | NT | + |
| 6 | 21/F | ND | IFN 48 | 48 | <100 | − | NT | <40 | − | NT | − | 250 | + | + |
| 7 | 42/F | 1b | IFN 48 | 48 | − | − | NT | <40 | − | NT | − | 300 | + | + |
| 8 | 55/M | 2b | IFN 12 and IFN/R 24 | 48 | 40 | <50 | + | <40 | − | NT | 10 | − | − | + |
| 9 | 69/M | 1a | IFN 48 | 60 | 200‡ | <50 | − | 100 | − | NT | − | 100 | − | + |
| 10 | 48/M | 1a | IFN 24 | 72 | <40 | <50 | − | <40 | − | NT | 100 | 300 | + | + |
| % Positivity | 7/8 (87.5) | 5/8 (62.5) | 3/5 (60) | 7/9 (77.8) | 0/10 (0) | 5/10 (50) | 7/9 (77.8) | 6/8 (75) | 10/10 (100) | |||||
| Group 2A | ||||||||||||||
| 11 | 41/F | 1a | NA | NA | − | − | NT | 100 | 50‡ | + | 103‡ | 5 | + | + |
| 12 | 37/F | 2b | IFN/R 48 | 24 | NA | NA | NA | 40 | 50‡ | + | 103‡ | − | + | + |
| 13 | 21/M | 3a | IFN/R 48 | 36 | <40 | − | NT | <40 | <5 | + | NA | 100 | + | + |
| 14 | 33/F | NT | IFN/R 48 | 36 | − | − | NT | <40 | 5 | − | − | 50 | + | + |
| 15 | 40/F | NT | IFN 48 | 36 | 40 | <5 | − | − | <5 | + | 50 | − | NA | + |
| 16 | 42/M | 1a | IFN 24 and IFN/R 48 | 48 | <40 | − | NT | <40 | 5 | − | 50 | 100‡ | − | + |
| 17 | 47/M | 1b | IFN 48 | 48 | <40 | <5 | NT | <40 | 50 | − | − | 500‡ | NT | + |
| Group 2B | ||||||||||||||
| 18 | 35/M | 2b | IFN/R 48 | 36 | 100 | − | NT | 40 | 103‡ | + | 140 | 140 | NA | + |
| 19 | 28/M | 1b | IFN 48 | 48 | <40 | 50 | + | <200 | 103‡ | + | − | − | NT | + |
| 20 | 47/M | 1b | IFN 48 | 60 | <100 | 100 | + | 100 | 7 × 103‡ | + | 6 × 103‡ | − | + | + |
| % Positivity | 7/9 (77.8) | 4/9 (44.4) | 2/3 (66.7) | 9/10 (90) | 10/10 (100) | 7/10 (70) | 6/9 (66.7) | 6/10 (60) | 5/6 (83.3) | 10/10 (100) | ||||
| Total%Positivity | 14/17 (82.3) | 9/17 (52.9) | 5/8 (62.5) | 16/19 (84.2) | 10/20 (50) | 7/10 (70) | 11/19 (57.9) | 13/19 (68.4) | 11/14 (78.6) | 20/20 (100) | ||||
Wk, week; SVR, sustained virological response; mo, month; HCV RNA+, HCV RNA positive strand; HCV RNA−, HCV RNA negative strand; C5, Combo 5; C5L, Combo 5 plus LPS; NA, not available; IFN, interferon‐alpha; R, Ribavirin; NT, not tested; ND, not determined.
†HCV load was estimated by densitometric analysis of RT‐PCR/NAH signals or, ‡when possible, by real‐time RT‐PCR using 5′ UTR‐specific primers and expressed as HCV vge/μg total RNA or vge/mL using dilutions of synthetic HCV UTR‐E2 as quantitative standards.
§Overall positivity defined as HCV RNA reactivity in serum and/or PBMC.
Paired serum and PBMC samples from 17 of the individuals were collected at two occasions approximately 12 months apart. The serum or PBMC from the first and second bleedings is herein referred to as the first sample and the second sample, respectively. Only the second PBMC samples were cryopreserved in a manner suitable for culture and ex vivo mitogen treatment experiments.
Study groups
Based on the status of HCV RNA reactivity determined by an RT‐PCR/NAH assay (sensitivity ≤10 vge/mL) [6] in untreated PBMC obtained during the second collection, the individuals were divided to two study groups (Table 1). Group 1 consisted of cases whose naive PBMC were HCV RNA negative, while PBMC of those in Group 2 were HCV RNA reactive prior to culture in the presence of mitogens. The latter group was further subdivided into Group 2A, if the HCV RNA load detected in untreated cells was lower than 50 vge/μg total RNA, and Group 2B, if the load was greater than 50 vge/μg total RNA (Table 1). As positive controls, HCV RNA‐positive PBMC from patients with documented CHC was used. The study was approved by the local Human Investigation Committees and the samples were collected after informed consent had been obtained.
Mitogen stimulations of lymphoid cells
Peripheral blood mononuclear cells were isolated from whole blood by gradient centrifugation on Ficoll‐Paque (Pharmacia Biotech, QC, Canada) and cultured as previously reported [6]. In the current study, different mitogen combinations were employed to upregulate HCV replication in various subsets of lymphoid cells. For this purpose, 1 × 107 PBMC were cultured for 72 h in the presence of Combo 5 (C5) cocktail [5], consisting of 5 μg/mL phytohemagglutinin (PHA; Sigma, Mississauga, ON, Canada), a T‐cell proliferation inducing mitogen, 5 μg/mL PWM (Sigma), a B‐ and T‐cell proliferation inducing lectin, IL‐2 (Roche Molecular Diagnostics, Laval, QC, Canada), a T‐cell supportive cytokine, and IL‐4 (Roche Molecular Diagnostics), a B‐cell supportive cytokine. In addition, lymphoid cells were stimulated with C5 plus 1 μg/mL LPS (Sigma), which is known to activate both B cells and monocytes [25]. This cocktail was designated as C5L. In each case, a sample of untreated cells was preserved for analysis.
