

Abstract
The Middle East respiratory syndrome coronavirus (MERS‐CoV) is an emerging virus that poses a major challenge to clinical management.
The 3C‐like protease (3CLpro) is essential for viral replication and thus represents a potential target for antiviral drug development. Presently, very few data are available on MERS‐CoV 3CLpro inhibition by small molecules. We conducted extensive exploration of the pharmacophoric space of a recently identified set of peptidomimetic inhibitors of the bat HKU4‐CoV 3CLpro. HKU4‐CoV 3CLpro shares high sequence identity (81%) with the MERS‐CoV enzyme and thus represents a potential surrogate model for anti‐MERS drug discovery. We used 2 well‐established methods: Quantitative structure‐activity relationship (QSAR)‐guided modeling and docking‐based comparative intermolecular contacts analysis. The established pharmacophore models highlight structural features needed for ligand recognition and revealed important binding‐pocket regions involved in 3CLpro‐ligand interactions. The best models were used as 3D queries to screen the National Cancer Institute database for novel nonpeptidomimetic 3CLpro inhibitors. The identified hits were tested for HKU4‐CoV and MERS‐CoV 3CLpro inhibition. Two hits, which share the phenylsulfonamide fragment, showed moderate inhibitory activity against the MERS‐CoV 3CLpro and represent a potential starting point for the development of novel anti‐MERS agents. To the best of our knowledge, this is the first pharmacophore modeling study supported by in vitro validation on the MERS‐CoV 3CLpro.
Highlights
MERS‐CoV is an emerging virus that is closely related to the bat HKU4‐CoV.
3CLpro is a potential drug target for coronavirus infection.
HKU4‐CoV 3CLpro is a useful surrogate model for the identification of MERS‐CoV 3CLpro enzyme inhibitors.
dbCICA is a very robust modeling method for hit identification.
The phenylsulfonamide scaffold represents a potential starting point for MERS coronavirus 3CLpro inhibitors development.
Keywords: 3CLpro inhibitors, coronavirus, dbCICA, MERS, pharmacophore modeling
3CLpro has been proposed as a potential target for the treatment of MERS‐CoV infection. The bat HKU4‐CoV 3CLpro has been exploited as a model for MERS‐CoV 3CLpro (81% sequence identity). We explored the pharmacophoric space of a set of peptidomimetic inhibitors of HKU4‐CoV 3CLpro. The established pharmacophores highlight structural features needed for ligand recognition and were used as 3D queries to screen the NCI database for novel nonpeptidomimetic 3CLpro inhibitors. The identified hits were tested for HKU4‐CoV and MERS‐CoV 3CLpro inhibition.
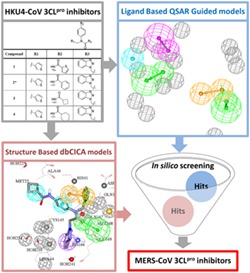
1. INTRODUCTION
Middle East respiratory syndrome coronavirus (MERS‐CoV; HCoV‐EMC/2012) is an emerging virus that causes severe pneumonia illness and exhibits a high mortality rate.1 The first known human MERS‐CoV cases occurred in Jordan in 2012, before the causative virus was detected and identified later during the same year in Saudi Arabia.2, 3 Since then, over 1900 laboratory‐confirmed cases have been reported to the WHO in 27 countries across the world.4
MERS‐CoV is an enveloped virus carrying a genome of positive sense RNA.5 The virus, which is considered primarily as a zoonotic virus, belongs to the lineage C of Betacoronavirus, thus is closely related to the bat coronaviruses HKU4 and HKU5.6, 7, 8 Several studies have shown that bats and camels are the most likely animal reservoir of MERS‐CoV.9, 10, 11 Accumulating evidence points to virus transmission from dromedary camels to humans.12, 13
As the case with many viral diseases, effective therapy against MERS is lacking and supportive care is the only available treatment option. Attempts to develop an effective vaccine against MERS‐CoV infection have led to promising results but are still in early stages.14, 15, 16 The high morbidity and mortality rates of MERS‐CoV as well as its potential to cause epidemics highlight the need for novel drug discovery to develop effective and safe anti‐MERS‐CoV therapeutics.
Several efforts have been undertaken to identify selective potent small molecules with anti–MERS‐CoV activity.17, 18, 19, 20, 21 Promising compounds were identified via screening of FDA‐approved drugs and drug‐like small molecules using cell‐based systems and in vitro screening.17, 18, 19, 20, 21, 22, 23, 24
Targets homologous to those identified in the severe acute respiratory syndrome coronavirus (SARS‐CoV) were investigated in MERS‐CoV (reviewed in Hilgenfeld and Peiris25).26, 27, 28, 29 Among these, MERS‐CoV main proteinase, also known as 3‐chymotrypsin‐like protease (3CLpro), is considered an important potential target due to its essential role in the viral life cycle.26, 29 The coronavirus genome encodes an 800‐kDa replicase polyprotein, which is processed by the 3CLpro to yield intermediate and mature nonstructural proteins responsible for many aspects of virus replication.5, 30, 31 The enzyme started to attract interest as a target for anti–MERS‐CoV drug development. However, data on the enzyme inhibition are scarce. The SARS‐CoV 3CLpro has been comprehensively explored as a drug target, and many potent enzyme inhibitors have been identified.1, 25, 32, 33 Elaborated structure‐ and ligand‐based in silico models obtained using the SAR‐CoV 3CLpro inhibitors proved fruitless for the identification of MERS‐CoV 3CLpro inhibitors (modeling studies conducted by our group, data not published). Interestingly, the 3CLpro enzymes from different CoV strains are known to share significant sequence and 3D structure homology providing a strong structural basis for designing wide‐spectrum anti‐CoV inhibitors.34, 35 Sequence alignment studies showed that the active site residues of the HKU4‐CoV 3CLpro that participated in inhibitor binding are conserved in the MERS‐CoV 3CLpro, which has 81.0% sequence identity36 to HKU4‐CoV 3CLpro (Figure 1). Therefore, the bat HKU4‐CoV 3CLpro has been investigated as a surrogate model for anti‐MERS development.36 Novel peptidomimetic inhibitors of MERS‐CoV 3CLpro have been identified by using the enzyme from HKU4‐CoV as a model.36
Figure 1.
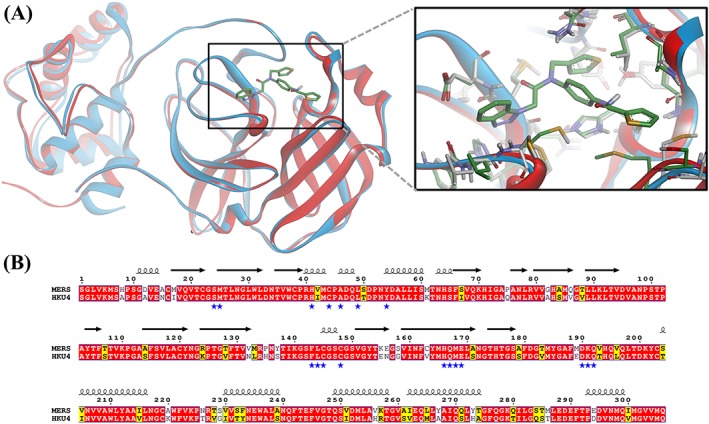
Comparison of the binding site of 3CLpro from HKU4‐CoV and MERS‐CoV. (A) A ribbon presentation of the superimposition of the HKU4‐CoV 3CLpro complex with a potent inhibitor (blue ribbons and green carbon atoms, 1.8 Å, PDB code 4YOI) and the MERS‐CoV enzyme (red ribbons and gray carbon atoms, 2.1 Å, PDB code 4YLU), showing the high similarity in protein folding and a close‐up view of the main residues interacting with inhibitors in HKU4‐CoV and MERS‐CoV 3CLpro binding pockets. The figure was prepared using the DS visualizer. (B) Amino acid sequence alignment of the 3CLpro from HKU4‐CoV and MERS‐CoV enzyme. The sequence alignment was generated by using Clustal Omega. Residues strictly conserved have a red background; similar residues are indicated by black bold letters with a yellow background according to a Risler matrix implemented in ESPript. The symbols above the sequence correspond to the secondary structure of MERS‐CoV3CLpro (PDB code 4YLU; Tomar et al30). The blue stars indicate residues in the binding pocket the enzymes. MERS‐CoV, Middle East respiratory syndrome coronavirus; PDB, Protein Data Bank
In this study, we used the set of peptidomimetic HKU4‐CoV 3CLpro inhibitors reported in St. John et al36 to conduct extensive computational modeling studies. These modeling efforts aim at establishing pharmacophore models to be used as 3D search queries for virtual screening of potential MERS‐CoV 3CLpro inhibitors. The methods used here were developed previously by our group: the QSAR‐guided pharmacophore modeling37, 38 and the docking‐based comparative intermolecular contacts analysis (dbCICA) pharmacophore modeling.39, 40 Both modeling approaches have been used successfully to identify potent inhibitors against several drug targets.37, 38, 39, 40, 41 The identified hits were tested in vitro for their inhibitory activity against the 3CLpro enzymes from HKU4‐CoV and MERS‐CoV.
2. MATERIAL AND METHODS
All chemicals and reagents were purchased from Sigma‐Aldrich (United States), unless otherwise stated.
2.1. QSAR‐guided pharmacophore modeling
2.1.1. Data preparation and pharmacophore exploration
The structures and biological data of 221 previously identified HKU4‐CoV 3CLpro inhibitors reported in St. John et al36 (1‐221, Table S1) were used in modeling.
The bioactivities of these inhibitors were expressed as the concentration of the test compound that inhibited the activity of HKU4‐CoV 3CLpro by 50% (IC50, μM). In cases of unavailable IC50 values (ie, 20‐25 and 48‐221, Table S1), the corresponding IC50 estimates were extrapolated based on reported inhibitory percentages at 100μM assuming linear dose‐response relationships. The logarithms of measured IC50 (μM) values were used in QSAR‐guided pharmacophore modeling to correlate bioactivity data linearly to free energy change. Chiral centers with unknown configuration were marked as “unknown” so that the inversion these chiral centers is sampled during conformation generation.
