

Abstract
Two natural products databases, the marine natural products database (MNPD) and the traditional Chinese medicines database (TCMD), were used to find novel structures of potent SARS‐CoV protease inhibitors through virtual screening. Before the procedure, the databases were filtered by Lipinski's ROF and Xu's extension rules. The results were analyzed by statistic methods to eliminate the bias in target‐based database screening toward higher molecular weight compounds for enhancing the hit rate. Eighteen lead compounds were recommended by the screening procedure. They were useful for experimental scientists in prioritizing drug candidates and studying the interaction mechanism. The binding mechanism was also analyzed between the best screening compound and the SARS protein. © 2005 Wiley Periodicals, Inc. J Comput Chem 26: 484–490, 2005
Keywords: SARS, virtual screening, molecular docking, marine natural products, traditional Chinese medicines
Introduction
Severe acute respiratory syndrome (SARS) is a serious epidemic disease dispersed in many countries during the period of the time from March to May 2003. In that extraordinary period, 5327 persons were infected, of whom 349 died (6.6%) in China (http://www.china.com.cn/chinese/zhuanti/feiyan/318261.htm). The main symptoms of SARS are hyperpyrexia, chilling, cough, and dyspnea.
Confronted with this new human coronavirus, many scientists devoted themselves to related research. Marra1 and his coworkers discovered the genome sequence of the SARS‐associated coronavirus, which lit a lamp for the perplexed investigators. Based on Marra's work, Anand et al.2 built a main proteinase structure using the homology modeling approach. Jenwitheesuk3 indicated that existing HIV‐1 protease inhibitors have high binding affinity to the SARS coronavirus (SARS‐CoV) proteinase. His findings may help scientists to design SARS inhibitors. De Groot4 also believed that there were some similarities between SARS‐CoV and HIV. Xiong et al. found 73 available protease inhibitors from the MDDR database (MDL Drug Data Report, http://www.mdl.com/) by virtual screening.5 Lee et al.6 identified four potent compounds taken from 16 antiviral drugs in the NCI database [National Cancer Institute (NCI) Database (http://cactus.nci.nih.gov/ncidb2/)]. In a recent report, Sirois et al. screened 3.6 million compounds through virtual screening using the MOE software package.7 Wu et al.8 also contributed their excellent work by a cell‐based assay, and 15 compounds with potent anti‐SARS‐CoV activity were found, including two existing drugs.
At present, there are still no effective SARS‐CoV protease inhibitors on the market, and the available ligand databases used for virtual screening are usually those of Western medicine databases, such as MDDR, NCI, ACD, etc.
Traditional Chinese medicine has been playing an important role in China for thousands of years, and now it is an valuable source of complements in Western medicines. During the SARS‐spreading period, people in China used TCM to prevent the disease, with positive results. The prescriptions focus on Honeysuckle [Lonicera japonica, Caprifoliaceae], Indigowoad Root [Isatis indigotica, Cruciferae], Forsythia [Forsythia suspensa, Oleaceae], Swordlike Atractylodes [Atractylodes lancea, Compositae], Licorice [Glycyrrhiza uralensis, Leguminosae], etc.
Along with the scale of isolating compounds from natural sources being bigger and bigger, many novel structures have been continually found. People are paying more attention to finding new effective drugs from natural sources, especially from medicinal plants and halobios.
Unlike land‐dwelling living beings, owing to their unique habitat such as high salinity, very little light, and high pressure, the marine organisms have different metabolism routes. These facts result in remarkable structural diversity of marine natural products. It is the character that makes marine natural products an invaluable treasure. There are more than 12,000 new compounds that have been isolated from sea living beings, adding 500 to 800 new compounds each year.9, 10 A new antineoplastic drug, ET‐743, has been synthesized by PharmaMar.11 The compound originally was isolated from tunicates. It is undergoing its clinical test period II in both Europe and America, and is expected to come onto the market this year.
In this article, we used two new natural products databases: the traditional Chinese medicines database (TCMD), and the marine natural products (MNPD), for virtual screening.
Database
Marine Natural Products Database
MNPD was constructed by our laboratory.12 There are 8078 compounds isolated from halobios, among them 3200 compounds are with bioactivity data, some 1200 with CAS Registry Numbers, and about 3700 with physical property data. This database runs on an ISIS/Base (MDL Information Systems, Inc.) platform.