HCV RNA detection by RT‐PCR/NAH
Typically, RNA from 2 × 106 PBMC or 250 μL serum was reversely transcribed to cDNA using MMLV‐RT (Invitrogen Life Technologies, Burlington, ON, Canada) [6]. The presence of HCV RNA positive strand was tested by nested PCR/NAH with primers specific for HCV 5′‐UTR or E2 region, as reported [6]. Ten‐fold serial dilutions of HCV synthetic RNA (HCV sRNA) positive strand fragment was used as quantitative standards. Sensitivity of this assay was ≤10 vge/mL for detection of the 5′ UTR and <10 vge/mL for that of E2 region. HCV RNA‐negative strand was determined by the strand‐specific RT‐PCR/NAH employing rTth DNA polymerase, as reported [6]. Ten‐fold serial dilutions of HCV sRNA negative strand amplified in parallel were used as semi‐quantitative standards. This assay detects approximately 100 vge of the correct (negative) strand, while nonspecifically identifying ≥ 106 vge of the positive strand [6]. A sample containing water instead of test cDNA and a mock were routinely included as contamination controls, and cDNA from PBMC of a healthy donor as a negative control in all reactions. Specificity of the amplified products and validity of the controls were routinely confirmed by NAH using recombinant HCV UTR‐E2 (rHCV UTR‐E2) as a probe [6].
HCV RNA quantification by real‐time RT‐PCR
When feasible, HCV RNA in serum and PBMC was quantified by real‐time RT‐PCR using the LightCycler Fast Start Master Hybridization Probes (Roche Diagnostics), as reported [6]. Ten‐fold serial dilutions of the rHCV UTR‐E2 were used for enumeration of viral load. A melting curve analysis was routinely performed to confirm specificity of the amplifications. It is of note that the sensitivity of this assay was approximately 10‐fold lower than that of the RT‐PCR/NAH assay with either 5′‐UTR or E2 specific primers.
Sequencing
For HCV genotyping, 5′‐UTR PCR fragments were sequenced in both directions by the Sanger dideoxy chain termination procedure using primers 5′‐GCAGAAAGCGTCTAGCCATGGC and 5′‐CAAGCAQCCCTATGAGGCAGT and the Beckmann CEQ 8000 sequencer (Beckmann Coulter, Fullerton, CA, USA). To assess possible genetic heterogeneity in HCV sequences in sera and lymphoid cells and to determine whether expansion of HCV variants occurred over time as a result of sustained replication in occult infection, 5′‐UTR amplicons were purified and cloned into the dual promoter pCRII vector using the TOPO‐TA cloning system (Invitrogen). Clones were expanded and the presence of the UTR insert confirmed by digestion with EcoRI enzyme (Invitrogen). An average of seven clones from each PCR product were sequenced by using the universal forward and reverse M13 primers (Invitrogen). The Sanger dideoxy nucleotide sequencing reaction was performed by using the CEQ DTCS Quick Start Kit (P/N 608120) and the CEQ 8000 sequencer (Beckman Coulter). The analysis of 196‐bp product sequences, devoid of the flanking UTR primer regions, was done with the aid of the CEQ 8000 genetic analysis system and nucleotide sequences aligned using clustal w software [26].
Quantification of cytokine mRNA
RNA samples from naive or mitogen‐treated lymphoid cells predestined for analysis of cytokine expression were treated with 2 U of DNAse I from the Sigma Deoxyribonuclease I kit for 10 min at ambient temperature and then incubated with the provided DNAse inactivating solution for 15 min at 72 °C. RNA was reversely transcribed by using MMLV‐RT. Expression of IFNγ and TNFα was quantified by real‐time RT‐PCR using SYBR and the Roche LightCycler (Roche Diagnostics). Reactions were performed in 20‐μL volumes (∼50 ng RNA) using sense primer 5′‐TTTTCAGCTCTGCATCGTTTTGGGT and antisense primer 5′‐CCTTGAAACAGCATCTGACTCCTT for detection of IFNγ and sense primer 5′‐TCTTCTCGAACCCCGAGTGA and antisense primer 5′‐CCTCTGATGGCACCACCAG for detection of TNFα. The expression of the genes was normalized against that of GAPDH, which was quantified in parallel in each set of test samples. Specificity of amplifications was confirmed by a melting curve analysis.
The presence of primer dimers hindered the use of real‐time RT‐PCR to accurately quantify IFNα5 and IFN‐inducible MxA. Therefore, the conventional RT‐PCR/NAH tests were applied. This was followed by densitometric analysis of the resulting hybridization signals using cloned IFNα5 and MxA DNA fragments as quantitative standards. Amplification of IFNα5 was done with sense primer 5′‐GGTCACTCAATCTCAACAGC and antisense primer 5′‐ATATGGATCCTGATTTCTGCTCTGACAACC, whereas that of MxA with sense primer 5′‐GCATCCCACCCTCTATTACT and antisense primer 5′‐TGTCTTCAGTTCCTTTGTCC. PCR mixtures were amplified for 40 cycles at 95 °C for 10 s, then at 58 °C (for IFNα5) or 47 °C (for MxA) for 30 s, and finally at 72 °C for 1 min. As a loading control, GAPDH was amplified by using cDNA derived from the same test samples using sense primer 5′‐CCATCACCATCTTCCAGGAG and antisense primer 5′‐CCTGCTTCACCACCTTCTTG for 27 cycles at 94 °C for 30 s, 52 °C for 1 min, and 72 °C for 1 min in a PTC‐200 thermocycler (MJR Research, Watertown, MA, USA). Specificity of IFN5α, MxA and GADPH amplicons, and validity of the controls were routinely verified by NAH using as probes respective gene recombinant DNA fragments labelled with 32P‐dCTP (GE Healthcare, QC, Canada).
