

Abstract
In the present study, 50 nasopharyngeal swabs from children with community‐acquired pneumonia (CAP) but negative for 18 common respiratory viruses, as measured by the Luminex xTAG Respiratory Viral Panel Assay, were subjected to multiplex metagenomic analyses using a next‐generation sequencing platform. Taxonomic analysis showed that all sequence reads could be assigned to a specific species. An average of 95.13% were assigned to the Bacteria kingdom, whereas, only 0.72% were potentially virus derived. This snapshot of the respiratory tract virome revealed most viral reads to be respiratory tract related, classified into four known virus families: Paramyxoviridae, Herpesviridae, Anelloviridae, and Polyomaviridae. Importantly, we detected a novel human parainfluenza virus 3 (HPIV 3) strain with a 32‐bp insertion in the haemagglutinin‐neuraminidase (HN) gene that produced a negative result in the Luminex assay, highlighting the strength of virome metagenomic analysis to identify not only novel viruses but also viruses likely to be missed by ordinary clinical tests. Thus, virome metagenomic analysis could become a viable clinical diagnostic method.
Keywords: community‐acquired pneumonia (CAP), human parainfluenza virus 3 (HPIV 3), Luminex assay, metagenomics analysis, virome
1. INTRODUCTION
Community‐acquired pneumonia (CAP) is a major cause of morbidity and mortality in children worldwide. CAP is caused by a variety of microbes, mainly including bacteria and viruses. Appropriate management of CAP requires identification of the potential aetiological agents. Clinical symptoms are often insufficient for making a clear diagnosis without clinical microbiology and/or molecular tests.
Molecular assays are increasingly popular for laboratory diagnosis because they often outperform traditional viral diagnostic methods (such as cell‐culture‐based testing and antigen detection) in efficiency, sensitivity, and specificity. In addition, several multiplex PCR assays and DNA microarray testing methods have been proposed to screen dozens to hundreds of pathogens simultaneously.1, 2, 3 High‐throughput next‐generation sequencing (NGS) technology has recently become a powerful tool for microbial community characterization and new pathogen identification because genetic materials are isolated directly from environmental/clinical samples and sequenced; thus, no culturing, cloning, or a priori knowledge of what pathogens may be present is required.
The application of NGS to the respiratory tracts of children is expected to provide comprehensive information about the respiratory tract virome and allow the characterization of specific viruses. Virome results for the pediatric respiratory tract have been reported by several groups.4, 5, 6, 7 We also previously compared respiratory tract viromes between children with severe acute respiratory infection (SARI) and those without SARI and showed that members of the Paramyxoviridae, Coronaviridae, Parvoviridae, Orthomyxoviridae, Picornaviridae, Anelloviridae, and Adenoviridae families represented the most abundant families identified in the respiratory tracts of children with SARI. The viral population in the respiratory tracts of children without SARI was less diverse and mainly dominated by the Anelloviridae family, with only a small proportion of common epidemic respiratory viruses.8
Although the agent most commonly detected in CAP is Streptococcus pneumoniae,9 new, highly sensitive techniques have allowed us to detect many respiratory viruses associated with CAP,9, 10 mostly influenza virus, respiratory syncytial virus, parainfluenza virus, and human metapneumovirus.10, 11, 12 Rhinovirus and human coronavirus have also been detected.10, 11, 13 Whether other still unidentified novel pathogens could cause CAP remains to be investigated. Comprehensive and detailed metagenomic analyses of the respiratory tracts of children with CAP but not carrying common respiratory viruses have not yet been reported.
Therefore, in the present study, clinical specimens from children with CAP but negative with common respiratory pathogens were screened using the FDA‐cleared Luminex xTAG Respiratory Viral Panel Assay. Fifty specimens that were negative for 18 common respiratory viruses were then subjected to multiplex metagenomic analyses using a next‐generation sequencing platform. We provide a picture of the viral content and diversity in the respiratory tracts of children with CAP but uninfected with common respiratory viruses.
2. MATERIALS AND METHODS
2.1. Study population and sample collection
We randomly enrolled 368 paediatric patients <6 years of age (mean age of child patients was 1.18 years) at Beijing Children's Hospital who met the child CAP case definition.14 All nasopharyngeal aspirate samples were collected from December 2014 to May 2015. This study was performed in strict accordance with the human subject protection guidance and was approved by the Ethical Review Committee of Beijing Children's Hospital. Written informed consent was obtained on the participants’ behalf from their parents or guardians.
2.2. Preparation of nucleic acids
Viral nucleic acids in clinical samples were extracted using the NucliSens easyMAG system (bioMérieux, Marcy‐l'Etoile, France) according to the manufacturer's instructions. RNA was reverse transcribed into cDNA using random primers and a SuperScript II reverse transcriptase (200 U) reaction mixture (20 µL) (Invitrogen, Carlsbad, CA).
2.3. Luminex respiratory viral panel assay
The Luminex xTAG respiratory viral panel (RVP) assay and instrument Luminex 200 (Luminex, Austin, TX) were to detect 18 common respiratory viral pathogens and subtypes in the nucleic acid samples, including influenza A, influenza A subtype H1, influenza A subtype H3, 2009 H1N1, influenza B, human adenoviruses (HAdV), human parainfluenza virus (HPIV) 1‐4, respiratory syncytial virus (RSV) A and B, human metapneumovirus (HMPV), enteroviruses and rhinoviruses (EV/Rh), human coronavirus (HCoV) HKU1, 229E, NL63 and OC43, and human bocavirus (HBoV).