Traditional Chinese Medicines Database
TCMD is a commercial database built by our laboratory (http://products.cambridgesoft.com/family.cfm?FID=57).13, 14, 15 It has 9127 entries. A typical entry includes detailed 3D molecular structures, English names and synonyms, physical properties, natural sources, and references information. Bioactivity data are available for 3000 of the entries. There are 3922 traditional Chinese medicine plant species including standard expression on TCM effects and indications.
There are many antivirus and anti‐HIV compounds in TCMD. According to Jenwitheesuk and De Groot's findings, they are helpful in finding SARS‐CoV protease inhibitors.3, 4 Honeysuckle, Indigowoad Root, Forsythia, Swordlike Atractylodes, and Licorice are also important effective components of the anti‐SARS TCM prescription. TCMD is run on an ISIS/Base platform.
All compound 2D structures in MNPD and TCMD were transformed to 3D molecules files by CONCORD standalone 4.016 at an SGI workstation.
Drug‐Like Filter
According to the Lipinski's Rule of Five (ROF), drug‐like compounds should have an appropriate molecular weight (MW), H‐bond donors, H‐bond acceptors, and LogP value.17 It has been difficult for molecules with larger molecular weight and lower LogP value to cross through the cell membrane. With larger LogP value, the drug will be difficult to dissolve in water, which is a necessary condition for drugs to be absorbed by an organism. Xu has extended that rule by definition of a drug‐like cluster center.18 The criteria we adopted here (see Table 1) for the filter of the two databases combined the ROF with Xu's regulations. Due to SARS‐CoV protease's larger active pocket, we expanded the MW to less than 900. Compounds with all their parameters meeting the drug‐like rules were picked out and written into a single molecules file. Then the databases were quickly narrowed to 3861 (MNPD) and 5454 (TCMD), respectively. LogP was calculated by the XLogP program.19
Table 1.
Drug‐Like Filter Rules.
| Parameters | Drug‐like values |
|---|---|
| Number of H‐bond donors (HDN) | 0–5 |
| Number of H‐bond acceptors (HAN) | 0–8 |
| Number of aromatic bonds (AB) | 0–28 |
| Number of smallest set of smallest rings (SSSRS) | 1–9 |
| Average atomic numbers (AZ) | 6–10 |
| Number of rotating bonds (RB) | 0–14 |
| Average electronegativity (AE) | 2.55–3.02 |
| Molecular weight (MW) | ≤900 |
| Octanol–water partition coefficient (LogP) | 0–5 |
Virtual Screening Calculation
Virus Target
Rao20 and his coworkers reported a 1.90‐Å crystal structure of the SARS‐CoV protease (PDB entry code: 1UJ1). It is a dimer, and the active pocket located at protomer A, which contains three domains and the substrate‐binding site, is in a cleft between domain I and II (residues 8–101 and 102–184, respectively). The active site has a Cys–His catalytic dyad, which is composed of Cys145 and His41. Also, the substrate‐binding pocket consists of the side chains of His163 and Phe140, and the main‐chain atoms of Met165, Glu166, and His172.
Dock Parameters
The Dock4.02 package of the Linux version was used in the first step of the virtual screening procedure,21, 22 and computations were carried out in the cluster of the PC servers. Each computational node has an HP/Compaq DL360 industrial standard server with dual pentium III 1.13‐GHz CPUs and a 512‐kb L2 cache. Residues around the sulfur atom of Cys145 at a radius of 13 Å were isolated for constructing the grids of the docking screening. Energy scoring grids were obtained using an all‐atom model and a distance‐dependent dielectric function with a 10‐Å cutoff. The macromolecule was a loaded Kollman charge, with Gasteiger–Huckel charges for small molecules on the SYBYL6.8.23 An anchor fragment orientation method was performed, and 25 conformations were produced per cycle.
AutoDock Study
The top 200 candidates filtered by the Dock procedure in both databases were then studied by the AutoDock3.05 program, respectively. The computations were processed on an SGI Octane 2 graphics workstation. The grid has the space of 0.375 Å and a size of about 60 Å × 49 Å × 63 Å. The macromolecule and the small molecules were loaded on Kollman and Gasteiger‐Huckel charges, respectively, on the SYBYL6.8. The GA‐LS method was adopted using the default settings. Compounds with better AutoDock scores and binding conformations will be selected as lead compounds of SARS for next project.