Results
Analysis of sequential serum and lymphoid cell samples enhances detection of occult HCV infection
Paired serum and PBMC samples were obtained from 17 of 20 individuals with follow‐up up to 72 months after SVR following IFNα and/or Ribavirin therapy. By testing the first set of the samples using the RT‐PCR/NAH assay, 82.3% of sera (14/17) and 52.9% of naive PBMC (9/17) were found reactive for HCV RNA positive strand (Table 1). Twelve months later, 84.2% of sera and 50% of naive PBMC were identified to be positive for HCV RNA by RT‐PCR/NAH. The estimated levels of HCV RNA were around 40–200 vge/mL in serum and 5–50 vge/μg of total RNA for most of the PBMC samples tested (Group 2a; Table 1). Surprisingly, the second lymphoid cell samples collected 3–5 years after SVR from three of the individuals carried HCV RNA at levels ranging from 1 × 103 to 7 × 103vge/μg total RNA (Group 2b; Table 1), which were comparable with those typically detected in PBMC from symptomatic CHC (Pham and Michalak, unpubl. data). Interestingly, HCV RNA levels in their sera were similar to those found in the other recovered persons. The presence of HCV RNA negative strand, indicative of active viral replication, was detected in 62.5% of the first and 70% of the second naive (mitogen‐untreated) lymphoid cells, which were found to be reactive for the viral RNA positive strand and were available for testing (Table 1). For the 17 cases with two samples collected, HCV RNA was detected at both occasions in sera of 12 of them (70.6%). Further, although in five individuals HCV RNA was detected in the first but not in the subsequent PBMC sample, the reverse was true in five other subjects.
In certain cases, when the material was sufficient to broaden the analysis, the presence of HCV RNA was also evaluated by using E2‐region‐specific primers. Figure 1 depicts detection of HCV UTR and E2 genomic regions in paired serum and PBMC samples collected from Patient 17 at 36 and 48 months after SVR. Taken together, it is important to emphasize that ultimately all individuals investigated were found to be HCV RNA positive when their sera and PBMC obtained at two 1‐year intervals of follow‐up, which were either naive or treated with C5 or C5L (see below) were examined (Table 1). This highlights the importance of testing multiple samples from individuals with SVR to identify with greater precision the existence of occult HCV persistence.
Figure 1.
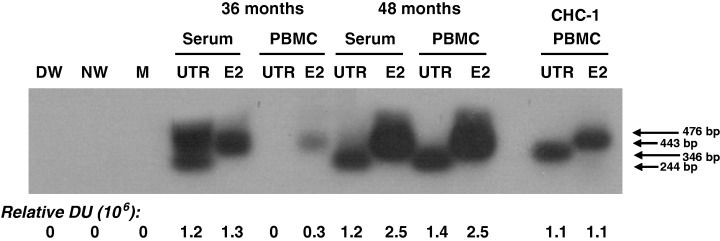
Detection of hepatitis C virus (HCV) RNA in paired serum and peripheral blood mononuclear cells (PBMC) samples collected a year apart from an individual with persistent occult HCV infection continuing after apparent complete recovery from hepatitis C. Samples from Patient 17 were obtained at 36 and 48 months (mo) after sustained virological response. RNA extracted from 250 μL serum or ∼2 × 106 naive (mitogen‐untreated) PBMC was amplified by nested reverse transcription‐polymerase chain reaction using 5′ untranslated region (5′‐UTR)‐ or E2‐specific primers. The specificity of the amplicons was verified by nucleic acid hybridization. RNA extracted from an equivalent amount of serum and the same number of naive PBMC from a patient with chronic hepatitis C (CHC‐1) were used as a positive control. Contamination controls included water added instead of cDNA and amplified by direct (DW) and nested (NW) reactions and mock (M) treated as test RNA. Positive signals showed the expected 346‐bp (direct) and 244‐bp (nested) 5′‐UTR fragments, as well as the 476‐bp (direct) and 443‐bp (semi‐nested) E2 amplicons. Numbers under the panel represent relative densitometric units given by hybridization signals.
HCV replication in lymphoid cells in occult infection is verified by sequence analysis
In an attempt to recognize whether HCV RNA sequences undergo modifications during occult infection and whether the sequences found in serum would differ from those in PBMC, we cloned and sequenced the 5′‐UTR fragments of HCV detected in paired serum and naive PBMC obtained at the second collection from Case 19 (Serum‐2 and PBMC‐2) and at the first (Serum‐1 and PBMC‐1) and the second (Serum‐2 and PBMC‐2) collections from Case 20 (Table 1). As illustrated in Fig. 2, consistent single nucleotide differences at specific positions were detected between serum and PBMC obtained at the same time point of follow‐up, as well as between serum and PBMC collected from the same individual at the two occasions. For example, while a guanidine at position 186 (G186) and an adenine at positions 192 (A192) and 231 (A231) were detected in all the clones derived from Serum‐2 of Case 19, a thymidine (T186), cytosine (C192), and guanidine (G231), respectively, were found in the clones from PBMC‐2 sample. In Case 20, two single nucleotide polymorphisms T186G and C192A were identified when HCV clones from Serum‐1 were compared with those from PBMC‐1. Interestingly, sequences of HCV detected in Serum‐2 obtained 1 year later from this individual, except for a few random mutations in some of the clones, were identical to those found in PBMC‐1 sample. Further, when sequences obtained from PBMC‐2 were compared with those of Serum‐2, three substitutions G186T, A192T and A231G were detected in all of the clones examined (Fig. 2). In addition, there were changes in the 5′‐UTR sequence when serum or PBMC from the first collection was compared with those obtained at the second collection. Specifically, relative to the sequences found in the respective samples from the first collection, T186G and C192A mutations were identified in the second serum sample, while G186T, A192T and A231G substitutions found in the second PBMC. Overall, these findings revealed the existence of minor but evident variations between the circulating virus and that residing in the PBMC. They also showed that over time the 5′‐UTR sequence underwent slight but clearly identifiable changes.
Figure 2.