These compounds were used to explore the pharmacophoric space of HKU4‐CoV 3CLpro through a series of established modeling steps as has been described previously.38, 42, 43, 44, 45, 46 The modeling workflow is detailed in Sections S1 to S5.
2.1.2. QSAR modeling
QSAR‐guided selection of optimal pharmacophores was conducted to find an optimal combination of pharmacophore models capable of explaining bioactivity variation across the whole set of collected training compounds (1‐221, Table S1).36 QSAR modeling was done using the genetic function algorithm (GFA) to generate combinations of descriptors (physicochemical and pharmacophores) (Sections S6 and S7). Subsequently, multiple linear regression (MLR) analyses were used to assess the qualities of selected descriptor combinations, ie, to explain bioactivity variations within collected inhibitors. This QSAR modeling was performed using a training set of 177 compounds of the total set of HKU4‐CoV 3CLpro inhibitors and validated using leave‐one‐out r 2 (r 2 LOO) and predictive r 2 (r 2 PRESS) against a randomly selected testing set of 44 inhibitors as described in Sections S6, S7, and S8. The test set was selected by ranking the total 221 inhibitors according to their IC50 values, and then every fifth compound was selected for the testing set starting from the high‐potency end.
2.2. Docking‐based comparative intermolecular contacts analysis
Docking studies were performed using a subset of 27 compounds of the peptidomimetic HKU4‐CoV 3CLpro inhibitors with known (absolute) stereochemistries (1‐27, Table S1). The 3D coordinates of HKU4‐CoV 3CLpro were retrieved from the Protein Data Bank (PDB code: 4YOI, 1.8 Å).36 The protein structure was modified by adding hydrogen atoms and Gasteiger‐Marsili charges to the protein atoms using the Discovery Studio (version 2.5.5; Accelrys Inc, San Diego). It was then used in subsequent docking experiments without energy minimization.
Docking was conducted using both LibDock47 and CDOCKER.48 LibDock is a site‐feature docking algorithm that docks ligands (after removing hydrogen atoms) into an active site guided by binding hotspots.47 While, CDOCKER is a CHARMm‐based simulated annealing/molecular dynamics method that implements simulated annealing to search for the most stable docked ligand poses.48 These docking engines consider the flexibility of the ligand while treat the receptor as rigid. Details of each docking engine and the corresponding docking settings are described in Sections S9 to S10. The highest‐ranking docked conformers/poses were scored using 7 scoring functions: Jain, LigScore1, LigScore2, PLP1, PLP2, PMF, and PMF04 (Section S11).49, 50, 51, 52, 53 The docking‐scoring cycles using both engines were repeated to cover all possible docking combinations resulting from the presence (or absence) of crystallographically explicit water molecules within the binding site.
Taking into account each scoring function in turn, the highest scoring docked conformer/pose of each inhibitor was chosen to be used in subsequent comparative intermolecular contacts analysis (dbCICA) modeling.39, 40 This step resulted in 7 docking/scoring combinations of the 27 compounds each of them scored with a corresponding scoring function. The docking and scoring cycle was repeated 2 times to cover all combinations of docking conditions, ie, the presence or absence of explicit water molecules. The resulting 14 docking/scoring sets were used in dbCICA modeling as described previously.39, 40 Sections S12 to S13 describe details of dbCICA modeling. Successful dbCICA models were used to guide the manual building of pharmacophores (Section S14).
2.3. Validation and steric refinement of pharmacophore models
Optimal pharmacophores (both structure and ligand based) were validated using the receiver operating characteristic (ROC) curve analysis to assess the ability of each model to correctly classify a group of compounds into actives and inactives (Section S15).39, 40, 54 Matthews correlation coefficient (MCC) was also undertaken as an additional validation.55 Additionally, exclusion spheres were added using HIPHOP‐REFINE module of Discovery Studio to improve the ROC properties of QSAR‐guided pharmacophore (Section S8).
2.4. Virtual screening for new HKU4‐CoV 3CLpro inhibitors
The selected pharmacophores were used as 3D search queries to screen the National Cancer Institute (NCI) database56 for new 3CLpro inhibitors.
Hits captured by the QSAR‐guided pharmacophore were filtered by the Lipinski criteria to ensure good pharmacokinetic properties57 and the SMILES arbitrary target specification (SMARTS) filter (Section S16) to remove reactive ligands (ie, alkyl halides or Michael acceptors).58 Remaining hits were fitted against the corresponding individual pharmacophores. The fit values were then substituted in the MLR‐based QSAR models to predict hits' bioactivities (−log(IC50)). The highest‐ranking hits were selected for in vitro testing using a voting system to minimize the influence of QSAR‐based predictions on hit prioritization. In this system, each hit fit value and the hit's overall QSAR predictions cast a vote of “one” if the value is within the top 20% of all captured hits, otherwise the vote is “zero.”
Similarly, hits captured from all successful dbCICA‐derived pharmacophores were pooled together and filtered according to the Lipinski criteria57 and SMARTS filter.58 The hits were then docked into HKU4‐CoV 3CLpro binding pocket (4YOI) using the same docking/scoring conditions of each successful dbCICA model. The resulting docked poses were then analyzed for critical contacts (according to successful dbCICA models), and the sums of critical contacts for each hit compound were used for the prediction of their corresponding IC50 values. The highest‐ranking hits were selected for in vitro testing using a similar voting system to that described above: Each docking solution casts a vote of “one” if the predicted value is within the top 10% of all captured hits, otherwise it casts a vote of “zero.”
2.5. Protein expression and purification
MERS‐CoV 3CLpro was expressed through auto‐induction in Escherichia coli BL21‐DE3 cells in the presence of 100 μg/mL of carbenicillin as described previously.30, 59 Cells were harvested by centrifugation at 5000g for 20 minutes at 4°C, and the pellets were stored at −80°C until further use. MERS‐CoV 3CLpro purification was performed using consecutive steps of hydrophobic‐interaction chromatography, DEAE anion‐exchange chromatography, Mono S cation‐exchange chromatography, and size‐exclusion chromatography as described previously.30 HKU4‐CoV 3CLpro was produced and purified using a modified protocol from Agnihothram et al.60 Final protein yield was calculated based on the measurement of total activity units (μM product/min), specific activity (units/mg), and milligrams of protein obtained (BioRad protein assay) after each chromatographic step.
2.6. Inhibition assays
Inhibition assays were conducted as described previously.36 Each of the acquired hits was screened for inhibition of HKU4 3CLpro and MERS 3CLpro at a concentration of 40μM in duplicate assays containing the following assay buffer (50mM HEPES, 0.1 mg/mL BSA, 0.01% TritonX‐100, 2mM DTT). Compound 1 (the most potent compound in the training set; Table S1; St. John et al 36,table 1A ) was used as a positive control. The assays were conducted in Costar 3694 EIA/RIA 96‐Well Half Area, Flat Bottom, Black Polystyrene plates (Corning, New York). A total of 1 μL of 100X inhibitor stock in dimethyl sulfoxide (DMSO) was added to 79 μL of enzyme in assay buffer, and the enzyme‐inhibitor mixture was incubated for 5 minutes. The reaction was initiated by the addition of 20 μL of 10μM UIVT3 substrate, a custom synthesized Förster resonance energy transfer substrate peptide with the following sequence: HilyteFluor 488‐ESATLQSGLRKAK‐QXL520‐NH2, producing final concentrations of 250nM HKU4‐CoV 3CLpro, 500nM MERS‐CoV 3CLpro, and 100μM UIVT3 substrate. The fluorescence intensity of the reaction was then measured over time as relative fluorescence units (RFUt) for a period of 10 minutes, using an excitation wavelength of 485 nm and bandwidth of 20 nm and monitoring emission at 528 nm and bandwidth of 20 nm using a BioTek Synergy H1 multimode microplate reader. The inhibition of the HKU4‐CoV 3CLpro and MERS‐CoV 3CLpro by hit compounds was monitored by following the change in RFUs over time, using the initial slope of the progress curve to determine the initial rate (Vi). The percent inhibition of each 3CLpro enzyme was determined using the following equation:
| (1) |
The IC50 values were determined at an ambient temperature from 100‐μL assays performed in triplicate in the following buffer: 50mM HEPES, 0.1 mg/mL BSA, 0.01% TritonX‐100, 2mM DTT. Kinetic assays were conducted in Costar 3694 EIA/RIA 96‐Well Half Area, Flat Bottom, Black Polystyrene plates (Corning, NY). Each inhibitor was tested at concentrations ranging from 2.5μM to 400μM. A total of 1 μL of 100X inhibitor stock in DMSO was added to 79 μL of enzyme in assay buffer, and the enzyme‐inhibitor mixture was incubated for 5 minutes. The reaction was initiated by the addition of 20 μL of 10μM UIVT3 substrate, producing final concentrations of 250nM HKU4‐CoV 3CLpro, 500nM MERS‐CoV 3CLpro, and 2μM UIVT3 substrate. The fluorescence intensity of the reaction was then measured over time as RFUt for a period of 10 minutes, using an excitation wavelength of 485 nm and bandwidth of 20 nm and monitoring emission at 528 nm and bandwidth of 20 nm using a BioTek Synergy H1 multimode microplate reader. The percent inhibition of the 3CLpro enzymes was then plotted as a function of inhibitor concentration. The SigmaPlot Enzyme Kinetics Wizard was used to fit the triplicate percent inhibition data and associated standard error to a nonlinear Michaelis‐Menten type regression model and determine the IC50 for each enzyme using the following equation:
| (2) |
where %Imax is the percent maximum inhibition of 3CLpro and the error in IC50 values was determined as the error in the fitted parameter.
Controls were performed, in which the enzyme, the substrate, or both was/were omitted. Fluorescence attenuation controls were carried by adding the inhibitors to the cleaved substrate in a reaction mixture identical to that used in the inhibition assays.