2.4. Viral enrichment and nucleic acid library construction
Briefly, samples were vortexed and centrifuged at 10 000g for 10 min at 4°C. The supernatant from each sample was filtered through a 0.45‐µm polyvinylidene difluoride filter (Millipore, Darmstadt, Germany), to remove eukaryotic and bacterial‐sized particles. The filtered samples were then centrifuged at 100 000g for 3 h at 4°C. The pellets were re‐suspended in Hank's balanced salt solution. To remove naked DNA and RNA, the re‐suspended pellet was digested in a cocktail of DNase and RNase enzymes, including 14 U of Turbo DNase (Ambion, Austin, TX), 20 U of benzonase (Novagen, Darmstadt, Germany), and 20 U of RNase One (Promega, Madison, WI) at 37°C for 2 h in 1 × DNase buffer (Ambion). The viral nucleic acids were then isolated using a QIAmp MinElute Virus Spin Kit (Qiagen, Valencia, CA). Viral first‐strand cDNA was synthesized using the primer K‐8N (5′‐GACCATCTAGCGACCTCCAC‐NNNNNNNN‐3′) and a Superscript III system (Invitrogen). To convert first‐strand cDNA into double‐stranded DNA, the cDNA was incubated at 37°C for 1 h in the presence of 5 U of Klenow fragment (NEB, Ipswich, MA) in 1 × NEB buffer 2 (final volume of 25 µL). Sequence‐independent PCR amplification was conducted using 1 µM primer K (5′‐GACCATCTAGCGACCTCCAC‐3′) and 0.5 U of Phusion DNA polymerase (NEB). The PCR products were analyzed by agarose gel electrophoresis. A DNA smear of larger than 500 bp was excised and extracted with a MinElute Gel Extraction Kit (Qiagen). Until then, each library was constructed from one specimen with a mixture of DNA and cDNA. Libraries for sequencing were made using Illumina's Nextera XT DNA Sample Preparation Kit (San Diego, CA) with 1 ng of input DNA. Each library was sequenced in an individual lane without indexing.
2.5. Next‐generation sequencing and taxonomic assignments
Amplified viral nucleic acid libraries were then analyzed using an Illumina HiSeq 4000 sequencer (Illumina, San Diego, CA) for a single read of 126 bp in length. The raw sequence reads were filtered using previously described criteria15 to obtain valid sequences: (i) reads filtered with Illumina's Consensus Assessment of Sequence and Variation (CASAVA) software, (ii) reads with no call sites, (iii) reads with similarity to the sequencing adaptor and the primer K sequence, and (iv) duplicate reads and low‐complexity reads. Only reads that passed the quality control were considered valid sequences. The viral origin of each read was evaluated by mapping to the NCBI nr database (February 25, 2016) by Diamond 0.7.11 and Megan 6.2.40 or NCBI nt database (January 28, 2016) by Blast 2.3.0 and Megan 6.2.40.
Reference sequences were simultaneously used in the CLC Genomic Workbench mapping analysis to determine the genome coverage and sequencing depth.
2.6. Nucleotide sequence and phylogenetic analysis
The obtained sequences were compared with representative sequences available in the GenBank database using BLAST analysis (NCBI BLAST server). The nucleic acid and deduced amino acid sequences were analyzed and aligned with BioEdit software (Tom Hall, North Carolina State University, Carolina). Phylogenetic analyses were performed (MEGA version 5.0) by the neighbor‐joining method, with the statistical significance of phylogenies estimated by bootstrap analysis with 1000 pseudoreplicate datasets. The newly identified sequences were deposited in GenBank.
3. RESULTS AND DISCUSSION
3.1. Luminex screening results
For the 368 nasopharyngeal aspirate samples collected from paediatric patients diagnosed with CAP, the Luminex xTAG respiratory viral panel (RVP) assay screening results for 18 common respiratory viral pathogens and subtypes showed 217 specimens (58.97%) to be positive for one or more viral pathogens, including influenza A, influenza A subtype H1, influenza A subtype H3, 2009 H1N1, influenza B, HAdV, HPIV 1‐4, RSV A and B, HMPV, EV/Rh, HCoV‐HKU1, 229E, NL63 and OC43, and HBoV. Of the remaining 151 negative specimens, we randomly selected 50 specimens for the following viral microbiome analyses.
3.2. Clinical characteristics of pediatric patients used in the metagenomic analysis
For the 50 specimens selected for viral microbiome analyses, clinical information, including the child's age and sex, history of repeated infection in the respiratory tract, and respiratory symptoms, is presented in Table 1. The mean age of the pediatric patients was 2.36 years, and 33 (66%) were boys. Among the patients, 3/50 (6%) had a history of repeated infection in the respiratory tract, and 44/50 (88%) had fever; 49/50 (98%) a cough and coughed up phlegm, and 9/50 (18%) wheezed. All 50 pediatric patients exhibited thoracalgia symptoms, and none had haemoptysis or rash.
Table 1.