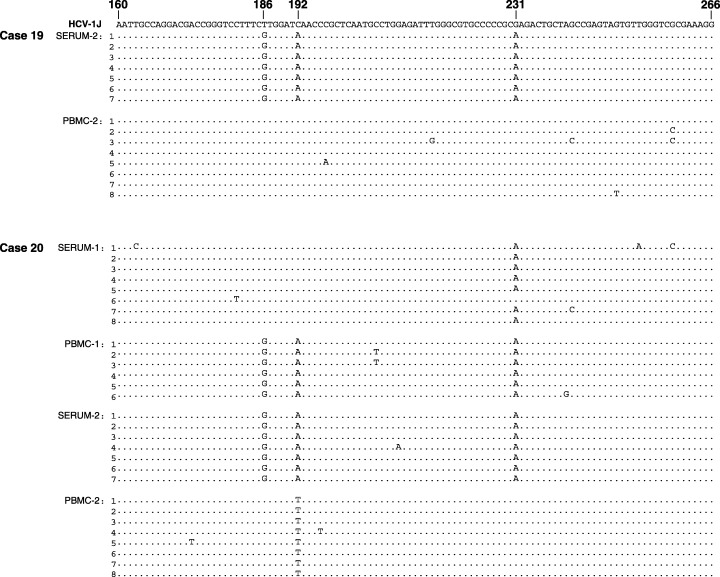
Hepatitis C virus (HCV) sequences identified in sera and lymphoid cells from individuals with occult HCV infection continuing after a sustained virological response. HCV 5′ untranslated region (5′‐UTR) sequences amplified from serum and peripheral blood mononuclear cells obtained at the second collection from Case 19 and from both collections from Case 20 were cloned, sequenced bidirectionally, and the sequences aligned with the prototype HCV genotype 1b sequence, HCV‐1J [27]. Sequences of 6 to 8 clones per test sample are shown. Nucleotides in the cloned sequences identical with the HCV reference (top line) are shown as dots and differences are identified by letters.
Ex vivo stimulation of PBMC with undetectable or low HCV loads augments virus replication
In our previous study, synergistic stimulation of T and B lymphocytes with appropriate mitogens augmented HCV replication leading to more frequent identification of the virus genome compared with PBMC treated with a single mitogen [5]. In the present work, to further increase likelihood of identification of HCV, viral RNA was examined after exposure of both lymphocytes and monocytes to mitogen cocktails. Thus, the second PBMC samples were treated with either C5, which activated T and B cells, or C5L, which also stimulated monocytes and macrophages. Both treatments led to enhanced expression of HCV RNA‐positive strand, despite the fact that some of these samples were apparently HCV RNA‐negative or carried very low HCV loads prior to mitogen stimulation, as illustrated in Fig. 3a and Table 1. It is of note that, considering densitometric values given by the respective hybridization signals, C5L was at least twice more effective in upregulating HCV RNA expression than C5 (Fig. 3a). The fact that C5L, but not C5, led to identification of viral RNA in PBMC from Patients 6 and 7 (Fig. 3a) suggested that monocytes were a predominant, if not the only reservoir of the persisting virus among circulating PBMC in these two individuals at the time of investigation. However, the synergistic activation of lymphocytes and monocytes occasionally led to almost complete inhibition of HCV RNA expression. This was evident in Patients 11 and 12 (Table 1) in which HCV RNA expression was increased by 20–100‐fold following stimulation of T and B lymphocytes with C5, while activation of both lymphocytes and monocytes with C5L resulted in a dramatic downregulation of HCV RNA presence. This observation implied that ex vivo treatments of lymphoid cells with both C5 and C5L were required to more accurately identify the existence of occult HCV infection.
Figure 3.
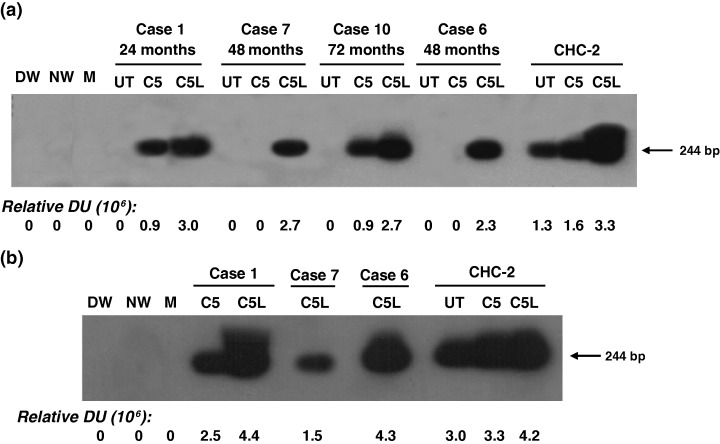
Upregulation of hepatitis C virus (HCV) RNA expression in lymphoid cells from individuals with occult infection following their ex vivo treatment with mitogen‐cytokine cocktails. RNA extracted from lymphoid cells which had been either untreated (UT) or cultured in the presence of C5 or C5L were analyzed for HCV RNA positive (a) and negative (b) strands by strand‐specific reverse transcription‐polymerase chain reaction/nucleic acid hybridization. Peripheral blood mononuclear cells samples from a patient with chronic hepatitis C (CHC‐2) treated under the same conditions as test cell samples were used as positive controls. Contamination controls were as described in the legends to Fig. 1. Numbers under the panel represent relative densitometric units given by hybridization signals.
Ex vivo stimulation of lymphoid cells with high HCV load inhibits virus replication and can completely eliminate virus
In the course of this study, we identified three cases with persistent occult HCV infection which were accompanied by high HCV RNA loads in naive lymphoid cells obtained at the time of the second sample collection (Group 2b, Table 1). When these cells were exposed to C5 or C5L, a dramatic decrease in HCV RNA expression was detected (Fig. 4). Specifically, while a nearly 90% reduction in the expression of HCV RNA positive strand was found in Case 18, as determined by densitometric analysis, a complete inhibition of the same strand was evident in Case 19, regardless of whether the cells had been treated with C5 or C5L (Fig. 4a). In Case 20, the level of HCV RNA positive strand was decreased by about 10% after C5 treatment, but became undetectable following the treatment with C5L (Fig. 4a). In all three cases, the drastic depletion of the HCV positive strand appeared to be a direct result of abrogation of HCV replication, as reflected by a concomitant inhibition of the viral RNA negative strand expression (Fig. 4b). Comparable levels of GAPDH mRNA detected in both untreated and mitogen‐treated cells (Fig. 4c) ruled out the possibility of unequal sample loading or that the treatment with mitogens might have been cytotoxic.