3. RESULTS AND DISCUSSION
3.1. Ligand‐based approach: QSAR‐guided pharmacophore modeling
The pharmacophoric space of 221 HKU4‐CoV 3CLpro inhibitors was extensively explored through 112 HYPOGEN automatic runs performed on 14 carefully selected training subsets comprising 14 to 22 compounds (Section 2.1 and Tables S1 and S2). The training compounds in each subset were selected in such a way to ensure that each set represent a common binding mode and guarantee that bioactivities differences among its members are attributable to the presence or absence of pharmacophoric features. Applying this strategy allows an effective exploration of the pharmacophoric space of HKU4‐CoV 3CLpro inhibitors and helps to identify pharmacophoric hypotheses representing all possible binding modes assumed by 3CLpro.38, 42, 43, 44, 45, 46 These runs resulted in 677 successful pharmacophore models, which were then clustered using the hierarchical average linkage method available in CATALYST. The best 68 representative models were used in subsequent QSAR modeling (Section 2.1).
The fit values obtained by mapping the 68 representative pharmacophores against the HKU4‐CoV 3CLpro inhibitors were enrolled together with a selection of 2D descriptors as independent variables in QSAR analysis.
Genetic function algorithm combined with MLR analyses was used to select different combinations of pharmacophores and 2D molecular descriptors that are capable of explaining bioactivity variation among collected inhibitors.
However, all attempts to achieve statistically successful QSAR models failed, prompting the use of ligand efficiency [LE = −log(IC50)/heavy atom count] as an alternative response variable instead of −log(IC50).61, 62, 63, 64 The best QSAR models are summarized in Equations (3) and (4). Figure 2A, B show the corresponding scatter plots of experimental versus estimated bioactivities for training and testing inhibitors.
Figure 2.
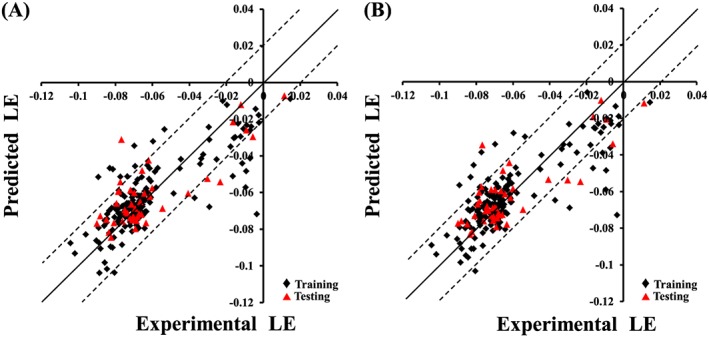
Experimental versus predicted bioactivities for the training and testing compounds. Predicted bioactivities calculated using the best QSAR models: (A) Equation (3) and (B) Equation (4). The solid line is the regression line for the fitted and predicted bioactivities of training and test compounds, respectively, whereas the dotted lines indicate arbitrary error margins.
| (3) |
| (4) |
where n is the number of training compounds used to generate this equation, F is Fisher statistic, r 2 LOO is the leave‐one‐out cross‐validation correlation coefficient, and r 2 PRESS is the predictive r 2 determined for 44 randomly selected test compounds. AromaticBonds is the number of aromatic bonds in the molecule, Dipole, DipoleX, and DipoleY are dipole moment descriptors that indicate the strength and orientation behavior of a molecule in an electrostatic field, LUMO is the energy of the lowest unoccupied molecular orbital,65 Hypo(L‐T5‐2), Hypo(K‐T5‐3), and Hypo(N‐T1‐1) represent the fit values of the training compounds against corresponding pharmacophores (see Table S3). Figure 3 shows the 3 pharmacophores and how they fit the most potent training compound (1, IC50 = 0.33μM36).
Figure 3.
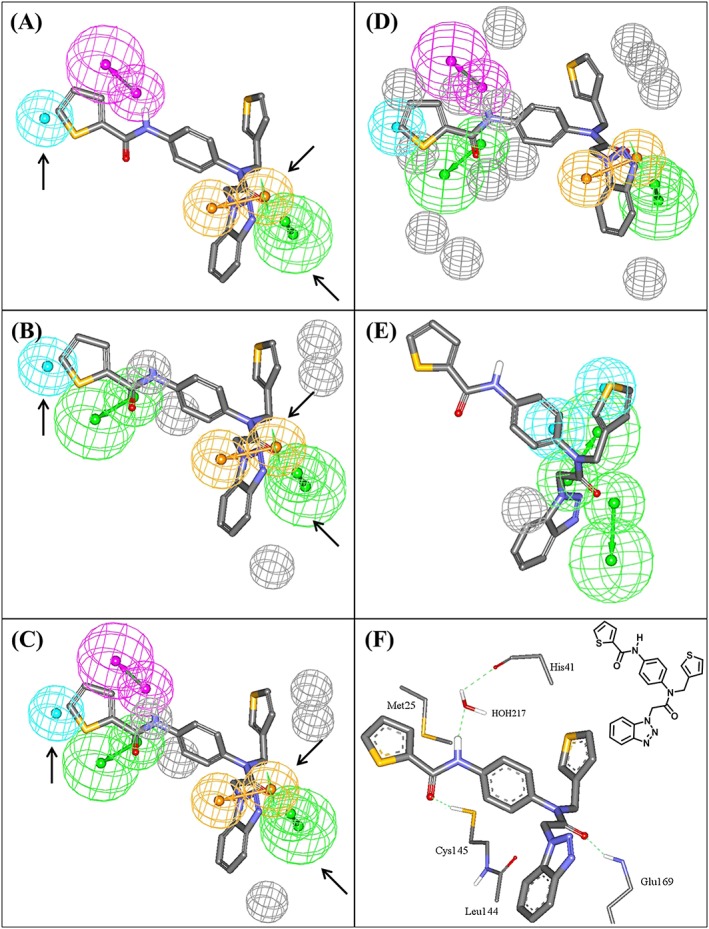
Pharmacophoric features of the QSAR‐guided pharmacophores and the corresponding merged model: green‐vectored spheres: HBA; blue spheres: Hbic; purple‐vectored spheres: HBD; and orange‐vectored spheres: RingArom, (A) Hypo(N‐T1‐1), (B) Hypo(K‐T5‐3), (C) Merged‐Hypo(K‐T5‐3/N‐T1‐1), (D) Refined Merged‐Hypo(K‐T5‐3/N‐T1‐1), and (E) Hypo(L‐T5‐2) fitted against co‐crystallized ligand within HKU4‐CoV 3CLpro (compound 1, IC50 = 0.33μM, PDB code 4YOI, 1.8 Ǻ). (F) Ligand co‐crystallized within HKU4‐CoV 3CLpro and the chemical structure of the co‐crystallized ligand. Arrows point to closely positioned common features in Hypo(N‐T1‐1) and Hypo(K‐T5‐3) allowing for merging. The 3D coordinates of these pharmacophores are shown in Table S6. HBA, hydrogen bond acceptor; HBD, hydrogen bond donor
The appearance of AromaticBonds descriptor combined with positive slopes in both QSAR equations indicates that HKU4‐CoV 3CLpro inhibitory activity is directly proportional to the number of aromatic rings in the inhibitor structure. This is to be expected, as the binding pocket is rich in aromatic amino acids (His41, His166, His175, Tyr54, and Phe143). Apparently, ligands' aromatic rings stack against these aromatic residues in the binding pocket is likely to lead to a high binding affinity. However, the emergence of several dipole moment descriptors (Dipole, DipoleX, and DipoleY) combined with positive and negative regression coefficients in Equations (3) and (4) is suggestive of an obscure role of ligands' dipole moments in binding within the enzyme‐binding pocket.
The emergence of LUMO in Equations (3) and (4) combined with negative slopes suggests that ligand/HKU4‐CoV 3CLpro affinity favors electrophilic ligands, perhaps due to a π‐stacking against certain electron‐rich aromatic centers in the binding pocket (eg, the aromatic rings of Tyr54 and Phe143).
The emergence of 3 pharmacophores—Hypo(K‐T5–3), Hypo(N‐T1–1), and Hypo(L‐T5–2)—in Equations (3) and (4) suggests possible multiple or complementary binding modes exhibited by ligands within the binding pocket. Receiver operating characteristic analysis of the 3 pharmacophores shows that Hypo(K‐T5‐3) and Hypo(N‐T1‐1) are significantly superior to Hypo(L‐T5‐2) (Table 1). Furthermore, MCC of the 3 pharmacophores reflects the very weak classification abilities of Hypo(L‐T5‐2) (Table 1).
Table 1.
ROC and MCC performances of QSAR‐guided pharmacophores
| Pharmacophore Model | ROC‐AUC | ACC | SPC | TPR | MCC |
|---|---|---|---|---|---|
| Hypo(L‐T5‐2) | 0.78 | 0.09 | 0.05 | 1.00 | 0.048 |
| Hypo(K‐T5‐3) | 0.78 | 0.52 | 0.50 | 0.74 | 0.099 |
| Hypo(N‐T1‐1) | 0.81 | 0.63 | 0.63 | 0.63 | 0.109 |
| Hypo(K‐T5‐3/N‐T1‐1) | 0.93 | 0.88 | 0.90 | 0.52 | 0.263 |
| Refined Hypo(K‐T5‐3/N‐T1‐1) | 0.94 | 0.89 | 0.91 | 0.48 | 0.262 |
Abbreviations: ACC, overall accuracy; AUC, area under the curve; MCC, Matthews correlation coefficient; ROC, receiver operating characteristic; SPC, overall specificity; TPR, overall true positive rate.
The very poor classification power of Hypo(L‐T5‐2) prompted us to exclude it from subsequent modeling efforts. However, Hypo(K‐T5‐3) and Hypo(N‐T1‐1) (Figure 3A,B) have 3 pharmacophoric features in common: hydrophobic (Hbic), ring aromatic (RingArom), and hydrogen bond acceptor (HBA) features. The close resemblance between these 2 pharmacophores combined with their equivalent contributions to bioactivity (as indicated by their slopes in QSAR Equations (3) and (4)) suggest that they might represent a common binding mode assumed by ligands within the HKU4‐CoV 3CLpro binding pocket. Therefore, these 2 pharmacophores were merged in a single binding model (Hypo(K‐T5‐3/N‐T1‐1) (Figure 3).