Clinical characteristics of the 50 children patients with CAP in this study
| Characteristic | All cases | |
|---|---|---|
| Age (years) | 2.36 ± 1.87 | |
| Male/Female | 33 (66%)/17 (34%) | |
| History of repeated infection in respiratory tract | 3 (6%) | |
| Fever | 44 (88%) | |
| Cough | 49 (98%) | |
| Cough up phlegm | 49 (98%) | |
| Hemoptysis | 0 (0%) | |
| Wheeze | 9 (18%) | |
| Rash | 0 (0%) |
3.3. Overview of sequencing data
The total number of raw reads per sample ranged from 34 280 310 (BCHNP11) to 54 722 470 (BCHNP18). Adapter and low‐quality sequences (QC) were trimmed, and the number of processed reads is listed in Supplementary Table S1. The proportion of reads related to the human genome and human cDNA ranged from 0.26% (BCHNP46) to 63.46% (BCHNP23), with an average of 14.30%. The percentage of reads that could be properly mapped to bacterial 16S, archaeal 16S, eukaryotic 18S, bacterial 23S, archaeal 23S, or eukaryotic 28S rRNA sequences in the SILVA database, RFAM 5S and RFAM 5.8S sequences ranged from 3.72% (BCHNP23) to 94.30% (BCHNP47), with an average of 71.23%. Therefore, only approximately 3.14% (BCHNP47) to 31.57% (BCHNP23) (average 13.42%) of the sequencing data from each specimen were utilized for further metagenomic analysis (Figure 1A and Supplementary Table S1).
Figure 1.
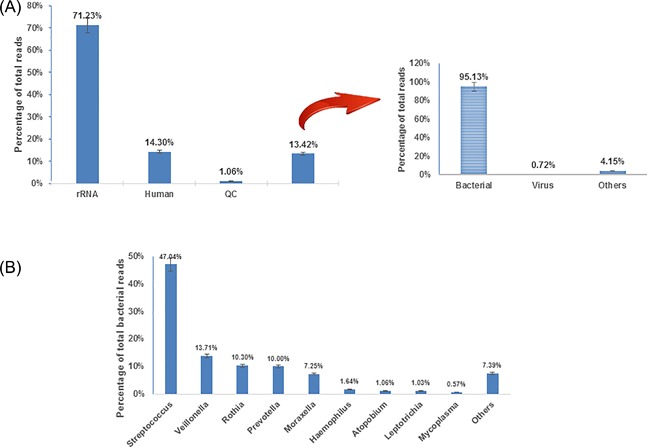
The distribution of sequencing reads from clinical specimens. A, The taxonomic distribution of total sequencing reads. B, The distribution of bacterial content sequencing reads from clinical specimens. Data are average values from 50 nasopharyngeal specimens
Most sequence reads could be assigned to a specific species, and taxonomic analysis showed that an average of 95.13% (range from 44.65% to 99.67) were assigned to the Bacteria kingdom and only 0.72% were potentially virus derived (Figure 1A and Supplementary Table S1). The latter data were valuable for further analyzing the CAP‐associated children respiratory virome. The remaining eukaryote‐like reads included unscreened human‐derived sequences and some animal or plant sequences, as food remains in the pharyngeal passage. Meanwhile, there were still some undefined sequences that lacked a known homolog, or for which almost equally close homologs were found in more than one category. These eukaryote‐like and undefined reads were excluded from further analysis.
In our data set, only 0.72% of the valid metagenomic sequences were useful for virus detection due to the large amount of host‐derived sequences (Supplementary Table S1). Human‐derived sequence contamination of greater than 90% has been reported by other researchers when using nasal specimens for metagenomic studies.16, 17 Hence, a high percentage of human‐derived sequences is likely an inherent problem of directly extracting RNA/DNA from respiratory samples. Although current computational subtraction methods can efficiently remove host contamination, additional sample‐filtering steps before sequencing to eliminate host cells and enrich for microbial genomes are undoubtedly required to detect microbial pathogens in a more efficient and cost‐effective manner.
3.4. Contigs showing bacterial homology
The bacterial contigs were split further into genera, as defined by their closest homology (Figure 1B). Streptococcus represented the largest portion (47.04%) among all bacterial‐related sequences, followed by Veillonella (13.71%), Rothia (10.30%), Prevotella (10.00%), Moraxella (7.25%), Haemophilus (1.64%), Atopobium (1.06%), Leptotrichia (1.03%), Mycoplasma (0.57%), and others (Capnocytophaga, Campylobacter, Fusobacterium, Neisseria, Actinomyces, Enterococcus, etc., 7.39%). Infection of these pathogens may be the main reason for CAP in these children, especially for Streptococcus.18, 19 Veillonella and Rothiataxa are generally considered part of the normal flora but have been identified in individuals with CAP.19, 20 Pigmented Prevotella have been detected in cases of aspiration pneumonia and empyemain children.21 For Moraxella, Haemophilus, Atopobium, Leptotrichia, and Mycoplasma, these bacteria frequently colonize the nasopharynx in healthy children and are also common pathogens in the respiratory tract.18, 19, 21 However, due to enrichment for viruses, the bacterial sequences found in this study are likely biased and not representative for these patients. These results provide a crude characterization of the bacterial content in these samples along with contigs of other origin.