Figure 4.

Inhibition of hepatitis C virus (HCV) replication in lymphoid cells from patients with occult HCV infection and high HCV RNA load after treatment with mitogens. Peripheral blood mononuclear cells with HCV RNA load of ∼103 vge/μg RNA were left untreated (UT) or cultured for 72 h in the presence of either C5 or C5L. RNA was extracted and evaluated for HCV RNA positive (a) and negative (b) strands or glyceraldehyde‐3‐phosphate dehydrogenase (c) by reverse transcription‐polymerase chain reaction/nucleic acid hybridization assays. Contamination controls were as described in the legend to Fig. 1. Numbers under the panel represent relative densitometric units of hybridization signals.
Abrogation of HCV replication in lymphoid cells is mediated by IFNα5
To learn as to why there was such a pronounced inhibition of HCV RNA expression following mitogen stimulation of the cells with initially high HCV load, we investigated whether induction of antiviral cytokines in these cells might be the underlying reason. For this purpose, the transcriptional activity of IFNα5, IFNγ, TNFα and MxA, an intracellular indicator of interferon activation [28], were examined whenever material permitted in both naive and mitogen‐treated PBMC from randomly selected cases in Groups 1 and 2a, as well as from all 3 patients in Group 2b (Table 1). In the preceding experiments, it was established that the levels of IFN5α mRNA in unstimulated human lymphoid cells were too low to be accurately measured by real‐time RT‐PCR. Therefore, a more sensitive RT‐PCR/NAH assay was employed to evaluate the level of this cytokine mRNA, while IFNγ and TNFα mRNAs were quantified by real‐time RT‐PCR. As depicted in Fig. 5a, lymphoid cells with initially undetectable or low HCV loads obtained from Cases 6 and 13, respectively (Table 1), which had exhibited augmented virus replication following treatment with C5L (Fig. 3), showed a decrease in the expression of IFN5α to a virtually nondetectable level after mitogen treatment. Interestingly, transcription of IFN5α in the cells from a CHC patient (CHC‐2), which carried <200 vge/μg total cellular RNA and had shown increased HCV RNA expression following mitogen stimulation (Fig. 3), was also inhibited. Conversely, transcription of the IFNα5 gene was evidently augmented in lymphoid cells from Cases 18 and 19 after treatment with C5 or C5L (Fig. 5). Intriguingly, these cells carried initially high HCV loads but the viral replication was completely or nearly completely abrogated after their exposure to the mitogens (Fig. 4). In sharp contrast, we found no association between the levels of IFNγ mRNA or TNFα mRNA and the rate of HCV replication in lymphoid cells (data not shown).
Figure 5.
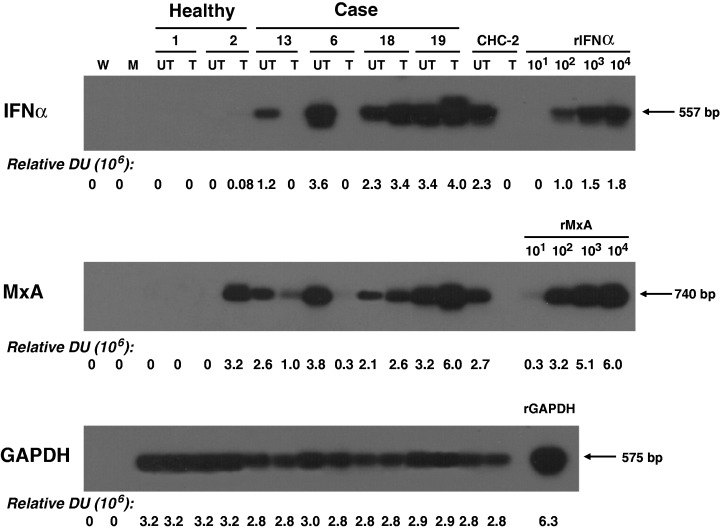
Expression of interferon 5 α (IFN5α) and MxA in naive and mitogen‐treated lymphoid cells which carried initially either low or high loads of hepatitis C virus (HCV). Lymphoid cells from two healthy persons, individuals with occult HCV infection carrying undetectable or low levels of HCV RNA in these cells (Cases 13 and 6), individuals with occult HCV infection and high HCV RNA load in the cells (Cases 18 and 19), and a patient with chronic hepatitis C (CHC‐2) were left untreated (UT) or treated with C5L as described in Materials and methods. RNA was examined for IFN5α, MxA and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) by specific reverse transcription‐polymerase chain reaction/nucleic acid hybridization assays. Ten‐fold serial dilutions of rIFNα and MxA (rMxA) gene fragments were used for enumeration of the test samples. Recombinant GAPDH gene fragment (rGAPDH) was amplified in parallel and used as a loading control. Contamination controls were described in legends to Fig. 1. Numbers under the panel represent relative densitometric units given by hybridization signals.
To confirm the inverse relation between the level of IFNα5 mRNA and the rate of HCV replication, the levels of MxA mRNA in the same untreated and mitogen‐treated lymphoid cells were assessed. As shown in Fig. 5b, an upregulation of HCV RNA following mitogen stimulation (Cases 6 and 13; Fig. 3 and Table 1) was associated with a down‐regulation of MxA in lymphoid cells. Similarly, in the two cases (Cases 18 and 19), where a significant inhibition of HCV replication after mitogen treatment was observed (Fig. 4 and Table 1), a marked increase in the amount of MxA transcripts was found. Such an inverse correlation between HCV load and MxA expression was consistent with that between HCV RNA and IFNα mRNA. However, it is of note that as MxA mRNA was detected with a relatively higher sensitivity than IFNα5 mRNA, mitogen‐treated lymphoid cells from one healthy individual were found to be positive for MxA mRNA despite the apparent absence of IFNα5 expression.