Interestingly, Hypo(K‐T5‐3/N‐T1‐1) showed noticeable improvement in distinguishing actives from decoys as indicated by the ROC analysis and MCC values (Table 1). Merging pharmacophores that share common features has been reported to improve the performance of pharmacophores in capturing active molecules.66 Additionally, Hypo(K‐T5‐3/N‐T1‐1) was further modified by adding exclusion spheres (Section S8 and Table S6) to further enhance its ROC profile (Table 1). Exclusion volumes resemble inaccessible regions within the binding site. Figure 3D shows the sterically refined version of Hypo(K‐T5‐3/N‐T1‐1) complemented with eight exclusion volumes.
Moreover, Hypo(K‐T5‐3/N‐T1‐1) maps the most potent ligand 1 (IC50 = 0.33μM) in a way that closely resembles the interactions observed in the co‐crystallized structure of the same compound with HKU4‐CoV 3CLpro (4YOI) (Figure 3). The close proximity between the ligand's thiophenoyl moiety and the sulfide of Met25 (Figure 3F) suggests the presence of a mutual hydrophobic interaction, which correlates with mapping the same ring against a Hbic feature in Hypo(K‐T5‐3/N‐T1‐1) (Figure 3C). Similarly, mapping the carbonyl of the same thiophenoyl moiety against HBA feature in Hypo(K‐T5‐3/N‐T1‐1) (Figure 3C) agrees with the hydrogen bonding interaction connecting this carbonyl to the thiol of Cys145 (Figure 3F). Likewise, the hydrogen bonding interaction connecting the amidic NH of the ligand to the peptidic carbonyl of His41 via bridging water molecule agrees with mapping the same NH against hydrogen bond donor (HBD) features in Hypo(K‐T5‐3/N‐T1‐1) (Figure 3F). Mapping the ligand's benzotriazole ring against RingArom feature in Hypo(K‐T5‐3/N‐T1‐1) (Figure 3C) correlates with stacking this ring system against the peptide amide connecting Cys145 and Leu144 in the binding pocket (Figure 3F). Finally, the hydrogen bonding interaction anchoring the ligand's tertiary amide carbonyl to the peptide NH of Glu169 corresponds to fitting the same carbonyl against HBA feature in Hypo(K‐T5‐3/N‐T1‐1) (Figure 3C). These findings showed that Hypo(K‐T5‐3/N‐T1‐1) represents a valid binding mode exhibited by the ligands within the binding pocket of HKU4‐CoV 3CLpro. These interactions, highlighted by the pharmacophoric features within this model, are very likely to be critical for ligand‐binding affinity.
3.2. Structure‐based approach: dbCICA modeling
Structure‐based pharmacophore models for HKU4‐CoV 3CLpro were obtained by using dbCICA. In this approach, a subset of inhibitors (1‐27, Table S1) were docked into the HKU4‐CoV 3CLpro binding pocket using LibDock,47 and CDOCKER48 (Section 2.2). The highest‐ranking conformers/poses based on each scoring function were aligned together to construct a corresponding dbCICA model. Genetic algorithm was then used to search for the best combination of ligand‐receptor intermolecular contacts capable of explaining bioactivity variation across the training compounds. Table 2 shows the contacts distance thresholds, number of positive and negative contacts, and statistical criteria of the best dbCICA models. Table 3 shows the critical binding site contact atoms proposed by optimal dbCICA models. The highest‐ranking dbCICA models exhibited excellent statistical criteria and were anticipated to act as good templates for building corresponding pharmacophore models (Table 2). Figure 4 shows how dbCICA model SB‐1 (Tables 2 and 3) was converted into its corresponding pharmacophore model Hypo(SB‐1) as an example. The emergence of significant positive contact atoms at Pro45 and HOH225 (Figure 4A) combined with the consensus among potent docked ligands to position hydrophobic alkyl, cycloalkyl, or aromatic rings nearby (within 3.5 Å from Pro45 and HOH225, Figure 4C) prompted us to place Hbic feature onto these ligand groups (Figure 4D). It is likely that hydrophobic fragments of the ligands interact with the side chain of Ala46.
Table 2.
The highest ranking dbCICA models and their corresponding parameters and statistical criteriaa
| Model | Docking Engine | Scoring Function | Positive Contactsb | Negative Contactsc | r 2 27 d | r 2 LOO e | r 2 5‐fold f | F statistic |
|---|---|---|---|---|---|---|---|---|
| SB‐1 | CDOCKER | PMF | 9 | 10 | 0.92 | 0.91 | 0.91 | 291.39 |
| SB‐2 | CDOCKER | PMF | 5 | 5 | 0.88 | 0.86 | 0.83 | 180.4 |
| SB‐3 | LibDock | PLP2 | 5 | 10 | 0.90 | 0.88 | 0.87 | 221.48 |
| SB‐4 | LibDock | PLP2 | 8 | 5 | 0.91 | 0.89 | 0.89 | 239.61 |
| SB‐5 | LibDock | Lig2 | 5 | 5 | 0.86 | 0.84 | 0.84 | 147.68 |
Abbreviation: dbCICA, docking‐based comparative intermolecular contacts analysis.
All successful models listed herein were generated by docking the ligands into the binding site in the presence of crystalographically explicit water molecules and at ligand/binding site contact distance thresholds of 3.5 Ǻ (Section S12).
Optimal number of combined (ie, summed) bioactivity‐enhancing ligand/binding site contacts.
Optimal number of bioactivity‐disfavoring ligand/binding site contacts.
Non–cross‐validated correlation coefficient for 27 training compounds.
Cross‐validation correlation coefficients determined by the leave‐one‐out technique.
Cross‐validation correlation coefficients determined by the leave‐20%‐out technique repeated 5 times.
Table 3.
Critical binding site contact atoms proposed by optimal dbCICA models
| dbCICA Modela | Favored Contact Atoms (Positive Contacts)b |
Disfavored Contact Atoms (Negative Contacts)e |
|
|---|---|---|---|
| Amino acids and atom identitiesc | Weightsd | ||
| ASP190:CB | 2 | CYS145:CB; CYS145:HB2; GLN167:O; GLN192:HA; GLN192:HG1; LEU144:C; LEU144:HD22; MET168:SD; HOH216:H1; HOH234:H1 | |
| SB‐1 | |||
| CYS145:HB1 | 1 | ||
| GLN192:HE21 | 2 | ||
| GLU169:HN | 2 | ||
| HIS166:NE2 | 3 | ||
| HIS41:CB | 1 | ||
| PRO45:CA | 1 | ||
| HOH225:H1 | 3 | ||
| HOH241:O | 3 | ||
| SB‐2 | PRO45:CA | 1 | LEU144:C; LYS191:HN; MET168:SD; MET25:SD; CYS145:HG |
| ASP190:O | 3 | ||
| GLU169:OE1 | 3 | ||
| HIS166:NE2 | 1 | ||
| PHE143:C | 2 | ||
| SB‐3 | ASP190:C | 3 | CYS44:HB1; CYS44:HB2; GLN195:HB1; HIS41:O; LYS191:C; LYS191:HN; MET25:CG; MET25:N; PRO52:HD1; HOH116:H1 |
| HIS194:HN | 1 | ||
| MET168:HB2 | 3 | ||
| PHE143:CA | 3 | ||
| SER24:HB2 | 2 | ||
| SB‐4 | ASP190:C | 3 | GLN192:CD; GLU169:O; LEU49:CG; LEU49:HB2; MET168:HE2 |
| HIS41:HD2 | 3 | ||
| LEU144:Ha | 2 | ||
| MET168:HB2 | 3 | ||
| MET168:SD | 2 | ||
| PHE143:C | 1 | ||
| THR193:N | 3 | ||
| HOH217:O | 2 | ||
| SB‐5 | ALA46:CB | 2 | ASP190:CB; CYS44:HB2; GLN167:O; HIS175:CD2; THR193:C |
| ASP190:C | 1 | ||
| PHE143:O | 2 | ||
| PRO52:HG2 | 3 | ||
| HOH401:H1 | 3 | ||
Abbreviation: dbCICA, docking‐based comparative intermolecular contacts analysis.
As in Table 2.
Bioactivity‐proportional ligand/binding site contacts.
Binding site amino acids and their atomic contacts. Atom codes are as provided by the PDB file except for hydrogen atoms, which were coded by Discovery Studio.
Degree of significance (weight) of corresponding contact atom. It points to number of times it emerged in the final dbCICA model (see Section S12).
Bioactivity‐disfavoring ligand/binding site contacts.
Figure 4.
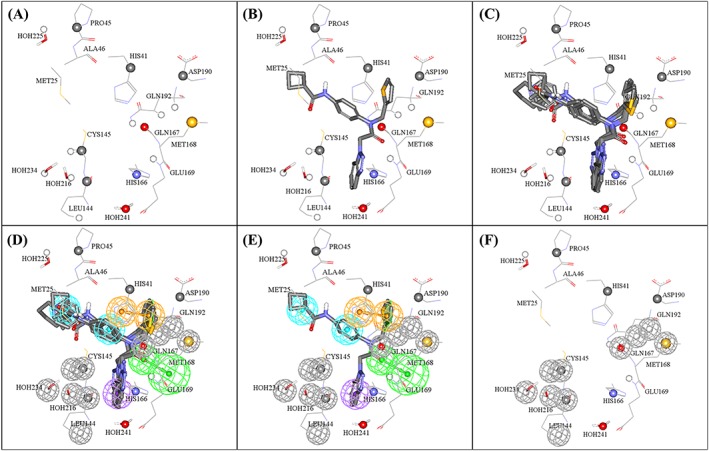
Steps used in the manual generation of binding model Hypo(SB‐1) as guided by dbCICA model SB‐1 (Tables 2 and 3): (A) The binding site moieties selected by dbCICA model SB‐1 with significant contact atoms shown as spheres. (B) The docked pose of the potent training compound 3 (IC50 = 1.2μM) within the binding pocket. (C) The docked poses of the potent compounds 3, 4, 5, 6, and 8. (D) Manually placed pharmacophoric features onto chemical moieties common among docked potent compounds 3, 4, 5, 6, and 8. (E) The docked pose of 3 and how it relates to the proposed pharmacophoric features. (F) Exclusion spheres fitted against binding site atoms showing negative correlations with bioactivity (dbCICA model SB‐1). Green vectored spheres: HBA, blue spheres: Hbic, violet spheres: HbicArom, and orange‐vectored spheres: RingArom. Exclusion spheres are shown in gray. dbCICA, docking‐based comparative intermolecular contacts analysis; HBA, hydrogen bond acceptor
Similarly, the emergence of the amidic NH of Gln192 as significant positive contact in SB‐1 combined with agreement among docked potent training compounds on placing their central benzene rings near to this contact suggested placing an Hbic feature onto these benzene ligand fragments. Clearly, these rings are involved in hydrophobic interaction with the nearby thiol of Cys145 instead of π‐stacking (as the nearest aromatic amino acid residue is His41 at about 4.5 Ǻ away). This explains our decision to place Hbic feature onto this region of the ligands (ie, rather than RingArom feature).