3.5. Contigs showing homology with respiratory‐related viruses
A total of 62 species of viruses (based on the NCBI taxonomy database) were identified with at least one specific sequence from the 50 samples, but 25 species belonged to bacteriophage. Most of the phage were Streptococcus phage, and Cronobacter phage, Lactobacillus phage, and Actinomyces phage were also observed. The variety of phage is an important part of the respiratory virome and may be associated with bacterial infections. The proportion of bacteriophage not only influences the viral composition but also reveals the presence of other pathogens in respiratory samples, such as various bacteria. Previous metagenomic analyses of respiratory samples from children with lower and upper respiratory tract infections have indicated that cases associated with bacterial infections accounted for a small proportion of SARI cases,22 especially those with unexplained fevers. More studies of the relationship between respiratory diseases and the diversity of bacteriophage are needed.
The viral metagenomic analyses revealed a variety of viruses in the respiratory tracts of children with CAP but negative for Luminex xTAG respiratory viral panel assay detection. Of these, most of the viral reads were respiratory tract related and could be classified into four virus families: Paramyxoviridae, Herpesviridae, Anelloviridae, and Polyomaviridae. The detailed results are summarized in Table 2.
Table 2.
Read numbers of respiratory tract‐related viruses derived from nasopharyngeal specimens, listed according to their similarities to virus groups
| Family | Paramyxoviridae | Herpesviridae | Anelloviridae | Polyomaviridae | |||
|---|---|---|---|---|---|---|---|
| Virus genus | Human parainfluenza virus 3 | Epstein‐Barr virus | Human cytomegalovirus | Torque teno virus | TTV‐like mini virus | SEN virus | WU Polyomavirus |
| BCHNP1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP6 | 0 | 649 | 0 | 0 | 0 | 0 | 0 |
| BCHNP7 | 0 | 75 | 1 | 0 | 0 | 0 | 0 |
| BCHNP8 | 0 | 0 | 3 | 0 | 0 | 0 | 0 |
| BCHNP9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP11 | 0 | 0 | 0 | 2 | 0 | 0 | 0 |
| BCHNP12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP13 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| BCHNP14 | 0 | 225 | 0 | 2 | 0 | 0 | 0 |
| BCHNP15 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| BCHNP17 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP18 | 0 | 0 | 0 | 5 | 0 | 0 | 0 |
| BCHNP19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP21 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP22 | 3 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP23 | 0 | 0 | 0 | 6 | 1 | 2 | 0 |
| BCHNP24 | 0 | 0 | 0 | 0 | 0 | 0 | 17 |
| BCHNP28 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP29 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| BCHNP32 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP33 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP34 | 18 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP35 | 72 | 0 | 11 | 0 | 0 | 0 | 0 |
| BCHNP36 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP37 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP38 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP39 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP40 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP41 | 28 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP42 | 29 | 0 | 0 | 68 | 34 | 12 | 0 |
| BCHNP43 | 75 487 | 0 | 2 | 0 | 0 | 0 | 0 |
| BCHNP44 | 0 | 0 | 0 | 3 | 0 | 0 | 0 |
| BCHNP45 | 80 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP47 | 23 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP48 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP49 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BCHNP50 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
3.5.1. Paramyxoviridae
Only one virus in the Paramyxoviridae family, HPIV 3, was detected by metagenomic analysis in these CAP specimens. Almost all children encounter HPIVs within the first few years after birth, but immunity is incomplete, and re‐infections occur throughout life.23 Approximately, two thirds of children are infected by HPIV3 in the first year of life, mainly result in bronchiolitis and pneumonia.24 In the United States of America, an estimated 7600‐48 000 children under the age of 1 and 8100‐42 600 children aged 1‐4 years are hospitalized with HPIV infection annually.25 In this study, 75 487 reads from specimen BCHNP43 matched HPIV 3. These sequences were used in the CLC Genomic Workbench mapping analysis to determine the genome coverage and sequencing depth. As shown in Figure 2A, the genome coverage for HPIV3 (reference seq: NC_001796.2) was 98.93% (15 297/15 462), and the average depth of sequencing was 1106.
Figure 2.