Discussion
In our previous studies, persistence of low levels of HCV RNA in individuals followed for years after they had been considered cured of hepatitis C, based on the results of standard laboratory assays for HCV RNA and normal liver function tests, was documented [5, 6]. Similar findings were subsequently reported by other groups by examining both lymphoid cells [7, 8, 21] and liver tissue fragments [8, 21, 22]. In this clinically silent form of HCV persistence, T and B lymphocytes were found to carry replicating HCV genomes [5]. In the present study, it has been demonstrated that cells of the monocytic lineage also are a reservoir of HCV replication in occult infection. This finding is consistent with a recent observation made in another laboratory [21], which showed the presence of HCV RNA in cultured monocytes from individuals with resolved hepatitis C. The current study, for the first time, documents the existence of a reverse relation between HCV load and expression of IFNα in lymphoid cells in the course of occult HCV infection. In addition, it was found that the level of HCV RNA in naive lymphoid cells during occult persistence is a predictor of whether ex vivo stimulation will enhance or abrogate viral replication in these cells. Specifically, when the level of HCV RNA in naive circulating lymphoid cells is equal to or >103 vge/μg total RNA (10–15 μg of RNA is typically recovered from 1 × 107 lymphoid cells), stimulation of the cells leads to drastic inhibition in HCV replication. However, under the same treatment conditions, augmentation of HCV replication is usually observed in cells carrying around 5–50 vge/μg total RNA. Subsequent analysis showed that the level of IFNα transcription, but not that of IFNγ or TNFα, is relevant to the control of HCV replication in lymphoid cells during occult infection.
In the current study, by employing the assays of superior sensitivity [5, 6], it was found that more than 80% of the individuals who had been deemed free of HCV by standard detection methods in fact carried small amounts of viral RNA in sera (≤102 vge/mL) and 50% of them were also positive for HCV RNA in naive lymphoid cells at the levels ranging between estimated 5 × 100 to 7 × 103 vge/μg total RNA. Importantly, HCV RNA replicative intermediate was identified in approximately 65% of the lymphoid cell samples positive for the HCV negative strand. Heterogeneity in viral sequences detected in serum and PBMC samples, collected at the same or at two different occasions from the same individuals, provided additional evidence for persistently progressing viral replication in lymphoid cells.
Interestingly, the nucleotide differences found in paired sera and lymphoid cells clustered around the same region of the 5′‐UTR where other groups of investigators had also observed HCV polymorphism [29, 30]. As such, our data support the notion of the compartmentalization of HCV variants, as it has been reported for patients with CHC [10, 31, 32, 33] and suggest that a similar event can occur in persistent silent infection continuing after resolution of a symptomatic disease.
Addition of LPS, which is a potent activator of monocytes and macrophages, to the previously designed mitogen‐cytokine cocktail activating T and B lymphocytes [5], further enhanced the detection of occult HCV infection in PBMC. In fact, PBMC treated with C5L showed higher levels of HCV RNA expression than those treated with C5 in a significant number of cases examined. The contribution of HCV residing in monocytes to the viral load identified in the total PBMC population was most pronounced in four individuals in whom HCV presence was only identified after treatment of the cells with C5L (Table 1 and Fig. 3). Our finding of monocytes being a reservoir of replicating HCV is in agreement with those suggested by others [3, 21]. Interestingly, susceptibility of monocytes to infection is not unique to HCV. The cells of this lineage have been shown to be permissive to a wide range of viruses [34], including dengue virus [35], feline coronaviruses [36] and human immunodeficiency virus [37].
Among the 20 individuals investigated in the current study, three patients were unique in that the levels of HCV RNA detected in their naive lymphoid cells were between 1 × 103 and 7 × 103 vge/μg total RNA, a viral load that we typically observe in lymphoid cells from patients with CHC (Pham and Michalak, unpubl. data). In all three cases, there was no apparent correlation between HCV level in lymphoid cells and in serum, which was in the range of 40–200 vge/mL. This indicates that relatively high HCV loads in lymphoid cells can occur in individuals with permanently low serum HCV RNA, albeit most likely transiently as PBMC samples collected a year before from the same persons contained low HCV loads, comparable with those detected in other individuals with persistent occult infection investigated in this study. A similar observation was made in occult hepadnaviral infection in the woodchuck model of hepatitis B where the number of circulating lymphoid cells carrying WHV genome was measured by in situ PCR followed by flow cytometric detection of positive cells [38]. The results of this study demonstrated that the number of virus‐reactive lymphoid cells in either primary or secondary occult WHV infection can be occasionally as high as that in symptomatic, serologically evident chronic hepatitis.
Ex vivo stimulation of lymphoid cells with high HCV loads obtained from the individuals with occult infection resulted in at least 90% inhibition of HCV RNA expression after treatment with C5 in two cases and virtually complete abrogation of viral expression in all three cases after exposure of the cells to C5L (Fig. 4). The fact that the levels of the housekeeping GAPDH gene remained similar in untreated and mitogen‐treated samples ruled out the possibility that the mitogen‐cytokine cocktails might have caused cell death or random changes to viral RNA expression. We subsequently assessed the transcriptional activity of selected antiviral cytokines in both naive and stimulated lymphoid cells to assess whether these cytokines may be of relevance to HCV replication. It is of note that IFNα5 was selected for this investigation as it has been reported that this isoform of IFNα, one of the 13 members of the IFNα family, was found to be predominantly induced in circulating lymphoid cells [39] but downregulated in hepatic tissue [40] of patients with CHC. The results of our analysis showed that a down‐regulation of HCV RNA was closely correlated with an increase in the level of IFNα5 expression. Conversely, augmentation of HCV replication paralleled a decrease in the intracellular IFNα5 mRNA level (Fig. 5). Demonstration of a similar relation between HCV RNA load and of IFN‐stimulated MxA confirmed that activation of the IFNα signalling pathway most likely plays a pivotal role in the control of HCV replication in lymphoid cells in the course of occult infection. In contrast, neither IFNγ nor TNFα expression appeared to have any direct association with the rate of HCV replication. Such an antiviral effect of endogenous IFNα against viral pathogens replicating in lymphoid cells is not exclusive to HCV, as similar observations have been made in both in vivo and in vitro settings for human herpesvirus‐8 [41], dengue virus [42] and West Nile virus [43]. Our findings and those reported for patients with CHC [39] collectively suggest that HCV infection does not seem to impede the production of IFNα in lymphoid cells, unlike that with hepatitis B virus [44] or HIV [45]. What however remains unknown, and will require a separate study, is the interplay between HCV and the host machinery mediating activation of lymphoid cells in response to mitogen treatment. Deciphering the underlying molecular mechanisms would be of importance as the amount of HCV residing in lymphoid cells appears to predetermine the outcome of mitogen stimulation in regard to IFNα expression and whether subsequently HCV replication is augmented, suppressed or even aborted in these cells. Understanding of this issue may have a direct impact not only on the designing practical approaches for detection of occult HCV infection, but also on individualized methods of induction on HCV clearance in particular virological situations.