Likewise, the appearance of His166 and HOH241 as positive contact points combined with agreement among potent hits to position their benzotriazoles close by suggested placing a hydrophobic aromatic (HbicArom) feature onto these benzotriazole moieties (Figure 4E). The reason for adding an HbicArom feature onto these rings instead of a vectored RingArom feature is because the benzotriazoles, although docked near to the imidazole of His166, it did not exhibit typical π‐stacking alignment with this residue. In contrast, the appearance of positive contacts at His41 and ASP190 combined with a consensus among docked potent inhibitors to project their thiophene rings close to the nearby imidazole of His41 suggests a mutual π‐stacking interaction involving the electron‐rich ligands' thiophenes and electron‐deficient His41 imidazole. This observation supported placing a RingArom feature onto the thiophene rings.
The emergence of positive contact on the amidic NH of GLN169 and agreement of docked compounds on placing their central amide oxygen close to the NH of GLN169 indicated the presence of hydrogen bonding interaction and suggested placing HBA feature onto the ligand amidic carbonyl groups (Figure 4E). This interaction is very likely to involve hydrogen bonding with the peptide amidic NH of GLU169.
Finally, all contacts points of negative correlation with bioactivity were assumed to represent areas of steric clashes with the bound ligand. Therefore, such contacts were used to define exclusion volumes within the vicinity of the binding pocket, as shown in Figure 4E.
The same strategy was used to translate all other optimal dbCICA models in Tables 2 and 3 into their corresponding pharmacophore models (Figure 5). The X, Y, and Z coordinates of the resulting pharmacophores are shown in Table S7. Subsequent validation using ROC analysis (Table 4).illustrated the excellent classification powers of these pharmacophores in distinguishing actives from decoys. Matthews correlation coefficient values indicate that the structure‐based dbCICA models are superior in their classification ability to the QSAR‐guided pharmacophores.
Figure 5.
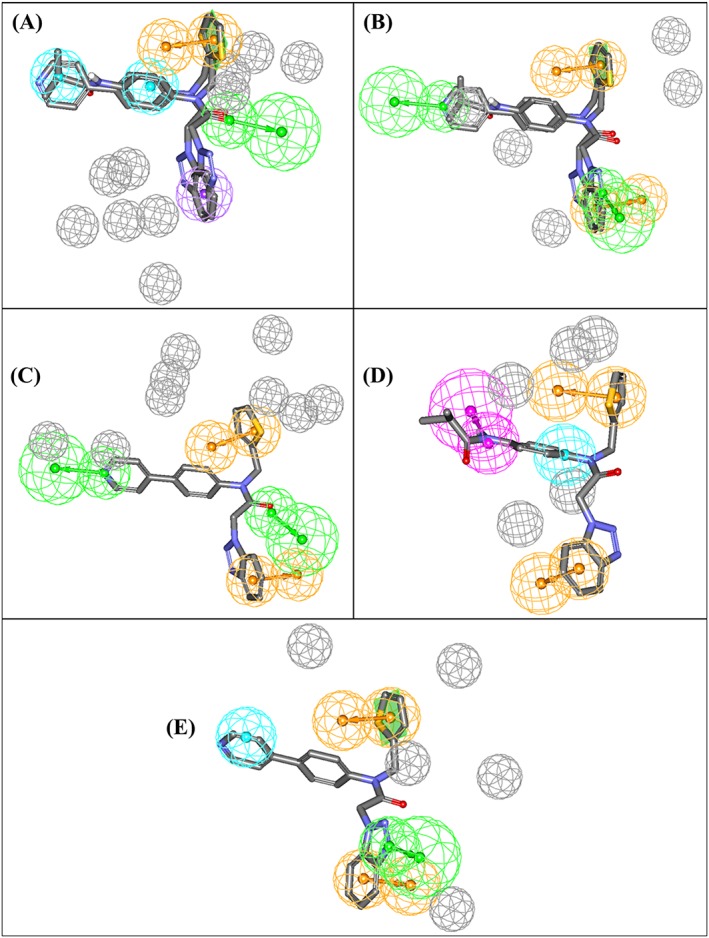
dbCICA pharmacophores derived from successful dbCICA models in Tables 2 and 3. (A) Hypo(SB‐1) mapped against training compounds 5 and 6 (IC50 = 1.5μM and 1.6μM, respectively, (Table S1), (B) Hypo(SB‐2) mapped against 5 and 6, (C) Hypo(SB‐3) fitted against 5, (D) Hypo(SB‐4) mapped against 6, and (E) Hypo(SB‐5) mapped against 5. Green vectored spheres: HBA, purple‐vectored spheres: HBD, blue spheres: Hbic, violet spheres HbicArom, and orange‐vectored spheres: RingArom. Exclusion spheres are shown in gray. dbCICA, docking‐based comparative intermolecular contacts analysis; HBA, hydrogen bond acceptor; HBD, hydrogen bond donor
Table 4.
ROC and MCC performances of the dbCICA‐based pharmacophores
| Pharmacophore Model | ROC‐AUC | ACC | SPC | TPR | MCC |
|---|---|---|---|---|---|
| Hypo(SB‐1) | 0.946 | 0.495 | 0.726 | 0.815 | 0.241 |
| Hypo(SB‐2) | 0.976 | 0.632 | 0.944 | 0.666 | 0.713 |
| Hypo(SB‐3) | 0.932 | 0.573 | 0.854 | 0.666 | 0.283 |
| Hypo(SB‐4) | 0.971 | 0.615 | 0.918 | 0.666 | 0.384 |
| Hypo(SB‐5) | 0.897 | 0.425 | 0.611 | 0.963 | 0.254 |
Abbreviations: ACC, overall accuracy; AUC, area under the curve; MCC, Matthews correlation coefficient; ROC, receiver operating characteristic; SPC, overall specificity; TPR, overall true positive rate.
3.3. In silico screening
The QSAR‐guided, sterically refined, merged pharmacophore Hypo(K‐T5‐3/N‐T1‐1) and 5 dbCICA‐based pharmacophores (Hypo(SB‐1) to Hypo(SB‐5)) were used as 3D search queries to screen the NCI virtual database for small molecule inhibitors of 3CLpro. Captured hits were filtered by the Lipinski criteria57 and SMARTS filter as described58 in Section 2.4.
The QSAR‐guided hits were fitted against component pharmacophores (ie, Hypo(K‐T5‐3), Hypo(N‐T1‐1), and Hypo(L‐T5‐2)) and their fit values were substituted in MLR‐QSAR Equations (3) and (4) to predict their bioactivities. The top 39 compounds (of the highest‐ranking hits; prioritized using the voting system described in Section 2.4) that were available in the NCI Open Chemicals Repository were acquired for in vitro testing.
On the other hand, filtered dbCICA‐derived hits were docked into HKU4‐3CLpro protein using the same docking conditions of each successful dbCICA model (SB1, SB‐2, SB‐3, SB‐4, and SB‐5, Tables 2 and 3) to predict their corresponding inhibitory IC50 values (Section 2.4). The hits were ranked and prioritized using the voting system described in Section 2.4, and the top 39 compounds were acquired for in vitro testing. Thus, the total of 78 compounds from the NCI Open Chemicals Repository were acquired for testing.
3.4. In vitro validation
A total of 78 NCI (Figure S1), 39 QSAR‐guided derived hits and 39 dbCICA derived hits, compounds were acquired and screened in vitro to determine their inhibitory activity against HKU4‐CoV‐3CLpro and MERS‐CoV‐3CLpro at 40μM hit concentration. The 3CLpro enzyme assay used in this study was carefully designed to avoid misleading false positives and to prevent wasted follow‐up on promiscuous compounds (by adding albumin, DTT, and triton‐100 to the reaction mixture). Tables S8 and S9 show the %inhibition against 3CLpro of the hits captured by the QSAR‐guided and the dbCICA derived pharmacophores, respectively.
Only a single compound (NCI code 134140) of the 39 tested hits, captured by the QSAR‐guided pharmacophores, showed inhibitory activity ≥50% against both HKU4‐CoV 3CLpro and MERS‐CoV 3CLpro. However, this compound has a molecular fragment known to cause pan assay interference (PAINS‐like; Baell67) and therefore was not considered as a hit in further characterizations. Three compounds of the same ligand‐based hits (NCI codes: 12156, 22906, and 28562; Table S8) showed unexpectedly high negative values of their activity against MERS‐CoV 3CLpro (−633.2%, −203.4%, and −662.6% at 40μM; Table S8). Several controls were performed in which either the substrate or the enzyme or both were omitted from the assay (data not shown). None of these hits showed evidence of fluorescence interference. It might be possible that these compounds act as activators of the enzyme. However, further evidence is still needed to support this hypothesis. It was previously observed that designed reversible peptidomimetic inhibitors acted as activators at a low compound concentration as a result of induced dimerization.30 Therefore, these 3 hits will not be discussed in the current publication.