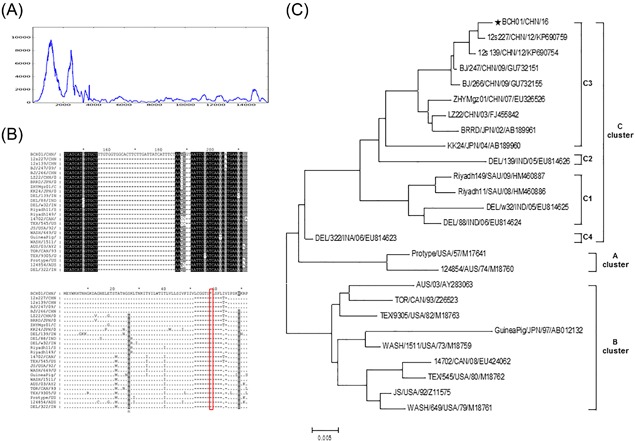
Sequence analysis of HPIV3/China/BCH01/2016 from specimen BCHNP43. A, Viral genome coverage and sequencing depth analysis. B, Nucleotide and amino acid sequence alignments of the HN gene of HPIV3 derived from specimen BCHNP43 in this study and other strains of HPIV‐3 from different geographic origins. A 32 bp insertion was found in the HPIV3/China/BCH01/2016 strain, which led to an advanced stop codon at residue 58 in hemagglutinin‐neuraminidase glycoprotein. C, Phylogenetic analysis based on the complete HN gene sequence ofHPIV3/China/BCH01/2016 and other strains of HPIV‐3 from different geographic origins
Notably, HPIV detection was included in the Luminex xTAG respiratory viral panel assay. Based on the random distribution of reads on the HPIV3 virus genome, the full‐length genome of HPIV3 virus from specimen BCHNP43 was successfully obtained using NGS methods and gap amplification. The newly identified virus was named HPIV3/China/BCH01/2016, and the genome sequence was deposited in GenBank under accession number KY234287. Interestingly, alignment of the sequence of the surface haemagglutinin‐neuraminidase (HN) gene of this strain with other HPIV3 HN gene sequences from isolates from seven countries (United States of America, Australia, India, Japan, Canada, Saudi Arabia, and China) in GenBank revealed a 32‐bp insertion in the HPIV3/China/BCH01/2016 strain. This insertion leads to an advanced stop codon at residue 58 in the haemagglutinin‐neuraminidase glycoprotein and has not been reported previously (Figure 2B). The presence of this insertion was further confirmed by PCR and direct Sanger sequencing. The influence of this insertion on the function of the HN protein and pathogenicity of the HPIV3 virus is unclear, and we are currently attempting to isolate this strain using human airway epithelial cell culture. Phylogenetic analysis showed that the HPIV3/China/BCH01/2016 strain belongs to Cluster C. Compared with Clusters A and B, which were isolated in the middle of the last century, most strains in Cluster C were isolated recently in Asia. The Cluster C isolates were further grouped into four sub‐clusters (C1, C2, C3, and C4). HPIV3/China/BCH01/2016 strain belongs to the C3 group together with previously reported isolates from Beijing, Zhejiang, Guangdong, and Gansu provinces of China and one isolate from Japan (Figure 2C). Because the primers used in the Luminex assays were patented, we could only deduced that the primers used for HPIV3 virus detection in the Luminex assay were annealing around the insertion area, leading to a negative result.
Specimen BCHNP43 was derived from a 3‐month‐old boy with congenital heart disease, and the admitting diagnoses were respiratory failure, dysentery, and moderate dehydration. He was diagnosed with severe pneumonia; he had been hospitalized for 14 days when 50 days old and underwent heart surgery when 100 days old. Although the patient has not admitted to the paediatric intensive care unit (PICU), an infant nasal continuous positive airway pressure (NCPAP) ventilator was used for 65 h. For respiratory pathogen IgM detection, positivity was found only for Mycoplasma pneumoniae. After 7 days of hospitalization in Beijing Children Hospital and treatment with antibiotics, cough‐relieving and sputum‐reducing, cardiac and diuresis, and anticoagulant, all symptoms were improved, and the patient was discharged.
In addition to specimen BCHNP43, we detected 3, 18, 72, 28, 29, 80, and 23 reads related to HPIV3 in specimens 22, 34, 35, 41, 42, 45, and 47, respectively. However, the genome coverage with HPIV3 was less than 3% for all specimens, and detection by conventional PCR was unsuccessful. It remains to be determined whether there was a cross‐contamination exist during the sample preparation or the sequencing step, or the virus loads in these samples were generally low, or mutations and recombinations happened in virus strains which making detection using specific PCR detection method less likely. However, what could be definitely confirmed is that no outbreak has happened, because all the positive samples related with HPIV3 by metagenomic analysis derived from patients with different disease time and geographic origins.
3.5.2. Herpesviridae
In the Herpesviridae family, Epstein‐Barr virus (EBV, human herpesvirus 4) and human cytomegalovirus (hCMV, human herpesvirus 5) were detected by metagenomic analysis. These two viruses are highly prevalent and ubiquitous. Primary infection occurs early in the life in the majority of cases and is followed by a lifelong latent infection, from which reactivation may occur with viral shedding in at least the saliva. The outcome of reactivation strongly depends on the host immunological status. Mostly, hCMV causes severe respiratory disease, whereas the role of EBV in pneumonia is debated.26 In this study, 5/50 patients were EBV positive by metagenomic analysis, with two patients (BCHNP6 and BCHNP14) having more than 200 reads.
3.5.3. Anelloviridae
At least three members of the Anelloviridae family were found in respiratory specimens of children with CAP, including the torque teno virus (TTV), TTV‐like mini virus (TTMV), and SEN virus. Very high TTV and TTMV prevalences (60‐98%) have been found in healthy populations around the world,27, 28, 29 and it was once hypothesized that TTV and TTMV are part of the normal human microflora.30, 31 We previously compared the differences in TTV genotypes between children with severe acute respiratory infection and a healthy group to analyze the relationship between acute respiratory infection and TTVs. The results suggested that there might be no direct link between the two and that children might be an important reservoir of TTVs.8 SEN virus (SENV) was first discovered in the serum of a human immunodeficiency virus type 1 (HIV‐1) infected patient. The prevalence of SEN‐V in the healthy population differs greatly. Vertical transmission of SENV, presumably at delivery, has been reported but may not induce persistent viraemia.32 Further studies are needed to define the pathogenesis and clinical importance of SENV infection.