Results and Discussion
Docking Data
Energy score (ES) is an important criterion to evaluate the binding affinity for the target protein with a ligand of certain orientation and conformation. With the shortcoming of scoring functions of the Dock program,24, 25, 26, 27, 28, 29, 30, 31, 32 the energy score is biased toward the selection of high molecular weight.33 Liu has provided a new arithmetic to eliminate this bias (unpublished data, Liu, Z. M; Shi, L; Lai, L. H. Considering Molecule Weight in Virtual Docking Screening: Implication for Inhibitors Selection). He found that most molecules with heavy atom number (HA) between 5 and 15 interact with the target protein in a proper binding mode. Those compounds usually represent the correct interacting mode of the whole data sets, whereas the energy scores of the molecules with a larger HA spread out partly because of more choices to escape from the “binding site” or pocket capacity limitation. HA numbers and the average energy scores have the relationship described as in eqs. (1) (MNPD) and (2) (TCMD).
The HA–AE exponential decay fit curve of MNPD:
| (1) |
The HA–AE exponential decay fit curve of TCMD:
| (2) |
Figures 1 and 2 give the distribution curve of heavy atoms and energy scores.
Figure 1.
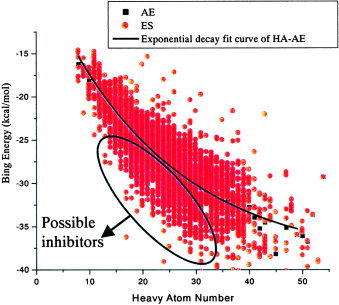
Dock energy score distribution of MNPD. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 2.
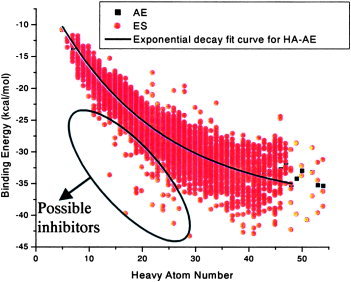
Dock energy score distribution of TCMD. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Best Compounds In Silico
After repetitive evaluation of by AutoDock, 18 compounds of high affinity in silico were selected (7 from MNPD and 11 from TCMD). They will be useful for experimental scientists in prioritizing drug candidates and studying the interaction mechanism. These structures and their drug‐like parameters are listed in Table 2.
Table 2.
Best 18 Compounds Found via Virtual Screening (Dock, kcal/mol; AutoDock, kcal/mol).
| Compound | HAN | HDN | AB | SSSRS | AZ | RB | AE | MW | LogP | Dock | AutoDock | Structure |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M3927 | 3 | 3 | 6 | 1 | 8.79 | 7 | 2.76 | 465.18 | 0.763 | −39.1 | −10.46 |
|
| M4367 | 4 | 5 | 12 | 2 | 6.44 | 12 | 2.75 | 469.54 | 1.553 | −41.07 | −10.7 |
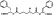
|
| M4890 | 7 | 6 | 6 | 3 | 6.48 | 14 | 2.77 | 642.75 | 3.42 | −44.24 | −11.76 |
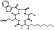
|
| M5410 | 6 | 5 | 12 | 2 | 6.49 | 11 | 2.77 | 512.56 | 1.464 | −41.71 | −11.21 |
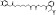
|
| M5789 | 7 | 3 | 6 | 4 | 6.49 | 7 | 2.77 | 578.7 | 3.615 | −43.43 | −10.3 |
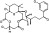
|
| M6601 | 1 | 2 | 6 | 1 | 9.65 | 4 | 2.71 | 370.13 | 3.39 | −36.23 | −9.67 |
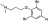
|
| M6602 | 3 | 5 | 6 | 2 | 8.88 | 5 | 2.8 | 461.11 | 2.565 | −35.53 | −7.06 |
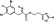
|
| T1434 | 7 | 2 | 6 | 4 | 6.43 | 9 | 2.74 | 580.67 | 2.83 | −37.54 | −12.36 |
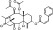
|