Only a single compound (222; NCI code 120178) of the dbCICA derived hits showed inhibitory activity ≥50% (at 40μM) against MERS‐CoV 3CLpro (51.9%; Table S9 and Figures 5). This activity is comparable to that of the positive control against the MERS‐CoV enzyme (compound 1; 63.8% Figure 6). However, the compound 222 failed to show significant inhibitory activity against HKU4‐CoV 3CLpro. The purity of 222 was confirmed using nuclear magnetic resonance and mass spectroscopy (Figure S2). Another compound, 223, was found to exhibit a bit lower activity against the MERS‐CoV enzyme (28% inhibition at 40μM). The purity of 223 was confirmed using nuclear magnetic resonance and mass spectroscopy (Figure S3). Compounds 222 and 223 (NCI code 128947) share a common phenylsulfonamide fragment, which is amenable to chemical modifications. Both compounds were captured by Hypo(SB‐3) and Hypo(SB‐5) pharmacophores (Table 4). Figure 7 shows how 222 hit maps the dbCICA pharmacophore models.
Figure 6.
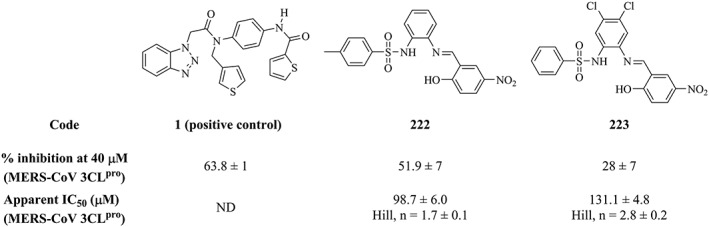
The chemical structures, inhibitory activities, and apparent IC50 values of the positive control 1, and the 2 tested hits captured by the dbCICA‐derived pharmacophores (222 and 223). dbCICA, docking‐based comparative intermolecular contacts analysis
Figure 7.
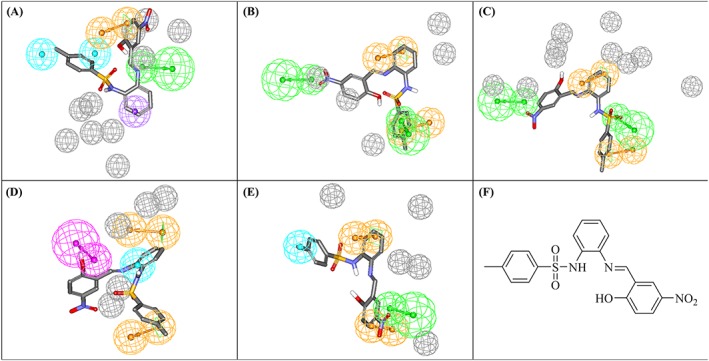
dbCICA‐based pharmacophores derived from successful dbCICA models (Tables 2 and 3) mapped against hit compound 222. (A) Hypo(SB‐1), (B) Hypo(SB‐2), (C) Hypo(SB‐3), (D) Hypo(SB‐4), and (E) Hypo(SB‐5). Green‐vectored spheres: HBA, purple‐vectored spheres: HBD, blue spheres: Hbic, violet spheres: HbicArom, violet spheres: HbicArom, and orange‐vectored spheres: RingArom. Exclusion spheres are shown in gray. dbCICA, docking‐based comparative intermolecular contacts analysis; HBA, hydrogen bond acceptor; HBD, hydrogen bond donor
Further controls were conducted (same as described above) to rule out fluorescence interference. None of these hits showed significant flourescence in the assay buffer (no enzyme and no substrate), in the presence of the enzyme (no substrates) or in the presence of the substrate (no enzyme) (data not shown). However, at concentrations >100μM, 222 showed approximately 10% attenuation of the cleaved substrate fluorescence (data not shown). Both 222 and 223 showed moderate apparent IC50 values against the MERS‐CoV 3CLpro of 98.7μM and 131.1μM, respectively (Figure 5). The shape of the activity curve of compound 222, where a linear inhibition of fluorescence up to a maximum inhibition, indicates the influence of the inner filter effect (Figure S4).68, 69 Inner filter effect is one of the major challenges usually encountered in FRET‐based enzyme assays.69
The low hit rate observed in this study can be justified by the limited availability of many of the top‐ranked hits in the NCI Open Chemicals Repository and hence limited number of tested hits (only 78 hits).
There was also a limitation in the availability of published potent MERS‐CoV 3CLpro inhibitors to be used as training set in modeling enzyme inhibition. Obviously, the prediction ability of the computational models is very much dependent on the compounds used in modeling. The training compounds used in the current study are all peptide‐like compounds, and only 14% of them exhibited IC50 values <10μM. The effect of the starting training set was prominent on the ligand‐based modeling (QSAR‐guided model), hence, explaining the poor quality of these models as indicated by their low MCC values. Clearly, the quality of the training set is a pivotal factor in determining the predictive validity of the obtained pharmacophores. It is also worth noting that the active site‐directed design of nonpeptidomimetic small molecule inhibitors of proteases is often challenging because of the unique chemistry of the peptide‐bond cleavage transition state and because some proteases cleave their substrates through an induced fit mechanism.70
4. CONCLUSIONS
Recently, special attention has been paid to bat coronaviruses. Two deadly emerging coronaviruses, which have caused unexpected human disease outbreaks, SARS‐CoV and MERS‐CoV, are suggested to be originated from bats. MERS‐CoV is now considered a threat to global public health. While its human‐to‐human transmission is so far limited, serious concerns over its pandemic potential have been raised. Therefore, there is an urgent need for the development of effective and safe anti–MERS‐CoV treatment.
In this study, we have explored the pharmacophoric space of the recently identified peptidomemic HKU4‐3CLpro inhibitors36 by 2 independent approaches; the QSAR‐guided pharmacophore modeling and the dbCICA‐based pharmacophore construction. Both approaches have successfully resulted in the identification of novel potent inhibitors on a wide variety of targets. QSAR‐guided pharmacophore modeling is a ligand‐based method, in which pharmacophores are derived by extensive exploration of the 3D space of a carefully selected variable small subset of the inhibitors. These pharmacophores are then allowed to compete within the context of classical QSAR using GFA and MLR to identify combinations that result in finest estimation of the bioactivities. dbCICA modeling, on the other hand, is a structure‐based pharmacophore construction method, which relies on the accurate selection of the most successful docking/scoring conditions combinations. The success criterion is the ability of the docking run to align potent ligands in a way that would allow them to form contacts unattainable by low‐potency ligands. dbCICA can be considered a 3D QSAR that correlates ligands' affinities to their contacts with certain binding site spots by using GFA and MLR. Successful dbCICA models can then be translated into binding models (pharmacophores) to be used as in silico screening tools of virtual databases.
We have applied these robust computational methods to model HKU4‐CoV 3CLpro inhibitors as a tool to identify inhibitors of MERS‐CoV 3CLpro. These models assisted the identification of 2 hit compounds with moderate apparent activity against MERS‐CoV 3CLpro. The identified inhibitors share a novel nonpeptidomimetic scaffold that is amenable to medicinal chemistry optimization efforts. Despite the fair inhibitory activity of this scaffold, it represents a potential starting point in the discovery of novel MERS‐CoV antivirals. There are several successful examples in the history of drug discovery in which the starting hits showed low‐to‐moderate enzyme inhibition. For example, the millimolar inhibitor Neu5Ac was the starting point in the development of zanamivir, the first influenza neuraminidase inhibitor introduced to the market.71
Most importantly, the established ligand‐based and structure‐based pharmacophore models aid as tools for advancing our understanding of small molecule recognition of the coronavirus 3CLpro enzymes. The pharmacophores obtained by modeling the HKU4‐CoV 3CLpro inhibitors revealed structural features needed for potent 3CLpro enzyme inhibitors design. While, dbCICA models (structure‐based models) highlighted potential hot‐spot regions in the 3CLpro pocket that could be targeted using small nonpeptidomimetic molecules. Such knowledge is valuable for the successful development of 3CLpro inhibitors as anti‐MERS drugs.
FUNDING
This publication is based on work supported by Awards No. 61557 and 61556 of the US Civilian Research & Development Foundation (CRDF Global) and by the National Science Foundation under Cooperative Agreement No. OISE‐9531011. Task Order No. OISE‐9531011‐NIH.
Any opinions, findings and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of CRDF Global.
Supporting information
Table S1. The structures of HKU4‐CoV 3CLpro inhibitors utilized in modeling. 25
Table S2: Training subsets (extracted from Table S1) employed in exploring the pharmacophoric space of HKU4‐CoV 3CLpro inhibitors. 48
Table S3: Training subsets and HYPOGEN run parameters employed for exploring HKU4‐CoV 3CLpro pharmacophoric space. 49
Table S4: The performance of the best representatives of clustered pharmacophore hypotheses generated for HKU4‐CoV 3CLpro. 54
Table S5: Pharmacophoric features and corresponding weights, tolerances and 3D coordinates of Hypo(L‐T5–2), Hypo(K‐T5–3),and Hypo(N‐T1–1) selected by QSAR‐guided method. 57
Table S6: Training subset used for adding excluded spheres for Hypo(K‐T5–3/N‐T1–1) using HIPHOP‐REFINE module of CATALYST. 58
Table S7: HKU4‐CoV 3CLpro based pharmacophore models selected by best dbCICA models. 59
Table S8: Captured hits by dbCICA derived pharmacophores. 60
Table S9: Captured hits by QSAR‐guided pharmacophores. 61
Figure S1: NMR and MS analysis of compound 222. 62
Figure S2: NMR and MS analysis of compound 223. 63
Figure S3: NMR and MS analysis of compound 224.
Figure S4: The dose–response curves of compounds 222 and 223 against MERS‐CoV 3CLpro
ACKNOWLEDGEMENT
The authors thank the CRDF Global (2014 US‐Qatar‐MENA Research Initiative) for the financial support of this project, the Deanship of Scientific Research and Hamdi‐Mango Center for Scientific Research at the University of Jordan for their generous funds, and the NCI Open Chemicals Repository for providing free samples of the virtual hits.