3.5.4. Polyomaviridae
Seventeen reads of WU Polyomavirus (WUPyV), which belongs to the Polyomaviridae family, were also detected by metagenomic analysis in specimen BCHNP24. This virus was identified in 2007 from respiratory tract samples following large‐scale molecular screening using high‐throughput DNA sequencing of random clones33 and named after the university (Washington University) where it was identified. Since its first identification, WUPyV sequences have been confirmed worldwide in respiratory samples from children with respiratory tract diseases, with a prevalence ranging from 1.1% to 7%.34, 35, 36, 37, 38 In this study, we reported that children with CAP could also be carrying WUPyV. However, WUPyV was found at similar frequencies in control groups without respiratory diseases.39 Thus, the link between WUPyV and acute respiratory diseases and CAP remains speculative.
However, in all above mentioned specimens which were positive for Paramyxoviridae, Herpesviridae, Anelloviridae, and Polyomaviridae, large amounts of sequence reads related to Streptococcus salivarius, Streptococcus parasanguinis, Streptococcus pneumoniae, Streptococcus mitis, Streptococcus oralis, and other pneumonia‐related pathogenic bacteria were also detected. Whether mixed infection with these respiratory‐related viruses with pathogenic bacteria exasperates symptoms of CAP requires further investigation.
3.6. Contigs showing homology with other viruses
In addition to these respiratory‐related viruses, sequences matching other eukaryotic viruses were also detected within the respiratory tract, albeit at much lower read abundances. These included hepatitis viruses B, human picobirnavirus, and human immunodeficiency virus 1 (Figure 3). The hepatitis virus B sequences might be derived from chronic hepatitis B carrier children. Picobirnavirus (PBV) is a novel group of viruses discovered in the late 1980s in clinical specimens from outbreaks of acute gastroenteritis in children.40, 41, 42 However, PBV is frequently detected in non‐diarrheic healthy hosts, and prolonged shedding has been observed in some individuals.43 Human PBV are poorly studied, and little is known about their pathogenicity. It is not even clear whether humans or intestinal microorganisms are hosts for these viruses. For human immunodeficiency virus 1 (HIV 1), because no more than 12 reads were detected and genome coverage of HIV were less than 1% for each four samples, one reasonable explanation was that the HIV related reads were due to contamination of the reagents used in sample process step, as murine leukemia virus (MLV) is commonly found in reverse transcriptase, and its shared extremely high homology with HIV in partial genome.
Figure 3.

Heatmap based on the read numbers of other viruses derived from nasopharyngeal specimens. The clinical specimens are listed in the top text row, and the virus names are presented in the left text column. The boxes colored from blue to red represent the metagenomic sequencing reads observed (reads varied between 20 and 212, respectively)
Interestingly, several sequences related to bovine diarrhea virus were also present in the respiratory tracts of one specimen (BCHNP23), albeit at a lower frequency (52 reads) (Figure 3). In addition to zoonotic viruses, more than 20 plant‐origin viruses were detected in the respiratory tracts of CAP children (Figure 3). The hosts of these plant viruses mainly include apple, cactus, cucumber, tomato, apricot, citrus, zucchini, watermelon, potato, fava bean, and pepper. These animal‐ and plant‐origin sequences may be due to food debris in the oral cavity.
Several other metagenomic analyses of the respiratory virome of children or adults have been conducted. As mentioned above, our previous study showed that members of the Paramyxoviridae, Coronaviridae, Parvoviridae, Orthomyxoviridae, Picornaviridae, Anelloviridae, and Adenoviridae families represented the mostabundant species identified in the respiratory tracts of children with severe acute respiratory infection.8 Lysholm et al5 used a metagenomic sequencing strategy to study nasopharyngeal aspirate samples from diagnosed respiratory tract infections and found most of the viral sequences were classified into one of three major families: Paramyxoviridae, Picornaviridae, or Orthomyxoviridae. Yang et al7 identified seven species of known respiratory viruses (HAdV, RSV, HRV, influenza virus, HPIV, HBoV, and HEV) from nasopharyngeal aspirate samples from patients with acute lower respiratory tract infections. Here, we conducted a metagenomic analysis of the virome of children with CAP but non‐common respiratory virus infections. This study is the first to perform a full, detailed analysis of the virome of respiratory tract samples among children with CAP but non‐common respiratory virus infection in childhood respiratory tracts, which will benefit the surveillance, prevention, and control of acute respiratory infections in children.
Besides a number of known human viruses were detected in these clinical specimens, putatively viruses which are not generally associated with human CAP were also detected, such as EBV, hCMV, anellovirus, and WUPyV. Some of these viruses have been mentioned on an individual level, and in this study, a more generalized observation of these viruses which may be affecting the trajectory of the disease progression have been drawn. This may extend our knowledge about the viral communities present in samples that were not infected by known respiratory pathogens in order to identify viruses that might be responsible for the unexplained cases of CAP. The fact that micro‐mass sequencing detected these canonical viruses in some of the specimens, despite conventional diagnostic testing by EIA and PCR, underscores the sensitivity limits of conventional diagnostics.