| T1441 | 6 | 2 | 6 | 4 | 6.39 | 9 | 2.72 | 566.69 | 3.75 | −36.73 | −11.67 |
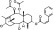
|
| T2826 | 7 | 3 | 6 | 7 | 6.36 | 4 | 2.71 | 575.71 | 1.83 | −39.20 | −11.21 |
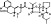
|
| T2831 | 7 | 3 | 6 | 7 | 6.38 | 3 | 2.72 | 547.65 | 1.17 | −37.98 | −12.49 |
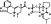
|
| T4744 | 4 | 2 | 24 | 7 | 6.24 | 3 | 2.66 | 449.53 | 2.66 | −38.38 | −12.41 |
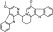
|
| T537 | 8 | 3 | 6 | 4 | 6.38 | 11 | 2.72 | 623.79 | 2.16 | −39.57 | −12.11 |
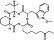
|
| T5656 | 8 | 2 | 6 | 7 | 6.35 | 5 | 2.71 | 589.73 | 2.35 | −38.03 | −12.31 |
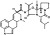
|
| T6791 | 5 | 3 | 18 | 6 | 6.38 | 8 | 2.72 | 566.61 | 1.94 | −39.28 | −11.71 |
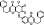
|
| T8593 | 5 | 3 | 24 | 7 | 6.33 | 2 | 2.7 | 580.68 | 2.16 | −31.64 | −11.61 |
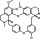
|
| T3091 | 3 | 5 | 6 | 2 | 6.52 | 9 | 2.78 | 434.49 | 2.571 | −37.51 | −6.91 |
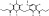
|
| T5242 | 7 | 3 | 12 | 4 | 6.36 | 5 | 2.71 | 575.71 | 1.52 | −31.78 | −12.51 |
|
Conformation Analysis
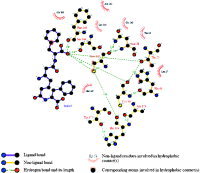
The coronavirus family exhibits one main protease, called 3CL, because of the nature of the catalytic site that acts a crucial role in the regulation of the virus life‐cycle.2 Cys145 and His41 residues are considered to be essential for the normal function of SARS protein. The docking simulation of compound M4367, which was isolated from Pseudomonas sp. or Alteromonas sp. in sponge Dysidea fragilis (Black Sea), showed that the inhibitor is folded into a ring‐like structure in the active site that was similar with that of Wu's compound 2 (Wu‐2).8 The K i value of Wu‐2 against the SARS‐CoV 3CL protease is 0.6 μm. One phenyl group of compound M4367 fits into the pocket defined by Leu27, Thr25, etc. One carbonyl instead of Wu‐2's phenyl fits into the pocket defined by the hydrophobic residues (Met165, Pro‐168, and Leu‐167). The M4367 groups interact with Cys145 and His41 directly by hydrogen bond interaction and hydrophobic contact. There are also four other hydrogen bonds between M4367 and Phe140, Ser144, Cys44, and Thr25, respectively. The complex was analyzed by the Ligplot 4.22 to identify some specific contacts (Fig. 3).34
Figure 3.
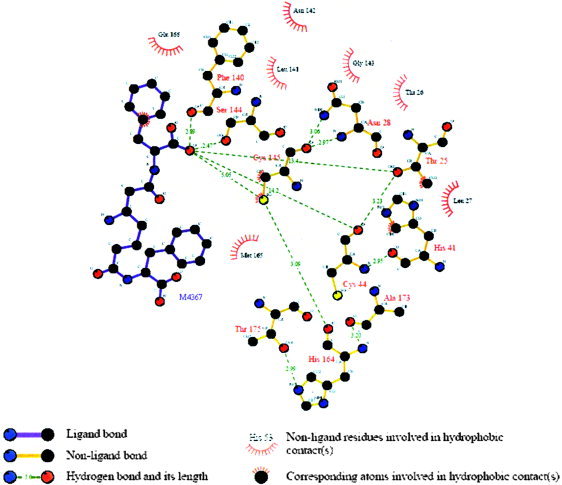
Schematic representation of SARS–M4367 interactions. The ligand atoms serving as the correspondence points in the subsequent structural alignment processes were marked with the atom type beside it. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The SARS target was so novel that there are still no effective inhibitors available in the market. Marra1 reports that the derivatives of AG7088 might be good starting points for the design of anticoronavirus drugs. AG7088 has already been clinically tested for treatment of the common cold. Its docking complex with SARS‐CoV protease also has multiple interactions, which is similar to that of our recommended compounds (Fig. 4).