Acknowledgements
The study was supported by operating grant MOP‐77544 awarded to T.I.M. from the Canadian Institutes of Health Research. T.N.Q.P. is supported by a Postdoctoral Fellowship Award from the National Canadian Research Training Program in Hepatitis C. P.M.M.‐C. was a recipient of a Doctoral Fellowship Award from the Canadian Blood Services. S.E.M. was supported by the Seved Soderquist Memorial Summer Studentship from the Canadian Liver Foundation. T.I.M. is the Canada Research Chair (Tier 1) in Viral Hepatitis/Immunology sponsored by the Canada Research Chair Program and funds from the Canadian Institutes of Health Research and the Canada Foundation for Innovation.
References
- 1. Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36: S35–S46. [DOI] [PubMed] [Google Scholar]
- 2. Morsica G, Tambussi G, Sitia R et al. Replication of hepatitis C virus in B lymphocytes (CD19+). Blood 1999; 94: 1138–1139. [PubMed] [Google Scholar]
- 3. Laskus T, Radkowski M, Piasek A et al. Hepatitis C virus in lymphoid cells of patients coinfected with human immunodeficiency virus type 1: evidence of active replication in monocytes/macrophages and lymphocytes. J Infect Dis 2000; 181: 442–448. [DOI] [PubMed] [Google Scholar]
- 4. Goutagny N, Fatmi A, De L et al. Evidence of viral replication in circulating dendritic cells during hepatitis C virus infection. J Infect Dis 2003; 187: 1951. [DOI] [PubMed] [Google Scholar]
- 5. Pham TNQ, MacParland SA, Coffin CS, Lee SS, Bursey FR, Michalak TI. Mitogen‐induced upregulation of hepatitis C virus expression in human lymphoid cells. J Gen Virol 2005; 86: 657–666. [DOI] [PubMed] [Google Scholar]
- 6. Pham TNQ, MacParland SA, Mulrooney PM, Cooksley H, Naoumov NV, Michalak TI. Hepatitis C virus persistence after spontaneous or treatment‐induced resolution of hepatitis C. J Virol 2004; 78: 5867–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee WM, Polson JE, Carney DS, Sahin B, Gale M. Jr Re‐emergence of hepatitis C virus after 8.5 years in a patient with hypogammaglobulinemia: evidence for an occult viral reservoir. J Infect Dis 2005; 192: 1088–1092. [DOI] [PubMed] [Google Scholar]
- 8. Carreno V, Pardo M, Lopez‐Alcorocho JM, Rodriguez‐Inigo E, Bartolome J, Castillo I. Detection of hepatitis C virus (HCV) RNA in the liver of healthy, anti‐HCV antibody‐positive, serum HCV RNA‐negative patients with normal alanine aminotransferase levels. J Infect Dis 2006; 194: 53–60. [DOI] [PubMed] [Google Scholar]
- 9. Radkowski M, Wilkinson J, Nowicki M et al. Search for hepatitis C virus negative strand RNA sequences and analysis of viral sequence in the central nervous system: evidence of replication. J Virol 2002; 76: 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forton DM, Karayianis P, Mahmud N, Taylor‐Robinson SD, Thomas HC. Identification of unique hepatitis C virus quasispecies in the central nervous system and comparative analysis of internal translational efficiency of brain, liver and serum variants. J Virol 2004; 78: 5170–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castillo I, Pardo M, Bartolome J et al. Occult hepatitis C virus infection in patients in whom the etiology of persistently abnormal results of liver‐function tests is unknown. J Infect Dis 2004; 189: 7–14. [DOI] [PubMed] [Google Scholar]
- 12. Castillo I, Rodriguez‐Inigo E, Bartolome J et al. Hepatitis C virus replicates in peripheral blood mononuclear cells in patients with occult hepatitis C virus infection. Gut 2005; 54: 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimizu YK, Iwamoto A, Hijikata M, Purcell RH, Yoshikura H. Evidence for in vitro replication of hepatitis C virus genome in a human T cell line. Proc Natl Acad Sci USA 1992; 89: 5477–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sung VM, Shimodaira S, Doughty AL et al. Establishment of B‐cell lymphoma cell lines persistently infected with hepatitis C virus in vivo and in vitro: the apoptotic effects of virus infection. J Virol 2003; 77: 2134–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Radkowski M., Bednarska A, Horban A et al. Infection of primary human macrophages with hepatitis C virus in vitro: induction of tumor necrosis factor‐alpha and interleukin 8. J Gen Virol 2004; 85: 47–59. [DOI] [PubMed] [Google Scholar]
- 16. MacParland SA, Pham TNQ, Gujar SA, Michalak TI. De novo infection and propagation of wild‐type hepatitis C virus in human T lymphocytes in vitro. J Gen Virol 2006; 87: 3577–3586. [DOI] [PubMed] [Google Scholar]
- 17. Oldstone MB. How viruses escape from cytotoxic T lymphocytes: molecular parameters and players. Virology 1997; 234: 179–185. [DOI] [PubMed] [Google Scholar]
- 18. Michalak TI. Occult persistence and lymphotropism of hepadnaviral infection: insights from the woodchuck viral hepatitis model. Immunol Rev 2000; 174: 98–111. [DOI] [PubMed] [Google Scholar]
- 19. Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005; 436: 967–972. [DOI] [PubMed] [Google Scholar]
- 20. Pawlotsky JM. Use and interpretation of virological tests for hepatitis C. Hepatology 2002; 36: 0S65–S73. [DOI] [PubMed] [Google Scholar]
- 21. Radkowski M, Gallegos‐Orozco JF, Jablonska J et al. Persistence of hepatitis C virus in patients successfully treated for chronic hepatitis C. Hepatology 2005; 41: 106–114. [DOI] [PubMed] [Google Scholar]
- 22. Carreno V, Castillo I, Rodriguez‐Inigo E et al. Hepatitis C virus persists and replicates in the liver of the majority of sustained responder patients to antiviral treatment. Hepatology 2005; 42: 284A. [Google Scholar]