Abuhammad A, Al‐Aqtash RA, Anson BJ, Mesecar AD, Taha MO. Computational modeling of the bat HKU4 coronavirus 3CLpro inhibitors as a tool for the development of antivirals against the emerging Middle East respiratory syndrome (MERS) coronavirus. J Mol Recognit. 2017;30:e2644 10.1002/jmr.2644
Contributor Information
Areej Abuhammad, Email: a.abuhammad@ju.edu.jo.
Mutasem O. Taha, Email: mutasem@ju.edu.jo.
REFERENCES
- 1. Coleman CM, Frieman MB. Coronaviruses: important emerging human pathogens. J Virol. 2014;88(10):5209‐5212. 10.1128/JVI.03488-13. PubMed PMID: 24600003; PubMed Central PMCID: PMCPMC4019136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814‐1820. 10.1056/NEJMoa1211721. PubMed PMID: 23075143 [DOI] [PubMed] [Google Scholar]
- 3. Dudley JP. Middle East respiratory syndrome coronavirus. Global Virus Network [Internet]. 2014.
- 4. WHO . Middle East respiratory syndrome coronavirus (MERS‐CoV) WHO MERS‐CoV global summary and risk assessment 2016.
- 5. Collier L, Oxford J, Kellam P. Human Virology. Fifth ed. Oxford: Oxford University Press; 2016. [Google Scholar]
- 6. Wang Q, Qi J, Yuan Y, et al. Bat origins of MERS‐CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe. 2014;16(3):328‐337. 10.1016/j.chom.2014.08.009. PubMed PMID: 25211075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang Y, Du L, Liu C, et al. Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat‐to‐human transmission of MERS coronavirus. Proc Natl Acad Sci U S A. 2014;111(34):12516‐12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu G, Wang Q, Gao GF. Bat‐to‐human: spike features determining ‘host jump’ of coronaviruses SARS‐CoV, MERS‐CoV, and beyond. Trends Microbiol. 2015;23(8):468‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reusken CB, Ababneh M, Raj VS, et al. Middle East respiratory syndrome coronavirus (MERS‐CoV) serology in major livestock species in an affected region in Jordan, June to September 2013. Euro Surveill. 2013;18(50):20662 PubMed PMID: 24342516 [DOI] [PubMed] [Google Scholar]
- 10. Nowotny N, Kolodziejek J. Middle East respiratory syndrome coronavirus (MERS‐CoV) in dromedary camels, Oman, 2013. Euro Surveill. 2014;19(16):20781 PubMed PMID: 24786259 [DOI] [PubMed] [Google Scholar]
- 11. Han H‐J, H‐l W, Zhou C‐M, et al. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 2015;205:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Memish ZA, Cotten M, Meyer B, et al. Human infection with MERS coronavirus after exposure to infected camels, Saudi Arabia, 2013. Emerg Infect Dis. 2014;20(6):1012‐1015. 10.3201/eid2006.140402. PubMed PMID: 24857749; PubMed Central PMCID: PMCPMC4036761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Azhar EI, El‐Kafrawy SA, Farraj SA, et al. Evidence for camel‐to‐human transmission of MERS coronavirus. N Engl J Med. 2014;370(26):2499‐2505. 10.1056/NEJMoa1401505. PubMed PMID: 24896817 [DOI] [PubMed] [Google Scholar]
- 14. Zhang N, Jiang S, Du L. Current advancements and potential strategies in the development of MERS‐CoV vaccines. Expert Rev Vaccines. \2014;13(6):761‐774. 10.1586/14760584.2014.912134. PubMed PMID: 24766432; PubMed Central PMCID: PMCPMC4241375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao G, Du L, Ma C, et al. A safe and convenient pseudovirus‐based inhibition assay to detect neutralizing antibodies and screen for viral entry inhibitors against the novel human coronavirus MERS‐CoV. Virol J. 2013;10(1):266 10.1186/1743-422X-10-266. PubMed PMID: 23978242; PubMed Central PMCID: PMCPMC3765664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Du L, Kou Z, Ma C, et al. A truncated receptor‐binding domain of MERS‐CoV spike protein potently inhibits MERS‐CoV infection and induces strong neutralizing antibody responses: implication for developing therapeutics and vaccines. PLoS One. 2013;8(12):e81587. 10.1371/journal.pone.0081587): PubMed PMID: 24324708; PubMed Central PMCID: PMCPMC3852489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dyall J, Coleman CM, Hart BJ, et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory coronavirus infection. Antimicrob Agents Chemother. 2014; 10.1128/aac.03036-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Law GL, Tisoncik‐Go J, Korth MJ, Katze MG. Drug repurposing: A better approach for infectious disease drug discovery? Curr Opin Immunol. 2013;25(5):588‐592. 10.1016/j.coi.2013.08.004. PubMed PMID: 24011665; PubMed Central PMCID: PMCPMC4015799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kilianski A, Baker SC. Cell‐based antiviral screening against coronaviruses: developing virus‐specific and broad‐spectrum inhibitors. Antiviral Res. 2014;101:105‐112. 10.1016/j.antiviral.2013.11.004. PubMed PMID: 24269477; PubMed Central PMCID: PMCPMC3931262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dyall J, Postnikova E, Zhou H, et al. Interferon‐β and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell‐based assays. J Gen Virol. 2014;95(Pt 3):571‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun Y, Wang Z, Tao J, et al. Yeast‐based assays for the high‐throughput screening of inhibitors of coronavirus RNA cap guanine‐N7‐methyltransferase. Antiviral Res. 2014;104:156‐164. 10.1016/j.antiviral.2014.02.002. PubMed PMID: 24530452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Wilde AH, Jochmans D, Posthuma CC, et al. Screening of an FDA‐approved compound library identifies four small‐molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother. 2014;58(8):4875‐4884. 10.1128/AAC.03011-14. PubMed PMID: 24841269; PubMed Central PMCID: PMCPMC4136071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. LaFemina RL. Alternative screening approaches for discovery of MERS coronavirus inhibitors. Antimicrob Agents Chemother. 2014:AAC;03406‐03414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adedeji AO, Singh K, Kassim A, et al. Evaluation of SSYA10‐001 as a replication inhibitor of severe acute respiratory syndrome, mouse hepatitis, and Middle East respiratory syndrome coronaviruses. Antimicrob Agents Chemother. 2014;58(8):4894‐4898. 10.1128/AAC.02994-14. PubMed PMID: 24841268; PubMed Central PMCID: PMCPMC4136041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hilgenfeld R, Peiris M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013;100(1):286‐295. 10.1016/j.antiviral.2013.08.015. PubMed PMID: 24012996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berry M, Fielding BC, Gamieldien J. Potential broad spectrum inhibitors of the coronavirus 3CLpro: a virtual screening and structure‐based drug design study. Virus. 2015;7(12):6642‐6660. 10.3390/v7122963. PubMed PMID: 26694449; PubMed Central PMCID: PMCPMC4690886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lei J, Mesters JR, Drosten C, Anemuller S, Ma Q, Hilgenfeld R. Crystal structure of the papain‐like protease of MERS coronavirus reveals unusual, potentially druggable active‐site features. Antiviral Res. 2014;109:72‐82. 10.1016/j.antiviral.2014.06.011. PubMed PMID: 24992731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu L, Liu Q, Zhu Y, et al. Structure‐based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat Commun. 2014;5:3067 10.1038/ncomms4067. PubMed PMID: 24473083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bacha U, Barrila J, Velazquez‐Campoy A, Leavitt SA, Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43(17):4906‐4912. 10.1021/bi0361766. PubMed PMID: 15109248 [DOI] [PubMed] [Google Scholar]
- 30. Tomar S, Johnston ML, St John SE, et al. Ligand‐induced dimerization of Middle East respiratory syndrome (MERS) coronavirus nsp5 protease (3CLpro): implications for nsp5 regulation and the development of antivirals. J Biol Chem. 2015;290(32):19403‐19422. 10.1074/jbc.M115.651463. PubMed PMID: 26055715; PubMed Central PMCID: PMCPMC4528106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomar S. Understanding the determinants for substrate recognition, regulation of enzymatic activity and the development of broad‐spectrum inhibitors of coronavirus 3‐chymotrypsin‐like proteases: Purdue University; 2015.