Figure 4.
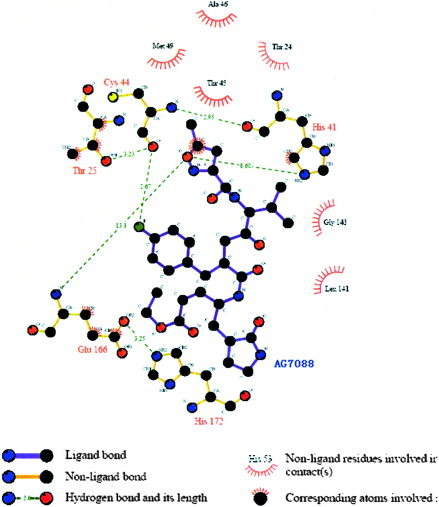
Schematic representation of the SARS–AG7088 interactions. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Conclusion
Eighteen novel‐structure compounds with best binding affinities and conformations were found via virtual screening and statistic methods. The interaction and binding mechanism were elucidated by the complex structure of SARS–M4367. The similarity of the protein binding mode between our screened compounds and Wu‐2, AG7088, which were reported as possible molecules of SARS inhibitors, showed certain values of our research for experimental scientists in prioritizing drug candidates. The results show that high‐affinity drugs for the SARS protein may have the characteristic of direct interaction with the functional residues, His41 and Cys145, which act as a crucial role in the regulation of the SARS life cycle.
Acknowledgements
We thank Dr. Zhenming Liu and Prof. Luhua Lai of Beijing University for many useful discussions in the result analysis. We also thank Hao He (ChemBay Technology Ltd., China), who helped to transform MNPD and TCMD to 3D molecules files with CONCORD.
References
- 1. Marra, M. A. ; Jones, S. J. ; Astell, C. R. ; Holt, R. A. ; Brooks–Wilson, A. ; Butterfield, Y. S. ; Khattra, J. ; Asano, J. K. ; Barber, S. A. ; Chan, S. Y. Science 2003, 300, 1399. [DOI] [PubMed] [Google Scholar]
- 2. Anand, K. ; Ziebuhr, J. ; Wadhwani, P. ; Mesters, J. R. ; Hilgenfeld, R. Science 2003, 300, 1763. [DOI] [PubMed] [Google Scholar]
- 3. Jenwitheesuk, E. ; Samudrala, R. Bioorganic Med Chem Lett 2003, 13, 3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Groot, A. S. D. Vaccine 2003, 21, 4095.14505885 [Google Scholar]
- 5. Xiong, B. ; Gui, C. S. ; Xu, X. Y. ; Luo, C. ; Chen, J. ; Luo, H. B. Acta Pharmacol Sin 2003, 24, 497. [PubMed] [Google Scholar]
- 6. Lee, V. S. ; Wittayanarakul, K. ; Remsungnen, T. ; Parasuk, V. ; Sompornpisut, P. ; Chantratita, W. ScienceAsia 2003, 29, 181. [Google Scholar]
- 7. Sirois, S. ; Wei, D. Q. ; Du, Q. S. ; Chou, K. C. J Chem Inf Comput Sci 2004, 44, 1111. [DOI] [PubMed] [Google Scholar]
- 8. Wu, C. Y. ; Jan, J. T. ; Ma, S. H. ; Kuo, C. J. ; Juan, H. F. ; Cheng, Y. S. ; Hsu, H. H. ; Huang, H. C. ; Wu, D. ; Brik, A. ; Liang, F. S. ; Liu, R. S. ; Fang, J. M. ; Chen, S. T. ; Liang, P. H. ; Wong, C. H. Proc Natl Acad Sci USA 2004, 101, 10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Konig, G. M. ; Wright, A. D. ; Sticher, O. ; Angerhofer, C. K. ; Pezzuto, J. Planta Med 1994, 60, 532. [DOI] [PubMed] [Google Scholar]
- 10. Faulkner, D. Nat Prod Rep 2001, 18, 1. [DOI] [PubMed] [Google Scholar]