In this study, a large proportion of the sequences from viruses and bacteria have been confirmed to be a part of the human microflora and many were in fact from known pathogens of the nasopharyngeal tract. However, the nasopharyngeal aspirates collected are likely to contain virus which mainly replicate in the upper respiratory tract. The mucosa may also contain temporary microorganisms that are not part of the normal microflora. They may come from the environment, for example, from dust and from food and water. It is likely that a proportion of the sequences came from such microorganisms. Viral metagenomics clearly provides a crucial tool for virus discovery. With this approach, not priori information is needed, in contrast to directed PCR or Luminex assays, and viruses that are difficult to propagate in cell culture can be discovered. In this study, we detected a novel HPIV3 strain with a 32‐bp insertion in the HN gene, highlighting the strength of the method to identify not only novel viruses but also viruses likely to be missed by ordinary clinical tests. Lysholm et al5 also identified one novel type of Rhinovirus C and a number of previously undescribed viral genetic fragments of unknown origin using a metagenomic sequencing strategy. Using the high‐throughput capacity of ultra‐deep sequencing, Yang et al7 detected a co‐infected case of HEV with HRV that was missed by regular PCR testing. As sequencing continuously becomes more available and inexpensive, viral metagenomic analysis could also become a viable clinical diagnostic method in the future.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Figure S1.A flow‐chart describing the entire process from viral enrichment of samples of the metagenomic analysis steps.
Table S1.Sequencing read information for each nasopharyngeal specimen.
ACKNOWLEDGMENTS
This work was supported by grants from the National Science and Technology Supported Projects (2013BAI09B11), the National Major S & T Research Projects for the Control and Prevention of Major Infectious Diseases in China (2014ZX10004‐001), and the National Natural Science Foundation of China (31370203), The Capital Health Research and Development of Special Project (2016‐2‐1142), Beijing Talents Fund (2014000021469G239), and Beijing Children's Hospital Young Investigator Program (BCHYIPB‐2016‐12).
CONFLICT OF INTEREST
The authors have no potential conflicts of interest.
Xu L, Zhu Y, Ren L, et al. Characterization of the nasopharyngeal viral microbiome from children with community‐acquired pneumonia but negative for Luminex xTAG respiratory viral panel assay detection. J Med Virol. 2017;89: 2098–2107. 10.1002/jmv.24895
Lili Xu and Yun Zhu contributed equally to this work.
Contributor Information
Zhengde Xie, Email: xiezhengde_bch@163.com.
Kunling Shen, Email: kunlingshen_bch@163.com.
REFERENCES
- 1. Lee WM, Grindle K, Pappas T, et al. High‐throughput, sensitive, and accurate multiplex PCR‐microsphere flow cytometry system for large‐scale comprehensive detection of respiratory viruses. J Clin Microbiol. 2007; 45:2626–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li H, McCormac MA, Estes RW, et al. Simultaneous detection and high‐throughput identification of a panel of RNA viruses causing respiratory tract infections. J Clin Microbiol. 2007; 45:2105–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang D, Coscoy L, Zylberberg M, et al. Microarray‐based detection and genotyping of viral pathogens. Proc Natl Acad Sci USA. 2002; 99:15687–15692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bogaert D, Keijser B, Huse S, et al. Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS ONE. 2011; 6:e17035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lysholm F, Wetterbom A, Lindau C, et al. Characterization of the viral microbiome in patients with severe lower respiratory tract infections, using metagenomic sequencing. PLoS ONE. 2012; 7:e30875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Willner D, Furlan M, Haynes M, et al. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non‐cystic fibrosis individuals. PLoS ONE. 2009; 4:e7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang J, Yang F, Ren LL, et al. Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J Clin Microbiol. 2011; 49:3463–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y, Zhu N, Li Y, et al. Metagenomic analysis of viral genetic diversity in respiratory samples from children with severe acute respiratory infection in China. Clin Microbiol Infect. 2016; 22:458 e1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marcos MA, Esperatti M, Torres A. Viral pneumonia. Curr Opin Infect Dis. 2009; 22:143–147. [DOI] [PubMed] [Google Scholar]
- 10. Johnstone J, Majumdar SR, Fox JD, Marrie TJ. Viral infection in adults hospitalized with community‐acquired pneumonia prevalence, pathogens, and presentation. Chest. 2008; 134:1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arancibia F, Cortes CP, Valdes M, et al. Importance of Legionella pneumophila in the etiology of severe community‐acquired pneumonia in Santiago, Chile. Chest. 2014; 145:290–296. [DOI] [PubMed] [Google Scholar]
- 12. Henrickson KJ. Parainfluenza viruses. Clin Microbiol Rev. 2003; 16:242–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berry M, Gamieldien J, Fielding BC. Identification of new respiratory viruses in the new millennium. Viruses‐Basel. 2015; 7:996–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nelson JD. Community‐acquired pneumonia in children: guidelines for treatment. Pediatr Infect Dis J. 2000; 19:251–253. [DOI] [PubMed] [Google Scholar]