- 32. Grum‐Tokars V, Ratia K, Begaye A, Baker SC, Mesecar AD. Evaluating the 3C‐like protease activity of SARS‐coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 2008;133(1):63‐73. 10.1016/j.virusres.2007.02.015. PubMed PMID: 17397958; PubMed Central PMCID: PMCPMC4036818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bacha U, Barrila J, Velazquez‐Campoy A, Leavitt SA, Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43(17):4906‐4912. 10.1021/bi0361766 [DOI] [PubMed] [Google Scholar]
- 34. Wang F, Chen C, Tan W, Yang K, Yang H. Structure of main protease from human coronavirus NL63: insights for wide spectrum anti‐coronavirus drug design. Sci Rep. 2016;6:22677 10.1038/srep22677. PubMed PMID: 26948040; PubMed Central PMCID: PMCPMC4780191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang H, Bartlam M, Rao Z. Drug design targeting the main protease, the Achilles' heel of coronaviruses. Curr Pharm Des. 2006;12(35):4573‐4590. PubMed PMID: 17168763 [DOI] [PubMed] [Google Scholar]
- 36. St. John SE, Tomar S, Stauffer SR, Mesecar AD. Targeting zoonotic viruses: Structure‐based inhibition of the 3C‐like protease from bat coronavirus HKU4—The likely reservoir host to the human coronavirus that causes Middle East respiratory syndrome (MERS). Bioorg Med Chem. 2015;23(17):6036‐6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taha MO. Mixing pharmacophore modeling and classical QSAR analysis as powerful tool for lead discovery. VIRTUAL SCREENING. 2012;1: [Google Scholar]
- 38. Abuhammad A, Taha MO. QSAR studies in the discovery of novel type‐II diabetic therapies. Expert Opin Drug Discov. 2016;11(2):197‐214. 10.1517/17460441.2016.1118046. PubMed PMID: 26558613 [DOI] [PubMed] [Google Scholar]
- 39. Taha MO, Habash M, Al‐Hadidi Z, Al‐Bakri A, Younis K, Sisan S. Docking‐based comparative intermolecular contacts analysis as new 3‐D QSAR concept for validating docking studies and in silico screening: NMT and GP inhibitors as case studies. J Chem Inf Model. 2011;51(3):647‐669. [DOI] [PubMed] [Google Scholar]
- 40. Al‐Sha'er MA, Taha MO. Application of docking‐based comparative intermolecular contacts analysis to validate Hsp90α docking studies and subsequent in silico screening for inhibitors. J Mol Model. 2012;18(11):4843‐4863. [DOI] [PubMed] [Google Scholar]
- 41. Abu Hammad AM, Afifi FU, Taha MO. Combining docking, scoring and molecular field analyses to probe influenza neuraminidase‐ligand interactions. J Mol Graph Model. 2007;26(2):443‐456. 10.1016/j.jmgm.2007.02.002. PubMed PMID: 17360207 [DOI] [PubMed] [Google Scholar]
- 42. Abu Hammad AM, Taha MO. Pharmacophore modeling, quantitative structure‐activity relationship analysis, and shape‐complemented in silico screening allow access to novel influenza neuraminidase inhibitors. J Chem Inf Model. 2009;49(4):978‐996. 10.1021/ci8003682. PubMed PMID: 19341295 [DOI] [PubMed] [Google Scholar]
- 43. Al‐Nadaf A, Abu Sheikha G, Taha MO. Elaborate ligand‐based pharmacophore exploration and QSAR analysis guide the synthesis of novel pyridinium‐based potent beta‐secretase inhibitory leads. Bioorg Med Chem. 2010;18(9):3088‐3115. 10.1016/j.bmc.2010.03.043. PubMed PMID: 20378363 [DOI] [PubMed] [Google Scholar]
- 44. Al‐Sha'er MA, Khanfar MA, Taha MO. Discovery of novel urokinase plasminogen activator (uPA) inhibitors using ligand‐based modeling and virtual screening followed by in vitro analysis. J Mol Model. 2014;20(1):1‐15. [DOI] [PubMed] [Google Scholar]
- 45. Alabed SJ, Khanfar M, Taha MO. Computer‐aided discovery of new FGFR‐1 inhibitors followed by in vitro validation. Future Med Chem. 2016;8(15):1841‐1869. 10.4155/fmc-2016-0056. PubMed PMID: 27643626 [DOI] [PubMed] [Google Scholar]
- 46. AlQudah DA, Zihlif MA, Taha MO. Ligand‐based modeling of diverse aryalkylamines yields new potent P‐glycoprotein inhibitors. Eur J Med Chem. 2016;110:204‐223. 10.1016/j.ejmech.2016.01.034. PubMed PMID: 26840362 [DOI] [PubMed] [Google Scholar]
- 47. Rao SN, Head MS, Kulkarni A, LaLonde JM. Validation studies of the site‐directed docking program LibDock. J Chem Inf Model. 2007;47(6):2159‐2171. 10.1021/ci6004299. PubMed PMID: 17985863 [DOI] [PubMed] [Google Scholar]
- 48. Wu G, Robertson DH, Brooks CL 3rd, Vieth M. Detailed analysis of grid‐based molecular docking: a case study of CDOCKER‐A CHARMm‐based MD docking algorithm. J Comput Chem. 2003;24(13):1549‐62. Epub 2003/08/20. 10.1002/jcc.10306. PubMed PMID: 12925999 [DOI] [PubMed] [Google Scholar]
- 49. Jain AN. Scoring noncovalent protein‐ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des. 1996;10(5):427‐440. PubMed PMID: 8951652 [DOI] [PubMed] [Google Scholar]
- 50. Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape‐directed rapid docking of ligands to protein active sites. J Mol Graph Model. 2003;21(4):289‐307. PubMed PMID: 12479928 [DOI] [PubMed] [Google Scholar]
- 51. Gehlhaar DK, Bouzida D, Rejto PA. Reduced dimensionality in ligand—protein structure prediction: covalent inhibitors of serine proteases and design of site‐directed combinatorial libraries. 1999.
- 52. Muegge I. A knowledge‐based scoring function for protein‐ligand interactions: probing the reference state. Virtual Screening: An Alternative or Complement to High Throughput Screening?: Springer; 2002. p. 99–114.
- 53. Muegge I, Martin YC. A general and fast scoring function for protein‐ligand interactions: a simplified potential approach. J Med Chem. 1999;42(5):791‐804. 10.1021/jm980536j. PubMed PMID: 10072678 [DOI] [PubMed] [Google Scholar]
- 54. Abuhamdah S, Habash M, Taha MO. Elaborate ligand‐based modeling coupled with QSAR analysis and in silico screening reveal new potent acetylcholinesterase inhibitors. J Comput Aided Mol Des. 2013;27(12):1075‐1092. 10.1007/s10822-013-9699-6. PubMed PMID: 24338032 [DOI] [PubMed] [Google Scholar]
- 55. Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. 1975;405(2):442‐451. PubMed PMID: 1180967 [DOI] [PubMed] [Google Scholar]
- 56. Milne GW, Nicklaus MC, Driscoll JS, Wang S, Zaharevitz D. National Cancer Institute Drug information system 3D database. J Chem Inf Comput Sci. 1994;34(5):1219‐1224. PubMed PMID: 7962217 [DOI] [PubMed] [Google Scholar]
- 57. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4‐17. [DOI] [PubMed] [Google Scholar]
- 58. Discovery Studio User Guide . Accelrys Software Inc. San Diego. 2005.
- 59. Báez‐Santos YM, Barraza SJ, Wilson MW, et al. X‐ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain‐like proteases. J Med Chem. 2014;57(6):2393‐2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Agnihothram S, Yount BL, Donaldson EF, et al. A mouse model for Betacoronavirus subgroup 2c using a bat coronavirus strain HKU5 variant. MBio. 2014;5(2):e00047‐e00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9(10):430‐431. 10.1016/S1359-6446(04)03069-7. PubMed PMID: 15109945 [DOI] [PubMed] [Google Scholar]
- 62. Abad‐Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discov Today. 2005;10(7):464‐469. [DOI] [PubMed] [Google Scholar]
- 63. Al‐Nadaf AH, Taha MO. Discovery of new renin inhibitory leads via sequential pharmacophore modeling, QSAR analysis, in silico screening and in vitro evaluation. J Mol Graph Model. 2011;29(6):843‐864. 10.1016/j.jmgm.2011.02.001. PubMed PMID: 21376648 [DOI] [PubMed] [Google Scholar]
- 64. Taha MO, Al‐Sha'er MA, Khanfar MA, Al‐Nadaf AH. Discovery of nanomolar phosphoinositide 3‐kinase gamma (PI3Kgamma) inhibitors using ligand‐based modeling and virtual screening followed by in vitro analysis. Eur J Med Chem. 2014;84:454‐465. 10.1016/j.ejmech.2014.07.056. PubMed PMID: 25050878 [DOI] [PubMed] [Google Scholar]
- 65. Todeschini R, Lasagni M, Marengo E. New molecular descriptors for 2D and 3D structures. Theory Journal of chemometrics. 1994;8(4):263‐272. [Google Scholar]
- 66. Taha MO, Tarairah M, Zalloum H, Abu‐Sheikha G. Pharmacophore and QSAR modeling of estrogen receptor beta ligands and subsequent validation and in silico search for new hits. J Mol Graph Model. 2010;28(5):383‐400. 10.1016/j.jmgm.2009.09.005. PubMed PMID: 19850503 [DOI] [PubMed] [Google Scholar]
- 67. Baell JB. Screening‐based translation of public research encounters painful problems. ACS Med Chem Lett. 2015;6(3):229‐234. 10.1021/acsmedchemlett.5b00032. PubMed PMID: 25941544; PubMed Central PMCID: PMCPMC4416424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lackowicz JR. Principles of Fluorescence Spectroscopy. New York: Plenum Press; 1983:111‐150. [Google Scholar]
- 69. Carroll SS, Inglese J, Mao S‐S, Olson DB. Drug Screening: assay development issues In: Prendergast GC, ed. editorMolecular Cancer Therapeutics: Strategies for Drug Discovery and Development. New Jersey: John Wiley & Sons; 2004:119‐140. [Google Scholar]
- 70. Deu E, Verdoes M, Bogyo M. New approaches for dissecting protease functions to improve probe development and drug discovery. Nat Struct Mol Biol. 2012;19(1):9‐16. 10.1038/nsmb.2203. PubMed PMID: 22218294; PubMed Central PMCID: PMCPMC3513415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pascolutti M, Thomson RJ, Itzstein MV. Development of Influenza Virus Sialidase Inhibitors In: Holenz J, ed. editorLead Generation: Methods and Strategies. Vol.68 Weinheim, Germany: Wiley; 2016:651‐686. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The structures of HKU4‐CoV 3CLpro inhibitors utilized in modeling. 25
Table S2: Training subsets (extracted from Table S1) employed in exploring the pharmacophoric space of HKU4‐CoV 3CLpro inhibitors. 48
Table S3: Training subsets and HYPOGEN run parameters employed for exploring HKU4‐CoV 3CLpro pharmacophoric space. 49
Table S4: The performance of the best representatives of clustered pharmacophore hypotheses generated for HKU4‐CoV 3CLpro. 54
Table S5: Pharmacophoric features and corresponding weights, tolerances and 3D coordinates of Hypo(L‐T5–2), Hypo(K‐T5–3),and Hypo(N‐T1–1) selected by QSAR‐guided method. 57
Table S6: Training subset used for adding excluded spheres for Hypo(K‐T5–3/N‐T1–1) using HIPHOP‐REFINE module of CATALYST. 58
Table S7: HKU4‐CoV 3CLpro based pharmacophore models selected by best dbCICA models. 59
Table S8: Captured hits by dbCICA derived pharmacophores. 60
Table S9: Captured hits by QSAR‐guided pharmacophores. 61
Figure S1: NMR and MS analysis of compound 222. 62
Figure S2: NMR and MS analysis of compound 223. 63
Figure S3: NMR and MS analysis of compound 224.
Figure S4: The dose–response curves of compounds 222 and 223 against MERS‐CoV 3CLpro