- 11. Guo, Y. Chin Marine Drugs 2002, 6, 53. [Google Scholar]
- 12. Lei, J. ; Zhou, J. J. J Chem Inf Comput Sci 2002, 42, 742. [DOI] [PubMed] [Google Scholar]
- 13. Zhou, J. ; Yan, X. ; Xie, G. ASHGATE: Burlington, VT, 1999. [Google Scholar]
- 14. Lu, A. J. ; Liu, B. ; Liu, H. B. ; Zhou, J. J. ; Xie, G. R. Internet Electron J Mol Des 2004, 3, 672. [Google Scholar]
- 15. Yan, X. J. ; Zhou, J. J. ; Xu, Z. J Chem Inf Comput Sci 1999, 39, 86. [DOI] [PubMed] [Google Scholar]
- 16. Pearlman, R. S. CONCORD User's Manual, Tripos, Inc.: St. Louis, MO, 1998. [Google Scholar]
- 17. Lipinski, C. A. ; Lombardo, F. ; Dominy, B. W. ; Feeney, P. J. AdvDrug Deliv Rev 1997, 23, 3. [DOI] [PubMed] [Google Scholar]
- 18. Xu, J. J Med Chem 2002, 45, 5311. [DOI] [PubMed] [Google Scholar]
- 19. Wang, R. X. ; Fu, Y. ; Lai, L. H. J Chem Inf Comput Sci 1997, 37, 615. [Google Scholar]
- 20. Yang, H. T. ; Yang, M. ; Ding, Y. ; Liu, Y. ; Lou, Z. ; Zhou, Z. ; Rao, Z. H. Proc Natl Acad Sci USA 2003, 100, 13190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuntz, I. D. Science 1992, 257, 1078. [DOI] [PubMed] [Google Scholar]
- 22. Meng, E. C. ; Shoichet, B. K. ; Kuntz, I. D. J Comp Chem 1992, 13, 505. [Google Scholar]
- 23. SYBYL Package, Tripos Associate Inc. St. Louis, MO, 63144, USA. [Google Scholar]
- 24. Kuntz, I. D. ; Meng, E. C. ; Shoichet, B. K. Acc Chem Res 1994, 27, 117. [Google Scholar]
- 25. Morris, G. M. ; Goodsell, D. S. ; Huey, R. ; Olson, A. J. J Comput‐Aided Mol Des 1996, 10, 293. [DOI] [PubMed] [Google Scholar]
- 26. Schnecke, V. ; Swanson, C. A. ; Getzoff, E. D. ; Tainer, J. A. ; Kuhn, L. A. Proteins 1998, 33, 74. [PubMed] [Google Scholar]
- 27. Charifson, P. S. ; Corkery, J. J. ; Murcko, M. A. ; Walters, W. P. J Med Chem 1999, 42, 5100. [DOI] [PubMed] [Google Scholar]
- 28. Knegtel, R. M. ; Wagener, M. Proteins 1999, 37, 334. [DOI] [PubMed] [Google Scholar]
- 29. Bissantz, C. ; Folkers, G. ; Rognan, D. J Med Chem 2000, 43, 4759. [DOI] [PubMed] [Google Scholar]
- 30. Stahl, M. ; Rarey, M. J Med Chem 2001, 44, 1035. [DOI] [PubMed] [Google Scholar]
- 31. Gohlke, H. ; Klebe, G. Curr Opin Struct Biol 2001, 11, 231. [DOI] [PubMed] [Google Scholar]
- 32. Doman, T. N. ; McGovern, S. L. ; Witherbee, B. J. ; Kasten, T. P. ; Kurumbail, R. ; Doman, T. N. J Med Chem 2002, 45, 2213. [DOI] [PubMed] [Google Scholar]
- 33. Pan, Y. ; Huang, N. ; Cho, S. ; Mackerell, A. D., Jr. J Chem Inf Comput Sci 2003, 43, 267. [DOI] [PubMed] [Google Scholar]
- 34. Wallace, A. C. ; Laskowski, R. A. ; Thornton, J. M. Protein Eng 1995, 8, 127. [DOI] [PubMed] [Google Scholar]