- 23. Pham TNQ, Michalak TI. Occult hepatitis C virus persistence: identification and characteristics. Med Lab Obser 2006; 38: 20–22. [PubMed] [Google Scholar]
- 24. Coffin CS, Pham TNQ, Urbanski SJ, Michalak TI, Lee SS. Persistent hepatic alterations in individuals with occult HCV infection following sustained virological response to antiviral therapy. J Hepatol 2006; 44: S196. [Google Scholar]
- 25. Triantafilou M, Triantafilou K. Lipopolysaccharide recognition: CD14, TLRs and the LPS‐activation cluster. Trends Immunol 2002; 23: 301–304. [DOI] [PubMed] [Google Scholar]
- 26. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position‐specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kato N, Hijikata M, Ootsuyama Y et al. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non‐A, non‐B hepatitis. Proc Natl Acad Sci USA 1990; 87: 9524–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samuel CE. Antiviral actions of interferons: interferon‐regulated cellular proteins and their surprisingly selective antiviral activities. Virology 1991; 183: 1–11. [DOI] [PubMed] [Google Scholar]
- 29. Laskus T, Radkowski M, Wilkinson J, Vargas H, Rakela J. The origin of hepatitis C virus reinfecting transplanted livers: serum‐derived versus peripheral blood mononuclear cell‐derived virus. J Infect Dis 2002; 185: 417–421. [DOI] [PubMed] [Google Scholar]
- 30. Nakajima N, Hijikata M, Yoshikura H, Shimizu YK. Characterization of long‐term cultures of hepatitis C virus. J Virol 1996; 70: 3325–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maggi F, Fornai C, Vatteroni ML et al. Differences in hepatitis C virus quasispecies composition between liver, peripheral blood mononuclear cells and plasma. J Gen Virol 1997; 78: 1521–1525. [DOI] [PubMed] [Google Scholar]
- 32. Navas S, Martin J, Quiroga JA, Castillo I, Carreno V. Genetic diversity and tissue compartmentalization of the hepatitis C virus genome in blood mononuclear cells, liver, and serum from chronic hepatitis C patients. J Virol 1998; 72: 1640–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roque‐Afonso AM, Ducoulombier D, Di Liberto G et al. Compartmentalization of hepatitis C virus genotypes between plasma and peripheral blood mononuclear cells. J Virol 2005; 79: 6349–6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mogensen SC. Role of macrophages in natural resistance to virus infections. Microbiol Rev 1979; 43: 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen YC, Wang SY. Activation of terminally differentiation monocytes/macrophages by dengue virus: productive infection, hierarchical production of innate cytokines and chemokines, and the synergistic effects of lipopolysaccharide. J Virol 2002; 76: 9877–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dewerchin HL, Cornelissen E, Nauwynck HJ. Replication of feline coronaviruses in peripheral blood monocytes. Arch Virol 2005; 150: 2483–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perno CF, Yarchoan R, Cooney DA et al. Replication of human immunodeficiency virus in monocytes. Granulocyte/macrophage colony‐stimulating factor (GM‐CSF) potentiates viral production yet enhances the antiviral effect mediated by 3′‐azido‐2′3′‐dideoxythymidine (AZT) and other dideoxynucleoside congeners of thymidine. J Exp Med 1989; 169: 933–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mulrooney PM, Michalak TI. Quantitative detection of hepadnavirus‐infected lymphoid cells by in situ PCR combined with flow cytometry: implications for the study of occult virus persistence. J Virol 2003; 77: 970–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larrea E, Alberdi A, Castelruiz Y, Boya P, Civeira M‐P, Prieto J. Expression of interferon‐ subtypes in peripheral blood mononuclear cells from patients with chronic hepatitis C: a role for interferon‐5. J Viral Hep 2001; 8: 103–110. [DOI] [PubMed] [Google Scholar]
- 40. Castelruiz Y, Larrea E, Boya P, Civeira MP, Prieto J. Interferon alpha subtypes and levels of type I interferons in the liver and peripheral mononuclear cells in patients with chronic hepatitis C and controls. Hepatology 1999; 29: 1900–1904. [DOI] [PubMed] [Google Scholar]
- 41. Monini P, Carlini F, Sturzl M et al. Alpha interferon inhibits human herpesvirus‐8 (HHV) reactivation in primary effusion lymphoma cells and reduces HHV‐8 load in cultured peripheral blood mononuclear cells. J Virol 1999; 73: 4029–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kurane I, Ennis FA. Production of interferon alpha by dengue virus‐infected human monocytes. J Gen Virol 1988; 69: 445–449. [DOI] [PubMed] [Google Scholar]
- 43. Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol 2005; 79: 13350–13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duan XZ, Wang M, Li HW, Zhuang H, Xu D, Wang FS. Decreased frequency and function of circulating plasmacytoid dendritic cells (pDC) in hepatitis B virus infected humans. J Clin Immunol 2004; 24: 637–646. [DOI] [PubMed] [Google Scholar]
- 45. Jiang W, Lederman MM, Salkowitz JR, Rodriguez B, Harding CV, Sieg SF. Impaired monocyte maturation in response CpG oligodeoxynucleotide is related to viral RNA levels in human immunodeficiency virus disease and is at least partially mediated by deficiencies in alpha/beta interferon responsiveness and production. J Virol 2005; 79: 4109–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]