- 15. Wu ZQ, Yang L, Ren XW, et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016; 10:609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greninger AL, Chen EC, Sittler T, et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS ONE. 2010; 5:e13381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura S, Yang CS, Sakon N, et al. Direct metagenomic detection of viral pathogens in nasal and fecal specimens using an unbiased high‐throughput sequencing approach. PLoS ONE. 2009; 4:e4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pettigrew MM, Gent JF, Kong Y, et al. Association of sputum microbiota profiles with severity of community‐acquired pneumonia in children. BMC Infect Dis. 2016; 16:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gunnarsson RK, Holm SE, Soderstrom M. The prevalence of potential pathogenic bacteria in nasopharyngeal samples from individuals with a respiratory tract infection and a sore throat—implications for the diagnosis of pharyngotonsillitis. Fam Pract. 2001; 18:266–271. [DOI] [PubMed] [Google Scholar]
- 20. Schiff MJ, Kaplan MH. Rothia dentocariosa pneumonia in an immunocompromised patient. Lung. 1987; 165:279–282. [DOI] [PubMed] [Google Scholar]
- 21. Brook I. Anaerobic pulmonary infections in children. Pediatr Emerg Care. 2004; 20:636–640. [DOI] [PubMed] [Google Scholar]
- 22. Taboada B, Espinoza MA, Isa P, et al. Is there still room for novel viral pathogens in pediatric respiratory tract infections? PLoS ONE. 2014; 9:e113570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hall CB. Respiratory syncytial virus and parainfluenza virus. N Engl J Med. 2001; 344:1917–1928. [DOI] [PubMed] [Google Scholar]
- 24. Glezen WP, Frank AL, Taber LH, Kasel JA. Parainfluenza virus type 3: seasonality and risk of infection and reinfection in young children. J Infect Dis. 1984; 150:851–857. [DOI] [PubMed] [Google Scholar]
- 25. Counihan ME, Shay DK, Holman RC, Lowther SA, Anderson LJ. Human parainfluenza virus‐associated hospitalizations among children less than five years of age in the United States. Pediatr Infect Dis J. 2001; 20:646–653. [DOI] [PubMed] [Google Scholar]
- 26. Friedrichs I, Bingold T, Keppler OT, Pullmann B, Reinheimer C, Berger A. Detection of herpesvirus EBV DNA in the lower respiratory tract of ICU patients: a marker of infection of the lower respiratory tract? Med Microbiol Immunol. 2013; 202:431–436. [DOI] [PubMed] [Google Scholar]
- 27. Vasconcelos HC, Cataldo M, Niel C. Mixed infections of adults and children with multiple TTV‐like mini virus isolates. J Med Virol. 2002; 68:291–298. [DOI] [PubMed] [Google Scholar]
- 28. Niel C, Lampe E. High detection rates of TTV‐like mini virus sequences in sera from Brazilian blood donors. J Med Virol. 2001; 65:199–205. [PubMed] [Google Scholar]
- 29. Biagini P, Gallian P, de Micco P, de Lamballerie X. TT virus and TT virus‐like mini‐virus infection in French blood donors. Transfusion. 2000; 40:1542–1542. [DOI] [PubMed] [Google Scholar]
- 30. Simmonds P, Prescott LE, Logue C, Davidson F, Thomas AE, Ludlam CA. TT virus‐part of the normal human flora? J Infect Dis. 1999; 180:1748–1750. [DOI] [PubMed] [Google Scholar]
- 31. Mushahwar IK. Recently discovered blood‐borne viruses: are they hepatitis viruses or merely endosymbionts? J Med Virol. 2000; 62:399–404. [DOI] [PubMed] [Google Scholar]
- 32. Pirovano S, Bellinzoni M, Ballerini C, et al. Transmission of SEN virus from mothers to their babies. J Med Virol. 2002; 66:421–427. [DOI] [PubMed] [Google Scholar]
- 33. Gaynor AM, Nissen MD, Whiley DM, et al. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007; 3:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neske F, Prifert C, Scheiner B, et al. High prevalence of antibodies against polyomavirus WU, polyomavirus KI, and human bocavirus in German blood donors. BMC Infect Dis. 2010; 10:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han TH, Chung JY, Koo JW, Kim SW, Hwang ES. WU polyomavirus in children with acute lower respiratory tract infections, South Korea. Emerg Infect Dis. 2007; 13:1766–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le BM, Demertzis LM, Wu G, et al. Clinical and epidemiologic characterization of WU polyomavirus infection, St. Louis, Missouri. Emerg Infect Dis. 2007; 13:1936–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abed Y, Wang D, Boivin G. WU polyomavirus in children, Canada. Emerg Infect Dis. 2007; 13:1939–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bialasiewicz S, Whiley DM, Lambert SB, et al. Presence of the newly discovered human polyomaviruses KI and WU in Australian patients with acute respiratory tract infection. J Clin Virol. 2008; 41:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Norja P, Ubillos I, Templeton K, Simmonds P. No evidence for an association between infections with WU and KI polyomaviruses and respiratory disease. J Clin Virol. 2007; 40:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gallimore CI, Green J, Casemore DP, Brown DWG. Detection of a picobirnavirus associated with Cryptosporidium positive stools from humans. Arch Virol. 1995; 140:1275–1278. [DOI] [PubMed] [Google Scholar]
- 41. Cascio A, Bosco M, Vizzi E, Giammanco A, Ferraro D, Arista S. Identification of picobirnavirus from faeces of Italian children suffering from acute diarrhea. Eur J Epidemiol. 1996; 12:545–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chandra R. Picobirnavirus, a novel group of undescribed viruses of mammals and birds: a minireview. Acta Virol. 1997; 41:59–62. [PubMed] [Google Scholar]
- 43. Ganesh B, Banyai K, Martella V, Jakab F, Masachessi G, Kobayashi N. Picobirnavirus infections: viral persistence and zoonotic potential. Rev Med Virol. 2012; 22:245–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Figure S1.A flow‐chart describing the entire process from viral enrichment of samples of the metagenomic analysis steps.
Table S1.Sequencing read information for each nasopharyngeal specimen.